

Abstract
We have shown that recombinant forms of VP8* domains of the human rotavirus outer capsid spike protein VP4 from human neonatal strains (N155(G10P[11]) and RV3(G3P[6]) and a bovine strain (B223) recognize unique glycans within the repertoire of human milk glycans. The accompanying study by Yu et al.2, describes a human milk glycan shotgun glycan microarray that led to the identification of 32 specific glycans in the human milk tagged glycan library that were recognized by these human rotaviruses. These microarray analyses also provided a variety of metadata about the recognized glycan structures compiled from anti-glycan antibody and lectin binding before and after specific glycosidase digestions, along with compositional information from mass analysis by matrix-assisted laser desorption ionization-mass spectrometry. To deduce glycan sequence and utilize information predicted by analyses of metadata from each glycan, 28 of the glycan targets were retrieved from the tagged glycan library for detailed sequencing using sequential disassembly of glycans by ion-trap mass spectrometry. Our aim is to obtain a deeper structural understanding of these key glycans using an orthogonal approach for structural confirmation in a single ion trap mass spectrometer. This sequential ion disassembly strategy details the complexities of linkage and branching in multiple compositions, several of which contained isomeric mixtures including several novel structures. The application of this approach exploits both library matching with standard materials and de novo approaches. This combination together with the metadata generated from lectin and antibody-binding data before and after glycosidase digestions provide a heretofore-unavailable level of analytical detail to glycan structure analysis. The results of these studies showed that, among the 28 glycan targets analyzed, 27 unique structures were identified, and 23 of the human milk glycans recognized by human rotaviruses represent novel structures not previously described as glycans in human milk. The functional glycomics analysis of human milk glycans provides significant insight into the repertoire of glycans comprising the human milk metaglycome.
Keywords: Human milk glycans, mass spectrometry, MSn, permethylation
Human milk contains an assortment of hundreds of soluble reducing glycans with many attributed and important functions (1–5). The human milk glycans (HMGs)1 are derived from lactose as the core component, and this is elongated through additions of N-acetylglucosamine, galactose, fucose, and sialic acid to generate the milk glycan repertoire. Although over 100 HMGs have been identified, our recent isolation of pooled HMGs, as described in the accompanying paper by Yu et al.2, led to the preparation of a human milk tagged glycan library (TGL) of 247 glycans, which had been derivatized with a fluorescent linker, 2-amino-N-(2-amino-ethyl)-benzamide (AEAB), and subsequently, covalently printed as a shotgun glycan microarray (SGM). The functional importance of these glycans was assessed by interrogating the SGM with recombinant forms of VP8* domains of the human rotavirus outer capsid spike protein VP4 from two human neonatal strains (N155(G10P[11]) and RV3(G3P[6])) and a bovine strain (B223), which led to the identification of 32 HMG targets recognized by different viruses. While much information about glycan structures recognized by different rotaviruses were predicted using a metadata-assisted glycan sequencing approach (3, 6), further definitions of glycan structure and sequence are required to confirm these predictions and provide a deeper understanding of the glycan determinants required for the unique rotavirus recognition.
Attempts to understand the diversity of such glycan structures brings multiple challenges. These include the stereo- and structural isomers of monomers which are compounded with additional structural isomers found in linkage and branching arrays of oligomers. However, these structural details can be resolved by the ion trap MS because of different monomer bonding energies (ketosidic, glycosidic, aminoglycosidic) that provides stepwise disassembly to smaller oligomer sets upon collisional activation (CA). Importantly, even with identical energies, CA on smaller sets, (fewer oscillators for energy dissipation), induces rupture of the more stable bonds exposing greater structural detail. The energies used in an ion trap are relatively low and slow, conditions that allow time for electron rearrangement and eliminations that expose reproducible details of former structure, a feature that is not available with higher CA energies. Multiple approaches to characterize and catalogue HMGs have been attempted (7, 8), and excellent reviews of compiled HMG structures are available (7, 9–11).
With regard to the chemical structure of glycans, it must be considered that monosaccharides (monomers) extending from opposing ends of an oligomer chain are significantly different (hemiacetal and hydroxyl linkages, respectively). Upon collision-induced dissociation (CID), two fragments (B- and A-ions) are the major sequencing fragments. The B-ions (or Y-ions, depending on charge localization) can only originate from a single bond rupture to the C1-carbon, providing monomer mass intervals, but such fragments fail to define linking connectivity. It is together with nonreducing-end A-ions that defines sequence. This critical feature of CID instability of O-linkage to the pyran/furan ring focuses the carbon position, and their different compositions that define point connectivity, (2-, 3-, 4-, or 6-carbon). The absence of these nonreducing end fragments (cross-ring) within a spectrum of higher molecular weight ions has been considered resolvable by imparting higher collision energies to precursor ions, (electron-transfer dissociation, electron-detachment dissociation, infrared multiphoton dissociation, photoionization), but these higher external energies are established in microseconds and fail to allow the time for electron rearrangement and eliminations that define A-ions. To approach such detail, we describe here a stepwise disassembly to obtain low molecular weight ion fragments where structural detail is exposed. Such fragment spectra are matched and compiled in a library that has been documented with synthetic standards (12).
We applied this approach here to selected HMGs that are shown in the accompanying report by Yu et al.2 to be recognized by different human rotaviruses. In conjunction with the previously established metadata-assisted glycan sequencing (MAGS) approach (3, 6), this represents a complementary, comprehensive approach to glycan structure determination. In this report, we describe the detailed analysis of 28 glycan targets from the 32 selected by screening on the HMG-SGM, and we identified 27 unique glycan structures, including 23 that had not been previously identified as free glycans in human milk.
EXPERIMENTAL PROCEDURES
Materials
The samples analyzed were glycan targets from a TGL of HMG obtained as described in the companion report (Yu et al.2) and selected for analysis based on their binding by one of three different recombinant VP8* domains from three strains of rotavirus as determined on an SGM. Acetic anhydride was from Fluka (St. Louis, MO). Sodium hydroxide (small beads), dimethyl sulfoxide, and trifluoroacetic acid were from Sigma (St. Louis, MO). Acetonitrile and dichloromethane were from EMD (Darmstadt, Germany). Sodium chloride was from Fisher (Fair Lawn, NJ). Graphitized carbon solid phase extraction cartridges were from Agilent (Santa Clara, CA). Sodium bicarbonate was from Mallinckrodt (St. Louis, MO). Spin columns were from Harvard Apparatus (Holliston, MA).
Preparation of AEAB-derivatized HMGs
The glycan targets selected for analysis had been labeled by reductive amination with AEAB as described (6, 13). Purified AEAB-derivatized HMGs retrieved from the TGL were acetylated using acetic anhydride in saturated sodium bicarbonate overnight (14). HMGs were then desalted via porous graphitized carbon solid phase extraction. Any esters formed during acetylation were hydrolyzed by incubation in 0.01 m sodium hydroxide at room temperature for approximately 4 h, followed by vacuum concentration. The samples were then permethylated using spin columns and purified by liquid-liquid extraction with dichloromethane and 0.5 m aqueous sodium chloride (15). The dried, permethylated samples were dissolved in 1:1 methanol/water for direct infusion.
Electrospray-ion trap MSn
The dissolved samples were placed onto a Triversa Nanomate (Advion, Ithaca, NY) mounted to an LTQ (ThermoFisher, San Jose, CA). Spray parameters on the Nanomate were generally set to 1.6–1.8 kV and 0.3 psi nitrogen gas pressure and were adjusted to achieve spray currents between 10–150 nA. Activation Q and activation time for CID were left at default values, 0.250 and 30 ms, respectively. Normalized collision energy was generally set to 35%. Precursor ion selection for MSn experiments was performed manually. Precursor m/z and isolation windows were generally set to capture an entire isotopic envelope. Signal averaging was performed to achieve suitable spectral quality. Typically, one isolation scan, with collision energy set to 0, was acquired in each CID data file to document the isolated isotopic envelope.
RESULTS
A total of 32 glycan targets were selected from the human milk TGL for further analysis, based on their identification as being recognized by recombinant forms of VP8* domains of the human rotavirus outer capsid spike protein VP4 from two human neonatal strains, N155(G10P[11]) and RV3(G3P[6]) and a bovine strain (B223) in the accompanying report by Yu et al.2 We focused on the structural analyses of these identified glycans because studies using the MAGS approach (3, 6) did not provide a complete definition of the selected glycan or indicated that the selected glycans had structures that were novel; i.e. not previously identified in the human milk metaglycome. To this end, AEAB-labeled glycans were acetylated and permethylated and were disassembled in the ion trap to determine structural detail. Specific targets were localization of fucose residues, discernment of Lewis and H antigen structures, and lactosamine (LacNAc) linkages. Overall branching and topology were also determined via MSn. In general, acetylation and permethylation of AEAB-derivatized HMGs produced three products, likely owing to the differing nucleophilicity of primary and secondary amines and amides. These products differed in the number of acetyl and/or methyl groups on the acetylated and permethylated derivatives as shown in Fig. 1. This variability was confined to the AEAB moiety (obvious upon disassembly) and was therefore not an impediment to data acquisition or interpretation. In general, the product with two acetyl groups and two methyl groups on the AEAB derivative, structure 1, which was the heaviest product, was chosen for MSn disassembly.
Fig. 1.
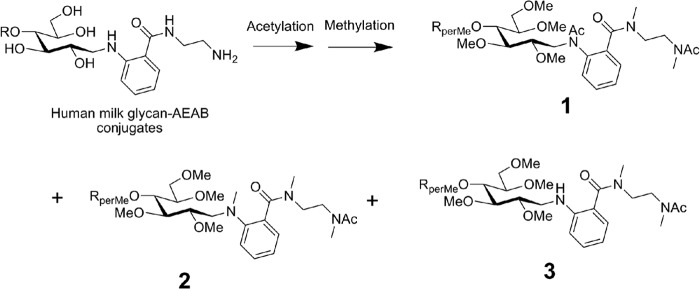
The acetylation and permethylation of AEAB-derivatized HMGs. The difficulties of analyzing permethylated glycan derivatives with accessible amino functional groups by MS can be overcome by first acetylating the primary and secondary amines which in the case of AEAB derivatives generates three major permethylated products, 1, 2, and 3.
The strategy for peak selection for disassembly was based on identifying fucosylated lactosamine trisaccharides (m/z 660 and 646), lactosamine units (m/z 486 and 472), and difucosylated lactosamine (m/z 834), if present. Combinations of these, forming larger diLacNAc, triLacNAc, and fucosylated versions of these, were also interrogated, if present. The concepts of permethylation and MSn are well documented (16–19), as is the ability to identify isomeric mixtures (14, 20–22). Data interpretation involved a combination of de novo and library-matching approaches. The evaluation of oligosaccharide standards, reproducibility, and comparison with biological samples is well appreciated (12). Where indicated, CID spectra of HMG fragment spectra were compared with those of standards for identification. In our experience, the visual inspection of spectra is often superior to blind computational spectral matching, but we also analyzed the spectra via an in-house algorithm to generate similarity scores.
Comprehensive MSn data sets and peak lists are shown in the supplemental material where each glycan target is identified by its HMG-number (see companion manuscript, Yu et al.2) that corresponds to the spectra (including spectral similarity scores, where applicable) and the peak lists for each spectrum. The glycan structures that were analyzed to identify the glycan-binding motif of each of the three rotavirus VP8* domains are shown in Figs. 2–4. The process of identifying the motif is complicated by the fact that a specific, high-affinity interaction might be elicited on the array by a minor component in the target glycan if it is a mixture. We therefore selected a relatively large number of glycans for analysis of the “binding glycans” or potential decoy receptors; and when the glycan preparation from the TGL contained two or more isomeric glycans, it was necessary to identify all of the isomeric structures in the mixture in order to identify the common motif recognized by the VP8* domain being evaluated. For example, the major component was taken to be the ligand detected by protein binding.
Fig. 2.

Structures of glycans comprising the glycan targets selected to identify the milk glycan determinants bound by VP8*N155. The glycan targets were selected from a TGL of HMG based on microarray screening of the corresponding HMG-SGM with the recombinant VP8* domain of spike protein VP4 from human neonatal rotavirus strain N155(G10P[11]) as described in the companion report (Yu et al.2). The detailed structural determinations by MSn are described in the supplemental materials. Linkages assigned by MSn analyses are indicated in green and those assumed based on being from milk glycans are indicated in blue. Trivial names of previously described glycans are indicated based on recent reviews (7, 10, 24), and NPI indicates not previously identified as a free glycan in human milk.
Fig. 3.

Structures of glycans comprising the glycan targets selected to identify the milk glycan determinants bound by VP8*RV3. The glycan targets were selected from a TGL of HMG based on microarray screening of the corresponding HMG-SGM with the recombinant VP8* domain of spike protein VP4 from human neonatal rotavirus strain RV3(G3P[6]) as described in the companion report (Yu et al.2). The detailed structural determinations by MSn are described in the supplemental materials. Linkages assigned by MSn analyses are indicated in green and those assumed based on being from milk glycans are indicated in blue. Trivial names of previously described glycans are indicated based on recent reviews (7, 10, 24), and NPI indicates not previously identified as a free glycan in human milk.
Fig. 4.

Structures of glycans comprising the glycan targets selected to identify the milk glycan determinants bound by VP8*B223. The glycan targets were selected from a TGL of HMG based on microarray screening of the corresponding HMG-SGM with the recombinant VP8* domain of spike protein VP4 from bovine rotavirus strain B223 as described in the companion report (Yu et al.2). The detailed structural determinations by MSn are described in the supplemental materials. Linkages assigned by MSn analyses are indicated in green and those assumed based on being from milk glycans are indicated in blue. Trivial names of previously described glycans are indicated based on recent reviews (7, 10, 24), and NPI indicates not previously identified as a free glycan in human milk.
The glycan targets selected for analysis of the HMG bound by VP8*N155 were HMG-13, -14, -18, -21, -27, -28, and 33. We also did the detailed analysis of HMG-29, -34, -51, and -60, which were identified as ligands for VP8*N155 after they were digested with a specific α1,2-fucosidase as described (Yu et al.2). The analyses of these structures revealed that the glycan targets selected were pure glycans or mixtures of two or three isomers, where all isomers were defined. As shown in Fig. 2, HMG-13 and -27 were pure (single component) glycans with identical structures, as were HMG-14 and -18. Identical structures may occur in two targets due to overlaps that occur during the two-dimensional chromatography used for their isolation. HMG-21 and -28 were also comprised of identical structures, but each possessed a different ratio of two isomers. HMG-51 and HMG-60 were mixtures of three and two isomeric structures, respectively. Thus, inspection of the glycan structures that were determined in these analyses indicated that all of the single component targets possessed the common motif identified as GlcNAcβ1–3Galβ1–4GlcNAc, and two single-component fractions (HMG-29 and -34) were converted to a corresponding binding glycan after specific removal of a terminal α1–2fucose. Among the glycan targets that were mixtures, all structures contained the common determinant except the novel structure in HMG-51 where the galactose was substituted at position 6 with a GlcNAc (Fig. 2).
The glycan targets selected for analysis of the HMG bound by VP8*RV3 were HMG-37, -41, -49, -55, -56, -62, -69, and -76. As shown in Fig. 3, all of these glycans were primarily di- or trifucosylated derivatives of octaoses; with the exception of HMG-76 that was a difucosylated decaose. HMG-37, -41, -49, and -56 were pure (single component) glycans while the other targets were comprised of two or three isomeric structures. All of the single component targets possessed the common pentasaccharide determinant, Galβ1–3GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAc where the fucose is a component of an “internal Lex” determinant. Inspection of the structures of the mixed glycan targets showed that all of these mixtures contained one glycan that possessed the pentasaccharide with the internal Lex structure. In HMG-55 the difucosylated lacto-N-octaose II, which did not possess the common pentasaccharide, was a minor component. In the mixture designated HMG-62, the glycan possessing the common motif was previously identified as a milk glycan, 5230a (7) and was one of the two major components; while the other two components had not been previously identified as milk glycans. Three novel glycan structures were identified in HMG-69 and one possessed the common pentasaccharide determinant. The structure shown for HMG-76 is representative and possesses two branches containing the common determinant; however, the detailed analysis indicated that this target contained contaminating structures with one or two terminal Galβ1–4GlcNAc.
The glycan targets selected for analysis of the HMG bound by VP8*B223 were HMG-16, -20, -31, -45, -47, -54, -65, -67, and 66. As shown in Fig. 4, only HMG-54 and -31 were mixtures of two and possibly five isomeric structures, respectively, while the remaining targets were comprised of single glycans, all possessing the common tetrasaccharide determinant selected as the motif for VP8*B223 Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAc. The predominant glycan in HMG-31 that contained up to five different structures was fucosylated-para-lacto-N-neohexaose II, which possessed the common tetrasaccharide. The other mixed glycan target, HMG-54, was comprised of two previously unidentified fucosylated octaoses, and both possessed the common tetrasaccharide motif for VP8*B223.
The overall strategy employed to determine HMG structure was to adopt a modular approach, using spectral library matching where appropriate. Using this approach, we identified lactosamine (or polylactosamine) units with or without substitutions and used both de novo and library-matching strategies to assign structure. Disassembly strategies can vary to some degree, with respect to selected fragmentation pathways, and more than one approach may afford the same information. In general, the approach employed was to isolate fragments of interest to generate informative MSn spectra. In addition, it is sometimes useful to successively disassemble a structure in stages to eventually produce a small, known substructure. For HMGs, the lactose core structure provided important information about branching. For the AEAB-derivatized HMGs, if the heaviest (two acetyl, three methyl) derivative was selected, an m/z 724 core fragment was considered indicative of a doubly branched core; an m/z 738 fragment was considered indicative of a linear, or singly branched, core. The combined use of permethylation and resulting methyl group (14 mu) differences enabled this interpretation. The latter is important because it provides empirical evidence of branching and is indicative of the importance of permethylation for oligosaccharide branching assignments. For larger structures, the combination of core AEAB-lactose fragment mass and the combination of terminal and internal LacNAc fragment masses facilitate the branching and topology assignments. Isomers differing in linkage and/or branching frequently give distinctly different mass spectra during disassembly. Frequently, this gives empirical evidence of the presence of multiple isomers in a given sample. In the case of physically separated compounds, the interrogation of these structures shows how MSn can elucidate these structure details and differences.
While human milk glycans have several fundamental structural differences from N- or O-glycans, the basic principles of structural assignment by mass spectrometry are the same. Typical milk glycans have a lactose core, which can be elaborated and/or modified by lactosamine extensions, fucosylation, etc. Fig. 5 shows a partial MSn data set for HMG-13, including assigned structures with putative fragment ion assignments. The MS2 spectrum shown in Fig. 5A shows fragments related to lactosamine units and the AEAB-derivatized lactose core. While acetylation and permethylation produced multiple products due to variable reactivity of the AEAB derivative, we typically chose the products with the mass and structure shown here. A singly substituted AEAB-derivatized lactose core will produce a singly charged, m/z 738 ion. This ion can be isolated and fragmented to generate an MS3 spectrum, as shown in Fig. 5E. The neutral loss, 204, of a singly substituted, internal hexose, producing the m/z 534 ion, positions the single core substitution at the galactose residue, leaving the C-type AEAB-derivatized glucose residue, as shown in the structure graphic.
Fig. 5.

Partial MSn data set for HMG-13. Panel A shows the MS2 spectrum, including core (m/z 738) and antennal fragments (m/z 935 and m/z 486). Panels B, C, and D show the successive disassembly of the diLacNAc extension, including linkage information for the terminal and internal LacNAc units. Panel E shows the fragmentation of the AEAB-lactose core fragment. These data are clearly indicative of a linear, or singly branched, structure.
For this particular HMG, we also see a diLacNAc fragment at m/z 935. The CID fragmentation of this ion is shown in Fig. 5B, indicating several informative fragments. There are the obvious terminal (m/z 486) and internal (m/z 472) lactosamine units, as expected. Additionally, the fragment at m/z 699, formed by neutral loss of 236, is typically found when terminal hexose residues are three-linked. We can isolate both of the lactosamine units to generate MS4 spectra, as shown in Fig. 5C and 5D. The fragmentation of the terminal LacNAc shown in Fig. 5C is typical of a three-linked LacNAc. The fragmentation of the internal LacNAc shown in Fig. 5D is typical of a four-linkage. Furthermore, the intensity pattern of the internal LacNAc is indicative of the -OH group at the 3-position of the galactose, rather than the 6-position, thus the assignment of the three-linkage between the LacNAc units. This last assignment was based on spectral comparisons with standard materials and was supported by computationally assigned spectral-matching scores (supplemental material HMG-13) (23).
Milk glycans with two core substitutions will have obviously different fragmentation patterns, though easily assigned. Figure 6 shows a partial MSn data set for HMG-16, which has the same composition as HMG-13. In this example, the AEAB-lactose core fragment is the singly charged m/z 724 ion. This is 14 mu lower than the singly substituted m/z 738 mass, accounting for one less methyl group and indicating a branched galactose. The m/z 534 ion is apparent at the MS2 level, suggesting that both substituents are located on the galactose residue, leaving the AEAB-glucose identical to that of HMG-13 in Fig. 5. These core fragment masses (m/z 724 and m/z 534) are also present in the MS3 spectrum in Fig. 6B, which was formed by loss of a single terminal LacNAc. In addition to the doubly substituted lactose core fragment ions, we also see through this fragmentation pathway the successive losses of two terminal LacNAc units, as the neutral loss of 232 between the doubly charged precursors 837 → 605. This is distinct from the losses of terminal and internal LacNAc units in HMG-13. This is a key feature of permethylation, as the distinction of terminal from internal LacNAc would have similar masses in unmethylated structures. Isolation of the terminal LacNAc unit, at both the MS4 and MS3 level in Fig. 6C and 6D, respectively, are clearly 4-linked with no hint of any additional linkage isomers. It should be stated that these particular data do not empirically position the linkages at the galactose residues of the core lactose.
Fig. 6.

Partial MSn data set for HMG-16. Panel A shows the MS2 spectrum, containing fragments consistent with a doubly branched core lactose (m/z 724). Panels B and C show that this structure contains two terminal LacNAc residues, consistent with the presence of the doubly branched core. Panels C and D show that the LacNAcs seem to be four-linked, with no evidence of additional linkage isomers.
Aside from the obvious core structure differences of milk glycans, N-glycans, glycosphingolipids, and O-glycans, the principles of fragmentation of permethylated glycans via CID in an ion trap are similar. Fucose localization, branching/topology, linkages, and other epitope structure assignments are similar for all (23).
One of the fundamental structural features is the linkage of lactosamine units, particularly terminal LacNAcs. Permethylation and MSn provide empirical data for these linkages (19). Selected terminal LacNAc spectra isolated from different HMGs are shown in Fig. 5C and Fig. 6C. HMG-13 and HMG-16 have the same composition but differ in branching, as previously discussed. Note the prominent differences in these spectra. The 3,5A cross-ring cleavage product, m/z 329 (Fig. 6C) is characteristic of a four-linked hexose. In this case, the three-linked structure produces a putative O,4A cleavage produce, m/z 412 (Fig. 5C). The four-linked structure could theoretically produce this ion, but empirical data show that it is very prominent in three-linkages but negligible in four-linkages. The analogous spectrum for HMG-55 (supplemental material), as the terminal LacNAc unit from a fucosylated diLacNAc, is indicative of a mixture, as both m/z 329 and m/z 412 ions are prominent (supplemental material). In some cases, MSn fragmentation of isomeric mixtures can effectively separate unique isomeric fragments, providing isomerically pure spectra. This approach was taken for several other HMGs, data for which are compiled under the corresponding HMG number in the supplemental material. For example, HMG-51 is a mixture of three isomers, two of which contained terminal LacNAc units. At the MS3 stage, the resulting spectrum indicated a mixture of three- and four-linked LacNAc units. By selecting fragmentation pathways unique to each of the different branching isomers, it was possible to isolate MS4 and MS5 spectra from each of these two isomers alone, providing an apparently pure three-linked terminal LacNAc and an apparently pure four-linked terminal LacNAc (supplemental material HMG-51, spectra K and O, respectively).
Several of the HMGs had diLacNAc motifs, with or without fucosylation. Localizing the fucose residues in these structures is important for defining the epitopes or determinants recognized by rotaviruses. For permethylated/sodiated structures, a monofucosylated diLacNAc composition will generally appear as a singly charged B-type ion of m/z 1109. A selection of CID spectra of this ion isolated from several HMGs shows the varying localization of the single fucose residue (Fig. 7). The first determination is to define whether the fucose is on the terminal LacNAc or an internal LacNAc; this is a relatively straightforward problem with permethylated samples. In HMG-28 (Fig. 7A), the important fragments are m/z 472 and 660; the m/z 660 is consistent with a terminal Fuc-LacNAc and the m/z 472 is consistent with an internal LacNAc. Further probing of these ions at the MS4 level was necessary to yield more detailed information (supplemental material HMG-28C and D), namely the Lewis A/X mixture and the linkage of the internal LacNAc. Of notable importance, the m/z 873 fragment is likely contributed by the Lewis A structure, not the Lewis X, due to the typical preferential loss of three-linked hexose residues from such structures. HMG-29 contained the same m/z 1109 ion, but disassembly reveals a slightly different fragmentation from that of HMG-28. As shown in Fig. 7B, while the m/z 660 and 472 ions localize the fucose to the terminal LacNAc, the fragments at m/z 433 and 699 suggest an H-antigen structure. Further, MS4 interrogation confirmed an H1-epitope for HMG-29 (supplemental material HMG-29C). The same ion was identified in the sample HMG-41 (Fig. 7C and supplemental material HMG-41B). Note that in HMG-41, the fucose is localized to the internal LacNAc, as evidenced by the 14 mu shift of the relevant ions; the m/z 486 is the terminal LacNAc and the m/z 646 is the internal Fuc-LacNAc. For internal Fuc-LacNAc units, spectrum matching with standards has indicated that these structures match very well with the B/Y-type Fuc-LacNAc trisaccharide fragment isolated for sialylated Lewis X standards. This similarity essentially indicates that this unit is an internal Lewis X structure, and the m/z 646 fragmentation matches that standard trisaccharide spectrum (12). The isomeric mixture spectrum isolated from HMG-51 is shown in Fig. 7D (supplemental material HMG-51H); this sample is a mixture of three isomers, two of which contain fucosylated di-LacNAc motifs. Note that this spectrum represents a mixture containing two structures, one with a terminal Fuc-LacNAc (m/z 660) and another with an internal Fuc-LacNAc (m/z 646), and are clearly distinguishable in this spectrum. Further, MS4 spectra clarified the details of structure and linkage for the m/z 472, 486, 646, and 660 fragments (supplemental material HMG-51L, K, J, and I, respectively).
Fig. 7.
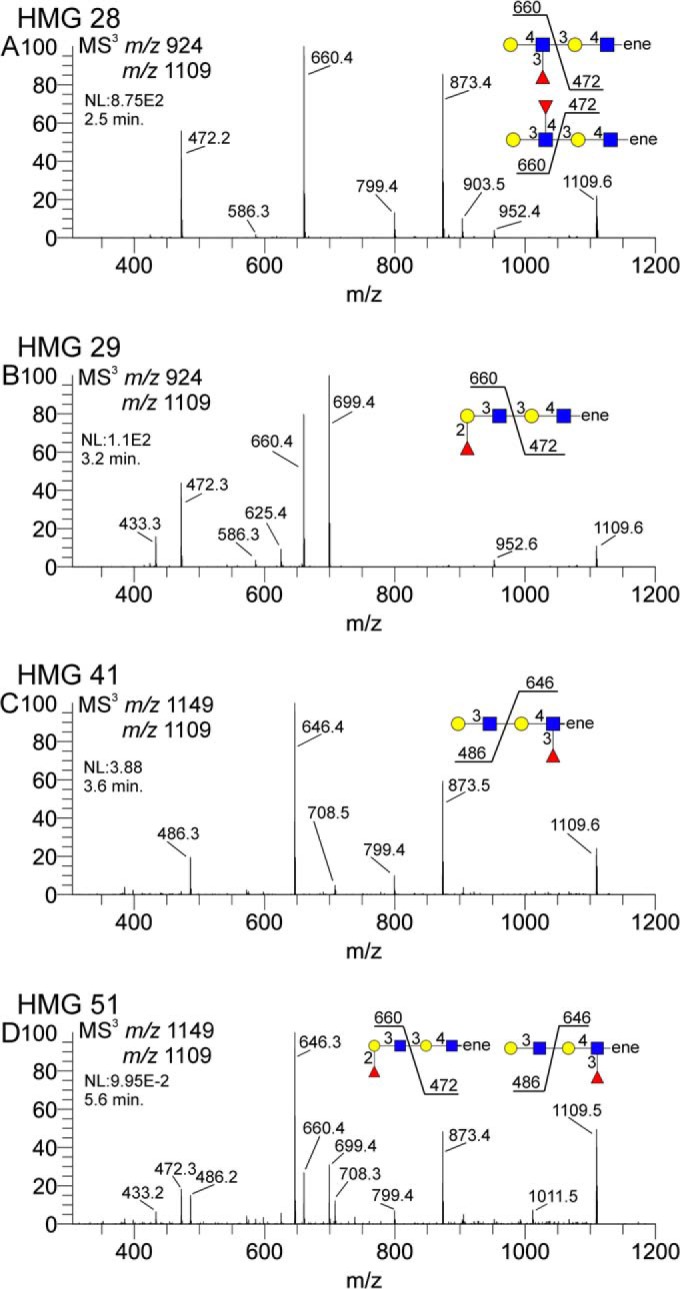
Isolated CID spectra of fucosylated diLacNAc structures from four different HMGs. Terminal fuc-diLacNAc (m/z 1109) fragments were isolated from various HMGs to show fucose localization. Panel A shows a terminal fuc-LacNAc coupled with an internal LacNAc, Panel B shows a terminal fuc-LacNAc and internal LacNAc, panel C shows a terminal LacNAc with an internal fuc-LacNAc, and panel D shows a mixture of fucose substitutions. Panels A and B differ in the terminal fuc-LacNAc structure; the specific terminal epitope was identified via MS4 m/z 660 CID (see supplemental materials).
Several HMGs had doubly-fucosylated di-LacNAc motifs. A selection of these from several samples is shown in Fig. 8. HMG-34 clearly shows both fucose residues attached to the terminal LacNAc unit, m/z 834, coupled with an unsubstituted internal LacNAc, m/z 472 (Fig. 8A, supplemental material HMG-34B). Subsequent MS4 and MS5 spectra confirmed the doubly fucosylated LacNAc as a Lewis B (supplemental material HMG-34C, D, and E). HMG-37 also contained this composition (Fig. 8B, supplemental material HMG-37B), but the MS3 spectrum is distinctly different and clearly indicates a terminal fucosylated LacNAc, m/z 660, coupled with an internal fucosylated LacNAc, m/z 646. Subsequent MS4 spectra confirmed the terminal H1 epitope and the internal Lewis X motif (supplemental material HMG-37C and D). HMG-62 contained a mixture of isomeric structures, and the MS3 spectrum shown (Fig. 8C, supplemental material HMG-62C) is clearly a composite of the two diFuc-diLacNAc topologies indicated in Fig. 8A and 8B. Again, MS4 and MS5 spectra of the relevant ions (supplemental material HMG-62D, E, F, G, and H) were obtained to confirm the indicated structures. In all three examples, the m/z 433 ion is indicative of the Fuc-1,2-Gal C-type fragment ion, present in both H1 and Lewis B structures.
Fig. 8.
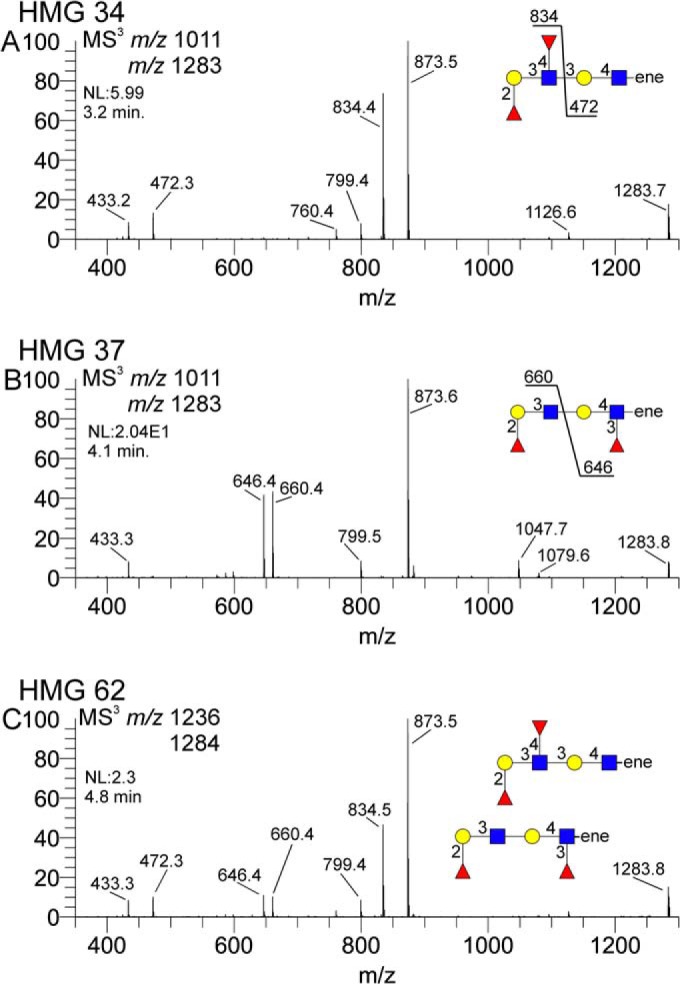
Difucosylated di-LacNAc motifs isolated from three different HMGs. Terminal difuc-diLacNAc (m/z 1283) fragments were isolated from various HMGs to determine fucose localization. Panel A shows a terminal difuc-LacNAc coupled with an internal LacNAc, panel B shows a terminal fuc-LacNAc coupled with an internal fuc-LacNAc, and panel C shows a mixture of these motifs.
Another structural feature, expected for some compositions but unexpected on others, was the branched tri-LacNAc motif depicted in Fig. 9. HMG-65, a decaose, is a previously reported structure, and this motif is expected. The octaose in HMG-45, however, is a structure that has not been previously reported, and this branched tri-LacNAc motif was not expected. For each structure, however, the same fragment ion could be isolated and disassembled via MSn to produce a set of fragment ion spectra to confirm this structural feature. Again, permethylation of the structures enables clear distinction of terminal (m/z 486), single-branched internal (m/z 472), and doubly branched (m/z 458) lactosamines, clearly establishing this structural motif.
Fig. 9.

Isolation of a tri-LacNAc branching motif isolated from a previously described lacto-N-decaose and a previously undescribed octaose. Panels A and B show the MS3 spectra of the tri-LacNAc ion, panels C and D show the internal di-LacNAc component, panels E and F show the terminal LacNAc unit, and panels G and H show the branched internal LacNAc unit. Each spectral pair displays a remarkable degree of similarity for each HMG, suggesting a common substructure to each.
Details of the analyses of the isomeric octaoses HMG-33 and HMG-45 shown in the supplemental materials indicate that these glycans are isomers with differing lactosamine linkages and differing topologies. Because these samples have the same mass and both have terminal lactosamine units, MS2 and some MS3 precursor selections are the same (supplemental material HMG-33B and C and HMG-45 B and C). While a significant number of fragment ion m/z values are similar, there are several significant differences between these spectra, indicating important structural differences. Most obviously, the AEAB-lactose core fragment masses are different and clearly indicative of core branching differences. These ions are m/z 724 for HMG-33 (supplemental material HMG-33B) and m/z 738 for HMG-45 (supplemental material HMG-45B and C). These masses indicate that HMG-33 has a doubly branched core and HMG -5 has a singly branched core and reflect an -OH versus -OCH3 mass difference. The mass of these compounds, coupled with the known AEAB-lactose cores, leaves three LacNAc residues to account for. For HMG-33, these will be distributed between two branches; for HMG-45, these will form a single branch, based on the AEAB-lactose core fragment masses (m/z 724 and m/z 738, respectively) for each. Both of these samples have clearly detectable terminal lactosamine fragments (m/z 486) at the MS2 and MS3 stages. The greater number of possible lactosamines relative to branches indicates that some polyLacNAc structures must be present. A diLacNAc B-type fragment at m/z 935 is present in HMG-33 (supplemental material HMG-33A and C). A triLacNAc fragment at m/z 1384 is detected for HMG-45 (supplemental material HMG-45A and D). The diLacNAc fragment in HMG-33 must be linear, while the triLacNAc fragment in HMG-45 could be linear or branched. For HMG-33, the second branch would be a single lactosamine unit. Selecting and disassembling these fragment ions will provide the information about how these branches are constructed. For HMG-33, the diLacNAc fragment (m/z 935) disassembly yields terminal (m/z 486) and internal (m/z 472) lactosamine units (supplemental material HMG-33C).
DISCUSSION
For structurally complex samples, multiple approaches to analysis can afford insights into the various sample components. Lectin/antibody binding strategies, using MAGS, as outlined in the companion manuscript (Yu et al.2), yield a great deal of information about the various binding determinants, so long as there exists suitable binding and nonbinding lectins and antibodies. Sequential mass spectrometry complements this approach in several important ways, particularly with respect to structural features that do not have available, reliable binders. A clear example of these in our study is glycans with the “internal Lewis X motif,” which is fairly common in HMGs. For the great majority of the single-component samples, especially the nonfucosylated samples, the MAGS approach made predictions in complete agreement with the MSn data. When dealing with certain fucosylated structures, particularly those samples containing multiple isomers, MSn data provided some key additional structural information that was able to clarify contradicting or incomplete lectin binding data.
The complementarity of the lectin/antiglycan antibody binding strategy and MSn analyses is also demonstrated for deciphering those isomeric structures that cannot be resolved by MSn analysis alone. For example, the location of the Lewis X determinant in HMG-20 to the 6-arm or 3-arm is not possible with the MSn analyses used in this report. However, it is possible to identify this particular isomeric structure based on differential lectin and anti-Lewis X antibody binding to this glycan before and after specific β-galactosidase digestion, as described in the companion paper (Yu et al.2).
The MSn data provided epitope assignments through library matching and scoring and provided the de novo structure analysis capability to determine linkage and branching details. Direct assessment of isomeric mixtures is made when fragment ions are present that could not arise from a single structure or are anomalous in some way. Distinct examples of apparently pure structure spectra contrasted with mixture spectra are shown in Figs. 7 and 8. In all cases, the mixture spectra are clearly superpositions of the various pure spectra and provide direct, empirical evidence of isomeric mixtures. It should be pointed out that, while these examples show various spectral comparisons, the mixture assignments could be made without reference to ostensibly pure standard spectra, using de novo spectral interpretation. We suspect that in these cases, sole reliance on binding assays using lectin/antibody approaches may lead one to miss such additional sample complexity.
In addition to the direct assessment of mixture spectra, MSn can also allow for the gas phase separation of ion fragments that select a particular isomer from a mixture of multiple isomers. For several HMG samples, including HMG-51 and HMG-54, fragmentation pathways could be selected that are unique to a particular isomer in the mixture, thus providing the ability to generate an isomerically pure fragment ion spectrum, even though the sample contained multiple isomeric precursor structures. In the case of HMG-51, several spectra (supplemental material HMG-51E and M) include contributions from two of the three precursor structures and a different pair of isomers for each. This feature is essential for assigning structures to such mixtures, as it provides some degree of simplification and reinforcement of the spectral interpretation. In addition, in some cases, this allows for a refined localization of certain motifs to particular isomers. In the case of HMG-51, while all three isomers contained terminal LacNAc units, there was a clear distinction among the linkage of this LacNAc to particular isomers. As shown in spectra K and O of the HMG-51 data set, the linear isomer clearly possessed a three-linked terminal LacNAc while the doubly branched isomers contained four-linked terminal lactosamines.
The branching motif of HMG-45 is a novel feature for this previously unreported octaose but is expected on certain decaose structures. Disassembly of this fragment ion shows an almost perfect comparability to the same fragment structure from HMG-65. HMG-64, -66, and- 67 differed in terminal lactosamine linkage, but otherwise all contained the branched tri-LacNAc motif. In this case, spectral comparability among samples provided valuable information about topology, even though none are true standard materials. We would suggest that this structure, while novel, should not be surprising, as the capacity to synthesize this branched tri-LacNAc motif clearly exists, given the previously reported decaose structures (24). The combination of permethylation and MSn seems to provide unambiguous determination of this branch point.
Our structural conclusions for HMG-76 also do not conform to expectations based on lectin and antibody binding, but the obtained MSn data appear to be unambiguous. One might have expected a difucosylated decaose to include a difucosylated branched triLacNAc motif, but the MSn data show no fragments consistent with this expectation. Our concluded structure includes a difucosylated lacto-N-neohexaose, which has been reported (24, 25). Our proposed structure has each arm extended by an additional LacNAc unit to form the difucosylated decaose. The terminal LacNAcs are a mixture of three- and four-linkages. Given the nature of this structure and the MSn analysis, it is difficult to conclude with certainty whether this a single structure with a different LacNAc linkage isomer on each arm or if there are multiple isomers, having the same branching topology, with differing terminal LacNAc linkage isomers.
Our results together with the companion paper by Yu et al.2, show our ability to combine data from lectin and antibody binding to glycans on the HM-SGM-v2 with sequential mass spectrometry approaches and provide structural definition for many novel HMGs recognized by several human rotaviruses. These results demonstrate the successful application of shotgun glycomics (26) for a functional glycomic analysis of the human milk free glycan metaglycome in regard to virus binding. The complementary approaches of defined lectin and antibody binding with the MSn analyses described here permit the compilation of metadata from a library of relatively pure fluorescent-labeled glycans that can be used to define their structures despite the AEAB derivative, which has been considered an impediment to MSn analyses. It is particularly noteworthy that, despite the large number of structural analyses directed to free human milk glycans in the past 50 years (7, 10, 11, 24, 27), this functional approach alone, which focused on identifying glycans bound by rotavirus adhesion proteins, led to the structural definition of 23 glycans that had not been previously discovered as components of human milk. Considering the large number of isomeric structures present in the free glycans from a pooled sample of human milk, this approach to the complex problem of determining glycan structures should be applicable to other metaglycomes and will permit the eventual conversion of the SGM to a defined glycan microarray, which will be available for screening other biologically relevant proteins.
Supplementary Material
Footnotes
Author contributions: D.J.A., R.D.C., D.F.S., and V.N.R. designed research; D.J.A. performed research; Y.Y., Y.L., X.S., L.H., S.R., B.P., M.K.E., R.D.C., and D.F.S. contributed new reagents or analytic tools; D.J.A., Y.Y., X.S., R.D.C., D.F.S., and V.N.R. analyzed data; D.J.A., R.D.C., D.F.S., and V.N.R. wrote the paper.
* This work was support by NIH Grants R01GM085448 (DFS), P41GM103694 (RDC), R01AI080656 (MKE), R01AI105101 (MKE), and R37AI36040 (BVVP) and the Robert A. Welch Foundation Grant Q1279 (BVVP). The MSn structural studies carried out were supported by a partnership arrangement between the UNH Glycomics Center and Glycan Connections, LLC, Lee, NH.
 This article contains supplemental material.
This article contains supplemental material.
2 Companion paper by Y. Yu, Y. Lasanajak, X. Song, L. Hu, S. Ramani, M.L. Mickum, D.J. Ashline, B.V.V. Prasad, M.K. Estes, V.N. Reinhold, R.D. Cummings, D.F. Smith.
1 The abbreviations used are:
- HMG
- human milk glycans
- TGL
- tagged glycan library
- AEAB
- 2-amino-N-(2-amino-ethyl)-benzamide
- SGM
- shotgun glycan microarray
- CID
- collision-induced dissociation
- MSn
- multistage mass spectrometry
- MAGS
- metadata-assisted glycan sequencing
- LacNAc
- lactosamine
- CA
- collisional activation
- NPI
- not previously identified.
REFERENCES
- 1. Bode L. (2006) Recent advances on structure, metabolism, and function of human milk oligosaccharides. J. Nutr. 136, 2127–2130 [DOI] [PubMed] [Google Scholar]
- 2. German J. B., Freeman S. L., Lebrilla C. B., Mills D. A. (2008) Human milk oligosaccharides: Evolution, structures and bioselectivity as substrates for intestinal bacteria. Nestle Nutr. Workshop Ser. Pediatr. Program 62, 205–218; discussion 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu Y., Mishra S., Song X., Lasanajak Y., Bradley K. C., Tappert M. M., Air G. M., Steinhauer D. A., Halder S., Cotmore S., Tattersall P., Agbandje-McKenna M., Cummings R. D., Smith D. F. (2012) Functional glycomic analysis of human milk glycans reveals the presence of virus receptors and embryonic stem cell biomarkers. J. Biol. Chem. 287, 44784–44799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morrow A. L., Ruiz-Palacios G. M., Jiang X., Newburg D. S. (2005) Human-milk glycans that inhibit pathogen binding protect breast-feeding infants against infectious diarrhea. J. Nutr. 135, 1304–1307 [DOI] [PubMed] [Google Scholar]
- 5. Newburg D. S., Ruiz-Palacios G. M., Morrow A. L. (2005) Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr. 25, 37–58 [DOI] [PubMed] [Google Scholar]
- 6. Smith D. F., Cummings R. D. (2013) Application of microarrays for deciphering the structure and function of the human glycome. Mol. Cell. Proteomics 12, 902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu S., Tao N., German J. B., Grimm R., Lebrilla C. B. (2010) Development of an annotated library of neutral human milk oligosaccharides. J. Proteome Res. 9, 4138–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amano J., Osanai M., Orita T., Sugahara D., Osumi K. (2009) Structural determination by negative-ion MALDI-QIT-TOFMSn after pyrene derivatization of variously fucosylated oligosaccharides with branched decaose cores from human milk. Glycobiology 19, 601–614 [DOI] [PubMed] [Google Scholar]
- 9. Kobata A. (2010) Structures and application of oligosaccharides in human milk. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 731–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Urashima T., Kitaoka M., Terabayashi T., Fukuda K., Ohnishi M., Kobata A. (2011) Milk oligosaccharides. In: Oligosaccharides: Sources, properties, and applications, Gordon N. S., ed., pp. 1–58, Nova Science Publishers, Inc., New York [Google Scholar]
- 11. Wu S., Grimm R., German J. B., Lebrilla C. B. (2011) Annotation and structural analysis of sialylated human milk oligosaccharides. J. Proteome Res. 10, 856–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ashline D. J., Hanneman A. J., Zhang H., Reinhold V. N. (2014) Structural documentation of glycan epitopes: Sequential mass spectrometry and spectral matching. J. Am. Soc. Mass Spectrom. 79, 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song X., Xia B., Stowell S. R., Lasanajak Y., Smith D. F., Cummings R. D. (2009) Novel fluorescent glycan microarray strategy reveals ligands for galectins. Chem. Biol. 16, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanneman A. J., Rosa J. C., Ashline D., Reinhold V. N. (2006) Isomer and glycomer complexities of core GlcNAcs in Caenorhabditis elegans. Glycobiology 16, 874–890 [DOI] [PubMed] [Google Scholar]
- 15. Kang P., Mechref Y., Klouckova I., Novotny M. V. (2005) Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass Spectrom. 19, 3421–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reinhold V. N., Reinhold B. B., Costello C. E. (1995) Carbohydrate molecular weight profiling, sequence, linkage, and branching data: ES-MS and CID. Anal. Chem. 67, 1772–1784 [DOI] [PubMed] [Google Scholar]
- 17. Reinhold V. N., Sheeley D. M. (1998) Detailed characterization of carbohydrate linkage and sequence in an ion trap mass spectrometer: glycosphingolipids. Anal. Biochem. 259, 28–33 [DOI] [PubMed] [Google Scholar]
- 18. Sheeley D. M., Reinhold V. N. (1998) Structural characterization of carbohydrate sequence, linkage, and branching in a quadrupole Ion trap mass spectrometer: neutral oligosaccharides and N-linked glycans. Anal. Chem. 70, 3053–3059 [DOI] [PubMed] [Google Scholar]
- 19. Ashline D., Singh S., Hanneman A., Reinhold V. (2005) Congruent strategies for carbohydrate sequencing. 1. Mining structural details by MSn. Anal. Chem. 77, 6250–6262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiskopf A. S., Vouros P., Harvey D. J. (1998) Electrospray ionization-ion trap mass spectrometry for structural analysis of complex N-linked glycoprotein oligosaccharides. Anal. Chem. 70, 4441–4447 [DOI] [PubMed] [Google Scholar]
- 21. Ashline D. J., Lapadula A. J., Liu Y. H., Lin M., Grace M., Pramanik B., Reinhold V. N. (2007) Carbohydrate structural isomers analyzed by sequential mass spectrometry. Anal. Chem. 79, 3830–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prien J. M., Huysentruyt L. C., Ashline D. J., Lapadula A. J., Seyfried T. N., Reinhold V. N. (2008) Differentiating N-linked glycan structural isomers in metastatic and nonmetastatic tumor cells using sequential mass spectrometry. Glycobiology 18, 353–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashline D. J., Hanneman A. J., Zhang H., Reinhold V. N. (2014) Structural documentation of glycan epitopes: sequential mass spectrometry and spectral matching. J. Am. Soc. Mass Spectrom. 25, 444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kobata A. (2010) Structures and application of oligosaccharides in human milk. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 731–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haeuw-Fievre S., Wieruszeski J. M., Plancke Y., Michalski J. C., Montreuil J., Strecker G. (1993) Primary structure of human milk octa-, dodeca- and tridecasaccharides determined by a combination of 1H-NMR spectroscopy and fast-atom-bombardment mass spectrometry. Evidence for a new core structure, the para-lacto-N-octaose. Eur. J. Biochem. 215, 361–371 [DOI] [PubMed] [Google Scholar]
- 26. Song X. Z., Lasanajak Y., Xia B. Y., Heimburg-Molinaro J., Rhea J. M., Ju H., Zhao C. M., Molinaro R. J., Cummings R. D., Smith D. F. (2011) Shotgun glycomics: a microarray strategy for functional glycomics. Nat. Methods 8, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kobata A. (2000) A journey to the world of glycobiology. Glycoconj. J. 17, 443–464 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.