

Abstract
The nuclear pore complex (NPC) is the sole passageway for the transport of macromolecules across the nuclear envelope. Nup133, a major component in the essential Y-shaped Nup84 complex, is a large scaffold protein of the NPC's outer ring structure. Here, we describe an integrative modeling approach that produces atomic models for multiple states of Saccharomyces cerevisiae (Sc) Nup133, based on the crystal structures of the sequence segments and their homologs, including the related Vanderwaltozyma polyspora (Vp) Nup133 residues 55 to 502 (VpNup13355–502) determined in this study, small angle X-ray scattering profiles for 18 constructs of ScNup133 and one construct of VpNup133, and 23 negative-stain electron microscopy class averages of ScNup1332–1157. Using our integrative approach, we then computed a multi-state structural model of the full-length ScNup133 and validated it with mutational studies and 45 chemical cross-links determined via mass spectrometry. Finally, the model of ScNup133 allowed us to annotate a potential ArfGAP1 lipid packing sensor (ALPS) motif in Sc and VpNup133 and discuss its potential significance in the context of the whole NPC; we suggest that ALPS motifs are scattered throughout the NPC's scaffold in all eukaryotes and play a major role in the assembly and membrane anchoring of the NPC in the nuclear envelope. Our results are consistent with a common evolutionary origin of Nup133 with membrane coating complexes (the protocoatomer hypothesis); the presence of the ALPS motifs in coatomer-like nucleoporins suggests an ancestral mechanism for membrane recognition present in early membrane coating complexes.
The Saccharomyces cerevisiae nuclear pore complex (NPC)1 is a large macromolecular assembly of ∼50 MDa made of at least 456 protein copies of ∼30 distinct proteins called nucleoporins (nups). The NPC is the sole passageway for the exchange of macromolecules across the nuclear envelope (NE) (1). Apart from its main function as the sole mediator of nucleocytoplasmic trafficking, the NPC plays additional roles in numerous essential cellular processes, such as gene expression and chromatin regulation (2), and defects in its components have been implicated in numerous major human diseases (3). The first description of the macromolecular architecture of the NPC (4) was determined via an integrative approach based on a wide variety of experimental data (5). The permeability barrier is formed by FG (phenylalanine-glycine repeat–containing) nups, which fill the central channel of the NPC and are anchored to the core scaffold (6). The NPC architectural core is formed by an 8-fold arrangement of symmetric units called spokes that connect to each other, forming coaxial rings: two outer rings (the nuclear and cytoplasmic rings), a membrane ring, and two inner rings (7). In S. cerevisiae, the membrane ring is mainly formed by the transmembrane nups Pom152, Pom34, and Ndc1; the two adjacent inner rings are formed by large nups Nup192, Nup188, Nup170, and Nup157; and the two outer rings are formed by a radial head-to-tail arrangement of eight copies of the Nup84 complex (4, 8, 9). The Nup84 complex is a conserved assembly formed by nine proteins in vertebrates (Nup107–160 complex) and by seven nups in yeast (Nup133, Nup120, Nup145c, Nup85, Nup84, Seh1, and Sec13). The yeast Nup84 complex arranges into a characteristic Y-shaped assembly (10, 11). The stalk of the Y is formed by a tail-to-tail connection between Nup133 and Nup84 and a head-to-center connection between Nup84 and the dimer Nup145c-Sec13 (8, 12, 13).
Nup133, a 133-kDa subunit of the Nup84 complex, consists of an N-terminal β-propeller and a C-terminal α-solenoid-like folds (14). Nup133 is located at the end of the stalk of the Nup84 complex through a connection with Nup84 (8). Nup133 has also been suggested to connect through the first 15 residues of its N-terminal domain to the Nup120 copy of an adjacent Nup84 complex heptamer (12). Nup133 is a highly conserved nup that plays key roles in interphase and post-mitotic NPC biogenesis (15, 16), as well as in efficient anchoring of the dynein/dynactin complex to tether centrosomes to the NE in prophase (17). A loop within the N-terminal β-propeller of human Nup133 was suggested to contain an ArfGAP1 lipid packing sensor (ALPS) motif (18), functioning as a membrane curvature sensor. This motif allows human Nup133 to interact with curved membranes both in vitro and in vivo (15, 18) and has been shown to be required for proper NPC biogenesis during interphase (15). However, previous studies have not been able to detect any membrane interaction motifs in yeast Nup133, leading to the suggestion that the ALPS motif in Nup133 is unique to organisms with open mitosis (18, 19), in turn implying that the ALPS motif is not even a part of the mechanism for membrane association of the NPCs in all eukaryotes. Interestingly, mutations in S. cerevisiae (Sc) Nup133 cause a characteristic phenotype that leads to clustering of the NPCs into discrete regions of the NE (20). Structure–function mapping of this NPC clustering phenotype suggests that ScNup133—as well as its ancient paralog ScNup120—is functionally involved in the stabilization of the NE membrane curvature (8), although the exact mechanism that drives the interaction of these proteins with the NE is unknown.
Multi-domain, full-length nucleoporins are generally not amenable to X-ray crystallographic structure determination, presumably because of their apparent flexibility. Indeed, the structures of the N- and C-terminal fragments of Nup133 in particular were determined only separately (19, 21–23); the full-length atomic structure has not yet been characterized. Consequently, the relative orientation of the N- and C-terminal domains was depicted only schematically (22). We therefore took an integrative approach to generate the structure and dynamics of full-length ScNup133, based on multiple types of data.
Here, we characterized the configuration of the individual domains, defining the shape and populations of the full-length ScNup133 conformations, based on template structures, X-ray crystallography, small angle X-ray scattering (SAXS), and electron microscopy (EM) data, and performed validation with mutational studies and a dataset from chemical cross-linking with mass spectrometric readouts. More specifically, we report the crystal structure of the Nup133 N-terminal domain (residues 55–502) from Vanderwaltozyma polyspora (Vp), as well as SAXS profiles for 18 constructs of ScNup133 and one VpNup133 construct and 23 negative-stain EM class averages of ScNup1332–1157. Using our integrative modeling approach described in this study, we then determined atomic models for multiple states of the full-length ScNup133, based on these new data as well as known structures of ScNup133944–1157 and a number of Nup133 homologs. The resulting model was subsequently validated by three sets of double point mutations at the ScNup133–ScNup84 interface and 20 disuccinimidyl suberate (DSS) and 25 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) chemical cross-links determined via mass spectrometry (24).
As a result, the model of the full-length ScNup133 allows us to annotate a potential ALPS motif in Sc- and VpNup133, suggesting that ALPS motifs are scattered throughout the NPC's scaffold in all eukaryotes and play a major role in the assembly and membrane anchoring of the NPC in the NE. Our results are consistent with a common evolutionary origin of Nup133 with membrane coating complexes (the protocoatomer hypothesis); the presence of the ALPS motifs in coatomer-like nucleoporins suggests an ancestral mechanism for membrane recognition present in early membrane coating complexes.
EXPERIMENTAL PROCEDURES
Construct Design, Cloning, Expression, and Purification of Sc- and VpNup133
Nup133 is divided into the N-terminal β-propeller and the C-terminal α-solenoid domains in an iterative manual process relying on predicted secondary structure, gaps in multiple sequence alignments, and sequence–structure alignment by threading (14).
We cloned, expressed, and purified the resulting 18 constructs of ScNup133: 7 constructs covering the N-terminal domain, 8 constructs covering the C-terminal domain, and 3 constructs covering both domains partially or entirely (supplemental Fig. S3 and supplemental Table S1). Cloning, expression, and purification were performed using a standard protocol as described previously (21) (supplemental “Experimental Procedures” section). The N-terminal domain of V. polyspora Nup133 covering residues 55 to 502 (VpNup13355–502) was also cloned, expressed, and purified following similar procedures (21).
Crystallization and Structure Determination of VpNup13355–502
The crystal used for structure determination via SeMET-SAD phasing was obtained by means of sitting-drop vapor diffusion (VpNup13355–502 concentration of 10.6 mg/ml) in the presence of 10% PEG3350, 100 mm ammonium sulfate, and 100 mm HEPES (pH 8.2) and flash-frozen in liquid nitrogen with 30% (v/v) glycerol. The diffraction dataset collected at the LRL-CAT 31-ID (Advanced Photon Source) beamline was processed with XDS (25) and AIMLESS (26), and structure solution was obtained using AutoSol (27) in Phenix (28). An initial model assembled using AutoBuild (29) was further extended with several cycles of density modification using Parrot (30) followed by automated model improvement with Buccaneer (31, 32), as implemented in CCP4 (33), and manual model building with COOT (34). The final stages of refinement were performed using Refmac5 (35). Illustrations were prepared using PyMol (36) and UCSF Chimera (37).
Small Angle X-ray Scattering
SAXS profiles of 18 constructs of ScNup133 and one construct of VpNup13355–502(Figs. 2B and 3B, supplemental Figs. S3 and S4, and supplemental Table S1) were measured at concentrations of 0.5, 1.0, and 2.0 mg/ml and the highest possible concentrations in the protein storage buffer at 10 °C to 15 °C, using up to 24 1-s to 10-s exposures at the SSRL (Menlo Park, CA) and ALS (Berkeley, CA) beamlines (supplemental “Experimental Procedures” section). The buffer SAXS profile was obtained in the same manner and subtracted from a protein SAXS profile. The merged experimental SAXS profile of VpNup13355–502 was compared with SAXS profiles calculated using FoXS (38, 39), for the crystal structure of VpNup13355–502 and the “complete” models in which disordered components and four Se-Met residues were built using MODELLER 9.13 (40) (Fig. 2B).
Fig. 2.

Crystal structure of VpNup13355–502. A, overall fold of VpNup13355–502 β-propeller domain is shown in cartoon representation with color shading in blue to red from the N to the C terminus. Terminal residues of the disordered segments are marked with gray spheres. The location of the DA34-loop containing a potential ALPS motif is indicated by an arrow. Secondary structure elements are shown as defined by the DSSP (86) program. Strands within each propeller blade are marked in pink. B, comparison of the merged experimental SAXS profile (black) of VpNup13355–502 with the calculated SAXS profiles from the complete dimer model (χ = 12.4, red) and the complete monomer model (χ = 1.14, blue). The lower plot presents the residuals (calculated intensity/experimental intensity) of each calculated SAXS profile. The upper inset shows the SAXS profiles in the Guinier plot with an Rg fit of 24.4 ± 0.3 Å. The maximum particle size (Dmax) was 77.8 Å (determined experimentally). C, superposition of VpNup13355–502 (shown as a pink cartoon) and HsNup13375–477 (PDB: 1XKS; shown as a light-blue cartoon) (19) structures performed using the SSM (87) routine as implemented in COOT (34). D, electrostatic potential of VpNup13355–502 plotted onto its solvent accessible surface. Missing side chains and charges were assigned for the VpNup13355–502 structure on the PDB2PQR Web server (88), and the electrostatic surface was calculated using APBS (89) within PyMOL. Negative (−4 kT/e) and positive (+4 kT/e) potentials are shown in red and blue, respectively. The positively charged surface located adjacent to the DA34-loop (annotated as the potential ALPS motif) is marked by an ellipse in yellow. Phenylalanines at positions 275 and 276 are shown in sticks as a reference for the location of the DA34-loop.
Fig. 3.

Structure and dynamics of ScNup133 revealed through integrative modeling approach. A, the minimal ensemble of four conformations (the multi-state model), comprising a single major extended conformation with a population weight of 0.506 (blue) and three minor compact conformations with weights of 0.242 (red), 0.202 (cyan), and 0.050 (yellow), is shown. The most populated conformation (blue) was used as a reference for rigid body least-squares superposition of the remaining three conformations. The ab initio shape (represented as a gray envelope) computed from the experimental SAXS profile was also superposed for comparison. B, comparison of the merged experimental SAXS profile (black) of ScNup1332–1157 with the calculated SAXS profiles from the ScNup1332–1157 comparative model (χ = 6.27, red) and the ensemble of four conformations (χ = 1.54, blue). The lower plot presents the residuals (calculated intensity/experimental intensity) of each calculated SAXS profile. The upper inset shows the SAXS profiles in the Guinier plot with an Rg fit of 48.3 ± 0.6 Å. The maximum particle size (Dmax) was 169.2 Å (determined experimentally). C, the 23 negative-stain EM class averages are shown along with the projections of each of the four conformations. 22 EM class averages were assigned to at least one of the four conformations with high confidence, as highlighted in colored boxes.
Negative-stain EM for ScNup1332–1157
A specimen of the full-length ScNup1332–1157 was prepared for negative-stain EM (41) (supplemental “Experimental Procedures” section). The 1976 individual particles were selected interactively from images using Boxer from EMAN (42) and windowed into individual images with a size of 120 × 120 pixels. The particles were centered and normalized and then subjected to the ISAC (iterative stable alignment and clustering) (43) technique to produce 23 stable two-dimensional class averages after 10 generations (these class averages comprised 1530 of the 1976 particles) (top rows in Fig. 3C and column 2 in supplemental Table S3).
Structure and Dynamics of ScNup133 Revealed through Integrative Modeling Approach—
We developed an integrative modeling approach that produces atomic models for multiple states of a protein based on EM images of the protein as well as SAXS profiles and crystal structures of the sequence segments and their homologs. We proceeded through three stages (Fig. 1): (i) gathering of data; (ii) conformational sampling and scoring to produce a minimal ensemble of conformations consistent with SAXS profiles, EM class averages, template structures, and chemical cross-links; and (iii) analysis of the ensemble. The integrative modeling protocol was scripted in Python, based on our open-source IMP (Integrative Modeling Platform) package, release 2.2 (44). Files for the input data, script, and output models are available online.
Fig. 1.

Integrative modeling approach for ScNup133. Our integrative modeling approach proceeds through three stages: (i) gathering of data; (ii) conformational sampling and scoring to produce a minimal ensemble of conformations consistent with SAXS profiles, EM class averages, template structures, and chemical cross-links; and (iii) analysis of the ensemble. The integrative modeling protocol was scripted in Python, based on our open-source IMP (Integrative Modeling Platform) package, release 2.2 (44).
Stage 1: Gathering of Data
19 SAXS profiles and 23 EM class averages were obtained as described above. The atomic structures of ScNup133944–1157 (PDB code 3KFO) (21); VpNup13355–502 (4Q9T; Fig. 2 and Table I); and human homologs HsNup13375–477 (1XKS) (19), HsNup133517–1156 (3I4R) (22), and HsNup133935–1156 (3CQC) (23) had been previously determined via X-ray crystallography. In addition, putative homologs of known structure were detected for the first 60 residues of the N-terminal domain by HHpred (45, 46) and ModWeb (47). Domain boundaries, secondary structure segments, and disordered regions were predicted by DomPRED (48), PSIPRED (49, 50), and DISOPRED (51), respectively. 18 DSS and 23 EDC intramolecular chemical cross-links for ScNup133 (Table II), as well as 2 DSS and 2 EDC intermolecular chemical cross-links spanning the ScNup133–ScNup84 interaction interface (Table III), were obtained from our companion study on the entire ScNup84 complex (24).
Table I. Crystallographic statistics.
| Data collection | VpNup13355–502 |
| PDB code | 4Q9T |
| Space group | C2221 |
| Unit-cell dimensions (Å) | a = 109.2, b = 133.8, c = 136.8 |
| Matthew's coefficient (Å3/Da) | 2.44 |
| Solvent content (%) | 49.6 |
| Wavelength (Å) | 0.9793 |
| Resolution (Å) | 38.00-3.00 (3.18-3.00)a |
| Number of unique reflections | 20,381 (3,224) |
| Completeness (%) | 99.7 (98.8) |
| Rmerge (%) | 18.9 (254.2) |
| Mean (I) half-set correlation CC(1/2) (91) | 0.990 (0.668) |
| Multiplicity | 14.6 (13.9) |
| <I/s(I)> | 14.2 (1.2) |
| Refinement | |
| Number of reflections | 19,268 |
| Number of reflections in test set | 1,042 |
| Rwork (%) | 21.5 |
| Rfree (%) | 26.8 |
| Root-mean-square deviations from ideal values | |
| Bond length (Å) | 0.0080 |
| Bond angles (°) | 1.3299 |
| Ramachandran plot | |
| MolProbity (92) residues in: | |
| Favored region (%) | 90.8 |
| Allowed region (%) | 99.6 |
a Values in parentheses correspond to the highest-resolution shell.
Table II. Validation of the multi-state model of ScNup1332–1157 with 41 chemical cross-links determined via mass spectrometry.
We validated the conformations of ScNup1332–1157 against each of the 18 DSS and 23 EDC chemical cross-links obtained from our companion study on the entire ScNup84 complex (24). As a result, 94.4% of DSS and 91.3% of EDC cross-links were satisfied by the multi-state model, within 35-Å and 25-Å thresholds, respectively. Therefore, the conformational dynamics of ScNup133 are consistent with 38 of the 41 intramolecular chemical cross-links, corresponding to the typical false-positive rate observed for chemical cross-linking (24).
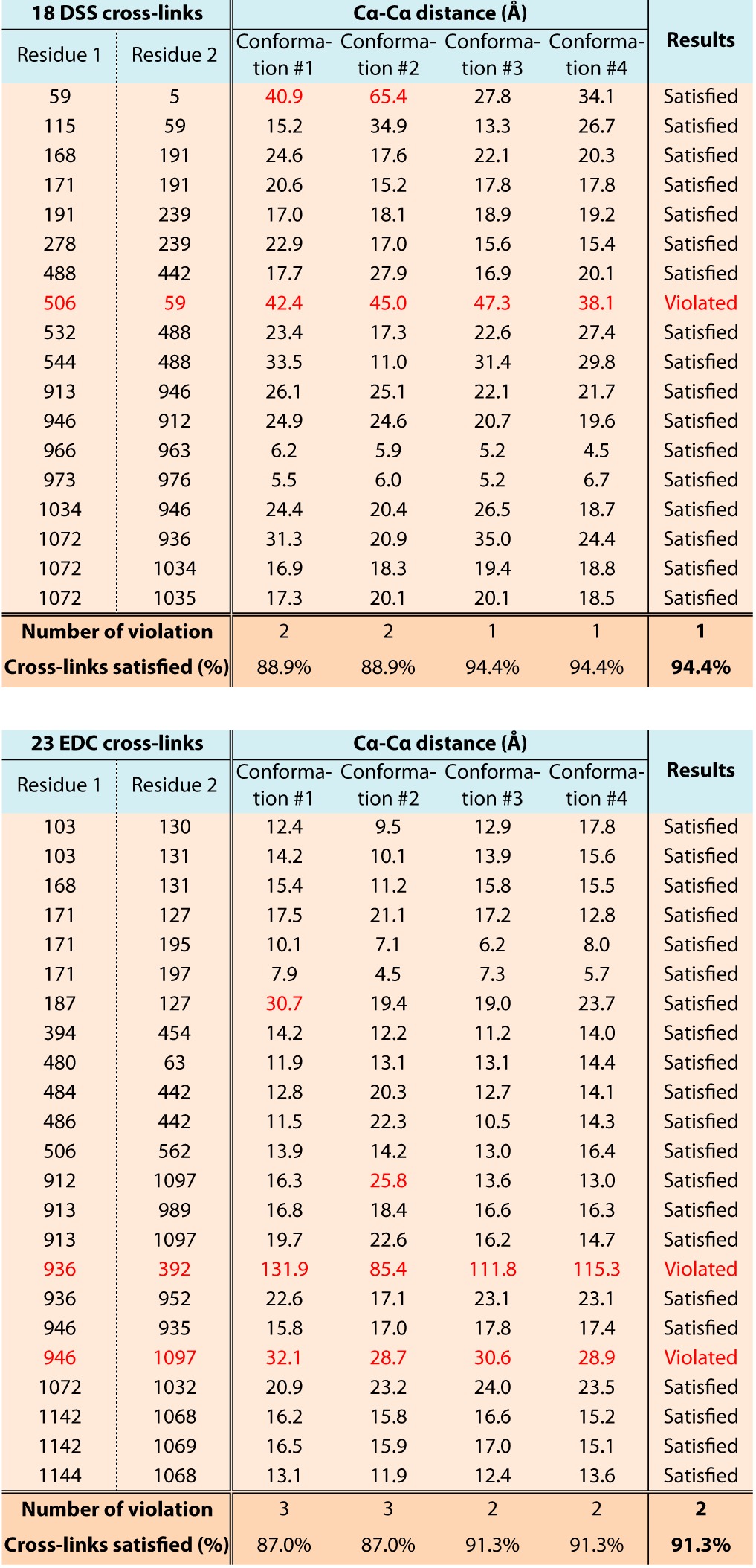
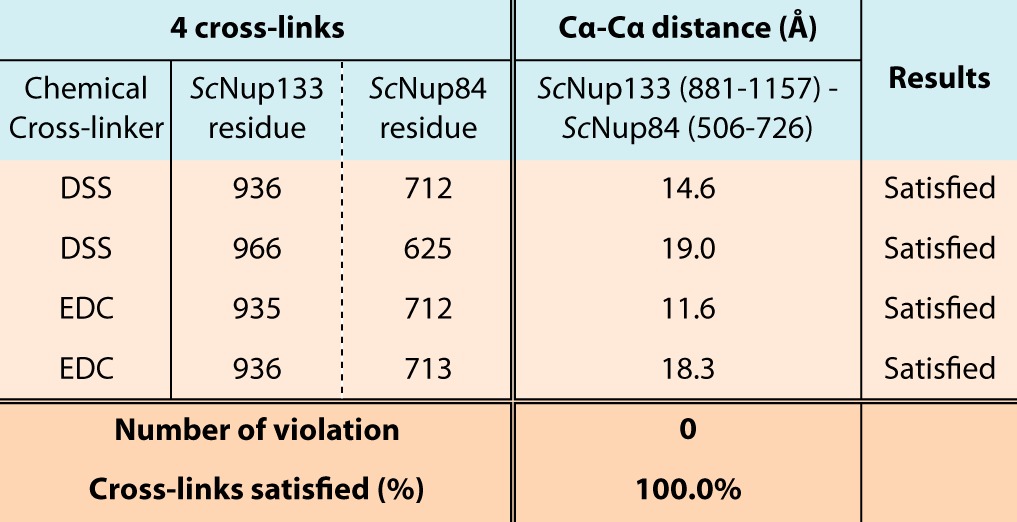
Table III. Validation of the ScNup133–ScNup84 interface with four chemical cross-links determined via mass spectrometry.
We validated the ScNup133881–1157–ScNup84506–726 interface model against each of the two DSS and two EDC chemical cross-links spanning the ScNup133–ScNup84 interface, which were obtained from our companion study on the entire ScNup84 complex (24). All the intermolecular chemical cross-links are fully consistent with the ScNup133881–1157–ScNup84506–726 interface model.
Stage 2: Conformational Sampling and Scoring to Produce a Minimal Ensemble of Conformations Consistent with SAXS Profiles, EM Class Averages, Template Structures, and Chemical Cross-links
(1) Building an Initial Atomic Model of ScNup1332–1157
The shape and size information contained in SAXS profiles can be used to improve the accuracy of atomic comparative models. An initial atomic model of the full-length ScNup1332–1157 was built based on template structures and SAXS profiles for 18 nested ScNup133 constructs (supplemental Figs. S3 and S4 and supplemental Table S1) as follows. First, we built 1000 atomic comparative models for the smallest construct (52–483), based on the crystal structure of VpNup13355–502 (Fig. 2) and the closest known structure detected by HHPred (45, 46) and ModWeb (47), using MODELLER 9.13 (40). The theoretical SAXS profile and the χ value of the fit to the experimental SAXS profile of the corresponding construct were calculated for each of the 1000 comparative models using FoXS (38, 39). Then, these 1000 models were ranked by the χ value of the fit to the experimental SAXS profile. Second, the best-scoring model was used as a template for comparative modeling of the next larger constructs (41–483 and 52–515), supplemented by the additional templates found by HHPred and ModWeb. For each of the two constructs, the resulting models were again ranked based on the corresponding SAXS profile fit. The entire process was repeated until the largest construct of ScNup1332–1157 was modeled, resulting in the initial model of the full-length ScNup1332–1157 (supplemental Figs. S3 and S4 and supplemental Table S1).
(2) Conformational Sampling Using AllosMod
The initial model of ScNup1332–1157 was subjected to molecular-dynamics-based conformational sampling using AllosMod (52), resulting in 7000 conformations, as follows. The AllosMod simulations were short, near-equilibrium trajectories based on an input sequence and the initial model of ScNup1332–1157. AllosMod constructed an energy landscape in which the atomic contacts from the input structure defined the major energetic minima (53), also generally known as a Gō model (54, 55). The energy landscape was then sampled using several constant temperature (at 300–350K) molecular dynamics simulations with short equilibration and a run time of 0.2 ns using 2-fs time steps and velocity rescaling every 200 steps (supplemental “Experimental Procedures” section).
(3) Scoring and Searching for a Minimal Ensemble of Conformations Consistent with the SAXS Profile, EM Class Averages, and Chemical Cross-links
The resulting 7000 AllosMod conformations of ScNup1332–1157 were pruned to identify a minimal ensemble of up to five conformations that reproduced both the experimental SAXS profile and EM class averages of ScNup1332–1157. The pruning was achieved by a MES (minimal ensemble search) program (56) that was modified to use a composite score defined as a weighted sum of the ensemble SAXS score and the ensemble em2D Z-score.
The ensemble SAXS score is the χ value for the comparison of the ensemble SAXS profile to the experimental profile; the ensemble SAXS profile is a weighted average of the theoretical SAXS profiles for the selected subset of conformations, calculated using FoXS (38, 39).
To compute the ensemble em2D Z-score, we first calculated individual em2D scores for each of the 7000 conformations matched against each of the 23 EM class averages, using the EMageFit application (57) of IMP (44) at 15 Å resolution; the em2D score is 1 minus the cross-correlation coefficient between a class average and the best-matching projection of a conformation (57). Each score was then normalized into a Z-score by using the average and standard deviation of the scores for the same class average. Finally, the ensemble em2D Z-score was obtained by summing the lowest individual em2D Z-scores determined for each of the 23 EM class averages in the subset.
Independent fitting of subsets ranging from one to five conformations showed that a minimal ensemble of four conformations was sufficient to explain both the experimental SAXS profile and EM class averages of ScNup1332–1157 (Fig. 3 and supplemental Table S3). The relative weight of −0.05 for the ensemble em2D Z-score in the composite score was determined by trial and error to balance the fit of the minimal ensemble to both SAXS and EM data.
As the final assessment step, we validated the conformations of ScNup1332–1157 against each of the 18 DSS and 23 EDC intramolecular chemical cross-links obtained from our companion study on the entire ScNup84 complex (24) (Table II).
Stage 3: Analysis of the Minimal Ensemble
The most populated conformation in the minimal ensemble of four conformations was used as a reference for rigid body least-squares superposition of the remaining three conformations. The ab initio shape of the full-length ScNup1332–1157 (a gray envelope in Fig. 3A) was generated from the experimental SAXS profile using DAMMIF (58) and DAMAVER (59). UCSF Chimera was used for visualization (37). The TM-scores between the conformations in the minimal ensemble were calculated on web-server (60).
We attempted to assign each of the 23 EM class averages to one or more of the four conformations in the minimal ensemble; an EM class average is assigned to a conformation when its em2D Z-score is less than −0.95 and the cross-correlation coefficient is greater than 0.82 or 0.85 (supplemental Table S3).
Validation of the ScNup133–ScNup84 Interface with Mutational Analysis and Chemical Cross-links
The interface between ScNup133881–1157 and ScNup84506–726 (in the stalk of the yeast Nup84 complex) was predicted by calculating the difference in solvent accessibility between the unbound and bound models of ScNup133881–1157 and ScNup84506–726. The ScNup133881–1157 model was extracted from the initial atomic model of ScNup1332–1157, and the ScNup84506–726 model was built based on the human Nup133-Nup107 complex structure (PDB code: 3CQC) (23) using MODELLER (40). The ScNup133881–1157–ScNup84506–726 interface model was built by structurally aligning each component into the corresponding chain of the human Nup133-Nup107 crystal structure. The unbound model of ScNup133881–1157 and ScNup84506–726 was obtained by separating the two components by 100 Å. Residue solvent accessibility was calculated using MODELLER. The residues with large solvent accessibility changes (residues of L922, L929, and L933) were identified as target interface residues (Fig. 4A). Finally, the ScNup133881–1157–ScNup84506–726 interface model was validated against the two DSS and two EDC intermolecular chemical cross-links spanning the ScNup133–ScNup84 interface, which were obtained from our companion study on the entire ScNup84 complex (24) (Table III).
Fig. 4.

Mutational analysis of the ScNup133–ScNup84 interface. The double mutations at the predicted ScNup133–ScNup84 interface disrupted the association of ScNup133 with the rest of the ScNup84 complex. A, alignment of the Nup133 amino acid sequences of different organisms (indicated on the left) corresponding to the predicted Nup84 interaction surface in S. cerevisiae. The alignment was performed with ClustalW2 (90). The amino acids affected by the designed point mutations are indicated by an asterisk. B, affinity purification of nucleoporins associated with the Nup133-PrA mutants indicated above each gel line. The identity of each protein is indicated in blue letters on the right. Marker protein molecular weights are indicated in kDa on the left side. Immunoglobulin contaminants are identified in gray letters.
To validate the predicted interface experimentally, we designed three sets of double point mutants located within and outside of the interface. Maximally disruptive mutations were predicted by the program EGAD (61) (supplemental Table S2), allowing us to propose two sets of double mutants affecting the binding interface (L922Y-L929Y and L929W-L933W), as well as a control mutation at a distal surface position (L1089Y-L1123Y) (Fig. 4B).
The three mutants were generated via PCR and cloned into a yeast centromeric expression plasmid under the control of the GAL inducible promoter. Protein A and GFP C-terminal tags were used for immunoprecipitation and subcellular localization, respectively. An ScNup133 null mutant was transformed with the different constructs, and the proteins were expressed by growth in yeast extract peptone (YP) media supplemented with glucose, in which the basal activity of the GAL promoter generated a close-to-wild-type expression of the constructs. Affinity purifications of the ScNup133 interacting proteins were performed using a buffer formulation that allowed exclusive purification of the Nup84 complex components (8). Fitness phenotypic analysis was also performed as described previously (8) (Fig. 5). GFP fusions were transformed into an ScNup133 null, Nup170-mCherry strain and analyzed via fluorescence microscopy. Cells grown in minimal media supplemented with glucose were visualized with a ×63 1.4-NA Plan-Apochromat objective (Carl Zeiss, Thornwood, NY) using a microscope (Axioplan 2, Carl Zeiss, Thornwood, NY) equipped with a cooled charge-coupled device camera (ORCA-ER, Hamamatsu Photonics, Bridgewater, NJ). The system was controlled with Openlab imaging software (PerkinElmer Life Sciences, Waltham, Massachusetts). Final images were assembled, and gamma-levels were adjusted to enhance contrast only using Photoshop software (Adobe Systems Inc., San Jose, CA).
Fig. 5.

Phenotypic analysis of the ScNup133 point mutants. A, fitness analysis of the Nup133 mutants. 10-fold serial dilutions of strains expressing the Nup133-PrA construct indicated on the left side or carrying an empty plasmid (controls ΔNup133 and parental w303 strains) were spotted in minimal media plates without tryptophan and grown at the indicated temperatures for 2 to 3 days. A semi-quantitative score, in arbitrary units (AU), normalized to the wild-type fitness, is shown on the right. B, Nup133 mutants associated with the NPC. NPC association of the Nup133–GFP constructs indicated on the left side was analyzed via live-cell direct fluorescence microscopy in a ΔNup133, Nup170-mCherry mutant background. Cells were grown to early log phase at 25 °C. Differential interference contrast (DIC, left) and single-channel fluorescence (center and right columns) are shown. Scale bar = 5 mm.
Annotating the Potential ALPS Motifs
We searched the sequences of ScNup133, VpNup133, and ScNup120 for potential amphipathic helix-forming patterns using the Membrane Protein Explorer (MPEx) package (62). Then, the resulting potential amphipathic helices were analyzed by the HeliQuest Web server (63) to annotate the potential ALPS motif as described previously (18, 64) (supplemental “Experimental Procedures” and supplemental Fig. S2).
RESULTS
Crystal Structure of VpNup13355–502
Although V. polyspora is considered to be a somewhat distant yeast relative of S. cerevisiae (65), both species diverged from a single ancestor that underwent a whole-genome duplication event. The sequence identity between Sc- and VpNup133, across their entire length, is 37%. As a part of the Protein Structure Initiative project, several fungal Nup133 proteins were screened for crystallization, and the N-terminal domain of V. polyspora Nup133 yielded diffraction-quality crystals. The construct encompassing residues 55 to 502, corresponding to the N-terminal domain of V. polyspora Nup133 (VpNup13355–502), yielded crystals (resolution of ∼3.0 Å) in the orthorhombic space group C2221 with two molecules in the asymmetric unit. The structure of VpNup13355–502, determined via SeMET-SAD, was refined to Rwork and Rfree values of 21.5% and 26.8%, respectively (Table I). As expected, the overall VpNup13355–502 adopts a disc-shaped, canonical β-propeller fold generated by radial arrangement of seven blades around a central channel, with each blade containing an anti-parallel β-sheet formed by four strands (Fig. 2A; PDB Code 4Q9T, chain B). Notably, disordered segments 86–98, 139–144, 157–180, and 202–214 occur within blades 1 and 2. Within each blade, strands A, B, C, and D are arranged in an innermost to outermost fashion, resulting in a top surface decorated with BC-loops (connecting BC strands within each blade) and DA-loops (loops connecting the D strand of the nth and the A strand of the n+1th blade) and a bottom surface decorated by the AB- and CD-loops. The β-sheets forming blades 4 and 5, which also have a helical insertion between them, show a greater degree of curvature than other blades. Interestingly, the seventh blade of the propeller has an additional strand with the A, B, and C strands from the C terminus of the domain interacting with a β-hairpin at the N terminus of the propeller (Fig. 2A). Residues of the E7 and D7 β-strands forming the N-terminal β-hairpin show a notable degree of conservation within fungal Nup133 sequences (supplemental Fig. S1). This type of “velcro” arrangement is important for stability of the circular, β-propeller architectures (66).
Analyses using PISA (67) suggested that the two monomers of VpNup13355–502 within the asymmetric unit of the crystals did not form a biological complex (total buried surface area = 1690 Å2) in solution (an important distinction when reconstructing the assembly state of the NPC in vivo). In addition, the DiMoVo score (68) of −0.140 also indicated that the two monomers were merely the crystallographic dimers. Accordingly, the merged experimental SAXS profile of VpNup13355–502 was well matched (χ = 1.14) to the SAXS profile calculated from the “complete” monomer model, generated by modeling the disordered residues of the crystal structure (blue in Fig. 2B and supplemental Table S1). In contrast, the SAXS profile for the “complete” dimer model, representing the crystallographic asymmetric unit, had an unacceptably high χ value of 12.4 and an Rg value of 35.7 Å (red in Fig. 2B). Further, the measured radius of gyration (Rg) of 24.4 ± 0.3 Å, determined from the experimental SAXS profile with AUTORG (69), was consistent with the Rg value of 24.8 Å calculated from the “complete” monomer model of VpNup13355–502. The composition of VpNup13355–502, estimated with OLIGOMER (70), based on the experimental SAXS profile was 100% monomer. Thus, our SAXS analyses of the solution behavior of VpNup13355–502 are fully consistent with the monomer of the X-ray crystal structure.
A superposition of human Nup133 (HsNup13375–477) (PDB code 1XKS) (19) and VpNup13355–502 crystal structures revealed that the two β-propeller domains showed overall similarity in their architecture in spite of low sequence identity. These two structures agreed well, resulting in a root-mean-square deviation of 2.2 Å over 293 superposed residues with sequence identity of only 15% (Fig. 2C). Notable differences in the architecture of the two structures included the following: (i) the seventh blade of the HsNup133 β-propeller had only four strands with the absence of a β-hairpin at its N-terminal region; (ii) the HsNup133 β-propeller had additional helical insertions between blades 7 and 1 and between blades 4 and 5 that were not present in the VpNup133; and (iii) importantly, the residues of the DA34-loop, connecting the D-strand of blade 3 to the A-strand of blade 4, which are disordered in HsNup133, adopted mostly a random-coil conformation with a 310-helix near the A4-strand (Figs. 2A and 2C). Interestingly, the disordered DA34-loop of HsNup133 has been shown to contain an ALPS motif, which is likely to facilitate membrane-curvature sensing and formation in the NPC. We have identified a potential ALPS motif within the DA34-loop VpNup133 β-propeller domain comprising the sequence 267-LIKPQNSFFFRNLDSSKEIISL-288 (Fig. 6B and supplemental Fig. S2B). Notably, an electrostatic potential map of VpNup13355–502 reveals a large positively charged surface adjacent to the DA34-loop (highlighted in yellow in Fig. 2D). This surface is decorated mainly by residues from the third and fourth blades, which show a notable degree of conservation (supplemental Fig. S1). It is plausible that this positively charged surface, being close to the DA34-loop, might play a role in facilitating interaction of the potential ALPS motif of VpNup133 with the membrane.
Fig. 6.

The potential ArfGAP1 lipid packing sensor (ALPS) motifs. Each of the potential ALPS motifs and sequences for HsNup133 (A), VpNup133 (B), ScNup133 (C), and ScNup120 (D, E) is presented. The potential ALPS motifs (red) were visualized in their corresponding structures (gray) using UCSF Chimera (37) and are highlighted by blue circles. The helical-wheel representations of the potential ALPS motifs are also shown, with an arrow in the center of the helical-wheel representing the direction and strength of the mean hydrophobic moment of the corresponding ALPS motif.
Structure and Dynamics of ScNup133 Revealed through an Integrative Modeling Approach—
An Initial Atomic Model of ScNup1332–1157
An initial atomic model of the full-length ScNup1332–1157 was built based on the nested template structures and 18 SAXS profiles (supplemental Figs. S3 and S4 and supplemental Table S1). The model contains a single “linker” region (residues 480–495) between the N- and C-terminal domains aligned with the template structures (supplemental Fig. S3), indicating potential variability in the relative orientation of the two domains. Elastic network model analyses using HingeProt (71) suggested a long-range motion of the N- and C- terminal domains about the hinge residues of Leu51-His52 (near the beginning of the β-propeller), Glu484-Thr485 (at the flexible linker residues), and Ser895-Tyr896 (near the beginning of the 3KFO template). This motion was also identified as one of the top-scoring normal modes by the Web server ElNemo (72). Such conformational dynamics might play a role in regulating the dynamic structure of the NPC. Therefore, we probed the structure and dynamics of ScNup133 in solution using an integrative modeling approach that benefits from both SAXS profiles and EM micrographs (Fig. 1).
Conformational Sampling and Minimal Ensemble Search
The experimentally measured SAXS profile of ScNup1332–1157 in solution (black in Fig. 3B) did not match the theoretical SAXS profiles computed from the comparative model (χ = 6.27; red in Fig. 3B) (supplemental Fig. S4C and supplemental Table S1), although each of the N- and C-terminal domains satisfied its corresponding SAXS profile (χ = 1.36 and 1.71, respectively) (supplemental Figs. S4A and S4B and supplemental Table S1). Further, the maximum particle size (Dmax) of 169.2 Å determined experimentally was 4.3% larger than the maximum dimension of 162.1 Å from the comparative model. EM analysis of ScNup1332–1157, using the iterative stable alignment and clustering method (43), revealed 23 stable class averages after 10 generations (top rows in Fig. 3C). Similarly to the SAXS results, a number of EM classes could not be fit by the comparative model of ScNup1332–1157.
To study the structure and dynamics of ScNup1332–1157 in solution, we carried out conformational sampling by molecular dynamics using AllosMod (52), starting from the initial atomic model of ScNup1332–1157. We tested whether the resulting 7000 conformations generated by AllosMod were consistent with the experimental SAXS profile and 23 EM class averages using a minimal ensemble search (56). As expected, both the SAXS profile and the 23 EM class averages were not explained simultaneously by any single sampled conformation of ScNup1332–1157, indicating that ScNup133 is heterogeneous in solution.
The Multi-state Structural Model of ScNup1332–1157
The analysis of the resulting 7000 conformations using the minimal ensemble search indicated that a minimal ensemble of four conformations (the multi-state model) was sufficient to explain the experimental SAXS profile (χ = 1.54; blue in Fig. 3B), most of the EM class averages (Fig. 3C), and most of the chemical cross-links determined via mass spectrometry (Table II). The multi-state model consists of a single major “extended” conformation with a population weight of 0.506 (blue) and three minor “compact” conformations with population weights of 0.242 (red), 0.202 (cyan), and 0.050 (yellow) (Fig. 3A). The maximum particle size (Dmax) of the extended conformation was measured as ∼180 Å, and Dmax of the three compact conformations ranged from ∼135 Å to ∼145 Å. In addition, the root-mean-square deviation and the TM-scores (60) of each pair from the three compact conformations ranged from 10.6 to 23.7 Å and from 0.44 to 0.64, respectively, indicating similar folds within the compact ones. In contrast, the root-mean-square deviation and the TM-scores of the extended conformation relative to the rest of the conformations ranged from 25.6 to 29.0 Å and from 0.38 to 0.49, respectively, indicating less similarity in folds between the extended and the compact conformations. Importantly, 22 EM class averages (out of the total of 23 class averages) could be assigned to at least one of the four conformations in the multi-state model with high confidence, covering 95.6% of the 23 EM class averages (Fig. 3C and supplemental Table S3). Moreover, 94.4% of the 18 DSS and 91.3% of the 23 EDC intramolecular chemical cross-links were satisfied by the multi-state model, within 35-Å and 25-Å thresholds, respectively, independently validating our modeling (Table II). Furthermore, the variability in the multi-state model supports the long-range motion indicated by HingeProt (71) and elNémo (72).
The conformational dynamics of ScNup133 appears to be dominated by the relative motions of the N- and C-terminal domains, connected by a relatively lengthy linker of 16 residues (480–495) (supplemental Fig. S3). The maximal displacement of the N-terminal β-propeller domains was measured as ∼110 Å in the multi-state model. In conclusion, it appears that ScNup133 is a highly dynamic molecule in solution, with the maximal dimension (Dmax) of the molecule changing from 135 Å to 180 Å in solution.
Validation of the ScNup133-ScNup84 Interface by Mutational Analysis and Chemical Cross-links
ScNup133 connects to the rest of the Nup84 complex through a tail-to-tail interaction with ScNup84 (8, 22, 23). The model of ScNup133 allowed us to predict the location and molecular details of this interaction surface (Fig. 4A). Accordingly, the intermolecular chemical cross-links spanning the ScNup133–ScNup84 interface (24) were fully consistent with the ScNup133881–1157–ScNup84506–726 interface model (Table III).
To further validate the predicted interface experimentally, we designed three sets of double point mutants located within and outside of the interface (Fig. 4). Combinations of two ScNup133 leucine residues within the interface were mutated to bulky hydrophobic (tryptophan; L929W-L933W) or bulky polar (tyrosine; L922Y-L929Y) residues in an effort to create steric incompatibility between ScNup133 and ScNup84. As a control, two ScNup133 leucine residues located at the distal surface positions were also mutated to tyrosine (L1089Y-L1123Y). The mutant proteins were expressed in an ScNup133 null background and tested for their ability to interact with ScNup84 by affinity purification (8). None of the mutant proteins affecting the ScNup133–ScNup84 interaction surface was able to copurify with any of the ScNup84 complex components (columns 2 and 3 in Fig. 4B), strongly suggesting that the binding between ScNup133 and ScNup84 had been disrupted.
Fitness analysis of the same mutant strains (Fig. 5A) showed that the mutants at the ScNup133–ScNup84 interface were not able to fully rescue the fitness phenotype caused by the deletion of ScNup133. The mutant proteins were also tagged with GFP so we could analyze their subcellular localization (Fig. 5B). All mutant proteins showed nuclear rim localization and co-localization with the NPC marker Nup170-mCherry, eliminating the possibility of mislocalization as the main cause of the loss of binding to Nup84. The wild type and the control mutant were able to effectively rescue the NPC clustering phenotype characteristic of the ScNup133 null mutation (Fig. 5B, upper right panel), but the mutants disrupting the ScNup133–ScNup84 interface still showed an NPC clustering defect similar to that of the ScNup133 deletion. In summary, our experimental tests validate the predicted ScNup84 binding sites on ScNup133. Moreover, ScNup133 interactions with other parts of the NPC scaffold are indicated by the relative independence of NPC localization on the ScNup133–ScNup84 interaction.
Identification of ALPS Motifs in Yeast Nup84 Complex Components
The seven-bladed β-propellers of Nup133s from V. polyspora (VpNup13355–502, PDB code: 4Q9T) and Homo sapiens (HsNup13375–477, PDB code: 1XKS) (19) share a high degree of similarity in their structural arrangement (Fig. 2C). A loop (245-LPQGQGMLSGIGRKVSSLFGILS-267) unresolved in the X-ray structure of HsNup13375–477 has been shown to act as a membrane-curvature-sensing ALPS motif in the vicinity of membranes (18, 64) (Fig. 6A and supplemental Fig. S2A). Notably, VpNup13355–502 contains a clearly resolved loop region (267-LIKPQNSFFFRNLDSSKEIISL-288) with a partial helical segment, including the DA34-loop between blades 3D and 4A at the position equivalent to the ALPS motif (245–267) in HsNup13375–477 (Figs. 1A, 1C, and 6B; supplemental Fig. S2B). According to the helical-wheel representation, the VpNup133267–288 loop region contains a polar face rich in serine residues and displays a high level of mean hydrophobicity and hydrophobic moments, similar to canonical ALPS motifs (18) (Fig. 6B and supplemental Fig. S2B). This observation suggests that VpNup133 might also contain an ALPS motif in its β-propeller domain.
To verify that the potential ALPS motif in VpNup133 is a conserved feature, we also analyzed the sequence of ScNup133 to look for an equivalent motif using the MPEx package (62) and the HeliQuest web server (63). Similar to VpNup133, we found that a loop region of ScNup133252–270 (252-FKLGIWSKIFNTNSSVVSL-270) at the position equivalent to the ALPS motif (245–267) in HsNup133 also contained a polar face very rich in serine and threonine residues and displaying a high level of mean hydrophobicity and hydrophobic moments (Fig. 6C and supplemental Fig. S2C). Its sequence also satisfies most of the biophysical criteria for identification as an ALPS motif (18).
Also, we annotated two potential ALPS motifs in the β-propeller N-terminal domain of ScNup120, an ancient paralog protein of ScNup133 in the yeast Nup84 complex: (i) the 2CD loop region ScNup120135–152 (135-LSFLFSSANTLNGEWFHL-152) and (ii) the 3D4A loop region ScNup120197–216 (197-LLFNDNSYLKSLTRFFSRSS-216) were identified as potential ALPS motifs (Figs. 6D and 6E; supplemental Figs. S2D and S2E), similarly to VpNup133267–288 and ScNup133252–270.
DISCUSSION
Structure and Dynamics of ScNup133 Revealed by Integrative Modeling Approach
Most proteins exist in multiple structural states. Conclusions based on only one state can therefore be incomplete or even misleading. In general, the structure of a protein is best determined based on all available data. Therefore, we developed an integrative structure determination approach for multiple states that relies on data from X-ray crystallography, SAXS, EM, point mutations, and chemical cross-linking for either component fragments or an entire target protein (or its homologs) (Fig. 1).
Components of the NPC have proven to be exemplars of proteins flexing between several states (73, 74), so we utilized this approach to determine the structure and dynamics of one such component, full-length ScNup1332–1157. We computed the minimal ensemble of four conformations (the multi-state model) of ScNup1332–1157 that are consistent with EM images of the protein as well as SAXS profiles and crystal structures of the sequence segments and their homologs (including VpNup13355–502 (Fig. 2), determined in this study) (Fig. 3). We validated the resulting model with three sets of double point mutations (Figs. 4 and 5) and 45 chemical cross-links determined via mass spectrometry (24) (Tables II and III).
The multi-state model consists of a single major extended conformation and three minor compact conformations (Fig. 3A). The conformational dynamics of ScNup133 appears to be dominated by the relative motions of the N- and C-terminal domains. Notably, the conformational dynamics of ScNup133 are consistent with 38 of the 41 intramolecular chemical cross-links (Table II), corresponding to the typical false-positive rate observed for chemical cross-linking (24). Finally, the ScNup133 dynamics is also consistent with the extreme flexibility shown for the HsNup133 region of the Nup107–160 complex by negative-stain EM (13).
Similar interdomain dynamics were shown for the N-terminal domain of Nup192 (73). These dynamics might contribute to the passage of bulky cargoes through the restricted central channel of the NPC. In addition, the conformational dynamics of ScNup133 might insulate the structure of the NPC from morphological changes of the NE during cell division and growth (73). Such flexibility in the NPC has been suggested by prior high-resolution EM studies (74, 75). Also, this flexibility might be required during the biogenesis of NPCs to interlock various nucleoporins in the Nup84 complex (76, 77) through the Nup133–Nup84 and Nup133–Nup120 interfaces (8, 12). A related possibility is that, once assembled into the mature NPC, Nup133 could be preferentially stabilized in one of the four conformations described in this study.
ScNup133 connects to the rest of the Nup84 complex through a tail-to-tail interaction with ScNup84 (8, 22, 23). The model of ScNup133 allowed us to predict the location and molecular details of this interaction surface, validated by three sets of double point mutations at the ScNup133–ScNup84 interface (Figs. 4 and 5). We also identified a number of intermolecular cross-links between ScNup133 and ScNup84 (24) that are consistent with our predicted interface (Table III). In addition, the predicted ScNup133–ScNup84 interface is consistent with that of HsNup133–HsNup107 identified via X-ray crystallography (22, 23).
The Presence of ALPS Motifs Indicates a Conserved Mechanism for Assembly and Membrane Anchoring of the NPC
In our previous structure–function analysis of the Nup84 complex (8), we identified the β-propeller regions of the paralog proteins Nup133 and Nup120 as hotspots for the NPC clustering phenotype, an abnormal distribution of NPCs into closely packed groups that occurs in response to mutations in certain nucleoporins. Based on this and additional functional data, we hypothesized that NPC clustering is caused by the inability of the Nup84 complex to interact with and stabilize the curvature of the NE membrane at the interface with the NPC. However, the mechanism used by the Nup84 complex to interact with the membrane was not clear, and previous studies have not been able to detect any membrane interaction motifs in yeast Nup133 (18), leading to the suggestion that the ALPS motif in Nup133 is restricted to organisms with open mitosis (18, 19).
In this study, we identified a potential ALPS motif located in the β-propeller domain of two related yeast Nup133 proteins (Figs. 6B and 6C), which is inconsistent with the previous suggestion (18, 19) because yeast have a fully closed mitosis. In both yeast species, each of the ALPS motifs closely matched the consensus previously established for this kind of membrane-curvature-sensing motif (supplemental Figs. S2B and S2C) (18). We identified the ALPS motif in a resolved loop in the crystal structure of the VpNup133 β-propeller (Fig. 6B); the equivalent region in the human Nup133 counterpart contains a functional ALPS motif (Fig. 6A) (15, 18). We also detected the ALPS motif in our model of the ScNup133 β-propeller (Fig. 6C). The presence of the ALPS motif in the domain that was previously identified as a hotspot for NPC clustering indicates a conserved mechanism used by yeast Nup133 to interact with the NE membrane and stabilize its curvature.
The β-propeller of the paralog protein ScNup120 was also identified as an NPC clustering hotspot, suggesting that a similar ALPS-motif-dependent mechanism could apply to ScNup120. Correspondingly, we detected two potential ALPS motifs in the ScNup120 β-propeller domain (Figs. 6D and 6E; supplemental Figs. S2D and S2E). Thus, the identification of the conserved ALPS motifs in the components of the yeast Nup84 complex strongly suggests that these motifs are a common and ancient eukaryotic feature, not restricted to open-mitosis organisms.
Here, we propose that the coordinated action of multiple copies of the ALPS motifs in the NPC (current stoichiometries of ScNup133 and ScNup120 (4, 5) would indicate the presence of at least 32 ALPS motifs per yeast NPC) results in a network of protein–membrane contacts around the NPC periphery that might help stabilize the NE membrane, both during early stages of the NPC biogenesis in interphase (15) and throughout the NPC life cycle. This hypothesis might explain why in certain organisms, such as the fungus Aspergillus nidulans, the presence of transmembrane nucleoporins is not required in order for a functional NPC to form, so long as an intact Nup84 complex is present (78). Other nups are predicted to carry similar membrane-interacting motifs; both the yeast (79) and the vertebrate Nup53 (80) contain membrane-interacting motifs necessary for correct NPC assembly. The presence of such a conserved network of membrane-interacting motifs spread across the inner face of the NPC's scaffold and facing the pore membrane might be a key factor driving the assembly and stable association of the NPC with the NE.
The protocoatomer hypothesis suggests that many of the eukaryotic membrane coating complexes originated from an ancestral coating complex through a series of duplication, specialization, and secondary loss processes (14, 81). The Nup84 complex shares a common evolutionary origin with the outer coats of vesicle coating complexes, including clathrin, COPI, and COPII (14, 81–83). Thus, our results suggesting that the ALPS membrane-interacting motifs are a common feature of the coat-like proteins Nup133 and Nup120 in the Nup84 complex allow us to refine the protocoatomer hypothesis. No ALPS-like motifs have been identified in the proteins that form the outer coat in clathrin, COPI, and COPII. However, the small GTPase components that mediate the first stages of vesicle coat formation commonly associate with membranes through a membrane anchor, which can be an amphipathic peptide (84, 85). Perhaps the Nup84 complex retains ancestral features, including the ALPS membrane-interacting motifs, that were lost from other outer coats during the development and specialization of vesicle coating complexes.
Supplementary Material
Acknowledgments
The authors are grateful to the scientists at Eli Lilly & Company, San Diego, CA, who contributed toward cloning and expression of the Sc- and VpNup133 constructs during PSI2. We thank Agustin Avila-Sakar and Yifan Cheng for helping with EM data collection at UCSF, and Paula Upla and William Rice at NYSBC for advice on analyzing the EM data. We thank Dina Schneidman-Duhovny at UCSF for a discussion on SAXS analysis and Roxana Mironska for her assistance with the functional experiments. We are also grateful to Drs. John Tainer and Michal Hammel for help in using the SIBYLS beamline 12.3.1 at ALS. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 Use of the LRL-CAT beamline facilities at Sector 31 of the APS was provided by Eli Lilly, which operates the facility. Authors would like to thank Dr. Stephen R. Wasserman and Sonal Sojitra for their help with X-ray diffraction data collection. Portions of this research were carried out at the SSRL, SLAC National Accelerator Laboratory operated for DOE by Stanford University. The SSRL SMBP is supported by the DOE Office of Biological and Environmental Research, by the National Institutes of Health, NCRR, Biomedical Technology Program (P41RR001209), and by NIGMS, National Institutes of Health (P41GM103393). Use of the SIBYLS beamline 12.3.1 at ALS was supported in part by the Office of Science, Office of Biological and Environmental Research, DOE, under Contract DE-AC02-05CH11231.
Footnotes
Author contributions: S.J.K., J.F.-M., P.S., H.T., T.M.W., J.B.B., J.M.S., S.K.B., B.T.C., S.C.A., M.P.R., and A.S. designed research; S.J.K., J.F.-M., P.S., A.M., T.M., Y.S., and A. M.-I. performed research; S.J.K., J.F.-M., P.S., M.P.R., and A.S. contributed new reagents or analytic tools; S.J.K., J.F.-M., and P.S. analyzed data; S.J.K., J.F.-M., P.S., S.C.A., M.P.R., and A.S. wrote the paper.
* Funding for the NYSGXRC and NYSGRC was provided by National Institutes of Health Grant Nos. U54 GM074945 (S.K.B.) and U54 GM094662 (S.C.A.), respectively. Additional funding for this work was provided by National Institutes of Health Grant Nos. R01 GM062427 (M.P.R.), R01 GM083960 (A.S.), U54 GM103511, and U01 GM098256 (A.S. and M.P.R.). The NYSGRC and NYSGXRC target identifiers are: NYSGRC-019383 and NYSGXRC-15133f for VpNup133; NYSGRC-019463 and NYSGXRC-15133a for ScNup133. Atomic coordinates and structure factors of VpNup133 (55-502) were deposited to the PDB on 01 May 2014 with accession code 4Q9T.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- NPC
- nuclear pore complex
- SAXS
- small angle X-ray scattering
- EM
- electron microscopy
- nup
- nucleoporin
- Sc
- Saccharomyces cerevisiae
- Vp
- Vanderwaltozyma polyspora
- Hs
- Homo sapiens
- ALPS
- ArfGAP1 lipid packing sensor
- GFP
- green fluorescent protein
- NE
- nuclear envelope
- DSS
- disuccinimidyl suberate
- EDC
- 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- SeMET-SAD
- Selenomethionine Single-wavelength Anomalous Dispersion.
REFERENCES
- 1. Aitchison J. D., Rout M. P. (2012) The yeast nuclear pore complex and transport through it. Genetics 190, 855–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wozniak R., Burke B., Doye V. (2010) Nuclear transport and the mitotic apparatus: an evolving relationship. Cell Mol. Life Sci. 67, 2215–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grossman E., Medalia O., Zwerger M. (2012) Functional architecture of the nuclear pore complex. Annu. Rev. Biophys. 41, 557–584 [DOI] [PubMed] [Google Scholar]
- 4. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Sali A., Rout M. P. (2007) The molecular architecture of the nuclear pore complex. Nature 450, 695–701 [DOI] [PubMed] [Google Scholar]
- 5. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Rout M. P., Sali A. (2007) Determining the architectures of macromolecular assemblies. Nature 450, 683–694 [DOI] [PubMed] [Google Scholar]
- 6. Peters R. (2009) Translocation through the nuclear pore: Kaps pave the way. Bioessays 31, 466–477 [DOI] [PubMed] [Google Scholar]
- 7. Rout M. P., Aitchison J. D., Suprapto A., Hjertaas K., Zhao Y., Chait B. T. (2000) The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148, 635–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandez-Martinez J., Phillips J., Sekedat M. D., Diaz-Avalos R., Velazquez-Muriel J., Franke J. D., Williams R., Stokes D. L., Chait B. T., Sali A., Rout M. P. (2012) Structure-function mapping of a heptameric module in the nuclear pore complex. J. Cell Biol. 196, 419–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Szymborska A., de Marco A., Daigle N., Cordes V. C., Briggs J. A., Ellenberg J. (2013) Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 341, 655–658 [DOI] [PubMed] [Google Scholar]
- 10. Lutzmann M., Kunze R., Buerer A., Aebi U., Hurt E. (2002) Modular self-assembly of a Y-shaped multiprotein complex from seven nucleoporins. EMBO J. 21, 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kampmann M., Atkinson C. E., Mattheyses A. L., Simon S. M. (2011) Mapping the orientation of nuclear pore proteins in living cells with polarized fluorescence microscopy. Nat. Struct. Mol. Biol. 18, 643–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seo H. S., Ma Y., Debler E. W., Wacker D., Kutik S., Blobel G., Hoelz A. (2009) Structural and functional analysis of Nup120 suggests ring formation of the Nup84 complex. Proc. Natl. Acad. Sci. U.S.A. 106, 14281–14286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bui K. H., von Appen A., DiGuilio A. L., Ori A., Sparks L., Mackmull M. T., Bock T., Hagen W., Andres-Pons A., Glavy J. S., Beck M. (2013) Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 14. Devos D., Dokudovskaya S., Williams R., Alber F., Eswar N., Chait B. T., Rout M. P., Sali A. (2006) Simple fold composition and modular architecture of the nuclear pore complex. Proc. Natl. Acad. Sci. U.S.A. 103, 2172–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doucet C. M., Talamas J. A., Hetzer M. W. (2010) Cell cycle-dependent differences in nuclear pore complex assembly in metazoa. Cell 141, 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walther T. C., Alves A., Pickersgill H., Loiodice I., Hetzer M., Galy V., Hulsmann B. B., Kocher T., Wilm M., Allen T., Mattaj I. W., Doye V. (2003) The conserved Nup107–160 complex is critical for nuclear pore complex assembly. Cell 113, 195–206 [DOI] [PubMed] [Google Scholar]
- 17. Bolhy S., Bouhlel I., Dultz E., Nayak T., Zuccolo M., Gatti X., Vallee R., Ellenberg J., Doye V. (2011) A Nup133-dependent NPC-anchored network tethers centrosomes to the nuclear envelope in prophase. J. Cell Biol. 192, 855–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drin G., Casella J. F., Gautier R., Boehmer T., Schwartz T. U., Antonny B. (2007) A general amphipathic alpha-helical motif for sensing membrane curvature. Nat. Struct. Mol. Biol. 14, 138–146 [DOI] [PubMed] [Google Scholar]
- 19. Berke I. C., Boehmer T., Blobel G., Schwartz T. U. (2004) Structural and functional analysis of Nup133 domains reveals modular building blocks of the nuclear pore complex. J. Cell Biol. 167, 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doye V., Wepf R., Hurt E. C. (1994) A novel nuclear pore protein Nup133p with distinct roles in poly(A)+ RNA transport and nuclear pore distribution. EMBO J. 13, 6062–6075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sampathkumar P., Gheyi T., Miller S. A., Bain K. T., Dickey M., Bonanno J. B., Kim S. J., Phillips J., Pieper U., Fernandez-Martinez J., Franke J. D., Martel A., Tsuruta H., Atwell S., Thompson D. A., Emtage J. S., Wasserman S. R., Rout M. P., Sali A., Sauder J. M., Burley S. K. (2011) Structure of the C-terminal domain of Saccharomyces cerevisiae Nup133, a component of the nuclear pore complex. Proteins 79, 1672–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Whittle J. R., Schwartz T. U. (2009) Architectural nucleoporins Nup157/170 and Nup133 are structurally related and descend from a second ancestral element. J. Biol. Chem. 284, 28442–28452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boehmer T., Jeudy S., Berke I. C., Schwartz T. U. (2008) Structural and functional studies of Nup107/Nup133 interaction and its implications for the architecture of the nuclear pore complex. Mol. Cell 30, 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shi Y., Fernandez-Martinez J., Tjioe E., Pellarin R., Kim S. J., Williams R., Schneidman-Duhovny D., Sali A., Rout M. P., Chait B. T. (2014) Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol Cell Proteomics mcp.M114.041673. First Published on August 26, 2014, 10.1074/mcp.M114.041673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evans P. R., Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., Hung L. W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cowtan K. (2010) Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 32. Cowtan K. (2008) Fitting molecular fragments into electron density. Acta Crystallogr. D Biol. Crystallogr. 64, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2002) The PyMOL User's Manual, DeLano Scientific, San Carlos, CA [Google Scholar]
- 37. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 38. Schneidman-Duhovny D., Hammel M., Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneidman-Duhovny D., Hammel M., Tainer J. A., Sali A. (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 105, 962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 41. Ohi M., Li Y., Cheng Y., Walz T. (2004) Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proc. Online 6, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 43. Yang Z., Fang J., Chittuluru J., Asturias F. J., Penczek P. A. (2012) Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 20, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Russel D., Lasker K., Webb B., Velazquez-Muriel J., Tjioe E., Schneidman-Duhovny D., Peterson B., Sali A. (2012) Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 10, e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soding J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960 [DOI] [PubMed] [Google Scholar]
- 46. Soding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pieper U., Webb B. M., Dong G. Q., Schneidman-Duhovny D., Fan H., Kim S. J., Khuri N., Spill Y. G., Weinkam P., Hammel M., Tainer J. A., Nilges M., Sali A. (2014) ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 42, D336–D346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marsden R. L., McGuffin L. J., Jones D. T. (2002) Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Sci. 11, 2814–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 50. Buchan D. W., Minneci F., Nugent T. C., Bryson K., Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ward J. J., McGuffin L. J., Bryson K., Buxton B. F., Jones D. T. (2004) The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 [DOI] [PubMed] [Google Scholar]
- 52. Weinkam P., Pons J., Sali A. (2012) Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U.S.A. 109, 4875–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wolynes P. G. (2005) Recent successes of the energy landscape theory of protein folding and function. Q. Rev. Biophys. 38, 405–410 [DOI] [PubMed] [Google Scholar]
- 54. Schug A., Whitford P. C., Levy Y., Onuchic J. N. (2007) Mutations as trapdoors to two competing native conformations of the Rop-dimer. Proc. Natl. Acad. Sci. U.S.A. 104, 17674–17679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li W., Wolynes P. G., Takada S. (2011) Frustration, specific sequence dependence, and nonlinearity in large-amplitude fluctuations of allosteric proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 3504–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pelikan M., Hura G. L., Hammel M. (2009) Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 28, 174–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Velazquez-Muriel J., Lasker K., Russel D., Phillips J., Webb B. M., Schneidman-Duhovny D., Sali A. (2012) Assembly of macromolecular complexes by satisfaction of spatial restraints from electron microscopy images. Proc. Natl. Acad. Sci. U.S.A. 109, 18821–18826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y., Skolnick J. (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 [DOI] [PubMed] [Google Scholar]
- 61. Pokala N., Handel T. M. (2005) Energy functions for protein design: adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J. Mol. Biol. 347, 203–227 [DOI] [PubMed] [Google Scholar]
- 62. Snider C., Jayasinghe S., Hristova K., White S. H. (2009) MPEx: a tool for exploring membrane proteins. Protein Sci. 18, 2624–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gautier R., Douguet D., Antonny B., Drin G. (2008) HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 [DOI] [PubMed] [Google Scholar]
- 64. Drin G., Antonny B. (2010) Amphipathic helices and membrane curvature. FEBS Lett. 584, 1840–1847 [DOI] [PubMed] [Google Scholar]
- 65. Kurtzman C. P. (2003) Phylogenetic circumscription of Saccharomyces, Kluyveromyces and other members of the Saccharomycetaceae, and the proposal of the new genera Lachancea, Nakaseomyces, Naumovia, Vanderwaltozyma and Zygotorulaspora. FEMS Yeast Res. 4, 233–245 [DOI] [PubMed] [Google Scholar]
- 66. Neer E. J., Smith T. F. (1996) G protein heterodimers: new structures propel new questions. Cell 84, 175–178 [DOI] [PubMed] [Google Scholar]
- 67. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 68. Bernauer J., Bahadur R. P., Rodier F., Janin J., Poupon A. (2008) DiMoVo: a Voronoi tessellation-based method for discriminating crystallographic and biological protein-protein interactions. Bioinformatics 24, 652–658 [DOI] [PubMed] [Google Scholar]
- 69. Petoukhov M. V., Konarev P. V., Kikhney A. G., Svergun D. I. (2007) ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40, S223–S228 [Google Scholar]
- 70. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 71. Emekli U., Schneidman-Duhovny D., Wolfson H. J., Nussinov R., Haliloglu T. (2008) HingeProt: automated prediction of hinges in protein structures. Proteins 70, 1219–1227 [DOI] [PubMed] [Google Scholar]
- 72. Suhre K., Sanejouand Y. H. (2004) ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sampathkumar P., Kim S. J., Upla P., Rice W. J., Phillips J., Timney B. L., Pieper U., Bonanno J. B., Fernandez-Martinez J., Hakhverdyan Z., Ketaren N. E., Matsui T., Weiss T. M., Stokes D. L., Sauder J. M., Burley S. K., Sali A., Rout M. P., Almo S. C. (2013) Structure, dynamics, evolution, and function of a major scaffold component in the nuclear pore complex. Structure 21, 560–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Akey C. W. (1995) Structural plasticity of the nuclear pore complex. J. Mol. Biol. 248, 273–293 [DOI] [PubMed] [Google Scholar]
- 75. Yang Q., Rout M. P., Akey C. W. (1998) Three-dimensional architecture of the isolated yeast nuclear pore complex: functional and evolutionary implications. Mol. Cell 1, 223–234 [DOI] [PubMed] [Google Scholar]
- 76. Melcak I., Hoelz A., Blobel G. (2007) Structure of Nup58/45 suggests flexible nuclear pore diameter by intermolecular sliding. Science 315, 1729–1732 [DOI] [PubMed] [Google Scholar]
- 77. Solmaz S. R., Chauhan R., Blobel G., Melcak I. (2011) Molecular architecture of the transport channel of the nuclear pore complex. Cell 147, 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu H. L., De Souza C. P., Osmani A. H., Osmani S. A. (2009) The three fungal transmembrane nuclear pore complex proteins of Aspergillus nidulans are dispensable in the presence of an intact An-Nup84–120 complex. Mol. Biol. Cell 20, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Marelli M., Lusk C. P., Chan H., Aitchison J. D., Wozniak R. W. (2001) A link between the synthesis of nucleoporins and the biogenesis of the nuclear envelope. J. Cell Biol. 153, 709–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vollmer B., Schooley A., Sachdev R., Eisenhardt N., Schneider A. M., Sieverding C., Madlung J., Gerken U., Macek B., Antonin W. (2012) Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J. 31, 4072–4084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Devos D., Dokudovskaya S., Alber F., Williams R., Chait B. T., Sali A., Rout M. P. (2004) Components of coated vesicles and nuclear pore complexes share a common molecular architecture. PLoS Biol. 2, e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Leksa N. C., Schwartz T. U. (2010) Membrane-coating lattice scaffolds in the nuclear pore and vesicle coats: commonalities, differences, challenges. Nucleus 1, 314–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Field M. C., Sali A., Rout M. P. (2011) Evolution: on a bender—BARs, ESCRTs, COPs, and finally getting your coat. J. Cell Biol. 193, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Faini M., Beck R., Wieland F. T., Briggs J. A. (2013) Vesicle coats: structure, function, and general principles of assembly. Trends Cell Biol. 23, 279–288 [DOI] [PubMed] [Google Scholar]
- 85. Antonny B. (2011) Mechanisms of membrane curvature sensing. Annu. Rev. Biochem. 80, 101–123 [DOI] [PubMed] [Google Scholar]
- 86. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 87. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 88. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., Baker N. A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Baker N. A., Sept D., Holst M. J., McCammon J. A. (2001) The adaptive multilevel finite element solution of the Poisson-Boltzmann equation on massively parallel computers. IBM J. Res. Dev. 45, 427–438 [Google Scholar]
- 90. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 91. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen V. B., Arendall W. B., III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.