

Abstract
Most cellular processes are orchestrated by macromolecular complexes. However, structural elucidation of these endogenous complexes can be challenging because they frequently contain large numbers of proteins, are compositionally and morphologically heterogeneous, can be dynamic, and are often of low abundance in the cell. Here, we present a strategy for the structural characterization of such complexes that has at its center chemical cross-linking with mass spectrometric readout. In this strategy, we isolate the endogenous complexes using a highly optimized sample preparation protocol and generate a comprehensive, high-quality cross-linking dataset using two complementary cross-linking reagents. We then determine the structure of the complex using a refined integrative method that combines the cross-linking data with information generated from other sources, including electron microscopy, X-ray crystallography, and comparative protein structure modeling. We applied this integrative strategy to determine the structure of the native Nup84 complex, a stable hetero-heptameric assembly (∼600 kDa), 16 copies of which form the outer rings of the 50-MDa nuclear pore complex (NPC) in budding yeast. The unprecedented detail of the Nup84 complex structure reveals previously unseen features in its pentameric structural hub and provides information on the conformational flexibility of the assembly. These additional details further support and augment the protocoatomer hypothesis, which proposes an evolutionary relationship between vesicle coating complexes and the NPC, and indicates a conserved mechanism by which the NPC is anchored in the nuclear envelope.
Macromolecular complexes are the building blocks that drive virtually all cellular and biological processes. In each eukaryotic cell, there exist many hundreds of these protein complexes (1–3), the majority of which are still poorly understood in terms of their structures, dynamics, and functions. The classical structure determination approaches of nuclear magnetic resonance, X-ray crystallography, and electron microscopy (EM)1 remain challenged in attempts to determine the high-resolution structures of large, dynamic, and flexible complexes in a living cell (4). Thus, additional robust and rapid methods are needed, ideally working in concert with these classical approaches, to allow the greatest structural and functional detail in characterizations of macromolecular assemblies.
Integrative modeling approaches help address this need, providing powerful tools for determining the structures of endogenous protein complexes (5, 6) by relying on the collection of an extensive experimental dataset, preferably coming from diverse sources (both classical and new) and different levels of resolution. These data are translated into spatial restraints that are used to calculate an ensemble of structures by satisfying the restraints, which in turn can be analyzed and assessed to determine precision and estimate accuracy (5, 7). A major advantage of this approach is that it readily integrates structural data from different methods and a wide range of resolutions, spanning from a few angstroms to dozens of nanometers. This strategy has been successfully applied to a number of protein complexes (8–16). However, it has proven difficult and time-consuming to generate a sufficient number of accurate spatial restraints to enable high-resolution structural characterization; thus, the determination of spatial restraints currently presents a major bottleneck for widespread application of this integrative approach. An important step forward is therefore the development of technologies for collecting high-resolution and information-rich spatial restraints in a rapid and efficient manner, ideally from endogenous complexes isolated directly from living cells.
Chemical cross-linking with mass spectrometric readout (CX-MS) (17, 18) has recently emerged as an enabling approach for obtaining residue-specific restraints on the structures of proteins and protein complexes (19–25). In a CX-MS experiment, the purified protein complex is chemically conjugated by a functional group-specific cross-linker, and this is followed by proteolytic digestion and analysis of the resulting peptide mixture by mass spectrometry (MS). However, because of the complexity of the peptide mixtures and low abundance of most of the informative cross-linked species, comprehensive detection of these cross-linked peptides has proven challenging. This challenge increases substantially in studies of endogenous complexes of modest to low abundance, which encompass the great majority of assemblies in any cell (26, 27). In addition, because most cross-linkers used for CX-MS target primary amines, comprehensive detection of cross-links is further limited by the occurrence of lysine, which constitutes only ∼6% of protein sequences, although these lysine residues are generally present on protein surfaces. The use of cross-linkers with different chemistries and reactive groups, especially toward abundant residues, would increase the cross-linking coverage and could be of great help for downstream structural analysis (28).
The nuclear pore complex (NPC) is one of the largest protein assemblies in the cell and is the sole mediator of macromolecular transport between the nucleus and the cytoplasm. The NPC is formed by multiple copies of ∼30 different proteins termed nucleoporins (Nups) that are assembled into discrete subcomplexes (8, 29). These building blocks are arranged into eight symmetrical units called spokes that are radially connected to form several concentric rings. The outer rings of the NPC are mainly formed by the Nup84 complex (a conserved complex, termed the Nup107–Nup160 complex in vertebrates). In budding yeast, the Nup84 complex is an essential, Y-shaped assembly of ∼600 kDa that is formed by seven nucleoporins (Nup133, Nup120, Nup145c, Nup85, Nup84, Seh1, and Sec13 in Saccharomyces cerevisiae) (30). The Nup84 complex has been shown to have a common evolutionary origin with vesicle coating complexes (VCCs), such as COPII, COPI, and clathrin (31, 32), but the evolutionary relationships between these VCCs have not been fully delineated. The Nup84 complex has been extensively characterized; several of its components have been analyzed via X-ray crystallography (33, 34), its overall shape has been defined by means of negative-stain electron microscopy (14, 30, 35, 36), and recently efforts were made to define the protein contacts in the Nup84 complex via CX-MS in humans (35) and a thermophilic fungus (37). Finally, we recently used an integrative modeling approach combining domain mapping, negative-stain electron microscopy (38), and publicly available crystal structures to generate a medium-resolution map of the native Nup84 complex (14). However, despite all these efforts, the fine features of the complex, and in particular the intricate domain orientations and contacts within the complex's hub, remain poorly described.
To address these issues, we present here an optimized CX-MS strategy for robust and in-depth structural characterization of endogenous protein complexes. To test the strategy, we generated a comprehensive high-quality CX-MS dataset on the endogenous Nup84 complex using two complementary cross-linkers, disuccinimidyl suberate (DSS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC). Using the resulting cross-linking restraints together with other sources of information (including electron microscopy, X-ray crystallography, and comparative modeling), we computed a detailed structure of the endogenous Nup84 complex. In addition to providing the overall architecture of the yeast Nup84 complex, the resulting structure reveals the previously unknown architecture of its pentameric structural hub. Our results demonstrate that the present approach provides a robust framework for the standardized generation and use of CX-MS spatial restraints toward the structural characterization of endogenous protein complexes.
EXPERIMENTAL PROCEDURES
Purification and Chemical Cross-linking of the Endogenous Nup84 Complex
To purify the native S. cerevisiae Nup84 complex, we used a procedure that we have optimized over the course of several years (14) (supplemental “Experimental Procedures”). The natively eluted Nup84 complex (200 μl) was cross-linked via the addition of isotopically labeled DSS (d0:d12 = 1:1, Creative Moleculesan an online company located at: http://creativemolecules.com/CM_Contact_Us.htm) to yield a final concentration of 1 mm and incubated for 45 min at 25 °C and 750 rpm of agitation. The reaction was then quenched using a final concentration of 50 mm ammonium bicarbonate.
In the case of cross-linking using EDC (Pierce), the sample was equilibrated and eluted in EDC cross-linking buffer (20 mm MES, pH 6.5, 500 mm NaCl, 2 mm MgCl2, 0.1% CHAPS, 1 mm DTT); EDC was added to the sample to yield a final concentration of 25 mm, and N-hydroxysulfosuccinimide (Sulfo-NHS, Pierce) was added to yield a final concentration of 0.5 mm (i.e. 2% molar ratio with respect to EDC). The sample was incubated for 45 min at 25 °C and 750 rpm of agitation. After the incubation, Tris-HCl was added to a final concentration of 50 mm and pH 8.0, and β-mercaptoethanol was added to a final concentration of 20 mm; the sample was then incubated at 25 °C for 15 min and 750 rpm of agitation to quench the reaction.
The cross-linked samples were either directly processed for in-solution digestion or precipitated using 90% cold methanol and resuspended in SDS-PAGE loading buffer for in-gel separation and digestion.
Proteolytic Digestion of Chemically Cross-linked Nup84 Complex
After cross-linking, the complex was reduced by 10 mm tris-(2-carboxyethyl)-phosphine (Invitrogen) at 55 °C, cooled to room temperature, and alkylated by 20 mm iodoacetamide for 20 min in the dark. The cross-linked complex was digested either in-solution or in-gel with trypsin to generate cross-linked peptides. For in-solution digestion, ∼10 to 20 μg of purified complex was digested using 0.5 μg trypsin (Promega, Madison, WI) in 1 m urea and 0.1% Rapigest (Waters, Etten-Leur, The Netherlands). After overnight (12 to 16 h) incubation, an additional ∼0.3-μg aliquot of trypsin was added to the digest, and it was incubated for a further 4 h. The resulting proteolytic peptide mixture was acidified and then centrifuged at 13,000 rpm for 5 min. The supernatant peptides were collected and desalted using a C18 cartridge (Sep-Pak, Waters), lyophilized in protein LoBind tubes (Eppendorf, Hamburg, Germany), and fractionated via peptide size exclusion chromatography (below). For in-gel digestion, ∼10 to 20 μg of purified complex was heated at 75 °C in 1× SDS loading buffer for 10 min. The sample was cooled at room temperature for cysteine alkylation and separated via electrophoresis in a 4–12% Bis-Tris SDS-PAGE gel (Invitrogen). The gel region above 220 kDa was sliced, crushed into small pieces, and digested in-gel by trypsin. After extraction and purification, the resulting proteolytic peptides were dissolved in 20 μl of a solution containing 30% acetonitrile and 0.2% formic acid and fractionated via peptide size exclusion chromatography (Superdex Peptide PC 3.2/30, GE Healthcare) using off-line HPLC separation with an autosampler (Agilent Technologies, Santa Clara, CA). Two to four size exclusion chromatography fractions covering the molecular mass range of ∼2.5 kDa to ∼8 kDa were collected and analyzed via LC/MS.
Mass Spectrometric Analysis of Cross-linked Peptides
To characterize the composition of our Nup84 complex preparation, ∼1 μg of the endogenously purified complex was in-gel digested, and 1/50 portion of the purified proteolytic peptides (corresponding to ∼20 ng of the purified complex) were loaded onto an EASY-Spray column (15 cm × 75 μm inner diameter, 3 μm) and analyzed with an Orbitrap Fusion mass spectrometer coupled on-line to an EASY-nLC 1000 nano-LC system (Thermo). A 10-min LC gradient was employed (8% B to 55% B, 0–7 min, followed by 55% B to 100% B, 7–10 min, where mobile phase A consisted of 0.1% formic acid and B consisted of 100% acetonitrile in 0.1% formic acid). A top-10 (high–low) method was used where the precursors (m/z = 300, 1700) were measured by the Orbitrap, isolated in the quadrupole mass filter (isolation window 1.8 Th), and fragmented within the higher energy collisional dissociation (HCD) cell (HCD normalized energy 28), and the product ions were analyzed in the low-resolution ion trap. Other instrumental parameters included a flow rate of 300 nl/min, spray voltage of 1.7 kv, S lenses (35%), and automatic gain control (AGC) targets of 5 × 105 (Orbitrap, West Palm, FL) and 1 × 104 (ion trap).
The raw data on the immunoprecipitation were converted to an mzXML file (by MM file conversion 3) and searched online by X! Tandem. Database search parameters included mass accuracies of MS1 < 10 ppm and MS2 < 0.4 Da, cysteine carbamidomethylation as a fixed modification, methionine oxidation, N-terminal acetylation, and phosphorylation (at S, T, and Y) as variable modifications. A maximum of one trypsin missed-cleavage site was allowed. The seven Nup84 components were identified as the top seven hits by spectral counting. The results are provided in supplemental Table S7.
For cross-link identification, the purified peptides were dissolved in the sample loading buffer (5% MeOH, 0.2% formic acid) and loaded onto a self-packed PicoFrit® column with an integrated electrospray ionization emitter tip (360 outer diameter, 75 inner diameter with 15-μm tip, New Objective, Woburn, MA). The column was packed with 8 cm of reverse-phase C18 material (3-μm porous silica, 200-Å pore size, Dr. Maisch GmbH, Entringen, Germany). Mobile phase A consisted of 0.5% acetic acid, and mobile phase B consisted of 70% acetonitrile with 0.5% acetic acid. The peptides were eluted in a 150-min LC gradient (8% B to 46% B, 0–118 min, followed by 46% B to 100% B, 118–139 min, and equilibrated with 100% A until 150 min) using an HPLC system (Agilent) and analyzed with an LTQ Velos Orbitrap Pro mass spectrometer (Thermo Fisher). The flow rate was ∼200 nL/min. The spray voltage was set at 1.9 to 2.3 kV. The capillary temperature was 275 °C, and ion transmission on Velos S lenses was set at 35%. The instrument was operated in the data-dependent mode, where the top eight most abundant ions were fragmented by HCD (39) (HCD energy 27–29, 0.1-ms activation time) and analyzed in the Orbitrap mass analyzer. The target resolution for MS1 was 60,000, and for MS2 it was 7500. Ions (370–1700 m/z) with a charge state of >3 were selected for fragmentation. A dynamic exclusion of (15 s/2/55 s) was used. Other instrumental parameters included the following: “lock mass” at 371.1012 Da, monoisotopic mass selection turning off, mass exclusion window of ±1.5 Th, and minimal threshold of 5000 to trigger an MS/MS event. Ion trap accumulation limits (precursors) were 1 × 105 and 1 × 106, respectively, for the linear ion trap and Orbitrap. For MS2, the Orbitrap ion accumulation limit was 5 × 105. The maximal ion injection times for the LTQ and Orbitrap were 100 ms and 500 to 700 ms, respectively.
The raw data were transformed to Mascot generic format by pXtract 2.0 and searched by pLink (version 1.16) (40) using a target-decoy search strategy with a concatenated FASTA protein sequence database containing the seven subunits of the Nup84 complex (accession numbers are YGL092W, YDL116W, YJR042W, YKL057C, YLR208W, and YGL100W) and bovine serum albumin (BSA). We also included the BSA sequence for the target-decoy database search to quickly pre-filter the false positive identifications containing the BSA sequence. An initial MS1 search window of 5 Da was allowed to cover all isotopic peaks of the cross-linked peptides. The data were automatically filtered using a mass accuracy of MS1 ≤ 10 ppm and MS2 ≤ 20 ppm of the theoretical monoisotopic (A0) and other isotopic masses (A+1, A+2, A+3, and A+4) as specified in the software. Other search parameters included cysteine carbamidomethylation as a fixed modification, methionine oxidation, and protein N-terminal methionine cleavage as a variable modification. A maximum of two trypsin missed-cleavage sites were allowed. The initial search results were obtained using a 5% false discovery rate, a default parameter estimated by the pLink software (40). We treated the 5% expected false discovery rate as an initial (permissive) filter of the raw data. We then manually applied additional stringent filters to remove potential false positive identifications from our dataset. For positive identifications, both peptide chains should contain at least five amino acid residues. For both peptide chains, the major MS/MS fragmentation peaks must be assigned and follow a pattern that contains a continuous stretch of fragments. The appearance of dominant fragment ions N-terminal to proline and C-terminal to aspartic acid and glutamic acid for arginine-containing peptides was generally expected (41, 42). The precise cross-linking site could not be determined for ∼20% of EDC cross-links because of the appearance of consecutive aspartic and glutamic acid in the tryptic peptide sequences (supplemental Fig. S5C and supplemental Table S2). Moreover, because cross-linking by EDC (hydrolysis) resembles peptide bond formation, intramolecular cross-link candidates (directly identified by the software) that are composed by two adjacent tryptic peptide sequences of the same protein are likely to be a single, missed cleavage peptide (supplemental Fig. S5D). Such ambiguous EDC cross-linking identifications were eliminated from further consideration. In essence, these additional filters represent a standard procedure for eliminating false positives at the cost of removing some true positives. The net result is a more conservative list of cross-linking restraints that are less likely to manifest as artifacts in the resulting structural models. The cross-link maps (Figs. 2A and 2C) were generated using AUTOCAD (educational version, Autodesk, Inc., New Yok, NY).
Fig. 2.

CX-MS analysis of the Nup84 complex. Cross-linking maps of the Nup84 complex with either DSS cross-linker (A) or EDC cross-linker (B). Straight lines in red connecting residues from different subunits represent DSS intersubunit cross-links, and straight lines in green represent EDC intersubunit cross-links. Curved, dotted lines represent intrasubunit cross-links. Only intrasubunit cross-links between residues more than 40 positions apart are shown. C, a high-resolution HCD MS/MS spectrum (m/z = 1096.169, z = 6) of the intersubunit DSS cross-link connecting Nup85 residue 30 (lysine) and Seh1 residue 1 (N-terminal methionine). The methionines are oxidized. F* indicates an immonium ion of phenylalanine that is frequently observed in HCD spectra. b and y ion series including their charge states are labeled. The fragment of y20 is zoomed, and the mass accuracy is labeled in parts per million. D, a high-resolution HCD MS/MS spectrum (m/z 1044.139, z = 6) of the intersubunit EDC cross-link connecting Nup85 residue 17 and Seh1 residue 1. The spectrum is labeled as in C. The fragment of y20 is zoomed, and the mass accuracy is labeled in parts per million. E, a high-resolution HCD MS/MS spectrum (m/z = 903.317, z = 7) of the intrasubunit DSS cross-link connecting Nup133 residue 506 and Nup133 residue 59. F* and Y* indicate immonium ions of phenylalanine and tyrosine that are common for HCD fragmentations. The fragment of b14 is zoomed, and the mass accuracy is labeled in parts per million. F, a high-resolution HCD MS/MS spectrum (m/z = 917.327, z = 6) of the intrasubunit EDC cross-link connecting Nup133 residue 506 and residue 562. F* and Y* indicate immonium ions of phenylalanine and tyrosine that are common to HCD fragmentations. The fragment of y16 is zoomed, and the mass accuracy is labeled in parts per million. Charge states of the fragments in the high-mass region of this spectrum (i.e. 1100–1300 m/z) are not resolved.
Molecular Architecture of the Endogenous Nup84 Complex Revealed by Integrative Modeling
Our integrative structure modeling proceeds through four stages (7, 13, 14, 43) (Fig. 4): (1) gathering of data, (2) representation of subunits and translation of the data into spatial restraints, (3) configurational sampling to produce an ensemble of models that satisfies the restraints, and (4) analysis and assessment of the ensemble. The modeling protocol (i.e. stages 2, 3, and 4) was scripted using the Python Modeling Interface, version be72c15, a library for modeling macromolecular complexes based on our open-source Integrative Modeling Platform package, version 829c3f0 (44). Files containing the input data, scripts, and output models are available online.
Fig. 4.

The four-stage scheme for integrative structure determination of the Nup84 complex. Our integrative approach to determining the Nup84 complex structure proceeds through four stages (7, 13, 14, 43): (1) gathering of data, (2) representation of subunits and translation of the data into spatial restraints, (3) configurational sampling to produce an ensemble of models that satisfies the restraints, and (4) analysis of the ensemble. The modeling protocol (i.e. stages 2, 3, and 4) was scripted using the Python Modeling Interface, version be72c15, a library for modeling macromolecular complexes based on our open-source Integrative Modeling Platform (IMP) package, version 829c3f0 (44).
Stage 1: Gathering of Data
163 unique DSS and 104 EDC cross-linking peptides were identified by MS (supplemental Tables S1 and S2; supplemental Figs. S9 and S10; 20% of the EDC cross-links were not determined unambiguously, resulting in 123 possible EDC cross-links). The atomic structures for some of the yeast Nup84 complex components and their close homologs had been previously determined via X-ray crystallography (supplemental Fig. S3) (31, 45–54). In addition, structurally defined remote homologs (PDB codes 2QX5 (chain B) and 4LCT (chain A)) (55, 56) were detected for the C-terminal domain in Nup85 by HHpred (57, 58) (supplemental Fig. S3). Secondary structure and disordered regions were predicted by PSIPRED (59, 60) and DISOPRED (61), respectively (supplemental Fig. S3). Domain mapping data (14), an EM class average (14), and a density map from single-particle EM reconstruction of the Nup84 complex (36) were also considered.
Stage 2: Representation of Subunits and Translation of the Data into Spatial Restraints
The domains of the Nup84 complex subunits were represented by beads of varying sizes, arranged into either a rigid body or a flexible string, based on the available crystallographic structures and comparative models (supplemental Fig. S3). In a rigid body, the beads have their relative distances constrained during configurational sampling, whereas in a flexible string the beads are restrained by the sequence connectivity, as described later in this section.
To maximize computational efficiency while avoiding using too coarse a representation, we represented the Nup84 complex in a multi-scale fashion, as follows.
First, the crystallographic structures of each Nup84 complex domain were coarse-grained using two categories of resolution, where beads represented either individual residues or segments of up to 10 residues. For the one-residue bead representation, the coordinates of a bead were those of the corresponding Cα atoms. For the 10-residue bead representation, the coordinates of a bead were the center of mass of all atoms in the corresponding consecutive residues (each residue was in one bead only). The crystallographic structures covered 54% of the residues in the Nup84 complex.
Second, for predicted non-disordered domains of the remaining sequences, comparative models were built with MODELLER 9.13 (62) based on the closest known structure detected by HHPred (57, 58) and the literature (supplemental Fig. S3). Similarly to the X-ray structures, the modeled regions were also coarse-grained using two categories of resolution, resulting in the 1-residue and 10-residue bead representations. The comparative models covered 30% of the residues in the Nup84 complex.
Finally, the remaining regions without a crystallographic structure or a comparative model (i.e. regions predicted to be disordered or structured without a known homolog) were represented by a flexible string of beads corresponding to up to 20 residues each. The residues in these beads corresponded to 16% of the Nup84 complex.
To improve the accuracy and precision of the model ensemble obtained through the satisfaction of spatial restraints (below), we also imposed crystallographic interface constraints based on three crystallographically defined interfaces: Nup85123–460–Seh1 (31, 53), Nup145c145–181–Sec13 (49, 50), and Nup145c145–181–Nup841–488 (49, 50). The three constrained dimers were simply represented as rigid bodies. These constraints are justified because their absence decreases the precision of the model ensemble but does not significantly change the average model (supplemental Fig. S7).
With this representation in hand, we next encoded the spatial restraints based on the information gathered in Stage 1, as follows.
First, the collected DSS and EDC cross-links were used to construct a Bayesian scoring function that restrained the distances spanned by the cross-linked residues (63), taking into account the ambiguity of some cross-linked residue identifications (supplemental “Experimental Procedures”). The cross-link restraints were applied to the 1-residue bead representation for the X-ray structures and comparative models and to the 20-residue bead representation for the remaining regions.
Second, the excluded volume restraints (7) were applied to the 10-residue bead representation for X-ray structures and comparative models and to the 20-residue bead representation for the remaining regions. The excluded volume of each bead was defined using the statistical relationship between the volume and the number of residues that it covered (7, 64).
Third, we applied the sequence connectivity restraint, using a harmonic upper-bound function of the distance between consecutive beads in a subunit, with a threshold distance equal to four times the sum of the radii of the two connected beads. The bead radius was calculated from the excluded volume of the corresponding bead, assuming standard protein density (7, 14, 64).
Finally, the EM two-dimensional restraint (65) was imposed on the highest resolution representation of each subunit, using the cross-correlation coefficient between the EM class average (14) and the best-matching projection of the model as the score (supplemental “Experimental Procedures”).
In summary, all information gathered in Stage 1 was encoded into a Bayesian scoring function (66) (supplemental “Experimental Procedures”). The likelihood function reflects the cross-linking data, and the prior depends on the sequence connectivity, excluded volume, and EM two-dimensional restraint. Most of the remaining information (crystallographic structures of the subunits, their homologs, and the three dimeric interfaces) is included in the representation, whereas the domain deletion data and the density map from single-particle EM reconstruction were used only for validating our final model. See the scripts at our group's website for details and parameter values.
Stage 3: Sampling the Configurations
Structural models of the Nup84 complex were computed using Replica Exchange Gibbs sampling, based on the Metropolis Monte Carlo algorithm (66). The Monte Carlo moves included random translation and rotation of rigid bodies (up to 2 Å and 0.04 radians, respectively) and random translation of individual beads in the flexible segments (up to 2 Å). 64 replicas were used, with temperatures ranging between 1.0 and 2.5. 20 independent sampling calculations were performed, each one starting with a random initial configuration. A model was saved every 10 Gibbs sampling steps, each consisting of a cycle of Monte Carlo steps that moved every rigid body and flexible bead once. The sampling produced a total of 15,000 models from the 20 runs. Models that had a score of less than 300 were included in an ensemble of 6520 solutions for subsequent analysis. The entire sampling procedure took ∼2 weeks on a cluster of 1280 central processing units (CPUs).
Stage 4: Analysis and Assessment of the Ensemble
First, we assessed the thoroughness of the configurational sampling by comparing a subset of 3413 solutions from runs 1–10 to another subset of 3107 solutions from runs 11–20. Each subset of solutions was converted into a density map that specified how often grid points of the map were occupied by a given protein (the “localization density map”) using VMD (67) (supplemental Fig. S6). The localization density map of a subunit was contoured at the threshold that resulted in 2.5 times its volume estimated from sequence (supplemental Table S6). Importantly, the two localization density maps were similar to each other, demonstrating that the Monte Carlo algorithm likely sampled well all solutions that satisfied the input restraints. The final localization density maps of the Nup84 subunits and the whole complex were computed from the complete ensemble of the 6520 solutions (Figs. 6A and 6B).
Fig. 6.
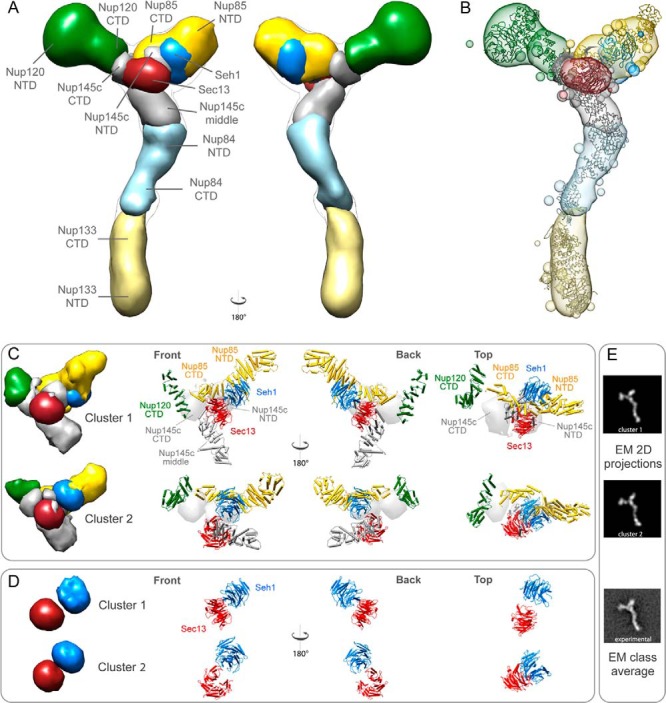
The Nup84 complex molecular architecture revealed by the CX-MS integrative pipeline. The localization density maps of the Nup84 subunits (solid contour surfaces) and the entire complex (transparent surfaces) were computed and contoured at the threshold of 2.5 times their volumes estimated from sequence (supplemental Table S6) (A through C). A, front and back views of the localization density maps of the Nup84 subunits and the entire complex. B, a representative single Nup84 complex structure (colored ribbon) is shown along with the localization density maps of the individual subunits. C, the localization density maps of the two dominant clusters computed on the hub region (Nup120-CTD, Nup85, Nup145c, Sec13, and Seh1) are shown, along with the representative single structures of the hub region for each of the two clusters, from multiple viewing points. D, the positions of Sec13 and Seh1 are presented for each of the two clusters. E, the representative model projections in each of the two clusters are shown, along with the EM class average (14).
Second, the ensemble of solutions was assessed in terms of how well they satisfied the data from which they were computed, including the cross-links, the excluded volume, sequence connectivity, and the EM two-dimensional restraints. We validated the ensemble of solutions against each of 163 DSS and 123 EDC cross-links (supplemental Tables S1 and S2; supplemental Figs. S9 and S10); a cross-link restraint was considered to be satisfied by the ensemble if the median distance between the surfaces of the corresponding beads was smaller than a distance threshold of 35 and 25 Å for the DSS and the EDC cross-links, respectively. The shape implied by the EM class average (14) was satisfied by the ensemble if the average EM two-dimensional cross-correlation was more than 0.8. The excluded volume and sequence connectivity were considered to be satisfied by an individual solution if their combined score was less than 20.
Third, to quantify the precision, the solutions were grouped by Cα root-mean-square deviation quality-threshold clustering (68), based on either the hub subunits (Nup145c, Nup85, Seh1, Sec13, and the C termini of Nup120) or all subunits, using the Cα root-mean-square deviation threshold of 40 Å. The precision of each cluster was calculated as the average Cα distance root-mean-square deviation (dRMSD) (69) between the individual solutions and the centroid solution, defined as the solution with the minimal sum of the dRMSDs to the other solutions in the cluster (supplemental Tables S4 and S5); distance difference terms involving a distance larger than 60 Å were omitted from the dRMSD calculation. The localization density maps of the clusters were computed as described above (Fig. 6C).
Finally, the proximities of any two residues in each cluster were measured by their relative “contact frequency,” which is defined by how often the two residues contact each other in the cluster (7); in the multi-scale representation, a pair of residues are in contact when the distance between the centers of the corresponding highest resolution beads is less than 30 Å (supplemental Fig. S4).
Correlation between the Number of Cross-links and the Accuracy of Dimer Models
As a benchmark, we modeled the Sec13-Nup145c dimer of known structure (Fig. 5A) using up to 10 DSS and 5 EDC intermolecular cross-links. Each of Sec13 and Nup145c was represented as a rigid body; we used the same representation, scoring function, and sampling method as described above. First, we modulated the sparseness of the cross-linking data by varying the number of DSS cross-links from 1 to 10, without considering the EDC cross-links. For each case, up to 10 different random cross-link sets were chosen. We also modeled the dimer with EDC cross-links alone and with all EDC and DSS cross-links. A total of 93 independent modeling runs were performed, producing 30,000 models each. To assess the accuracy of the solutions, the Cα dRMSD (Figs. 5B and 5C; supplemental Fig. S2) was calculated between every produced model and the crystallographic dimer (PDB code 3IKO (50)).
Fig. 5.

Correlation between the number of cross-links and the accuracy of dimer models. A, localization density maps of the Nup84 subunits (solid contour surfaces; Fig. 6A) and position of the Nup145c–Sec13 crystallographic dimer (PDB code 3IKO (50)). B, total scores (i.e. the sum of excluded volume and cross-link restraint scores) are plotted as a function of the Cα dRMSD of the Nup145c–Sec13 dimer models with respect to the crystallographic dimer; as the number of cross-links increases, the ensemble of models is enriched in accurate structures (i.e. low dRMSD models, left of dotted line). C, accuracy of dimer models as a function of the number and type of cross-links. Each symbol displays the first and third quartile (lower and upper side of the boxes), median (red line), and minimum and maximum (lower and upper limit of the dashed whiskers, respectively) of the Cα dRMSD with respect to the crystallographic structure for the 100 best-scoring models. The median and the spread (i.e. difference between the first and third quartile) of the distribution are measures of the accuracy and precision of the ensemble of models, respectively.
RESULTS
Development of a Workflow for CX-MS of Endogenous Protein Complexes
Our goal was to develop a robust and sensitive methodology for determining chemical cross-links for the structural characterization of endogenous protein complexes. The overall strategy is summarized in Fig. 1. The endogenous protein assembly is isolated from a cryogenically milled whole cell lysate by means of single-step affinity purification using antibody-coupled magnetic beads (14, 70). This approach produces highly enriched, relatively homogenous complexes with high recovery, in buffers that can be optimized for diverse downstream applications (14, 70). The complex is natively eluted from the affinity matrix and cross-linked in solution using two complementary chemical cross-linkers, DSS and EDC. DSS is primary amine specific (lysines and amino termini of proteins), with a spacer arm of 11.4 Å (α-carbon), whereas EDC cross-links amines to carboxylic acids (aspartic acids, glutamic acids, and carboxyl termini of proteins) and is generally considered as a “zero” length cross-linker. The relatively high prevalence of carboxylic acid and lysine residues in the protein sequences (∼12% and ∼6%, respectively, with these charged residues generally present on protein surfaces) makes EDC a useful complement to the strictly amine-specific cross-linkers. The cross-linked complex is digested in solution and separated by SDS-PAGE for in-gel proteolysis. In both cases, the digested peptides are then fractionated via peptide size exclusion chromatography (71), and peptides corresponding to the large molecular weight fractions (∼2.5 to 8 kDa) are analyzed via LC-MS. Putative cross-linked peptides are identified by pLink (40), after which the spectra are filtered using additional stringent criteria and manually verified (“Experimental Procedures”). Finally, each of the resulting cross-links is translated into a spatial restraint for integrative modeling (Fig. 4).
Fig. 1.

CX-MS integrative pipeline for the structural characterization of endogenous complexes. The workflow of the cross-linking and mass spectrometric analysis of endogenous Nup84 complex is summarized. The endogenously tagged protein complex is affinity purified from cell cryolysis, natively eluted, and cross-linked by two complementary cross-linkers, DSS and EDC (14, 70). The cross-linked complex is then subjected to in-solution and in-gel digestion, and the resulting peptide mixtures are fractionated using peptide size exclusion chromatography (71). The cross-linked peptides were analyzed by a Velos Orbitrap mass spectrometer using high-resolution HCD and searched by pLink (40). All software-produced identifications were manually inspected (see “Experimental Procedures”) (44).
Chemical Cross-linking and MS Analysis of the Nup84 Complex
The workflow described above was applied to the endogenous heptameric Nup84 complex. Both DSS and EDC cross-linking reactions were optimized for the downstream CX-MS analysis as follows (also see supplemental Fig. S1). We controlled the cross-linking reaction such that the majority of the proteins migrated as diffusely staining regions into the uppermost part (>220 kDa) of an SDS-PAGE gel. The protein samples isolated from these uppermost regions of a gel were enriched in cross-linked peptides. We generated a high-quality cross-linking dataset that included 163 unique DSS and 104 EDC cross-linking peptides (supplemental Tables S1 and S2; supplemental Figs. S9 and S10). The overall connectivity patterns of the DSS and EDC cross-links were similar (Figs. 2A and 2B), showing that they provide complementary information on similar conformers. Even though 15% to 20% of the cross-linked peptides were in the high-molecular-mass range (4500–7000 Da), the use of high-resolution and high-mass-accuracy MS allowed us to pinpoint the cross-linking sites for virtually all the DSS cross-links and the majority of EDC cross-links, including those for large peptide species (Figs. 2C–2F and supplemental Tables S1 and S2). We were unable to unambiguously locate the conjugation sites for ∼20% of the EDC cross-links (supplemental Table S2), because many result from peptides containing consecutive and/or adjacent carboxylic acids (supplemental Table S2 and supplemental Fig. S5C). These ambiguities were accounted for in our modeling calculations.
Importantly, all previously identified interfaces of the complex (identified using a variety of different approaches including X-ray crystallography, domain deletions, and affinity purifications (14, 33)) were recapitulated by the cross-links (Figs. 2A and 2C). As expected, we observed a high correlation between the coverage of cross-links and the size of interfaces within the complex; the majority of the cross-links were mapped to the large, intricate, yet previously poorly described region of the pentameric structural hub (Nup145c, Nup85, Seh1, Sec13, and the C termini of Nup120), and relatively fewer were identified in the stalk of the complex (e.g. between Nup84 and Nup133 (46, 47)) where smaller interfaces were expected.
To further evaluate the fidelity of the cross-linking data, we mapped the Euclidean Cα–Cα distances between the cross-linked residues onto several domains of the Nup84 complex proteins where high-resolution crystal structures had previously been determined (49, 50) (Fig. 3A). The distributions of distance differences between either DSS or EDC cross-links and the crystal structure were narrow, and were notably distinct from the distributions of randomly connected residues of the same type (Fig. 3A). All the measured DSS cross-links fell within the expected maximum reach threshold of ∼30 Å (72). Although the great majority of the EDC cross-links fell within the expected reach threshold of 17 Å (= 6 Å for the lysine side chain + 5 Å for the carboxylic acid side chain + 6 Å for flexibility of backbones (20)), interestingly, the distances for ∼30% of EDC cross-links were longer than expected (Fig. 3A). These discrepancies might have arisen from differences in the flexibility of the subunits at the optimum pH values for EDC versus DSS, the longer times of incubation for the EDC experiments, or some other as yet unknown cause. Nevertheless, the EDC and DSS cross-links provide mutually confirmatory and complementary spatial information (Fig. 3B).
Fig. 3.

Distance distribution of the cross-links and their mapping on the available crystal structures. A, we mapped the Euclidean Cα–Cα distances between the cross-linked residues onto several domains of the Nup84 complex proteins with available crystal structures (49, 50). The Cα–Cα distance distributions of the cross-linked residues are shown for both DSS and EDC cross-links. All the measured DSS cross-links fall within the expected maximum threshold of ∼30 Å (72), and the great majority of EDC cross-links fall within the expected threshold of 17 Å. B, both DSS (in red) and EDC (in green) cross-links are mapped on the crystallographic structure of the Sec13–Nup145c–Nup84 (PDB code 3IKO (50)). The EDC and DSS cross-links provide complementary spatial information.
Correlation between the Number of Cross-links and the Accuracy of Dimer Models
We estimated the number of cross-links required in order to model a subunit dimer with the required accuracy. The estimate was possible because we had a large number of experimentally determined cross-links for several heterodimers of known atomic structure. We chose to focus on the Nup145c–Sec13 dimer (49, 50, 73) because of the abundance of intermolecular DSS and EDC cross-links identified for this dimer. By varying the number of DSS cross-linking restraints used to model the dimer (Fig. 5), we found that at least four or five cross-links (either DSS or EDC) are needed to determine the dimer structure with an accuracy better than 10 Å Cα dRMSD. Increasing the number of cross-links beyond five (up to 15), irrespective of their type, did not further improve the accuracy, although it increased the precision of the resulting ensemble of solutions (Fig. 5C and supplemental Fig. S2).
Molecular Architecture of the Endogenous Nup84 Complex Revealed by Integrative Modeling
We computed the configuration of the seven subunits of the budding yeast Nup84 complex (Fig. 6) using crystallographic structures of seven constituent domains (supplemental Fig. S3) as well as 163 DSS and 123 EDC cross-links (supplemental Tables S1 and S2; supplemental Figs. S9 and S10) and one negative-stain EM class average (14) (Fig. 4). To improve the precision of the resulting models, we included the three crystallographically defined interfaces, namely, Nup85–Seh1 (31, 53), Nup145c–Sec13 (49, 50), and Nup145c–Nup84 (49, 50), as constraints in the modeling calculation; the constrained dimers were simply represented as rigid bodies in the configurational sampling.
The molecular architecture of Nup84 complex was computed from an ensemble of 6520 solutions, shown as a localization density map representing the probability of any volume element being occupied by a given protein (Fig. 6A). The sampling procedure was thorough, as indicated by the similarity of two ensembles of solutions independently calculated using the same cross-link dataset (supplemental Fig. S6).
Next, we validated the solutions against a previously published molecular architecture of the Nup84 complex. The current solutions were in agreement with the Nup84 complex structure determined primarily using the domain mapping data and the EM class average in our earlier study (14) (supplemental Fig. S8), as well as the density map from the single-particle EM reconstruction (36) (supplemental Fig. S8).
We also validated the ensemble of solutions against the data used to compute it. First, the ensemble satisfied 86.5% of the DSS cross-links and 83.6% of the EDC cross-links (“Experimental Procedures,” supplemental Fig. S4, and supplemental Table S3). Second, 99% of the 6520 solutions satisfied the excluded volume and sequence connectivity restraints (“Experimental Procedures”). Finally, the solutions also fit the EM class average, with an average cross-correlation coefficient of 0.9.
Next, we quantified the precision of the ensemble of solutions (“Experimental Procedures”). The clustering analysis identified two dominant clusters of similar structures in the hub, including 1257 and 1010 solutions, respectively (Figs. 6C and 6D; supplemental Table S4). The precision of the ensemble was sufficient to pinpoint the locations and orientations of the constituent proteins and even domains (Fig. 6; supplemental Tables S4 and S5), as described in “Discussion.” The hub region was determined at the highest precision of 12.7 Å (supplemental Table S4). Furthermore, even the independent modeling calculation without crystallographic interface constraints (supplemental Fig. S7) was able to recapitulate the structures of the Nup145c–Sec13, Nup85–Seh1, and Nup84–Nup145c crystallographic interfaces with an accuracy of 4.0, 12.0, and 7.5 Å, respectively, in the most populated cluster, cluster 1 (Table I). These accuracies demonstrate the quality of the cross-link data and validate the application of the three crystallographic interfaces as constraints. Notably, the accuracy of the Nup145c–Sec13 dimer (Cα dRMSD of 4.0 Å) in the model of the entire complex was greater than the accuracy of modeling the Nup145c–Sec13 dimer on its own (Cα dRMSD of 6.5 Å; Figs. 5B and 5C). This observation underscores the synergy between orthogonal data, thus demonstrating the premise of integrative modeling.
Table I. Accuracy of determining the crystallographic interfaces.
| Cluster index | Accuracy of crystallographic interface (Å) |
||
|---|---|---|---|
| Nup145c–Sec13 | Nup85–Seh1 | Nup84–Nup145c | |
| 1 | 4.0 (min 2.4) | 12.4 (min 6.5) | 7.5 (min 2.0) |
| 2 | 4.0 (min 2.7) | 12.4 (min 4.4) | 6.2 (min 2.2) |
The accuracy of modeling each crystallographic interface was calculated as the average Cα dRMSD between the models in the cluster and the corresponding crystallographic interface. The minimum (min) value of Cα dRMSD in the cluster is indicated in parentheses.
Finally, the proximities of any two residues within each of the two dominant solution clusters were measured by their relative “contact frequency,” which is defined by how often the two residues contact each other in the cluster (7). The contact frequencies were in remarkable agreement with the cross-link dataset (supplemental Fig. S4).
DISCUSSION
Integrative Structural Characterization of the Endogenous Nup84 Complex Based on CX-MS Data
We present here an optimized CX-MS workflow for integrative structural characterization of native protein complexes. Importantly, this pipeline generates structures of complexes with near-atomic resolution and in a fraction of the time that, in our experience, previous approaches have taken to achieve similar accuracy. To achieve this goal, we have developed an improved CX-MS pipeline for robust and comprehensive analysis of endogenous protein complexes available in limited amounts (10 to 20 μg), as demonstrated for both the Nup84 complex and the SEA complex (in an accompanying paper (74)). Our approach also allows rapid and efficient integration of structural information from various sources at different levels of resolution, including complementary cross-linking restraints, crystallographic structures, comparative models, electron microscopy class averages and density maps, and affinity purification data. This approach also provides information concerning the heterogeneity and structural flexibility of endogenous complexes, which have been poorly investigated before.
We found that at least four or five cross-links between two crystallographic structures is sufficient to relatively accurately model the corresponding dimer (Fig. 5C and supplemental Fig. S2). Additional cross-links further increase the precision of the final solutions, although they only marginally improve their accuracy. We demonstrate the benefits of using two complementary, commercially available cross-linkers (DSS and EDC) targeting amine and carboxylic groups, respectively. Further efforts are needed to design cross-linkers of varying length and/or type (28).
We empirically observed that in-gel digestion generally yielded more cross-links than in-solution digestion (supplemental Fig. S12). We speculate that one reason for this higher yield is the presence of SDS in the gel, which greatly helps to denature the otherwise rigidified cross-linked complex; efficient proteolysis by trypsin thus becomes of great importance for a successful experiment. In addition, the use of a gel makes it easy to clean up contaminants such as detergents and elution peptides.
Conformational Heterogeneity of Endogenous Protein Complex Inferred from Non-self-consistency of Structural Information
Like many native protein assemblies, the Nup84 complex has been shown to exist in multiple conformational states (14, 30, 35, 36). We noticed that not all cross-links could readily fit into our ensemble of structures calculated via integrative modeling (supplemental Fig. S4 and supplemental Table S3). We suggest that such inconsistency largely reflects the heterogeneity, flexibility, and/or disorder, as seen previously via EM (14, 30, 35, 36). Examples include the N termini of Nup85 and Nup145c, which are located in the Nup85–Seh1 arm that has been suggested to be flexible based on negative-stain EM data (14). Other notable examples are the Nup133(253)–Nup84(340) and Nup133(936)–Nup133(392) cross-links, connecting the β-propeller N-terminal region of Nup133 to the N-terminal region of Nup84 and the C-terminal region of Nup133, respectively. It has been reported that the Nup133 region of the stalk is flexible in yeast (14, 30, 36), and in an accompanying paper we describe how the S. cerevisiae Nup133 molecule shows significant flexibility in the hinge between its N and C termini (91). Also, recently, negative-stain EM analysis of the human counterpart (Nup107–160 complex (35)) suggested extreme flexibility for the Nup133 homolog. Our cross-linking restraints connecting the base of the stalk with its middle region would imply a similar bending of the yeast Nup133 molecule. Other constituents of the NPC core have been shown to present significant flexibility (76). This flexibility might allow for the dilations of the NPC when accommodating large cargoes and the tensions that the nuclear envelope suffers during cell growth.
Molecular Architecture of the Endogenous Nup84 Complex Revealed by Integrative Modeling
Our CX-MS Nup84 complex structure recapitulates most of the features already described in our previous structure (14) and by others (30, 36, 77), including the Y-shape, the kinked stalk, the key role of the α-solenoid interfaces in establishing the overall architecture, and peripheral locations of the β-propeller domains (Fig. 6). The stalk of the Y was shown to be formed by a tail-to-tail connection between Nup133 and Nup84 and a head-to-middle-region connection between Nup84 and Nup145c. Nup120 and the Nup85–Seh1 dimer form the two arms of the Y, respectively. The pentameric hub of the complex is formed by an intricate connection between Nup145c, the C termini of Nup120 and Nup85, and the β-propeller Nups of Seh1 and Sec13. This arrangement is consistent with several previous structures and models (14, 35, 77) but does not agree with docking of crystal structures into a density map from single-particle EM reconstruction of the entire complex, where the Nup85–Seh1 arm is oriented the other way around (36), or with the fence model in which homodimeric interactions connect different copies of the complex (53, 73). The overall topology of the complex is conserved across the different clusters of solutions (Figs. 6C and 6D). The exact positions of the domains vary slightly across the clusters, reflecting either intrinsic flexibility or a lack of sufficient restraints. The main variability is detected in two regions: the Nup85–Seh1 arm and the stalk of the complex. The Nup85–Seh1 arm adopts slightly different arrangements, depending on whether the three crystallographic interface constraints are considered (Fig. 6A and supplemental Fig. S7). This variability of the arm configuration is consistent with the apparent flexibility observed in our negative-stain raw EM particle images (not shown) and the class averages obtained in an independent analysis (36). The stalk of the complex is kinked in our structure (Fig. 6), consistent with the shape of the crystallized Sec13–Nup145c–Nup84 trimer (49, 50). However, solutions calculated without the EM two-dimensional restraint and crystallographic interface constraints (data not shown) include structures compatible with an alternative, extended conformation of the stalk of the complex (30, 36). Structural characterization of the Nup84 complex in the context of the whole NPC might be able to address which of the complex conformations is the predominant one in vivo.
The main benefit of the newly determined structure is the higher level of detail in the hub region of the complex (Figs. 6C and 6D), indicating a more intricate arrangement than previously described (49). This region has proved refractory to classical high-resolution methods because of its heterogeneity and/or flexibility. Our approach, however, was able to overcome these challenges. The hub arrangement involves the β-propeller proteins Sec13 and Seh1; the C-terminal α-helical domains of Nup85, Nup120, and Nup145c; and the N-terminal region of Nup145c. The C-terminal domain of Nup85 wraps around the Seh1 β-propeller and projects into the hub to interact with the C terminus of Nup120. The resulting interface serves as a platform for the interaction with the C-terminal domain of Nup145c. The N-terminal domain of Nup145c is inserted between Nup85, Sec13, and Seh1, thereby bridging the interaction between the dimers Nup145c–Sec13 and Nup85–Seh1. A remarkable feature is the position of the β-propeller proteins Seh1 and Sec13 (Figs. 6C and 6D). The position of Sec13 is consistent among the different clusters of solutions, forming the lower part of the hub. In contrast, Seh1 moves between two configurations: a distal position, away from Sec13 (Fig. 6D, bottom row), and a proximal one close to Sec13 (Fig. 6D, top row). Both configurations are supported by the input data, and they may identify alternative configurations of the Nup85–Seh1 arm. The significant flexibility shown by EM data for the Nup85–Seh1 arm (discussed above) is consistent with the different configurations suggested by our clustering analysis (Figs. 6C and 6D). The proximal localization of Seh1 (Fig. 6D, top row) likely implies an interaction with Sec13 that was already indicated by our previous structure (14). The fact that Sec13 and Seh1 are not required for the formation of the complex (14) suggests that the interaction between them is weak, allowing the conformational changes observed for the Nup85–Seh1 arm. Such transient β-propeller arrangements are common in the vertices of evolutionarily related VCCs, such as COPII and COPI, and have been suggested to be important for the assembly of the coats (54, 78). None of our experimental evidence points to a vertex-like arrangement between Nup84 complex copies, but one interesting possibility is that—in the context of the whole NPC—the Seh1–Sec13 pair is arranged in close contact with other β-propellers in the NPC. There are four other Nups not in the Nup84 complex that contain β-propeller domains (Nup82, Nup159, Nup157, and Nup170). We have already suggested that all of them are in close proximity to the Nup84 complex, based on the coarse structure of the entire NPC (8).
The Nup84 Complex as a Membrane Coating Module
The protocoatomer hypothesis suggests that the NPC shares a common evolutionary origin with the VCCs clathrin, COPI, and COPII (32, 79, 80), as well as other membrane-associated assemblies such as the SEA (74, 81) and IFT (82) complexes. The Nup84 complex forms part of the core scaffold of the NPC, and in many ways this scaffold resembles that within the outer coats of VCCs (7, 8). However, although shared components, common protein domain arrangements (32), and structural features (31, 54, 73) clearly reveal this ancient relationship, evolution appears to have shaped the overall architecture of the Nup84 coat and VCC outer coats in distinct ways subsequent to their evolutionary divergence (83). Moreover, the present structure of the Nup84 complex reinforces our previous observations (76) that the NPC carries a mixture of the architectural motifs found among the other VCCs. In addition to previously described COPII-like features such as the Nup145c–Nup84 connection (31, 49) and the Nup145c–Sec13 and Nup85–Seh1 dimers (31, 54, 73), our structure also reveals a trimeric interaction between the helical C-terminal domains of Nup145C, Nup120, and Nup85 in the hub (Figs. 6C and 6D). This arrangement might have an ancient evolutionary relationship with the trimeric C-terminal interaction of the clathrin triskelion (84), and indeed the first description of the hub region of the Nup84 complex noted its triskelion-like shape (85). Also within this hub, we detected for the first time a heterodimeric interaction between the β-propeller proteins Sec13 and Seh1 (Figs. 6C and 6D). Similar β-propeller arrangements are key for both COPI and COPII coat assembly (83). This mixture of motifs, found scattered among the different VCCs, is consistent with our hypothesis that different architectures previously postulated to be discrete (31, 86) actually evolved together within a single ancestral protocoatomer before the divergence of VCCs and NPCs (76). Recent evidence further underscores this idea by providing a possible “missing link” between COPI and the adaptin complexes that can mediate VCC interaction with membrane-bound receptors (87).
The protocoatomer hypothesis also suggests that NPCs and VCCs arose through a series of gene duplications to generate architectural complexity from a simpler precursor set of only a few proteins. In the case of the eight-fold NPC, we found that each octagonal symmetry unit could be divided into two parallel columns, in which every Nup in a given column contains a similarly positioned homolog in the adjacent column (7, 8). This pattern can be seen in the Nup84 complex. Our structure indicates a similarity in the connections of the clathrin/COPI-like paralogs Nup133 and Nup120, through a region proximal to their C termini, with the C termini of their respective cognate COPII-like partners, Nup84 and Nup145c. Such similarities agree with our previous suggestion that the Nup84 complex is the result of an ancient duplication of a hub-like triskelion module followed by selective loss of proteins and domains from the Nup133-containing copy of the hub (14).
Putative Membrane Interacting Motifs Are Localized to the Periphery of the Nup84 Complex
Human Nup133 has been shown to contain an ArfGAP1 lipid packing sensor (ALPS) motif (residues 245–267) in its N-terminal β-propeller (45, 88, 89). An ALPS motif generally functions as a membrane curvature sensor and may help anchor the protein to the pore membrane (88). Previous analyses were not able to detect such motifs in fungal Nup133 homologs (88), but an accompanying paper by Kim et al. annotates the presence—and solves the atomic structure—of one putative ALPS motif in the β-propeller domain of ScNup133 (252–270) and annotates the presence of two ALPS motifs in the β-propeller domain of its paralog ScNup120 (135–152 and 197–216).2 We mapped the positions of these motifs in our Nup84 complex ensemble (Fig. 7). All the putative ALPS motifs were localized to peripheral positions of the Nup84 complex and showed similar orientations (although the conformational variability of the complex precluded a precise localization). The positions of these putative ALPS motifs are consistent with the Nup133 and Nup120 tips of the Nup84 complex contacting the nuclear envelope membrane at the interface with the NPC (8, 14), forming clusters of ALPS motifs that might enhance their membrane binding as a result of the head-to-tail arrangement of the different Nup84 complex copies in the NPC. In addition, the predicted ALPS location is also entirely consistent with the functional role suggested for Nup120 and Nup133 in our previous work as key hotspots for the stabilization of the NPC membrane curvature (14). We suggest that similar ALPS motif arrangements and mechanisms are conserved between yeast and vertebrates, as supported by recent cryo-EM tomography observations (35) of close proximity between the equivalent Nup120 and Nup133 tips of the human Nup107–160 (HsNup107–160) complex and the NPC membrane.
Fig. 7.
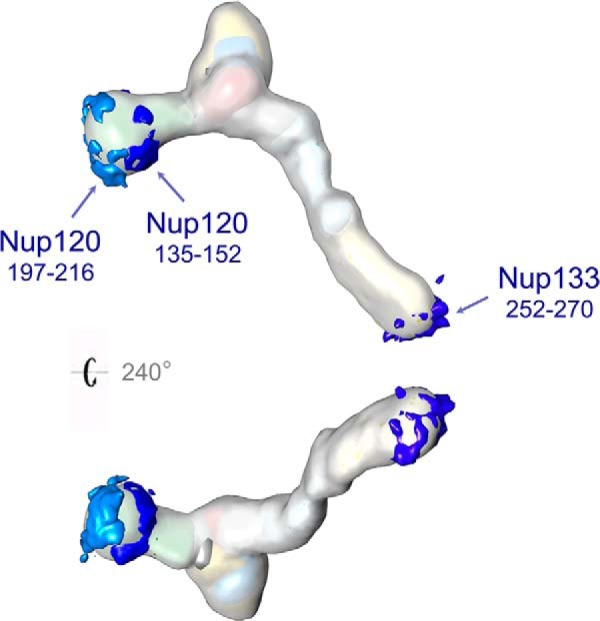
The putative membrane-interacting ALPS motifs. One putative ALPS motif in Nup133 (252–270) and two in Nup120 (135–152 and 197–216) are mapped on the Nup84 complex. All three putative ALPS motifs are localized to peripheral positions of the Nup84 complex.
Structural Basis for the Difference in Size and Architecture between Yeast and Human NPCs
Our groups have previously shown that the yeast Nup84 complex is present in 16 copies per NPC (8, 29), organized into two head-to-tail rings at the cytoplasmic and nuclear sides of the NPC (8, 14). In a recent study (35), cryo-EM tomography revealed that the human NPC contains 32 copies of the ScNup84 complex counterpart (HsNup107–160 complex) arranged head-to-tail into two concentric rings on each side of the NPC. The resulting model for the architecture of the HsNup107–160 complex suggests a conserved arrangement for the seven components that are common to the Nup84 complex and localizes the two additional β-propeller proteins specific to the HsNup107–160 complex, HsNup43 and HsNup37, at the Nup85–Seh1 and Nup120 arms, respectively. The cryo-EM map indicates that the main contacts between the concentric rings of HsNup107–160 complexes are established through the hub and arms of the complex, potentially involving both HsNup43 and HsNup37. We speculate that the absence of these two β-propeller proteins in the ScNup84 complex would not allow an oligomerization similar to the one described in the human NPC and goes a long way toward explaining the difference in the Nup84 complex copy number and in the overall size and mass between the yeast and human NPCs (8, 29, 75, 90). This scenario is also compatible with our previous hypothesis suggesting that the Nup84 complex evolved through a series of duplication, divergence, and secondary loss events (14). We suggest that a wider picture of the NPC composition and arrangement coming from distantly related organisms is the best way to trace the evolution of this molecular machine and the origin of the eukaryotic nuclei, as well as reveal its varied functional roles in NPCs between different organisms.
Supplementary Material
Acknowledgments
We thank Paula Upla and Ruben Diaz-Avalos at NYSBC for their help with the analysis of the EM data. We are also grateful to Daniel Russel, Charles Greenberg, and Benjamin Webb at UCSF for their help with and support of the PMI library and the Integrative Modeling Platform (IMP) package. We are grateful to Andrew N. Krutchinsky and Julio C. Padovan at Rockefeller University and to Shengbo Fan at the Institute of Computational Technology, CAS, for their suggestions and comments.
Footnotes
Author contributions: Y.S., J.F., E.T., R.P., S.J.K., A.S., M.P.R., and B.T.C. designed research; Y.S., J.F., E.T., R.P., S.J.K., and R.W. performed research; Y.S., J.F., E.T., R.P., S.J.K., D.S., A.S., M.P.R., and B.T.C. contributed new reagents or analytic tools; Y.S., J.F., E.T., R.P., S.J.K., A.S., M.P.R., and B.T.C. analyzed data; Y.S., J.F., E.T., R.P., S.J.K., A.S., M.P.R., and B.T.C. wrote the paper.
* Funding for this work was provided by National Institutes of Health Grant Nos. P41 GM103314 (B.T.C.), R01 GM062427 (M.P.R.), R01 GM083960 (A.S.), U54 GM103511 (A.S., M.P.R., and B.T.C.), and U01 GM098256 (A.S. and M.P.R.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- EM
- electron microscopy
- CX-MS
- chemical cross-linking with mass spectrometric readout
- LC-MS
- liquid chromatography–mass spectrometry
- DSS
- disuccinimidyl suberate
- EDC
- 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- NPC
- nuclear pore complex
- Nup
- nucleoporin
- VCC
- vesicle coating complex
- HCD
- higher energy collisional dissociation
- dRMSD
- distance root-mean-square deviation
- ALPS
- ArfGAP1 lipid packing sensor.
REFERENCES
- 1. Gavin A. C., Aloy P., Grandi P., Krause R., Boesche M., Marzioch M., Rau C., Jensen L. J., Bastuck S., Dumpelfeld B., Edelmann A., Heurtier M. A., Hoffman V., Hoefert C., Klein K., Hudak M., Michon A. M., Schelder M., Schirle M., Remor M., Rudi T., Hooper S., Bauer A., Bouwmeester T., Casari G., Drewes G., Neubauer G., Rick J. M., Kuster B., Bork P., Russell R. B., Superti-Furga G. (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636 [DOI] [PubMed] [Google Scholar]
- 2. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrin-Alvarez J. M., Shales M., Zhang X., Davey M., Robinson M. D., Paccanaro A., Bray J. E., Sheung A., Beattie B., Richards D. P., Canadien V., Lalev A., Mena F., Wong P., Starostine A., Canete M. M., Vlasblom J., Wu S., Orsi C., Collins S. R., Chandran S., Haw R., Rilstone J. J., Gandi K., Thompson N. J., Musso G., St Onge P., Ghanny S., Lam M. H., Butland G., Altaf-Ul A. M., Kanaya S., Shilatifard A., O'Shea E., Weissman J. S., Ingles C. J., Hughes T. R., Parkinson J., Gerstein M., Wodak S. J., Emili A., Greenblatt J. F. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 3. Malovannaya A., Lanz R. B., Jung S. Y., Bulynko Y., Le N. T., Chan D. W., Ding C., Shi Y., Yucer N., Krenciute G., Kim B. J., Li C., Chen R., Li W., Wang Y., O'Malley B. W., Qin J. (2011) Analysis of the human endogenous coregulator complexome. Cell 145, 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robinson C. V., Sali A., Baumeister W. (2007) The molecular sociology of the cell. Nature 450, 973–982 [DOI] [PubMed] [Google Scholar]
- 5. Alber F., Forster F., Korkin D., Topf M., Sali A. (2008) Integrating diverse data for structure determination of macromolecular assemblies. Annu. Rev. Biochem. 77, 443–477 [DOI] [PubMed] [Google Scholar]
- 6. Ward A. B., Sali A., Wilson I. A. (2013) Biochemistry. Integrative structural biology. Science 339, 913–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Rout M. P., Sali A. (2007) Determining the architectures of macromolecular assemblies. Nature 450, 683–694 [DOI] [PubMed] [Google Scholar]
- 8. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Sali A., Rout M. P. (2007) The molecular architecture of the nuclear pore complex. Nature 450, 695–701 [DOI] [PubMed] [Google Scholar]
- 9. Duan Z., Andronescu M., Schutz K., McIlwain S., Kim Y. J., Lee C., Shendure J., Fields S., Blau C. A., Noble W. S. (2010) A three-dimensional model of the yeast genome. Nature 465, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boura E., Rozycki B., Herrick D. Z., Chung H. S., Vecer J., Eaton W. A., Cafiso D. S., Hummer G., Hurley J. H. (2011) Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 9437–9442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalhor R., Tjong H., Jayathilaka N., Alber F., Chen L. (2012) Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lander G. C., Estrin E., Matyskiela M. E., Bashore C., Nogales E., Martin A. (2012) Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lasker K., Forster F., Bohn S., Walzthoeni T., Villa E., Unverdorben P., Beck F., Aebersold R., Sali A., Baumeister W. (2012) Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fernandez-Martinez J., Phillips J., Sekedat M. D., Diaz-Avalos R., Velazquez-Muriel J., Franke J. D., Williams R., Stokes D. L., Chait B. T., Sali A., Rout M. P. (2012) Structure-function mapping of a heptameric module in the nuclear pore complex. J. Cell Biol. 196, 419–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tosi A., Haas C., Herzog F., Gilmozzi A., Berninghausen O., Ungewickell C., Gerhold C. B., Lakomek K., Aebersold R., Beckmann R., Hopfner K. P. (2013) Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell 154, 1207–1219 [DOI] [PubMed] [Google Scholar]
- 16. Greber B. J., Boehringer D., Leitner A., Bieri P., Voigts-Hoffmann F., Erzberger J. P., Leibundgut M., Aebersold R., Ban N. (2014) Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505, 515–519 [DOI] [PubMed] [Google Scholar]
- 17. Cohen S. L., Chait B. T. (2001) Mass spectrometry as a tool for protein crystallography. Annu. Rev. Biophys. Biomol. Struct. 30, 67–85 [DOI] [PubMed] [Google Scholar]
- 18. Young M. M., Tang N., Hempel J. C., Oshiro C. M., Taylor E. W., Kuntz I. D., Gibson B. W., Dollinger G. (2000) High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 97, 5802–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Z. A., Jawhari A., Fischer L., Buchen C., Tahir S., Kamenski T., Rasmussen M., Lariviere L., Bukowski-Wills J. C., Nilges M., Cramer P., Rappsilber J. (2010) Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 29, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F. U., Ban N., Malmstrom L., Aebersold R. (2012) Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352 [DOI] [PubMed] [Google Scholar]
- 21. Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., Aebersold R. (2010) Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol. Cell. Proteomics 9, 1634–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinz A. (2006) Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom. Rev. 25, 663–682 [DOI] [PubMed] [Google Scholar]
- 23. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A. L., Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kao A., Chiu C. L., Vellucci D., Yang Y., Patel V. R., Guan S., Randall A., Baldi P., Rychnovsky S. D., Huang L. (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics 10, M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weisbrod C. R., Chavez J. D., Eng J. K., Yang L., Zheng C., Bruce J. E. (2013) In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J. Proteome Res. 12, 1569–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 27. Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., Herzog F., Rinner O., Ellenberg J., Aebersold R. (2011) The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leitner A., Joachimiak L. A., Unverdorben P., Walzthoeni T., Frydman J., Forster F., Aebersold R. (2014) Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proc. Natl. Acad. Sci. U.S.A. 111, 9455–9460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rout A., Aitchison J. D., Suprapto A., Hjertaas K., Zhao Y., Chait B. T. (2000) The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 635–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lutzmann M., Kunze R., Buerer A., Aebi U., Hurt E. (2002) Modular self-assembly of a Y-shaped multiprotein complex from seven nucleoporins. EMBO J. 21, 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brohawn S. G., Leksa N. C., Spear E. D., Rajashankar K. R., Schwartz T. U. (2008) Structural evidence for common ancestry of the nuclear pore complex and vesicle coats. Science 322, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Devos D., Dokudovskaya S., Alber F., Williams R., Chait B. T., Sali A., Rout M. P. (2004) Components of coated vesicles and nuclear pore complexes share a common molecular architecture. PLoS Biol. 2, e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bilokapic S., Schwartz T. U. (2012) 3D ultrastructure of the nuclear pore complex. Curr. Opin. Cell Biol. 24, 86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoelz A., Debler E. W., Blobel G. (2011) The structure of the nuclear pore complex. Annu. Rev. Biochem. 80, 613–643 [DOI] [PubMed] [Google Scholar]
- 35. Bui K. H., von Appen A., DiGuilio A. L., Ori A., Sparks L., Mackmull M. T., Bock T., Hagen W., Andres-Pons A., Glavy J. S., Beck M. (2013) Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 36. Kampmann M., Blobel G. (2009) Three-dimensional structure and flexibility of a membrane-coating module of the nuclear pore complex. Nat. Struct. Mol. Biol. 16, 782–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thierbach K., von Appen A., Thoms M., Beck M., Flemming D., Hurt E. (2013) Protein interfaces of the conserved Nup84 complex from Chaetomium thermophilum shown by crosslinking mass spectrometry and electron microscopy. Structure 21, 1672–1682 [DOI] [PubMed] [Google Scholar]
- 38. Velazquez-Muriel J., Lasker K., Russel D., Phillips J., Webb B. M., Schneidman-Duhovny D., Sali A. (2012) Assembly of macromolecular complexes by satisfaction of spatial restraints from electron microscopy images. Proc. Natl. Acad. Sci. U.S.A. 109, 18821–18826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsen J. V., Macek B., Lange O., Makarov A., Horning S., Mann M. (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 4, 709–712 [DOI] [PubMed] [Google Scholar]
- 40. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., Dong M. Q. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 41. Qin J., Chait B. T. (1995) Preferential fragmentation of protonated gas-phase peptide ions adjacent to acidic amino-acid-residues. J. Am. Chem. Soc. 117, 5411–5412 [Google Scholar]
- 42. Michalski A., Neuhauser N., Cox J., Mann M. (2012) A systematic investigation into the nature of tryptic HCD spectra. J. Proteome Res. 11, 5479–5491 [DOI] [PubMed] [Google Scholar]
- 43. Lasker K., Phillips J. L., Russel D., Velazquez-Muriel J., Schneidman-Duhovny D., Tjioe E., Webb B., Schlessinger A., Sali A. (2010) Integrative structure modeling of macromolecular assemblies from proteomics data. Mol. Cell. Proteomics 9, 1689–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Russel D., Lasker K., Webb B., Velazquez-Muriel J., Tjioe E., Schneidman-Duhovny D., Peterson B., Sali A. (2012) Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 10, e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berke I. C., Boehmer T., Blobel G., Schwartz T. U. (2004) Structural and functional analysis of Nup133 domains reveals modular building blocks of the nuclear pore complex. J. Cell Biol. 167, 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whittle J. R. R., Schwartz T. U. (2009) Architectural nucleoporins Nup157/170 and Nup133 are structurally related and descend from a second ancestral element. J. Biol. Chem. 284, 28442–28452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boehmer T., Jeudy S., Berke I. C., Schwartz T. U. (2008) Structural and functional studies of Nup107/Nup133 interaction and its implications for the architecture of the nuclear pore complex. Mol. Cell 30, 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sampathkumar P., Gheyi T., Miller S. A., Bain K. T., Dickey M., Bonanno J. B., Kim S. J., Phillips J., Pieper U., Fernandez-Martinez J., Franke J. D., Martel A., Tsuruta H., Atwell S., Thompson D. A., Emtage J. S., Wasserman S. R., Rout M. P., Sali A., Sauder J. M., Burley S. K. (2011) Structure of the C-terminal domain of Saccharomyces cerevisiae Nup133, a component of the nuclear pore complex. Proteins 79, 1672–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brohawn S. G., Schwartz T. U. (2009) Molecular architecture of the Nup84-Nup145C-Sec13 edge element in the nuclear pore complex lattice. Nat. Struct. Mol. Biol. 16, 1173–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nagy V., Hsia K. C., Debler E. W., Kampmann M., Davenport A. M., Blobel G., Hoelz A. (2009) Structure of a trimeric nucleoporin complex reveals alternate oligomerization states. Proc. Natl. Acad. Sci. U.S.A. 106, 17693–17698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seo H. S., Ma Y., Debler E. W., Wacker D., Kutik S., Blobel G., Hoelz A. (2009) Structural and functional analysis of Nup120 suggests ring formation of the Nup84 complex. Proc. Natl. Acad. Sci. U.S.A. 106, 14281–14286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leksa N. C., Brohawn S. G., Schwartz T. U. (2009) The structure of the scaffold nucleoporin Nup120 reveals a new and unexpected domain architecture. Structure 17, 1082–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Debler E. W., Ma Y., Seo H. S., Hsia K. C., Noriega T. R., Blobel G., Hoelz A. (2008) A fence-like coat for the nuclear pore membrane. Mol. Cell 32, 815–826 [DOI] [PubMed] [Google Scholar]
- 54. Fath S., Mancias J. D., Bi X., Goldberg J. (2007) Structure and organization of coat proteins in the COPII cage. Cell 129, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 55. Jeudy S., Schwartz T. U. (2007) Crystal structure of nucleoporin Nic96 reveals a novel, intricate helical domain architecture. J. Biol. Chem. 282, 34904–34912 [DOI] [PubMed] [Google Scholar]
- 56. Lee J. H., Yi L., Li J., Schweitzer K., Borgmann M., Naumann M., Wu H. (2013) Crystal structure and versatile functional roles of the COP9 signalosome subunit 1. Proc. Natl. Acad. Sci. U.S.A. 110, 11845–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Soding J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960 [DOI] [PubMed] [Google Scholar]
- 58. Soding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 60. Buchan D. W., Minneci F., Nugent T. C., Bryson K., Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ward J. J., McGuffin L. J., Bryson K., Buxton B. F., Jones D. T. (2004) The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 [DOI] [PubMed] [Google Scholar]
- 62. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 63. Erzberger J., Stengel F., Pellarin R., Zhang S., Schaefer T., Aylett C., Cimermancic P., Boehringer D., Sali A., Aebersold R., Ban N. (2014) Molecular architecture of the 40S-eIF1-eIF3 Translation Initiation Complex. Cell 158 (5), 1123–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shen M. Y., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schneidman-Duhovny D., Rossi A., Avila-Sakar A., Kim S. J., Velazquez-Muriel J., Strop P., Liang H., Krukenberg K. A., Liao M., Kim H. M., Sobhanifar S., Dotsch V., Rajpal A., Pons J., Agard D. A., Cheng Y., Sali A. (2012) A method for integrative structure determination of protein-protein complexes. Bioinformatics 28, 3282–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rieping W., Habeck M., Nilges M. (2005) Inferential structure determination. Science 309, 303–306 [DOI] [PubMed] [Google Scholar]
- 67. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38, 27–38 [DOI] [PubMed] [Google Scholar]
- 68. Heyer L. J., Kruglyak S., Yooseph S. (1999) Exploring expression data: identification and analysis of coexpressed genes. Genome Res. 9, 1106–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Levitt M. (1983) Molecular dynamics of native protein. II. Analysis and nature of motion. J. Mol. Biol. 168, 621–657 [DOI] [PubMed] [Google Scholar]
- 70. Oeffinger M., Wei K. E., Rogers R., DeGrasse J. A., Chait B. T., Aitchison J. D., Rout M. P. (2007) Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Methods 4, 951–956 [DOI] [PubMed] [Google Scholar]
- 71. Leitner A., Reischl R., Walzthoeni T., Herzog F., Bohn S., Forster F., Aebersold R. (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteomics 11, M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Merkley E. D., Rysavy S., Kahraman A., Hafen R. P., Daggett V., Adkins J. N. (2014) Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 23, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hsia K. C., Stavropoulos P., Blobel G., Hoelz A. (2007) Architecture of a coat for the nuclear pore membrane. Cell 131, 1313–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Algret R., Fernandez-Martinez J., Shi Y., Kim S. J., Pellarin R., Cimermancic P., Cochet E., Sali A., Chait B. T., Rout M. P., Dokudovskaya S. (2014) Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway. Mol. Cell. Proteomics, mcp.M114.039388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yang Q., Rout M. P., Akey C. W. (1998) Three-dimensional architecture of the isolated yeast nuclear pore complex: functional and evolutionary implications. Mol. Cell 1, 223–234 [DOI] [PubMed] [Google Scholar]
- 76. Sampathkumar P., Kim S. J., Upla P., Rice W. J., Phillips J., Timney B. L., Pieper U., Bonanno J. B., Fernandez-Martinez J., Hakhverdyan Z., Ketaren N. E., Matsui T., Weiss T. M., Stokes D. L., Sauder J. M., Burley S. K., Sali A., Rout M. P., Almo S. C. (2013) Structure, dynamics, evolution, and function of a major scaffold component in the nuclear pore complex. Structure 21, 560–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brohawn S. G., Partridge J. R., Whittle J. R., Schwartz T. U. (2009) The nuclear pore complex has entered the atomic age. Structure 17, 1156–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee C., Goldberg J. (2010) Structure of coatomer cage proteins and the relationship among COPI, COPII, and clathrin vesicle coats. Cell 142, 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Devos D., Dokudovskaya S., Williams R., Alber F., Eswar N., Chait B. T., Rout M. P., Sali A. (2006) Simple fold composition and modular architecture of the nuclear pore complex. Proc. Natl. Acad. Sci. U.S.A. 103, 2172–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Field M. C., Sali A., Rout M. P. (2011) Evolution: on a bender—BARs, ESCRTs, COPs, and finally getting your coat. J. Cell Biol. 193, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dokudovskaya S., Waharte F., Schlessinger A., Pieper U., Devos D. P., Cristea I. M., Williams R., Salamero J., Chait B. T., Sali A., Field M. C., Rout M. P., Dargemont C. (2011) A conserved coatomer-related complex containing Sec13 and Seh1 dynamically associates with the vacuole in Saccharomyces cerevisiae. Mol. Cell. Proteomics 10, M110.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. van Dam T. J., Townsend M. J., Turk M., Schlessinger A., Sali A., Field M. C., Huynen M. A. (2013) Evolution of modular intraflagellar transport from a coatomer-like progenitor. Proc. Natl. Acad. Sci. U.S.A. 110, 6943–6948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Faini M., Beck R., Wieland F. T., Briggs J. A. (2013) Vesicle coats: structure, function, and general principles of assembly. Trends Cell Biol. 23, 279–288 [DOI] [PubMed] [Google Scholar]
- 84. Fotin A., Cheng Y., Sliz P., Grigorieff N., Harrison S. C., Kirchhausen T., Walz T. (2004) Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature 432, 573–579 [DOI] [PubMed] [Google Scholar]
- 85. Siniossoglou S., Lutzmann M., Santos-Rosa H., Leonard K., Mueller S., Aebi U., Hurt E. (2000) Structure and assembly of the Nup84p complex. J. Cell Biol. 149, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Leksa N. C., Schwartz T. U. (2010) Membrane-coating lattice scaffolds in the nuclear pore and vesicle coats: commonalities, differences, challenges. Nucleus 1, 314–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hirst J., Schlacht A., Norcott J. P., Traynor D., Bloomfield G., Antrobus R., Kay R. R., Dacks J. B., Robinson M. S. (2014) Characterization of TSET, an ancient and widespread membrane trafficking complex. eLife 3, e02866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Drin G., Casella J. F., Gautier R., Boehmer T., Schwartz T. U., Antonny B. (2007) A general amphipathic alpha-helical motif for sensing membrane curvature. Nat. Struct. Mol. Biol. 14, 138–146 [DOI] [PubMed] [Google Scholar]
- 89. Drin G., Antonny B. (2010) Amphipathic helices and membrane curvature. FEBS Lett. 584, 1840–1847 [DOI] [PubMed] [Google Scholar]
- 90. Akey C. W., Radermacher M. (1993) Architecture of the Xenopus nuclear pore complex revealed by three-dimensional cryo-electron microscopy. J. Cell Biol. 122, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kim S. J., Fernandez-Martinez J., Sampathkumar P., Martel A., Matsui T., Tsuruta H., Weiss T., Shi Y., Markina-Inarrairaegui A., Bonanno J. B., Sauder J. M., Burley S. K., Chait B. T., Almo S. C., Rout M. P., Sali A. (2014) Integrative structure-function mapping of the nucleoporin Nup133 suggests a conserved mechanism for membrane anchoring of the nuclear pore complex. Mol. Cell. Proteomics, mcp.M114.040915 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.