

Abstract
Congenital muscular dystrophy with laminin α2 chain deficiency (MDC1A) is one of the most severe forms of muscular disease and is characterized by severe muscle weakness and delayed motor milestones. The genetic basis of MDC1A is well known, yet the secondary mechanisms ultimately leading to muscle degeneration and subsequent connective tissue infiltration are not fully understood. In order to obtain new insights into the molecular mechanisms underlying MDC1A, we performed a comparative proteomic analysis of affected muscles (diaphragm and gastrocnemius) from laminin α2 chain–deficient dy3K/dy3K mice, using multidimensional protein identification technology combined with tandem mass tags. Out of the approximately 700 identified proteins, 113 and 101 proteins, respectively, were differentially expressed in the diseased gastrocnemius and diaphragm muscles compared with normal muscles. A large portion of these proteins are involved in different metabolic processes, bind calcium, or are expressed in the extracellular matrix. Our findings suggest that metabolic alterations and calcium dysregulation could be novel mechanisms that underlie MDC1A and might be targets that should be explored for therapy. Also, detailed knowledge of the composition of fibrotic tissue, rich in extracellular matrix proteins, in laminin α2 chain–deficient muscle might help in the design of future anti-fibrotic treatments. All MS data have been deposited in the ProteomeXchange with identifier PXD000978 (http://proteomecentral.proteomexchange.org/dataset/PXD000978).
Congenital muscular dystrophy with laminin α2 chain deficiency, also known as MDC1A,1 is a severe muscle wasting disease for which there is no cure. MDC1A is caused by mutations in the LAMA2 gene that lead to complete or partial deficiency of laminin α2 chain (1–3). Although the primary defect in MDC1A is known, the secondary molecular mechanisms eventually leading to muscle degeneration are not fully understood. In normal muscle, laminin α2 chain binds to the cell surface receptors dystroglycan and integrin α7β1, which both indirectly bind the cytoskeleton (4–7). Both of these adhesion complexes are important for normal skeletal muscle function, and laminin α2 chain binding to dystroglycan contributes to the maintenance of sarcolemmal integrity and protects muscles from damage (8), whereas laminin α2 chain binding to integrin α7β1 promotes myofiber survival (9, 10). In MDC1A, laminin α2 chain is absent or severely reduced, and the expression of dystroglycan and α7β1 is also dysregulated in MDC1A (9, 11, 12). Thus, the structural link is broken, and the yet to be determined downstream intracellular signaling pathways are also interrupted. Consequently, laminin α2 chain–deficient muscle fibers undergo degeneration–regeneration cycles, but rather quickly regeneration fails and muscle fibers die by apoptosis/necrosis followed by a major replacement of muscle tissue with connective tissue (3, 7). In order to unravel novel secondary molecular mechanisms, which could indicate new therapeutic targets, we decided to evaluate the protein expression profile in laminin α2 chain–deficient dy3K/dy3K muscle. Several proteomic profiling studies of dystrophin-deficient muscles (Duchenne muscular dystrophy) have been performed (13–20), as well as some with dysferlin-deficient muscles (Limb-girdle muscular dystrophy type 2B, Miyoshi myopathy) (21, 22). They all showed a great number of proteins that were differentially expressed in different dystrophic muscles and at different ages (13–22). However, proteomic analyses of laminin α2 chain–deficient muscle have not yet been performed. We here used multidimensional protein identification technology with tandem mass tags (TMT), a powerful shotgun label-based proteomic method that separates peptides in two-dimensional liquid chromatography (23, 24). We identified around 100 proteins that were differentially expressed in laminin α2 chain–deficient gastrocnemius and diaphragm muscles relative to the corresponding wild-type muscles, and the differential expression of selected proteins was verified with Western blot analysis or immunofluorescence.
EXPERIMENTAL PROCEDURES
Animals
Four-week-old laminin α2 chain–deficient dy3K/dy3K mice and wild-type littermates (n = 15 each) were used (25). Mice were maintained in the animal facilities of BMC (Lund) according to the animal care guidelines. All mouse experimentation was approved by the Malmö/Lund (Sweden) ethical committee for animal research (permit number M62-09).
Protein Extraction, Digestion, TMT Labeling, and Strong Cation Exchange Fractionation
Animals were sacrificed by means of cervical dislocation, and the diaphragm and gastrocnemius muscles were collected, frozen in liquid nitrogen, and pulverized using a mortar and pestle. Subsequently, we used the same experimental setup as previously described (18). Three different pools for each group (dy3K/dy3K and wild-type mice) were made, each composed of muscles from five animals (Table I). Protein was extracted in lysis buffer (10 mm NaHCO3, 5% SDS containing freshly added protease and phosphatase inhibitors (Roche)) via ultrasonication (3 × 5 s at 4 °C). Samples were centrifuged for 5 min (15,000 × g), and supernatant was collected. The protein concentration was determined using a BCA Protein Assay Kit (Pierce). Subsequently, samples were processed according to the instructions for the TMT isobaric mass tagging kits and reagents (Pierce). Briefly, 100 μg of proteins per condition were placed in an Eppendorf tube. Forty-five microliters of 100 mm triethyl ammonium bicarbonate were added to the sample, and the sample was adjusted to a final volume of 100 μl with ultrapure water. Five microliters of 200 mm TCEP (reducing agent) were added and incubated at 55 °C for 1 h. Five microliters of 375 mm iodoacetamide was added, and samples were incubated for 30 min while protected from light. After this, samples were precipitated overnight with six volumes of prechilled (−20 °C) acetone. Finally, protein pellets were resuspended in 100 μl of 100 mm triethyl ammonium bicarbonate. At this point trypsin was added, and proteolytic digestion was allowed overnight at 37 °C. Label reagents were prepared at room temperature, and 41 μl of each reagent were used to label 100 μg of protein. Besides our four samples to be analyzed, a standard one, composed of equal fractions of the four others, was labeled. This standard sample was our reference, used to define the relative protein amounts for each analyzed sample. Our labeling design allowed a label swap, in order to avoid possible bias due to technical errors (Table I). The label reaction proceeded for 1 h at room temperature, and subsequently reactions were quenched with 8 μl of 5% hydroxylamine for 15 min, mixed, and stored at −80 °C. Pooled TMT-labeled samples were fractionated by strong cation exchange (Applied Biosystems, Foster City, CA) using 500 μl of elution buffer (KH2PO4, 25 mm acetonitrile, pH 2.9) containing increasing concentrations of KCl (30, 60, 90, 120, 240, 300, 420, and 500 mm KCl) and collected as fractions 1–8, respectively. The fractions were cleaned on Ultra Microspin C18 columns (The Nest Group, Southborough, MA), dried, and resuspended in 30 μl of 0.1% formic acid.
Table I. Experimental setup for gastrocnemius (GAST) and diaphragm (DIA) muscles under two conditions (dy3K/dy3K and wild-type (WT)) and with three biological replicates (pools 1, 2, and 3). The internal standard was a mixture of all samples.

LC-MS/MS Analysis on LTQ-OrbitrapXL
Samples were analyzed at the Gothenburg Proteomics Core, Gothenburg University, following the protocol described below. Desalted and dried fractions were reconstituted into 0.1% formic acid and analyzed on an LTQ-OrbitrapXL (Thermo Fisher Scientific) interfaced with an in-house-constructed nano-LC column. Two-microliter sample injections were made with an HTC-PAL autosampler (CTC Analytics AG, Zwingen, Switzerland) connected to an Agilent 1200 binary pump (Agilent Technologies, Santa Clara, CA). The peptides were trapped on a pre-column (40 × 0.075 mm inner diameter) and separated on a reversed phase column (200 × 0.050 mm). Both columns were packed in-house with 3-μm Reprosil-Pur C18-AQ particles. The flow through the analytical column was reduced by a split to ∼100 nl/min, and the gradient was as follows; 0–6 min, 0.1% formic acid; 6–76 min, 7% to 35% acetonitrile, 0.1% formic acid; 76–79 min, 40% to 80% acetonitrile, 0.1% formic acid. LTQ-OrbitrapXL settings were as follows: spray voltage, 1.4 kV; 1 microscan for MS1 scans at 60,000 resolution (m/z 400); full MS mass range, m/z 400–2000. The LTQ-OrbitrapXL was operated in a data-dependent mode with one MS1 Fourier transform MS scan of precursor ions followed by collision-induced dissociation and high-energy collision dissociation MS2 scans of the three most abundant doubly, triply, and quadruply protonated ions in each Fourier transform MS scan. The settings for the MS2 were as follows: 1 microscan for high-energy collision dissociation MS2 at 7500 resolution (at m/z 400), mass range of m/z 100–2000 with a collision energy of 50%, and 1 microscan for collision-induced dissociation MS2 with a collision energy of 30%. Dynamic exclusion of a precursor selected for MS2 was used for 120 s after one repeat, allowing most of the co-eluting precursors to be selected for MS2. All samples were analyzed a second time as described above, and also a third time using an exclusion list of all m/z within a 3-min retention window, already passing the identification criteria within the TMT set (eight fractions) in the database search.
Database Search and TMT Quantification
MS raw data files from all eight strong cation exchange fractions per one TMT set and three MS runs were merged for relative quantification and identification using Proteome Discoverer version 1.3 (Thermo Fisher Scientific). The database search was performed with the Mascot search engine using the following criteria: Mus musculus in Swiss-Prot protein database from April 2012 (535,698 entries); MS peptide tolerance of 10 ppm; MS/MS tolerance of 0.5 Da; trypsin digestion allowing one missed cleavage with variable modifications (methionine oxidation, cysteine methylthiol) and fixed modifications (N-terminal TMT6-plex label, lysine TMT6-plex label). The detected protein threshold in the software was set to a confidence using the 1% false discovery rate method, and identified proteins were grouped by shared sequence to minimize redundancy. For quantification, the ratios of TMT reporter ion intensities in MS/MS spectra (m/z 126.12, 127.13, 128.13, 129.14, 130.14) from raw datasets were used to calculate fold changes between samples via the relative ratio to the reference pool. Only peptides unique for a given protein were considered for relative quantitation, excluding those common to other isoforms or proteins of the same family. Only peptides with a score of >10 and below the Mascot significance threshold filter of p = 0.05 were included. Single peptide identifications required a score equal to or above the Mascot identity threshold. Normalization of protein median was used, and the median of peptides was used for determining protein ratio; the resulting ratios were exported into Excel for manual data interpretation. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (26) with the dataset identifier PXD000978. Please see the supplemental material for annotated spectra and Table I for the experimental setup.
Data Analysis
To determine which proteins were differentially expressed, only proteins quantified in all three runs (see Table I for the experimental setup) were used; average values were compared, and Student's t test was performed to validate differences (p < 0.05) (supplemental Tables S1–S3). Still, compared proteins should present a variation coefficient of less than 25% and a protein ratio less than 1.25 or greater than 1.25. In order to avoid multiple hypothesis test error, q-values were estimated (27, 28). Also, to ensure the reproducibility of our findings, the retrospective statistical power was calculated using the free statistical software package R, according to Ref. 29. Only results for β ≥ 0.8 were validated as true.
Western Blot Analysis
Muscles were collected and proteins were extracted as described above, but from a different animal cohort (n = 4 for each genotype). Thirty micrograms of total protein were applied per well. SDS-PAGE was performed according to Laemmli's protocol using 12% pre-casted gels in a Mini Protean Tetracell Electrophoresis System (Bio-Rad), followed by electrophoretic transfer. PVDF membranes were blocked in 5% non-fat dry milk in TBS with 0.05% Tween-20 overnight at 4 °C. Membranes were incubated with primary antibodies (rabbit anti-isocitrate dehydrogenase, 1:1000, NBP1–31599, Novus (Littleton, CO); mouse anti-SERCA1 ATPase, 1:4000, ab2819, Abcam; mouse anti-calsequestrin 1 and 2, 1:1000, ab3516, Abcam (Cambridge, UK); goat anti-annexin A1, 1:1000, NBP1–18842) for 1 h at room temperature and subsequently washed three times for 10 min in TBS with 5% Tween-20. Membranes were once again blocked in 5% non-fat dry milk for 1 h and then incubated with secondary antibodies (1 h at room temperature). After three 10-min washes with TBS with 5% Tween-20, membranes were incubated in ECL chemiluminiscence solution (Amersham Biosciences), exposed to Hyperfilm (Amersham Biosciences), and developed (Curix 60, AGFA, Mortsel, Belgium). We applied the Mann–Whitney U test to assess whether the difference in protein expression was statistically significant. Statistical significance was accepted for p < 0.05.
Immunofluorescence
Skeletal muscle (quadriceps and diaphragm) sections of 7 μm were processed for immunofluorescence analyses following standard procedures (30) with rabbit polyclonal antibodies directed against periostin (1:200, NBP1–30042, Novus) and galectin-1 (1:100, NBP1–89791, Novus). Sections were analyzed using a Zeiss Axioplan fluorescence microscope. Images were captured using an ORCA 1394 ER digital camera with the Openlab 3 software.
RESULTS
Shotgun Proteomic Analysis of Laminin α2 Chain–deficient Gastrocnemius and Diaphragm
Two-dimensional polyacrylamide gel electrophoresis has mainly been used as the proteomic approach to analyze dystrophic muscle (13–17). To overcome several drawbacks associated with this technique, we decided to use a label-based proteomic approach combining TMT labels and the multidimensional protein identification technology method. In this kind of analysis, whole samples are digested with a protease prior to identification and quantification of proteins in the sample. In our case we used trypsin, and peptides were labeled with an isobaric tag (TMT) for quantification (24). Subsequently, samples were fractionated using biphasic capillary columns packed with strong cation exchange and reversed phase material to separate digested and TMT-labeled whole protein mixtures. Labeled peptides were thus separated based on their charge and hydrophobicity, and the identification and quantification of peptides in eluted fractions was achieved using a mass spectrometer (peptides were identified through database searching and quantified by evaluation of reporter ion intensities from labels) (31, 32).
We analyzed gastrocnemius and diaphragm muscles from 4-week-old laminin α2 chain–deficient dy3K/dy3K and age-matched wild-type littermates. At this age, dy3K/dy3K muscles display typical pathological hallmarks such as small or rounded fibers (which indicate de-/regeneration), variation in muscle fiber diameter, centralized nuclei (indicative of regeneration), and the presence of inflammatory cells or adipose and fibrotic tissue (25, 33). With the multidimensional protein identification technology approach, we identified around 700 proteins in gastrocnemius and diaphragm muscles (supplemetal Table S4). Among these, 113 and 101 proteins, respectively, were found to be differentially altered in their levels by at least a factor of ±1.25 in dy3K/dy3K gastrocnemius and diaphragm muscle relative to corresponding wild-type muscles. In laminin α2 chain–deficient gastrocnemius muscle, there were 82 down-regulated and 31 up-regulated proteins, and in diaphragm muscle, 77 proteins were down-regulated and 24 were up-regulated. Around 62% of the down-regulated proteins and ∼67% of the up-regulated proteins in the two muscles were identical (Tables II–V). Overall, a vast majority of the differentially expressed proteins were intracellular proteins, but also several extracellular and plasma membrane proteins were identified. We used the PANTHER classification system to group the proteins with differential expression into different biological processes. Among the down-regulated proteins were proteins involved in metabolic processes, generation of precursor metabolites and energy, cellular processes, system processes, transport processes, and developmental processes. Among the up-regulated proteins were proteins involved in cellular processes, metabolic processes, cellular component organization, cell communication, developmental processes, and system processes (Figs. 1A–1D). Oxidoreductases, transferases, cytoskeletal proteins, and calcium-binding proteins were among the most underrepresented proteins in dy3K/dy3K muscles, whereas receptors, calcium-binding proteins, cytoskeletal proteins, and extracellular matrix proteins were among the most overrepresented proteins (Tables II–V).
Table II. Underrepresented proteins in laminin α2 chain–deficient gastrocnemius versus wild-type gastrocnemius.
| Accession | Description | Σ Coverage | p value | Σ Number of peptides | Fold change |
|---|---|---|---|---|---|
| P32848 | Parvalbumin α | 66.36 | 6.13E-01 | 11 | −3.59 |
| O88990 | α-Actinin-3 | 45.33 | 7.11E-01 | 33 | −2.98 |
| Q5XKE0 | Myosin-binding protein C, fast-type | 33.19 | 9.80E-01 | 32 | −2.97 |
| Q5SX39 | Myosin-4 | 61.84 | 9.50E-01 | 140 | −2.96 |
| Q3TJD7 | PDZ and LIM domain protein 7 | 4.16 | 8.09E-01 | 2 | −2.65 |
| P07310 | Creatine kinase M-type | 61.42 | 4.90E-01 | 25 | −2.56 |
| P05064 | Fructose-bisphosphate aldolase A | 43.96 | 8.75E-01 | 18 | −2.40 |
| Q9WUB3 | Glycogen phosphorylase, muscle form | 42.87 | 9.41E-01 | 35 | −2.35 |
| O70250 | Phosphoglycerate mutase 2 | 34.39 | 7.39E-01 | 9 | −2.33 |
| P58771 | Tropomyosin α-1 chain | 74.30 | 6.96E-01 | 31 | −2.21 |
| P21550 | β-Enolase | 40.09 | 8.30E-01 | 18 | −2.10 |
| Q9D6R2 | Isocitrate dehydrogenase [NAD] subunit α, mitochondrial | 11.20 | 5.59E-01 | 3 | −2.06 |
| P06151 | l-lactate dehydrogenase A chain | 37.35 | 7.17E-01 | 16 | −2.03 |
| Q8R429 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | 32.39 | 6.11E-01 | 34 | −2.01 |
| Q9QZ47 | Troponin T, fast skeletal muscle | 30.15 | 5.16E-01 | 9 | −2.00 |
| P13707 | Glycerol-3-phosphate dehydrogenase [NAD+], cytoplasmic | 25.50 | 9.96E-01 | 7 | −1.97 |
| P47857 | 6-phosphofructokinase, muscle type | 22.69 | 7.64E-01 | 14 | −1.93 |
| O08532 | Voltage-dependent calcium channel subunit α-2/δ-1 | 3.81 | 5.37E-01 | 2 | −1.92 |
| P13412 | Troponin I, fast skeletal muscle | 28.57 | 7.49E-01 | 6 | −1.89 |
| P17751 | Triosephosphate isomerase | 34.78 | 7.51E-01 | 8 | −1.89 |
| P20801 | Troponin C, skeletal muscle | 47.50 | 6.61E-01 | 6 | −1.89 |
| P28650 | Adenylosuccinate synthetase isozyme 1 | 17.94 | 7.11E-01 | 6 | −1.89 |
| P09411 | Phosphoglycerate kinase 1 | 27.34 | 9.91E-01 | 11 | −1.89 |
| Q9D0F9 | Phosphoglucomutase-1 | 15.48 | 9.27E-01 | 8 | −1.89 |
| O09165 | Calsequestrin-1 | 23.21 | 7.28E-01 | 10 | −1.87 |
| P16858 | Glyceraldehyde-3-phosphate dehydrogenase | 46.25 | 8.51E-01 | 12 | −1.84 |
| Q9R0Y5 | Adenylate kinase isoenzyme 1 | 57.73 | 6.36E-01 | 12 | −1.82 |
| P62631 | Elongation factor 1-α 2 | 31.97 | 9.43E-01 | 11 | −1.76 |
| P52480 | Pyruvate kinase isozymes M1/M2 | 36.35 | 9.38E-01 | 15 | −1.75 |
| P56392 | Cytochrome c oxidase subunit 7A1, mitochondrial | 16.25 | 6.95E-01 | 1 | −1.75 |
| Q9JK37 | Myozenin-1 | 39.19 | 6.91E-01 | 8 | −1.72 |
| Q6P8J7 | Creatine kinase S-type, mitochondrial | 33.65 | 5.68E-01 | 14 | −1.72 |
| Q60932 | Voltage-dependent anion-selective channel protein 1 | 47.97 | 6.69E-01 | 11 | −1.71 |
| P06745 | Glucose-6-phosphate isomerase | 17.38 | 8.76E-01 | 9 | −1.71 |
| Q91YE8 | Synaptopodin-2 | 2.58 | 6.30E-01 | 3 | −1.70 |
| A2ASS6 | Titin | 19.75 | 6.91E-01 | 550 | −1.70 |
| P63038 | 60 kDa heat shock protein, mitochondrial | 17.63 | 7.56E-01 | 9 | −1.68 |
| Q9CR68 | Cytochrome b-c1 complex subunit Rieske, mitochondrial | 46.35 | 5.81E-01 | 7 | −1.67 |
| Q9WUM5 | Succinyl-CoA ligase [GDP-forming] subunit α, mitochondrial | 14.74 | 4.92E-01 | 5 | −1.67 |
| Q9D855 | Cytochrome b-c1 complex subunit 7 | 20.72 | 6.22E-01 | 2 | −1.66 |
| P97457 | Myosin regulatory light chain 2, skeletal muscle isoform | 71.60 | 9.22E-01 | 16 | −1.66 |
| P05977 | Myosin light chain 1/3, skeletal muscle isoform | 76.06 | 8.05E-01 | 18 | −1.66 |
| P68134 | Actin, α skeletal muscle | 58.62 | 5.40E-01 | 23 | −1.66 |
| P08249 | Malate dehydrogenase, mitochondrial | 46.45 | 4.87E-01 | 13 | −1.65 |
| Q61425 | Hydroxyacyl-coenzyme A dehydrogenase, mitochondrial | 17.83 | 5.72E-01 | 4 | −1.63 |
| Q9CQC7 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 4 | 27.13 | 6.62E-01 | 2 | −1.59 |
| Q62234 | Myomesin-1 | 16.08 | 6.80E-01 | 22 | −1.59 |
| Q06185 | ATP synthase subunit e, mitochondrial | 35.21 | 7.80E-01 | 2 | −1.59 |
| Q9Z1E4 | Glycogen [starch] synthase, muscle | 1.08 | 5.65E-01 | 1 | −1.59 |
| P14152 | Malate dehydrogenase, cytoplasmic | 25.75 | 6.77E-01 | 6 | −1.56 |
| Q9Z2I9 | Succinyl-CoA ligase [ADP-forming] subunit β, mitochondrial | 11.23 | 6.86E-01 | 6 | −1.53 |
| Q9D051 | Pyruvate dehydrogenase E1 component subunit β, mitochondrial | 29.53 | 6.87E-01 | 8 | −1.53 |
| Q9D3D9 | ATP synthase subunit δ, mitochondrial | 41.07 | 4.95E-01 | 4 | −1.52 |
| O08749 | Dihydrolipoyl dehydrogenase, mitochondrial | 14.93 | 6.08E-01 | 5 | −1.51 |
| Q9CPQ1 | Cytochrome c oxidase subunit 6C | 21.05 | 9.04E-01 | 2 | −1.50 |
| P01942 | Hemoglobin subunit α | 71.13 | 7.73E-01 | 9 | −1.50 |
| Q99KI0 | Aconitate hydratase, mitochondrial | 31.79 | 4.90E-01 | 21 | −1.49 |
| P58774 | Tropomyosin β chain | 79.23 | 5.15E-01 | 34 | −1.48 |
| P12787 | Cytochrome c oxidase subunit 5A, mitochondrial | 39.04 | 6.12E-01 | 8 | −1.48 |
| Q9CZU6 | Citrate synthase, mitochondrial | 34.70 | 5.37E-01 | 11 | −1.46 |
| Q9DCX2 | ATP synthase subunit d, mitochondrial | 69.57 | 9.78E-01 | 9 | −1.45 |
| P54071 | Isocitrate dehydrogenase [NADP], mitochondrial | 28.32 | 6.62E-01 | 11 | −1.45 |
| Q9DCW4 | Electron transfer flavoprotein subunit β | 42.35 | 6.38E-01 | 11 | −1.44 |
| Q8VEM8 | Phosphate carrier protein, mitochondrial | 14.57 | 6.39E-01 | 6 | −1.44 |
| P56480 | ATP synthase subunit β, mitochondrial | 64.46 | 5.28E-01 | 22 | −1.43 |
| Q91WD5 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, mitochondrial | 12.31 | 6.08E-01 | 4 | −1.43 |
| Q9JKS4 | LIM domain-binding protein 3 | 17.01 | 5.78E-01 | 9 | −1.42 |
| P19783 | Cytochrome c oxidase subunit 4 isoform 1, mitochondrial | 20.71 | 7.65E-01 | 5 | −1.41 |
| Q07417 | Short-chain specific acyl-CoA dehydrogenase, mitochondrial | 7.77 | 5.59E-01 | 3 | −1.40 |
| P56391 | Cytochrome c oxidase subunit 6B1 | 20.93 | 5.00E-01 | 3 | −1.40 |
| Q9DB20 | ATP synthase subunit O, mitochondrial | 41.31 | 4.98E-01 | 9 | −1.39 |
| P35486 | Pyruvate dehydrogenase E1 component subunit α, somatic form, mitochondrial | 19.74 | 9.28E-01 | 6 | −1.39 |
| Q9CZ13 | Cytochrome b-c1 complex subunit 1, mitochondrial | 19.17 | 8.11E-01 | 9 | −1.38 |
| P05202 | Aspartate aminotransferase, mitochondrial | 26.98 | 6.77E-01 | 10 | −1.37 |
| Q8R1I1 | Cytochrome b-c1 complex subunit 9 | 39.06 | 5.25E-01 | 3 | −1.37 |
| Q03265 | ATP synthase subunit α, mitochondrial | 54.97 | 5.37E-01 | 27 | −1.33 |
| Q91YT0 | NADH dehydrogenase [ubiquinone] flavoprotein 1, mitochondrial | 19.61 | 7.55E-01 | 7 | −1.32 |
| Q9D2G2 | Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial | 12.33 | 5.87E-01 | 5 | −1.31 |
| P70670 | Nascent polypeptide-associated complex subunit α, muscle-specific form | 1.19 | 7.92E-01 | 3 | −1.28 |
| P97807 | Fumarate hydratase, mitochondrial | 25.44 | 5.55E-01 | 12 | −1.27 |
| Q7TQ48 | Sarcalumenin | 21.76 | 7.18E-01 | 12 | −1.27 |
| P11404 | Fatty acid–binding protein, heart | 54.14 | 5.67E-01 | 7 | −1.26 |
Table III. Overrepresented proteins in laminin α2 chain–deficient gastrocnemius versus wild-type gastrocnemius.
| Accession | Description | Σ Coverage | p value | Σ Number of peptides | Fold change |
|---|---|---|---|---|---|
| Q62009 | Periostin | 4.30 | 1.96E-05 | 2 | 4.95 |
| Q9Z1T2 | Thrombospondin-4 | 3.63 | 2.84E-02 | 2 | 3.75 |
| P10107 | Annexin A1 | 10.40 | 1.87E-03 | 3 | 3.18 |
| P62962 | Profilin-1 | 27.14 | 4.90E-04 | 3 | 2.82 |
| P62806 | Histone H4 | 52.43 | 1.25E-02 | 7 | 2.38 |
| P04117 | Fatty acid–binding protein, adipocyte | 29.55 | 8.51E-03 | 5 | 2.31 |
| P16045 | Galectin-1 | 17.78 | 5.24E-04 | 2 | 2.27 |
| P20152 | Vimentin | 53.86 | 2.25E-02 | 26 | 2.20 |
| P51885 | Lumican | 25.15 | 1.40E-02 | 7 | 2.18 |
| P28653 | Biglycan | 28.18 | 1.56E-02 | 8 | 2.12 |
| P15864 | Histone H1.2 | 33.02 | 1.45E-04 | 9 | 2.05 |
| P48036 | Annexin A5 | 22.88 | 7.68E-04 | 7 | 1.98 |
| P48678 | Prelamin-A/C | 26.32 | 2.03E-03 | 14 | 1.97 |
| Q9D0J8 | Parathymosin | 11.88 | 1.30E-03 | 1 | 1.87 |
| P43276 | Histone H1.5 | 8.97 | 2.17E-03 | 2 | 1.79 |
| P97447 | Four and a half LIM domains protein 1 | 11.07 | 1.65E-02 | 3 | 1.78 |
| O88569 | Heterogeneous nuclear ribonucleoproteins A2/B1 | 9.63 | 9.16E-03 | 3 | 1.75 |
| P10854 | Histone H2B type 1-M | 47.62 | 3.04E-03 | 7 | 1.73 |
| P13542 | Myosin-8 | 48.89 | 1.57E-02 | 111 | 1.66 |
| P68433 | Histone H3.1 | 16.91 | 1.34E-02 | 3 | 1.64 |
| P23927 | α-Crystalline B chain | 20.00 | 2.58E-02 | 3 | 1.60 |
| Q8VHX6 | Filamin-C | 9.98 | 5.25E-03 | 18 | 1.55 |
| Q9CZM2 | 60S ribosomal protein L15 | 16.18 | 1.69E-02 | 3 | 1.54 |
| Q8CGP6 | Histone H2A type 1-H | 32.03 | 8.67E-03 | 4 | 1.53 |
| P17742 | Peptidyl-prolyl cis-trans isomerase A | 27.44 | 8.84E-04 | 5 | 1.51 |
| P62301 | 40S ribosomal protein S13 | 23.18 | 8.58E-04 | 3 | 1.44 |
| Q8BH64 | EH domain-containing protein 2 | 15.47 | 2.39E-02 | 6 | 1.39 |
| Q04857 | Collagen α-1(VI) chain | 12.20 | 2.79E-02 | 11 | 1.38 |
| O09161 | Calsequestrin-2 | 9.88 | 2.14E-02 | 4 | 1.37 |
| P10922 | Histone H1.0 | 26.80 | 1.55E-02 | 5 | 1.36 |
| P07724 | Serum albumin | 31.09 | 1.44E-02 | 22 | 1.35 |
Table IV. Under-represented proteins in laminin α2 chain deficient-diaphragm versus wild-type diaphragm.
| Accession | Description | Σ Coverage | p value | Σ Number of peptides | Fold change |
|---|---|---|---|---|---|
| P32848 | Parvalbumin α | 66.36 | 8.62E-03 | 11 | −4.12 |
| P04247 | Myoglobin | 57.14 | 6.47E-03 | 10 | −1.99 |
| Q9WUB3 | Glycogen phosphorylase, muscle form | 42.87 | 3.09E-03 | 35 | −1.98 |
| P56392 | Cytochrome c oxidase subunit 7A1, mitochondrial | 16.25 | 7.82E-03 | 1 | −1.78 |
| Q5SX40 | Myosin-1 | 61.79 | 7.96E-03 | 141 | −1.71 |
| O09165 | Calsequestrin-1 | 23.21 | 8.62E-03 | 10 | −1.71 |
| P16125 | l-lactate dehydrogenase B chain | 25.15 | 6.29E-04 | 10 | −1.68 |
| P13707 | Glycerol-3-phosphate dehydrogenase [NAD+], cytoplasmic | 25.50 | 3.10E-03 | 7 | −1.66 |
| Q9D023 | Brain protein 44 | 22.83 | 6.95E-03 | 4 | −1.64 |
| P13412 | Troponin I, fast skeletal muscle | 28.57 | 8.36E-03 | 6 | −1.64 |
| P16015 | Carbonic anhydrase 3 | 46.92 | 4.44E-03 | 10 | −1.63 |
| P07310 | Creatine kinase M-type | 61.42 | 5.35E-03 | 25 | −1.62 |
| Q9CQC7 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 4 | 27.13 | 1.04E-02 | 2 | −1.61 |
| Q9R0Y5 | Adenylate kinase isoenzyme 1 | 57.73 | 4.19E-03 | 12 | −1.59 |
| Q9CXZ1 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial | 8.57 | 1.86E-02 | 1 | −1.58 |
| Q60932 | Voltage-dependent anion-selective channel protein 1 | 47.97 | 2.76E-02 | 11 | −1.56 |
| Q8R429 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | 32.39 | 1.49E-02 | 34 | −1.55 |
| P21550 | β-enolase | 40.09 | 1.56E-02 | 18 | −1.54 |
| P45952 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 19.48 | 4.87E-04 | 8 | −1.53 |
| Q9DCS9 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 10 | 14.20 | 1.63E-02 | 2 | −1.53 |
| Q9D6J6 | NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial | 12.10 | 4.23E-03 | 2 | −1.53 |
| Q99JY0 | Trifunctional enzyme subunit β, mitochondrial | 25.26 | 6.01E-03 | 11 | −1.53 |
| Q924X2 | Carnitine O-palmitoyltransferase 1, muscle isoform | 9.59 | 1.34E-02 | 6 | −1.52 |
| P19783 | Cytochrome c oxidase subunit 4 isoform 1, mitochondrial | 20.71 | 1.43E-03 | 5 | −1.52 |
| P97457 | Myosin regulatory light chain 2, skeletal muscle isoform | 71.60 | 1.63E-02 | 16 | −1.51 |
| Q9Z2I9 | Succinyl-CoA ligase [ADP-forming] subunit β, mitochondrial | 11.23 | 1.95E-02 | 6 | −1.50 |
| P41216 | Long-chain-fatty-acid–CoA ligase 1 | 17.31 | 3.95E-03 | 11 | −1.50 |
| Q6P8J7 | Creatine kinase S-type, mitochondrial | 33.65 | 4.52E-03 | 14 | −1.50 |
| P20801 | Troponin C, skeletal muscle | 47.50 | 2.44E-02 | 6 | −1.48 |
| Q9DCT2 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 3, mitochondrial | 20.53 | 1.07E-02 | 5 | −1.48 |
| P05977 | Myosin light chain 1/3, skeletal muscle isoform | 76.06 | 5.77E-03 | 18 | −1.47 |
| Q91VD9 | NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial | 16.78 | 2.11E-02 | 9 | −1.47 |
| Q9WUM5 | Succinyl-CoA ligase [GDP-forming] subunit α, mitochondrial | 14.74 | 8.82E-03 | 5 | −1.47 |
| P48962 | ADP/ATP translocase 1 | 41.28 | 1.39E-02 | 14 | −1.47 |
| P05201 | Aspartate aminotransferase, cytoplasmic | 32.20 | 5.97E-03 | 11 | −1.47 |
| P28650 | Adenylosuccinate synthetase isozyme 1 | 17.94 | 2.70E-02 | 6 | −1.46 |
| P62897 | Cytochrome c, somatic | 50.48 | 2.11E-03 | 6 | −1.46 |
| P05064 | Fructose-bisphosphate aldolase A | 43.96 | 1.17E-02 | 18 | −1.46 |
| P11404 | Fatty acid–binding protein, heart | 54.14 | 1.42E-04 | 7 | −1.46 |
| P54071 | Isocitrate dehydrogenase [NADP], mitochondrial | 28.32 | 7.79E-03 | 11 | −1.46 |
| Q8BMS1 | Trifunctional enzyme subunit α, mitochondrial | 41.28 | 2.86E-02 | 23 | −1.46 |
| Q9CZU6 | Citrate synthase, mitochondrial | 34.70 | 3.01E-02 | 11 | −1.45 |
| P08249 | Malate dehydrogenase, mitochondrial | 46.45 | 1.70E-03 | 13 | −1.44 |
| Q9QZ47 | Troponin T, fast skeletal muscle | 30.15 | 5.33E-03 | 9 | −1.44 |
| Q3TJD7 | PDZ and LIM domain protein 7 | 4.16 | 5.81E-03 | 2 | −1.44 |
| P12787 | Cytochrome c oxidase subunit 5A, mitochondrial | 39.04 | 2.12E-02 | 8 | −1.42 |
| Q9CQ62 | 2,4-dienoyl-CoA reductase, mitochondrial | 28.06 | 4.55E-03 | 7 | −1.42 |
| Q60936 | Chaperone activity of bc1 complex-like, mitochondrial | 9.61 | 6.42E-03 | 5 | −1.42 |
| P97807 | Fumarate hydratase, mitochondrial | 25.44 | 2.82E-03 | 12 | −1.42 |
| Q9DCW4 | Electron transfer flavoprotein subunit β | 42.35 | 1.05E-02 | 11 | −1.41 |
| Q9CPQ1 | Cytochrome c oxidase subunit 6C | 21.05 | 2.78E-02 | 2 | −1.41 |
| P58774 | Tropomyosin β chain | 79.23 | 1.19E-02 | 34 | −1.41 |
| P56391 | Cytochrome c oxidase subunit 6B1 | 20.93 | 4.53E-03 | 3 | −1.40 |
| Q9DCX2 | ATP synthase subunit d, mitochondrial | 69.57 | 1.25E-02 | 9 | −1.39 |
| Q91VR2 | ATP synthase subunit γ, mitochondrial | 25.17 | 1.51E-02 | 9 | −1.39 |
| Q99LC3 | NADH dehydrogenase [ubiquinone] 1 α subcomplex subunit 10, mitochondrial | 14.08 | 1.38E-02 | 4 | −1.39 |
| P56480 | ATP synthase subunit β, mitochondrial | 64.46 | 6.68E-03 | 22 | −1.39 |
| P14152 | Malate dehydrogenase, cytoplasmic | 25.75 | 2.26E-04 | 6 | −1.39 |
| Q60597 | 2-oxoglutarate dehydrogenase, mitochondrial | 12.71 | 2.77E-02 | 11 | −1.38 |
| Q8CAQ8 | Mitochondrial inner membrane protein | 15.59 | 1.73E-02 | 10 | −1.37 |
| Q9D3D9 | ATP synthase subunit δ, mitochondrial | 41.07 | 4.05E-03 | 4 | −1.37 |
| P47857 | 6-phosphofructokinase, muscle type | 22.69 | 1.46E-02 | 14 | −1.36 |
| Q9CQQ7 | ATP synthase subunit b, mitochondrial | 23.05 | 5.29E-03 | 5 | −1.36 |
| P58771 | Tropomyosin α-1 chain | 74.30 | 1.78E-02 | 31 | −1.36 |
| Q99KI0 | Aconitate hydratase, mitochondrial | 31.79 | 1.05E-02 | 21 | −1.35 |
| Q9DB20 | ATP synthase subunit O, mitochondrial | 41.31 | 1.26E-02 | 9 | −1.35 |
| Q62234 | Myomesin-1 | 16.08 | 2.91E-02 | 22 | −1.35 |
| Q9CQ69 | Cytochrome b-c1 complex subunit 8 | 34.15 | 1.23E-02 | 3 | −1.34 |
| Q9CR68 | Cytochrome b-c1 complex subunit Rieske, mitochondrial | 46.35 | 2.41E-02 | 7 | −1.34 |
| Q03265 | ATP synthase subunit α, mitochondrial | 54.97 | 1.74E-02 | 27 | −1.33 |
| P35486 | Pyruvate dehydrogenase E1 component subunit α, somatic form, mitochondrial | 19.74 | 1.04E-02 | 6 | −1.31 |
| Q9CZ13 | Cytochrome b-c1 complex subunit 1, mitochondrial | 19.17 | 1.58E-02 | 9 | −1.31 |
| P05202 | Aspartate aminotransferase, mitochondrial | 26.98 | 6.45E-03 | 10 | −1.30 |
| Q921G7 | Electron transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial | 1.14 | 1.18E-02 | 1 | −1.29 |
| A2ASS6 | Titin | 19.75 | 2.43E-02 | 550 | −1.29 |
| Q8BWT1 | 3-ketoacyl-CoA thiolase, mitochondrial | 25.19 | 9.29E-03 | 9 | −1.28 |
| P56382 | ATP synthase subunit ϵ, mitochondrial | 28.85 | 1.16E-02 | 2 | −1.27 |
Table V. Overrepresented proteins in laminin α2 chain–deficient diaphragm versus wild-type diaphragm.
| Accession | Description | Σ Coverage | p value | Σ Number of peptides | Fold change |
|---|---|---|---|---|---|
| P13541 | Myosin-3 | 23.09 | 2.54E-02 | 50 | 4.36 |
| Q62009 | Periostin | 4.30 | 1.58E-03 | 2 | 3.93 |
| P48678 | Prelamin-A/C | 26.32 | 2.18E-03 | 14 | 1.87 |
| P51885 | Lumican | 25.15 | 2.35E-02 | 7 | 1.83 |
| P62962 | Profilin-1 | 27.14 | 5.01E-03 | 3 | 1.83 |
| P16045 | Galectin-1 | 17.78 | 4.82E-03 | 2 | 1.77 |
| P13542 | Myosin-8 | 48.89 | 6.58E-03 | 111 | 1.70 |
| P10107 | Annexin A1 | 10.40 | 5.55E-03 | 3 | 1.65 |
| P19123 | Troponin C, slow skeletal and cardiac muscles | 23.60 | 2.21E-02 | 3 | 1.60 |
| P68433 | Histone H3.1 | 16.91 | 1.61E-02 | 3 | 1.58 |
| P17742 | Peptidyl-prolyl cis-trans isomerase A | 27.44 | 1.71E-02 | 5 | 1.54 |
| Q9DBJ1 | Phosphoglycerate mutase 1 | 15.35 | 7.96E-03 | 4 | 1.52 |
| P20152 | Vimentin | 53.86 | 5.22E-04 | 26 | 1.49 |
| P43276 | Histone H1.5 | 8.97 | 3.60E-03 | 2 | 1.48 |
| P28653 | Biglycan | 28.18 | 1.45E-02 | 8 | 1.47 |
| P48036 | Annexin A5 | 22.88 | 7.78E-03 | 7 | 1.43 |
| Q9D0J8 | Parathymosin | 11.88 | 2.75E-02 | 1 | 1.42 |
| P15864 | Histone H1.2 | 33.02 | 4.90E-03 | 9 | 1.41 |
| P10854 | Histone H2B type 1-M | 47.62 | 9.23E-03 | 7 | 1.39 |
| P14131 | 40S ribosomal protein S16 | 11.64 | 8.48E-03 | 2 | 1.34 |
| P47963 | 60S ribosomal protein L13 | 2.84 | 2.48E-04 | 1 | 1.32 |
| P43274 | Histone H1.4 | 31.96 | 2.36E-02 | 8 | 1.31 |
| P07356 | Annexin A2 | 17.99 | 1.59E-02 | 6 | 1.31 |
| P51881 | ADP/ATP translocase 2 | 34.23 | 1.15E-03 | 11 | 1.27 |
Fig. 1.

Pie charts of differentially expressed proteins in laminin α2 chain–deficient muscles grouped according to different biological processes using PANTHER classification. A, down-regulated proteins in laminin α2 chain–deficient gastrocnemius. B, up-regulated proteins in laminin α2 chain–deficient gastrocnemius. C, down-regulated proteins in laminin α2 chain–deficient diaphragm. D, up-regulated proteins in laminin α2 chain–deficient diaphragm.
Altered Metabolism in Laminin α2 Chain–deficient Muscle
A clear majority of the down-regulated proteins belonged to the class of metabolic processes/generation of precursor metabolites and energy, such as glycolysis, fatty acid β-oxidation, acyl CoA metabolic processes, pyrimidine and purin base metabolic processes, tricarboxylic acid cycle, oxidative phosphorylation, and respiratory electron transport chain (Tables II and IV). Each of these pathways is complex, involving numerous proteins. For example, the following proteins were down-regulated in laminin α2 chain–deficient muscles: fructose-biphosphate aldolase A, 6-phosphofrucokinase, l-lactate dehydrogenase, and glucose-6-phosphate isomerase (glycolysis-related); short-chain specific acyl-CoA dehydrogenase, medium-chain specific acyl-CoA dehydrogenase, and 2,4-dienoyl-CoA reductase (involved in fatty acid β-oxidation and acyl coA metabolic processes); adenylate kinase isoenzyme 1, phosphoglutamase-1, and various subunits of ATP synthase (pyrimidine and purin base metabolic processes); isocitrate dehydrogenase, succinyl-CoA ligase, malate dehydrogenase, and citrate synthase (tricarboxylic acid cycle–related); and various subunits of cytochrome c oxidase, electron transfer flavoprotein, and various subunits of ATP synthase (involved in oxidative phosphorylation and respiratory electron transport chain). Also, in a previous gene expression profiling study we noted that several down-regulated genes were assigned to metabolic themes (34). We next performed Western blot analysis to validate the proteomic findings, and indeed isocitrate dehydrogenase was significantly down-regulated in laminin α2 chain–deficient muscle (Fig. 2).
Fig. 2.

Western blot analysis of selected proteins. Quantification of isocitrate dehydrogenase (IDH1), SERCA1, and calsequestrin (CASQ) by Western blot analysis of extracts of gastrocnemius muscle from wild-type and laminin α2 chain–deficient dy3K/dy3K mice. α-actinin (α-ACTN) was used as a loading control. n = 4 and p < 0.05, Mann–Whitney.
Impaired Calcium Homeostasis in Laminin α2 Chain–deficient Muscle
An increase in cytosolic calcium concentration has been connected to the pathomechanism of muscular dystrophy (35), but it is not yet clear whether impaired calcium homeostasis is pathogenic in MDC1A. In some muscular dystrophies (e.g. Duchenne muscular dystrophy), the muscle cell membrane is easily damaged, and this could lead to an influx of calcium in the cell; alternatively, enhanced calcium entry may occur through a class of stretch-activated channels. The increased calcium leads to necrosis and/or apoptosis of the muscle fiber through distinct mechanisms (36–38). Also, the expression levels of proteins involved in the regulation of intracellular calcium concentration could be altered in muscular dystrophy (39). In MDC1A, there is little evidence of sarcolemmal disruption (40, 41), but we noted that parvalbumin, a protein that acts as a soluble intracellular calcium buffer (and is present in high concentrations in fast-contracting skeletal muscles), was significantly down-regulated in laminin α2 chain–deficient muscles (Table II and IV, first protein in the lists). Also, SERCA1 (which resides in the sarcoplasmic reticulum and transfers calcium from cytosol to sarcoplasmic reticulum lumen) was down-regulated in laminin α2 chain–deficient muscles, as was calsequestrin-1 (which also resides in sarcoplasmic reticulum and helps hold calcium in sarcoplasmic reticulum) (Tables II and IV; Fig. 2). The expression of calsequestrin-2, in contrast, was up-regulated, at least in gastrocnemius muscle (Table III; Fig. 2).
Inflammation and Composition of Fibrotic Tissue in Laminin α2 Chain–deficient Muscle
We detected increased levels of annexins A1, A2, and A5 in laminin α2 chain–deficient muscle (Tables III and V). The annexins are phospholipid- and calcium-binding proteins involved in numerous physiological processes (42). Several members of the annexin family (e.g. annexins A1, A2, and A5) are differentially expressed in normal muscle relative to Duchenne muscular dystrophy, and the three annexins are highly expressed in macrophages and T cells in dystrophin-deficient muscle (18, 43). The annexins could therefore have a role in the inflammatory response in muscular dystrophy, and MDC1A is indeed characterized by early-onset transient inflammation (44).
Furthermore, several of the up-regulated proteins are extracellular matrix proteins. Previous gene expression profiling experiments have demonstrated that up-regulation of genes encoding extracellular matrix components is more pronounced in congenital muscular dystrophies (including MDC1A) than in Duchenne muscular dystrophy (45). Yet the composition and properties of fibrotic lesions in laminin α2 chain–deficient chain muscle have not been extensively studied. The expression of periostin, galectin-1, collagen VI α2 chain, biglycan, lumican, and thrombospondin-4 was significantly increased in laminin α2 chain–deficient skeletal muscles (Tables III and V). Immunostaining of selected proteins (periostin and galectin-1) was performed to validate the proteomics data. Periostin expression was dramatically increased in laminin α2 chain–deficient muscle (Fig. 3), and galectin-1 was moderately augmented (Fig. 3).
Fig. 3.
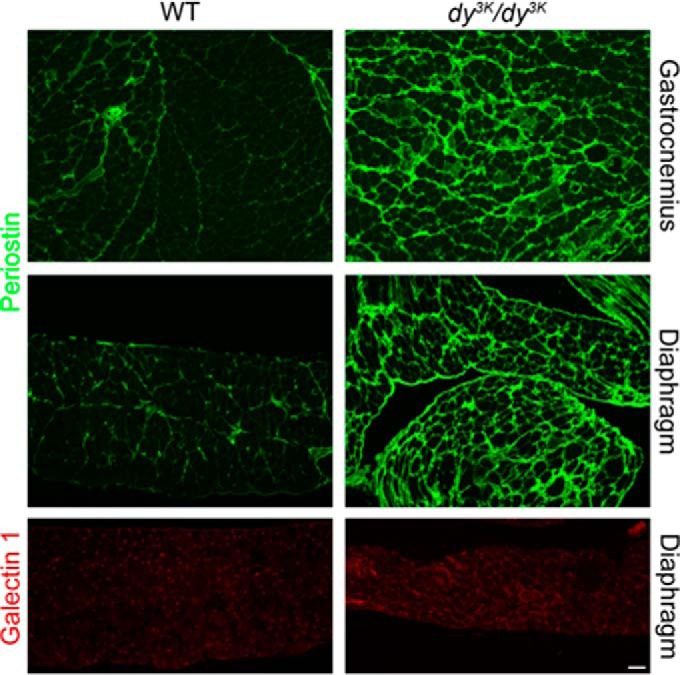
Periostin and galectin-1 are up-regulated in laminin α2 chain–deficient muscle. Cross-sections of gastrocnemius and diaphragm muscles were stained with antibodies against periostin and galectin-1. Bar, 50 μm.
DISCUSSION
Knowledge of the differences between normal and laminin α2 chain–deficient muscles is crucial for designing future treatment of MDC1A. Although several different approaches to combat muscular dystrophy in laminin α2 chain–deficient mouse models have been tested, and although these approaches include correction of both primary and secondary manifestations (11, 12, 25, 30, 41, 46–55), the clinical appliance is still years away. Thus, we performed a comparative proteomic analysis of dy3K/dy3K muscles, and we report an extensive catalogue of differentially expressed proteins in dystrophic muscle. A majority of the differentially expressed proteins are involved in metabolic processes (of which a majority take place in the mitochondria), calcium handling, or extracellular remodeling, making them valuable candidates for potential drug targets for MDC1A.
Similar metabolic crises and mitochondrial abnormalities have been reported in gene and/or proteome expression profiling of different types of muscular dystrophy, including Duchenne muscular dystrophy and sarcoglycanopathy (56, 57), and biochemical studies unveiled limitations in glycolytic and tricarboxylic acid cycle pathways and defective regulation of energy metabolism in mdx mice (58, 59). Also, metabolic defects in β-sarcoglycan-deficient mice have been reported (60). Thus, the metabolic crisis and mitochondrial abnormalities most likely represent secondary effects of laminin α2 chain deficiency and of muscular dystrophy in general, but we cannot entirely rule out that these are unique features of MDC1A. It will be important to investigate whether metabolic alterations are present before the dystrophic pathology becomes visible and/or in myoblasts that generally are not affected by disease. For example, energy metabolism was shown to be affected in mdx myoblasts (61). Despite prevalent metabolic alterations in many muscular dystrophies, it is interesting to note that only few attempts to restore energy metabolism have been made (62), and future efforts should therefore be considered.
We further showed that the expression of several calcium-binding proteins was altered in laminin α2 chain–deficient muscles, indicating their role as well as a role for calcium in disease pathology. In line with the hypothesis that calcium dysregulation could be a primary event in the onset of muscular dystrophy, several treatment approaches aimed at normalizing the calcium homeostasis in dystrophic muscle have been evaluated in mice, and in particular in the mdx mouse model for Duchenne muscular dystrophy (63–65). Notably, it has been demonstrated that overexpression of SERCA1 is beneficial in the mdx mouse, as well as in a mouse model for sarcoglycanopathy (65). However, whether SERCA1 overexpression attenuates laminin α2 chain–deficient muscular dystrophy remains to be demonstrated. Ablation of cyclophilin D encoding gene (which prevents mitochondrial calcium overload) reduced muscular dystrophy in mdx and sarcoglycan-deficient mice, as well as in laminin α2 chain–deficient animals, but whether pharmacological inhibition of cyclophilin diminishes pathology in laminin α2 chain–deficient animals (like in mdx and sarcoglycan-deficient mice) is not known (37). Thus, further approaches aimed at modulating calcium regulation in laminin α2 chain–deficient muscles should be tested.
Finally, it was recently demonstrated that anti-fibrotic therapy is beneficial in mouse models for MDC1A. Losartan and an analog derivative inhibited TGF-β signaling and reduced fibrosis and inflammation in skeletal muscle of two different mouse models (54, 55). Yet, TGF-β1 has a very broad spectrum of action. Therefore, it becomes important to elucidate the composition of the inflammatory/fibrotic tissue in laminin α2 chain–deficient muscle in more detail, as such exhaustive information might be helpful for the design of more specific anti-fibrotic treatments. For example, the expression of periostin, which has been suggested to function upstream but also downstream of TGF-β (66), was significantly increased in laminin α2 chain–deficient muscles. Thus, it is tempting to speculate that the removal of periostin would reduce the pathogenesis of laminin α2 chain–deficient muscular dystrophy by decreasing fibrosis, just like the deletion of periostin significantly improved sarcoglycan-deficient muscle structure and function (67).
In summary, we have here mapped alterations in the proteomic signature of laminin α2 chain–deficient mouse muscle. Although we used a powerful tandem mass spectrometry methodology with TMT isobaric labels, it also has its weaknesses, including the problem of ratio distortion, and we acknowledge that the expression ratio changes might have been underestimated (68, 69). Nevertheless, we identified differences in around 100 proteins, and a major part of these proteins are involved in metabolic, calcium, and extracellular matrix homeostasis, indicating that these processes could be targets for MDC1A therapy.
Supplementary Material
Acknowledgments
We gratefully acknowledge SCIBLU Proteomics Resource Centre at Lund University (especially Karin Hansson) and the Proteomics Core Facility at Gothenburg University (in particular Carina Sihlbom) for excellent help with protein analysis. We also acknowledge the PRIDE team (Attilla Csordas) for the deposition of our data to the ProteomeXchange Consortium. The MS data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000978. To access the data, please visit http://tinyurl.com/ojhrmot with username reviewer64343@ebi.ac.uk and password P07lsY9k.
Footnotes
Author contributions: B.M. and M.D. designed research; B.M., C.Y.M., C.C.F., K.I.G., and H.A. performed research; B.M., P.W., and M.D. analyzed data; B.M. and M.D. wrote the paper.
* This work was supported by the Swedish Research Council. C.Y.M. was the recipient of a fellowship from CAPES (Grant No. 2014–10−6). The instrument at the Proteomics Core Facility at Gothenburg University was funded by the Knut and Alice Wallenberg Foundation (Grant No. KAW2007.0118 to Gunnar C. Hanson).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MDC1A
- merosin congenital muscular dystrophy type 1A
- TMT
- tandem mass tags.
REFERENCES
- 1. Allamand V., Guicheney P. (2002) Merosin-deficient muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of laminin). Eur. J. Hum. Genet. 10, 91–94 [DOI] [PubMed] [Google Scholar]
- 2. Helbling-Leclerc A., Zhang X., Topaloglu H., Cruaud C., Tesson F., Weissenbach J., Tomé F. M. S., Schwartz K., Fardeau M., Tryggvason K., Guicheney P. (1995) Mutations in the laminin α2 chain gene (LAMA2) cause merosin-deficient muscular dystrophy. Nat. Genet. 11, 216–218 [DOI] [PubMed] [Google Scholar]
- 3. Voit T., Tomé F. S. (2004) The congenital muscular dystrophies. In Myology (Angel A., Franzini-Armstrong C., eds) Vol. 2, pp. 1203–1238, McGraw-Hill, New York [Google Scholar]
- 4. Ibraghimov-Beskrovnaya O., Ervasti J. M., Leveille C. J., Slaughter C. A., Sernett S. W., Campbell K. P. (1992) Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702 [DOI] [PubMed] [Google Scholar]
- 5. Talts J. F., Andac Z., Gohring W., Brancaccio A., Timpl R. (1999) Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO J. 18, 863–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. von der Mark H., Williams I., Wendler O., Sorokin L., von der Mark K., Pöschl E. (2002) Alternative splice variants of α7β1 integrin selectively recognize different laminin isoforms. J. Biol. Chem. 277, 6012–6016 [DOI] [PubMed] [Google Scholar]
- 7. Gawlik K. I., Durbeej M. (2011) Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet. Muscle 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han R., Kanagawa M., Yoshida-Moriguchi T., Rader E. P., Ng R. A., Michele D. E., Muirhead D. E., Kunz S., Moore S. A., Iannaccone S. T., Miyake K., McNeil P. L., Mayer U., Oldstone M. B. A., Faulkner J. A., Campbell K. P. (2009) Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of α-dystroglycan. Proc. Natl. Acad. Sci. U.S.A. 106, 12573–12579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vachon P. H., Xu H., Liu L., Loechel F., Hayashi Y., Arahata K., Reed J. C., Wewer U. M., Engvall E. (1997) Integrins (α7β1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J. Clin. Invest. 10, 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mayer U. (2003) Integrins: redundant or important players in skeletal muscle and beyond. J. Biol. Chem. 279, 14587–14590 [DOI] [PubMed] [Google Scholar]
- 11. Gawlik K. I., Mayer U., Blomberg K., Sonnenberg A., Ekblom P., Durbeej M. (2006) Laminin α1 chain mediated reduction of laminin α2 chain deficient muscular dystrophy involves integrin α7β1 and dystroglycan. FEBS Lett. 580, 1759–1765 [DOI] [PubMed] [Google Scholar]
- 12. Moll J., Barzaghi P., Lin S., Bezakova G., Lochmuller H., Engvall E., Muller U., Ruegg M. A. (2001) An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 413, 302–307 [DOI] [PubMed] [Google Scholar]
- 13. Doran P., Martin G., Dowling P., Jockusch H., Ohlendieck K. (2006) Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 6, 4610–4621 [DOI] [PubMed] [Google Scholar]
- 14. Gardan-Salmon D., Dixon J. M., Lonergan S. M., Selsby J. T. (2011) Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur. J. Appl. Physiol. 111, 2763–2773 [DOI] [PubMed] [Google Scholar]
- 15. Carberry S., Zweyer M., Swandulla D., Ohlendieck K. (2012) Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. Int. J. Mol. Med. 30, 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge Y., Molloy M. P., Chamberlain J. S., Andrews P. C. (2003) Proteomic analysis of mdx skeletal muscle: great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics 3, 1895–1903 [DOI] [PubMed] [Google Scholar]
- 17. Guevel L., Lavoie J. R., Perez-Iratxeta C., Rouger K., Dubreil L., Feron M., Talon S., Brand M., Megeney L. A. (2010) Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 10, 2465–2449 [DOI] [PubMed] [Google Scholar]
- 18. Matsumura C. Y., Menezes de Oliveira B., Durbeej M., Marques M. J. (2013) Isobaric tagging-based quantification for proteomic analysis: a comparative study of spared and affected muscles from mdx mice at the early phase of dystrophy. PLoS One 8, e65831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rayavarapu S., Coley W., Cakir E., Jahnke V., Takeda S., Aoki Y., Grodish-Dressman H., Jaiswal J. K., Hoffman E. P., Brown K. J., Hathout Y., Nagaraju K. (2013) Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteomics 12, 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lewis C., Carverry S., Ohlendieck K. (2009) Proteomic profiling of x-linked muscular dystrophy. J. Muscle Res. Cell. Motil. 30, 267–279 [DOI] [PubMed] [Google Scholar]
- 21. De la Torre C., Illa I., Faulkner G., Soria L., Robles-Cedeno R., Pereles-Dominguez-Perles R., De Luna N., Gallardo E. (2009) Proteomics identification of differentially expressed proteins in the muscle of dysferlin myopathy patients. Proteomics Clin. Appl. 3, 486–497 [DOI] [PubMed] [Google Scholar]
- 22. de Morrée A., Hensbergen P. J., van Haagen H. H., Dragan I., Deelder A. M., ‘t Hoen P. A., Frants R. R., van der Maarel S. M. (2010) Proteomic analysis of the dysferlin protein complex unveils its importance for sarcolemmal maintenance and integrity. PLoS One 5, e13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Washburn M., Wolters D., Yates J., III (2001) Large scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 24. Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–904 [DOI] [PubMed] [Google Scholar]
- 25. Carmignac V., Quere R., Durbeej M. (2011) Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum. Mol. Genet. 20, 541–552 [DOI] [PubMed] [Google Scholar]
- 26. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karp N. A., McCormick P. S., Russell M. R., Lilley K. S. (2007) Experimental and statistical considerations to avoid false conclusions in proteomics studies using differential in-gel electrophoresis. Mol. Cell. Proteomics 6, 1354–1364 [DOI] [PubMed] [Google Scholar]
- 28. Storey J. D., Tibshirani R. (2003) Statistical significance for genome wide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levin Y. (2011) The role of statistical power analysis in quantitative proteomics. Proteomics 11, 2565–2567 [DOI] [PubMed] [Google Scholar]
- 30. Gawlik K., Miyagoe-Suzuki Y., Ekblom P., Takeda S., Durbeej M. (2004) Laminin α1 chain reduces muscular dystrophy in laminin α2 chain deficient mice. Hum. Mol. Genet. 13, 1775–1784 [DOI] [PubMed] [Google Scholar]
- 31. Kline K. G., Wu C. C. (2009) MudPIT analysis: application to human heart tissue. Methods Mol. Biol. 528, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martins de Souza D., Oliveira B. M., Castro-Dias E., Winck F. V., Horiuchi R. S., Baldasso P. A., Caetano H. T., Pires N. K., Marangoni S., Novello J. C. (2008) The untiring search for the most complete proteome representation: reviewing the methods. Brief Funct. Genomic. Proteomic. 7, 312–321 [DOI] [PubMed] [Google Scholar]
- 33. Miyagoe Y., Hanaoka K., Nonaka I., Hayasaka M., Nabeshima Y., Arahata K., Nabeshima Y., Takeda S. (1997) Laminin α2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 415, 33–39 [DOI] [PubMed] [Google Scholar]
- 34. Häger M., Bigotti M. G., Meszaros R., Carmignac V., Holmberg J., Allamand V., Åkerlund M., Kalamajski S., Brancaccio A., Mayer U., Durbeej M. (2008) Cib2 binds integrin α7Bβ1D and is reduced in laminin α2 chain-deficient muscular dystrophy. J. Biol. Chem. 283, 24760–247695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deconinck N., Dan B. (2007) Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr. Neurol. 26, 1–7 [DOI] [PubMed] [Google Scholar]
- 36. Vandebrouck C., Martin D., Colson-Van Schoor M., Debaix H., Gailly P. (2002) Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 158, 1089–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Millay D. P., Sargent M. A., Osinska H., Baines C. P., Barton E. R., Vuagniaux G., Sweeney H. L., Robbins J., Molkentin J. D. (2008) Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 14, 442–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Millay D. P., Goonasekera S. A., Sargent M. A., Maillet M., Aronow B. J., Molkentin J. D. (2009) Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 19023–19028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Culligan K. G., Ohlendieck K. (2002) Abnormal calcium handling in muscular dystrophy. Basic Appl. Myol. 12, 147–157 [Google Scholar]
- 40. Straub V., Rafael J. A., Chamberlain J. S., Campbell K. P. (1997) Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 139, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gawlik K. I., Åkerlund M., Carmignac V., Elamaa H., Durbeej M. (2010) Distinct roles for laminin globular domains in laminin α1 chain mediated rescue of murine laminin α2 chain deficiency. PLoS One 5, e11549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rescher U., Gereke V. (2004) Annexins—unique membrane binding proteins with diverse functions. J. Cell Sci. 117, 2631–2639 [DOI] [PubMed] [Google Scholar]
- 43. Probst-Cousinm S., Berghoff C., Neundörfer B., Heuss D. (2004) Annexin expression in inflammatory myopathies. Muscle Nerve 30, 102–110 [DOI] [PubMed] [Google Scholar]
- 44. Jeudi S., Wardrop K. E., Alessi A., Dominov J. A. (2011) Bcl-2 inhibits the innate immune response during early pathogenesis of murine congenital muscular dystrophy. PLoS One 6, e22369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taniguchi M., Kurahashi H., Noguchi S., Sese J., Okinaga T., Tsukahara T., Guicheney P., Ozono K., Nishino I., Morishita S., Toda T. (2006) Expression profiling from Fukuyama-type congenital muscular dystrophy and laminin-α2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem. Biophys. Res. Commun. 342, 489–502 [DOI] [PubMed] [Google Scholar]
- 46. Kuang W., Xu H., Vachon P. H., Engvall E. (1998) Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J. Clin. Invest. 102, 844–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meinen S., Barzaghi P., Lin S., Lochmuller H., Rüegg M. A. (2007) Linker molecules between laminins and dystroglycan ameliorate laminin-α2-deficient muscular dystrophy at all disease stages. J. Cell Biol. 176, 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Doe J. A., Wuebbles R. D., Allred E. T., Rooney J. E., Elorza M., Burkin D. J. (2011) Transgenic overexpression of the α7 integrin reduces muscle pathology and improves viability in the dy(W) mouse model of merosin-deficient congenital muscular dystrophy type 1A. J. Cell Sci. 124, 2287–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Girgenrath M., Dominov J. A., Kostek C. A., Miller J. B. (2004) Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J. Clin. Invest. 114, 1635–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Erb M., Meinen S., Barzaghi P., Sumanovski L. T., Courdier-Fruh I., Rüegg M. A., Meier T. (2009) Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-α2 deficiency. J. Pharmacol. Exp. Ther. 331, 787–795 [DOI] [PubMed] [Google Scholar]
- 51. Kumar A., Yamauchi J., Girgenrath T., Girgenrath M. (2011) Muscle-specific expression of insulin-like growth factor 1 improves outcome in Lama2Dy-w mice, a model for congenital muscular dystrophy type 1A. Hum. Mol. Genet. 20, 2333–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carmignac V., Svensson M., Körner Z., Elowsson L., Matsumura C., Gawlik K., Allamand V., Durbeej M. (2011) Autophagy is increased in laminin α2 chain-deficient muscle and inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 20, 4891–4902 [DOI] [PubMed] [Google Scholar]
- 53. Rooney J. E., Knapp J. R., Hodges B. L., Wuebbles R. D., Burkin D. J. (2011) Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am. J. Pathol. 180, 1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Elbaz M., Yanay N., Aga-Mizrachi S., Brunschwig Z., Kassis I., Ettinger K., Barak V., Nevo Y. (2012) Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in the dy2J/dy2J mouse. Ann. Neurol. 71, 699–708 [DOI] [PubMed] [Google Scholar]
- 55. Meinen S., Lin S., Rüegg M. A. (2012) Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy (MDC1A). Skelet. Muscle 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Turk R., Sterrenburg E., van der Wees C. G., de Meijer E. J., de Menezes R. X., Groh S., Campbell K. P., Noguchi S., van Ommen G. J., den Dunnen J. T., 't Hoen P. A. (2006) Common pathological mechanisms in mouse models for muscular dystrophies. FASEB J. 20, 127–129 [DOI] [PubMed] [Google Scholar]
- 57. Chen Y. W., Zhao P., Borup R., Hoffman E. P. (2000) Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J. Cell Biol. 151, 1321–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chinet A. E., Even P. C., Decrouy A. (1994) Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 59, 602–605 [DOI] [PubMed] [Google Scholar]
- 59. Even P. C., Decrouy A., Chinet A. (1994) Defective regulation of energy metabolism in the mdx-mouse skeletal muscles. Biochem. J. 304, 649–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Groh S., Zong H., Goddeeris M. M., Lebakken C. S., Venzke D., Pessin J. E., Campbell K. P. (2009) Sarcoglycan complex: implications for metabolic defects in muscular dystrophies. J. Biol. Chem. 284, 19178–19182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Onopiuk M., Brutkowski W., Wierzbicka K., Wojciechowska S., Szczepanowska J., Fronk J., Lochmuller H., Gorecki D. C., Zablocki K. (2009) Biochem. Biophys. Res. Commun. 386, 463–466 [DOI] [PubMed] [Google Scholar]
- 62. Le Borgne F., Guyot S., Logerot M., Beney L., Gervais P., Demarquoy J. (2012) Exploration of lipid metabolism in relation with plasma membrane properties of Duchenne muscular dystrophy cells: influence of L-carnitine. PLoS One 7, e49346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsumura C. Y., Pertille A., Albuquerge T. C., Santo Neto H., Marques M. J. (2009) Diltiazem and verapamil protect dystrophin-deficient muscle fibers of MDX mice from degeneration: a potential role in calcium buffering and sarcolemmal stability. Muscle Nerve 39, 167–176 [DOI] [PubMed] [Google Scholar]
- 64. Jørgensen L. H., Blain A., Greally E., Lavala S. H., Blamire A. M., Davison B. J., Brinkmeier H., MacGowan G. A., Schrøder H. D., Bushby K., Straub V., Lochmüller H. (2011) Long-term blocking of calcium channels in mdx mice results in differential effects on heart and skeletal muscle. Am. J. Pathol. 178, 273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goonasekera S. A., Lam C. K., Millay D. P., Sargent M. A., Hajjar R. J., Kranias E. G., Molkentin J. D. (2011) Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Horiuchi K., Amizuka N., Takeshita S., Takamatsu H., Katsuura M., Ozawa H., Toyama Y., Bonewald L. F., Kudo A. (1999) Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor β. J. Bone Miner. Res. 14, 1239–1249 [DOI] [PubMed] [Google Scholar]
- 67. Lorts A., Schwanekamp J. A., Baudino T. A., McNally E. M., Molkentin J. D. (2012) Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-β pathway. Proc. Natl. Acad. Sci. U.S.A. 109, 10978–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ting L., Rad R., Gygi S. P., Haas W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Christoforou A., Lilley K. S. (2011) Taming the isobaric tagging elephant in the rooms in quantitative proteomics. Nat. Methods 8, 911–913 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.