

Abstract
Cancer is responsible for many deaths and is a major source of healthcare expenditures. The identification of new, non-invasive biomarkers might allow improvement of the direct diagnostic or prognostic ability of already available tools. Here, we took the innovative approach of interrogating the activity of exopeptidases in the serum of cancer patients with the aim of establishing a distinction based on enzymatic function, instead of simple protein levels, as a means to biomarker discovery. We first analyzed two well-characterized mouse models of prostate cancer, each with a distinct genetic lesion, and established that broad exopeptidase and targeted aminopeptidase activity tests reveal proteolytic changes associated with tumor development. We also describe new peptide-based freeze-frame reagents uniquely suited to probe the altered balance of selected aminopeptidases, as opposed to the full array of exopeptidases, and/or their modulators in patient serum or plasma. One particular proteolytic activity was impaired in animals with aggressive disease relative to cancer-free littermates. We identified the protease in question as dipeptidyl peptidase 4 (DPP4) by analyzing selected knockout mice and evaluating the effect of specific inhibitors. DPP4 activity was also reduced in the sera of patients with metastatic prostate cancer relative to patients with localized disease or healthy controls. However, no significant differences in DPP4 serum levels were observed, which established the loss of activity as the result of impaired enzymatic function. Biochemical analysis indicated that reduced activity was the result not of post-translational modifications or allosteric changes, but instead of a low-molecular-weight inhibitor. After we adjusted for age and total prostate-specific antigen, reduced DPP4 activity remained a significant predictor of cancer status. The results of this proof-of-principle study suggest that DPP4 activity might be a potential blood-based indicator of the presence of metastatic cancer of prostatic origin, either by itself or, more likely, as a means to improve the sensitivity and specificity of existing markers.
Biomarkers have featured prominently in tests designed to aid in medical decision making, such as establishing a diagnosis, determining prognosis, and assessing the effects of treatment. In clinical oncology practice, biomarkers are required to address relevant questions related not only to patients with early stage disease, but also to those with metastatic and, in some cases, incurable cancer. An ideal marker for cancer diagnosis and surveillance is one that is noninvasive and reproducible, with high sensitivity and specificity. The classic path to cancer biomarker discovery involves measuring differential levels of proteins in the blood or tissue of interest using immunohistochemical- or mass spectrometry (MS)-based screens. This approach has not been a great success, mainly because the complexity of the blood proteome precludes the detection of proteins and peptides at low levels without time-consuming prefractionation. As a result, disappointingly few assays have been translated into clinical practice so far (1, 2), a regrettable disconnect that advocates conceptually novel biomarker discovery and validation schemes. An example of an alternate approach is examination of the activity of proteins, in particular enzyme families, that are relevant with respect to the disease of interest. In the case of cancer, proteases are one such class, as several of its members have been implicated in promoting both tumor progression and suppression (3–6).
It has been suggested that the cumulative exopeptidase activity in blood can provide accurate class discrimination between patients with solid tumors and controls without cancer (7, 8). Initial assessments were made either by carefully measuring and identifying a subset of the endogenous serum peptide metabolome—a notoriously difficult process—or by monitoring the degradation of spiked, synthetic peptide substrates using a method that allows straightforward yet accurate quantitation of the breakdown products on a whole serum proteome background. This method, termed the sequence-specific exopeptidase activity test (SSEAT),1 provides an aggregate read-out of protease activities and has the important advantage of all but eliminating reproducibility problems related to sample collection, storage, and handling that have beset serum oncopeptidomic studies of the past (8–11). From a classical proteomics point of view, some of these proteases may also be exceedingly low abundant in serum and therefore “invisible” in traditional MS-based discovery schemes. However, given enough substrate, time, and optimal assay conditions, catalytic product may accumulate to such a level that it becomes readily detectable in any type of mass spectrometer. To date, SSEAT assays have never been applied to study well-characterized animal models of cancer to determine whether they may reveal proteolytic changes associated with tumor development or whether such changes are relevant to human cancer.
Prostate cancer (PCa) is the most prevalent malignancy in men and the second leading cause of cancer death in North America, with one in six men having a lifetime risk of being diagnosed and a 3.4% chance of death (12). It is a heterogeneous disease, with some patients diagnosed at an early stage who either do not require treatment or are cured following surgery, and some diagnosed with advanced disease or who suffer recurrence despite initial, apparently effective treatment (13, 14). Serum prostate-specific antigen (PSA) is the only protein biomarker routinely used for the detection and management of a common cancer, but it is not a reliable intermediate indicator of overall survival (15–18). As an example, metastatic castration-resistant prostate cancer (mCRPC) is generally associated with poor outcomes, but precise survival times are hard to predict at present (14, 19–21). A newly developed biomarker used independently is unlikely to surpass the accuracy of the current gold standards for diagnosis, but a goal of discovery would be to integrate a new marker in the process of clinical decision making to improve upon the diagnostic or prognostic ability of already existing tools.
The current investigation sought to exploit the merits of analyzing mouse models of PCa to establish whether SSEAT assays may reveal proteolytic changes with tumor development and whether such changes are relevant to human disease. We also describe new peptide-based reagents uniquely suited to probe the altered balance of selected aminopeptidases, as opposed to the full array of exopeptidases, and/or their modulators in serum or plasma of cancer patients. Using suitable animal models and individualized assays, we found that DPP4 activity was markedly reduced in serum of mCRPC patients relative to that of patients with localized disease and healthy control individuals. Biochemical analysis suggests the existence of a low-molecular-weight inhibitor of circulating DPP4 that is either uniquely present or at elevated levels in patients with advanced disease. After we adjusted for age and total PSA, DPP4 activity remained a significant predictor of cancer status.
EXPERIMENTAL PROCEDURES
Animal Studies
The various wild-type, transgenic, and gene knockout mouse strains used in this study (details in supplemental Table S1) were gifts from Drs. Charles Sawyers (c-Myc overexpressing mice and corresponding wild types), Pier Paolo Pandolfi (Pten and p53 knockouts, and wild types), and Didier Marguet (DPP4 knockouts and wild types). All animal care and use procedures were in strict accordance with the standing committees on the use of animals in research and teaching at Memorial Sloan Kettering Cancer Center and Harvard University and the National Institutes of Health guidelines for the humane treatment of laboratory animals. Prostate-cancer-bearing PB-MYC (∼12 months), Pten prostate conditional null (∼12 months), Pten/p53 prostate conditional null (∼6 months), and DPP4 null mice and age-matched wild-type littermate controls were euthanized, and blood was collected into a BD microtainer tube (BD Biosciences) via intracardiac aspiration. Prostates were also harvested for pathologic evaluation. Serum was isolated through centrifugation and stored at −80 °C. Serum pools were prepared by taking equal-volume aliquots from multiple animals of each mouse model or control that were combined into a cryovial to then generate a series of 20-μl aliquots that were stored at −80 °C until use. In general, each serum sample had been frozen and thawed twice before it was subjected to solid-phase peptide extraction for exopeptidase activity assays.
Patient Samples
Human reference serum was purchased from Sigma (catalog number S-7023, lot 034K8937). Blood samples from male volunteers with no known malignancies (I) and samples from patients diagnosed with either localized (II) or metastatic (III) prostate carcinoma were collected following informed consent, under a protocol approved by the Memorial Sloan Kettering Cancer Center Institutional Review and Privacy Board. Forty-eight samples were selected from each group to yield the best possible age match. All 144 samples were collected at Memorial Sloan Kettering Cancer Center according to the same standard operating procedure (9). Patient clinical data are given in supplemental Table S2, except for ages and serum PSA levels, which are listed together with the serum aminopeptidase activities measured for each individual in this study (see “Results” and associated supplemental Table S5). Following the standard operating procedure, each serum sample was frozen and thawed twice before it was subjected to solid-phase peptide extraction and mass spectrometric analysis. Serum pools were obtained for the control and each of the two patient groups by mixing 50-μl aliquots from, respectively, each of the 48 individual controls, the 48 localized, and the 48 metastatic PCa patient samples. Each pool was divided into 20-μl aliquots and stored at −80 °C until further use. Healthy volunteer (control) pools were used for analytical studies and assay-optimization purposes.
Synthetic Peptides
All peptides were synthesized, purified (>98% purity), and quality/purity-controlled at the Memorial Sloan Kettering Cancer Center microchemistry and proteomics core facility, as described elsewhere (8). Peptide stocks of degradable substrates (supplemental Table S3, top panel) and the corresponding, non-degradable, reference peptide ladders (supplemental Table S3, bottom panel) were reconstituted in 30% acetonitrile/0.1% formic acid and stored at −80 °C. Quantitation was done via amino acid analysis at the Keck Biotechnology core facility (Yale University, New Haven, CT), and stocks were adjusted to 100 pmol/μl for all substrates and to a final concentration of 10 pmol/μl for each peptide contained in each of the three reference mixtures (five peptides in the “A1AT-derived ladder” mixture, five in the “C3F-ladder,” and five in the “CLUS2-ladder”). Designer peptide substrates (the design concept is explained in the “Results” section, and the sequences are listed in supplemental Table S4) and corresponding, non-degradable reference peptides were similarly prepared and quantitated; final concentrations were 100 pmol/μl, except for the [EP]-substrate (200 pmol/μl).
Sequence-specific Exopeptidase Activity Test
Assays were performed as described elsewhere (8, 22). Briefly, individual or pooled serum samples (frozen 20-μl aliquots for one-time use) were thawed on ice, and 5 μl was transferred to 0.2-ml polypropylene tubes (eight-tube strips from USA Scientific, Ocala, FL). Substrate and reference peptide stock solutions were separately diluted into water (2 to 10 μl final volume each) to yield the selected concentrations and were then added to the sera. The tubes were capped and incubated at room temperature for the relevant period of time. The amounts of added peptides per reaction and incubation times were as follows: 100 pmol degradable C3f plus 2 pmol each of the C3f-derived, non-degradable (all-D) reference peptides incubated for 1 h; 100 pmol degradable A1AT plus 10 pmol each reference peptide incubated for 15 h; and 100 pmol degradable CLUS2 plus 2 pmol each reference peptide incubated for 2 h. Upon completion of the reactions, serum peptide profiling was done by means of magnetic particle-assisted peptide extraction and processing using 0.3 mg of Dynabeads RPC-18 (Life technologies, Carlsbad, CA, catalog no. 102.01) per reaction followed by a MALDI-TOF mass spectrometric read-out (Bruker, Bellerica, MA, AutoFlex) (8, 22–25). Spectra were taken in reflectron mode geometry under 20 kV (16.45 kV during delayed extraction) of ion accelerating and −1.4 kV multiplier potentials and with gating of mass ions set to m/z = 500, as described. Delayed extraction was maintained for 80 ns to give time lag focusing after each laser shot. Daily robotic and mass spectrometric performance tests were done using Sigma human reference serum, and the effective laser energy delivered to the target was adjusted as necessary. Ratios were then calculated of the normalized ion intensities of the degradation product (“degradable”) peaks over the normalized ion intensities of the corresponding reference (“reference”) peptide peaks for each rung of the ladder (i.e. each of the individual peptides constituting the five-member nested set) (22, 25). Analyses were performed in triplicate or more, as indicated in “Results” and the figure legends.
Freeze-frame Analysis of Aminopeptidase Activity Using Designer Peptide Substrates
Pooled serum samples (frozen 20-μl aliquots for one-time use) were thawed on ice, and 5 μl was transferred to 0.2-ml polypropylene tubes (eight-tube strips). Experiments were always performed in triplicate or more. Substrate and reference peptide stock solutions were separately diluted into water (10 μl final volume each) to yield the selected concentrations and were then added to the sera and incubated at room temperature for 1 h. 100 pmol degradable peptide ([RP]-G-S-SRSRSRSRSR or any others listed in supplemental Table S4) and 2 pmol non-degradable CLUS2-D8 reference peptide (supplemental Table S3) were typically used. Upon completion of the reactions, serum peptide profiling was done using magnetic particle-assisted peptide extraction and processing with MALDI-TOF mass spectrometric read-out as described above. Ratios were then calculated of the normalized ion intensities of the degradation product (P) peak over the normalized ion intensities of the product peak plus the remaining substrate (S) peak. This ratio (P/P+S) denotes substrate conversion (as a percentage or as a fraction of 1.0) over a given time period.
Multiplexed assays were carried out sometimes, wherein three or more different designer substrates were incubated in pooled serum samples simultaneously for product read-out. Group 1 included 100 pmol each of [RP]- and [LP]-peptides and 200 pmol [EP]-peptide, incubated for 30 min; group 2 contained 100 pmol each of [A]-, [F]-, [L]-, [M]-, [R]-, and [Y]-peptides, incubated for 30 min; group 3 consisted of 100 pmol each of [E]-, [K]-, and [DS]-peptides, incubated for 3 h. When used as a substrate, [G]-peptide was incubated separately for 4 h.
Serum samples from individual patients and healthy controls were processed and spotted on the MALDI target plates with the aid of a Genesis Freedom 100 liquid handler (Tecan, Morrisville, NC) (23). Three complete sets (48 control, 48 primary PCa, and 48 metastatic PCa patient samples) were assayed after sample randomization on two 96-well plates per replicate, with identical randomization on the MALDI target plates. Samples (frozen 20-μl aliquots for one-time use) were thawed on ice, and 5 μl of each sample was transferred to the wells of 0.2-ml polypropylene Template III PCR half-skirted, 96-well microtiter plates (USA Scientific). A mixture of three different non-degradable peptides containing 1 pmol CLUS-D8, 1 pmol CLUS-D10, and 3 pmol A1AT-D21 was prepared, and 1.5 μl of the mix was added to each well. [RP]-peptide substrate (supplemental Table S4) stock solution was separately diluted into water (10 μl final volume) to yield a final concentration per sample of 100 pmol peptide. Upon completion of the reactions, magnetic particle-assisted peptide extraction and processing before MALDI-TOF mass spectrometric analysis was performed, as described above. Ratios (P/P+S) were calculated also as described above.
Assaying Activity of Immuno-immobilized DPP4 from Serum
DPP4 was immuno-captured from various serum pools using immobilized anti-DPP4 antibody and assayed while bound to the beads after extensive washing to remove all other serum components. To this end, 10 μg of polycloncal antibody (R&D Systems, Minneapolis, MN, catalog no. AF1180) was bound to 1.5 mg of Dynabeads® Protein G coated superparamagnetic beads (Invitrogen, catalog no. 10007D) for 30 min while rotating. This was followed by washing and resuspension in 200 μl of binding buffer, as per the manufacturer's instructions. 40 μl of serum and 60 μl of 1× PBS were then added to 40 μl of the antibody-coated bead suspension and incubated for 2.5 h at 4 °C on a rotating wheel. Beads were collected with a magnet, extensively washed, and resuspended in 100 μl of 1× PBS containing 1 mg/ml of bovine serum albumin; 10 μl of bead suspension was then taken, and 100 pmol degradable [RP]-peptide was added. This was followed by a 30-min incubation at room temperature, solid-phase peptide extraction/processing, and a MALDI-TOF MS read-out, all as described above. Ratios (P/P+S) were calculated also as described above.
Assaying Recombinant DPP4 Activity in Immuno-depleted Serum
DPP4 was immuno-depleted from various serum pools (40 μl each) by two sequential rounds of capture and removal using immobilized anti-DPP4 antibody. Immuno-capture was done exactly as described above, but the beads were discarded, and a second round of capture followed. DPP4-immuno-depleted serum was then assayed for DPP4 activity as described (i.e. degradation of [RP]-peptide) and taken for reconstitution experiments only when the remaining activity was ≤5% of the original. 20 ng of recombinant (rec) DPP4 (R&D Systems, catalog no. 1180-S.E.-010), or the same volume of rec DPP4-free assay buffer, was then added to 40 μl of depleted serum and pre-incubated for 30 min at room temperature. To each sample, we added 100 pmol degradable [RP]-peptide; samples then underwent 30 min of incubation at room temperature, solid-phase peptide extraction/processing, and a MALDI-TOF MS read-out, all as described above. Ratios (P/P+S) were calculated also as described above.
Assaying Recombinant Aminopeptidase Activities in Serum Filtrates
Each of the PCa pools (control, primary, and metastatic) was thawed on ice, and a 40-μl aliquot was transferred to a 0.5-ml polypropylene tube containing 460 μl of 1× PBS. Each 500-μl sample was then transferred to an Amicon, Bellerica, MA, Ultra-0.5 centrifugal filter with a 10K membrane (catalog no. UFC501024) and spun in a centrifuge at 14,000g for ∼5 min at 4 °C, until the volume was concentrated down to ∼100 μl and the filtrate (∼400 μl) was concentrated down to ∼100 μl using a Savant speed vacuum system (SC11A/UVS400). Each concentrate was divided into two 40-μl aliquots, and we added 10 ng of rec DPP4, rec DPP9, or rec ANPEP (R&D Systems, catalog nos. 5419-S.E.-10 and 3815-ZN-010) to one and the same volume of protease-free reaction buffer to the other. Both samples (plus and minus rec aminopeptidase) were incubated for 30 min at room temperature in the presence of 100 pmol [RP]-peptide or another designer peptide substrate, as needed. Upon completion of the reactions, serum peptide profiling was done using magnetic particle-assisted peptide extraction and processing with MALDI-TOF mass spectrometric read-out as described above. Ratios (P/P+S) were calculated also as described above.
Serum DPP4 and PSA Levels
Total DPP4 measurements blinded to outcome were performed using the Human DPPIV/CD26 Quantikine ELISA Kit (R&D Systems, catalog no. DC260), as per the manufacturer's instructions. Total PSA measurements blinded to outcome were performed using the dual-label DELFIA® Prostatus total/free PSA assay (26) from PerkinElmer Life Sciences (Turku, Finland), which was calibrated against the WHO 96/670 standard.
Statistical Analysis
In order to determine whether protease activity was associated with the risk of primary prostate cancer or metastatic prostate cancer after adjusting for total PSA, we utilized multivariable logistic regression. Regression analyses were conducted using Stata 12 (Stata Corp., College Station, TX).
RESULTS
Mouse Serum Aminopeptidase Activity Changes Associated with Prostate Tumor Development
Owing to low or nonexistent variability in genetics, rearing conditions, age, disease and treatment histories, and many other confounding factors that are frequent sources of bias in biomarker discovery and validation studies, we believed that mouse models of cancer that carry defined molecular lesions in an otherwise uniform genetic background might be a better source for biomarker studies. As increased Myc gene copy numbers have been observed in human prostate cancer, we decided to analyze proteolytic activities in serum samples from transgenic mice expressing human c-Myc in the mouse prostate under control of a composite probasin promoter (PB-MYC; see supplemental Table S1), resulting in malignancies that share molecular features with human prostate tumors (27). Transgene c-Myc expression results in complete penetrance of mouse prostatic intraepithelial neoplasia, which progresses to invasive adenocarcinoma when the mice are within 6 to 12 months of age.
We used three isotopically labeled peptide substrates (see supplemental Table S3), modeled after abundant serum peptides (7) and previously shown to be uniquely degraded in sera from patients with metastatic thyroid cancer relative to matched controls (8), to measure activities of exopeptidase panels in 12-month-old PB-MYC mice and wild-type (WT) littermates of the same age. The assays were carried out under strictly controlled conditions using semi-automated MALDI-TOF mass spectrometric analysis as a read-out of the resulting patterns. Each fragment was quantitated through comparison with fixed amounts of double-labeled, non-degradable internal standards (all-d-amino acid peptides; see supplemental Table S3) spiked into the serum samples at the same time as the substrates (8, 22). Following incubation in pooled sera from the PB-MYC or WT groups, degradative patterns of the CLUS2 and C3F peptides were calculated (shown in Figs. 1A and 2A) and indicated ≥2-fold quantitative differences for fragments CLUS-12 and C3F-15 between cancer (red bars) and controls (blue bars). No such differences between disease and control groups were observed for any of the A1AT degradation products (supplemental Fig. S1).
Fig. 1.

Sequence-specific exopeptidase activity test (SSEAT) assays of sera from mice with prostate cancer and corresponding wild-type strains using a clusterin fragment as substrate. A, PB-MYC, 12-month-old mice expressing human c-Myc in the prostate under control of a composite probasin promotor. B, PTEN−/−, 12-month-old Pten prostate conditional null (knockout) mice; PTEN−/−;p53−/−, 6-month-old Pten and p53 conditonal null (double knockout) mice. WT, wild-type strains with the same genetic background as the corresponding transgenic or knockout mouse strains. CLUS2 is a clusterin fragment (sequence listed at the top): NCBI Protein accession NP-001822 (precursor residues 267–278; C terminus of β-chain, minus Arg-279). Sera were pooled from 18 mice each (PB-MYC or corresponding WT) or 5 mice each (PTEN−/− or PTEN−/−;p53−/− or corresponding WT), and 5-μl samples were incubated with substrate (100 pmol) and 2 pmol of each of the CLUS2-derived, non-degradable (all-D) reference peptides (supplemental Table S3: CLUS2-D8, FPKSRIV; CLUS2-D9, FFPKSRIV; CLUS2-D10, FFFPKSRIV; CLUS2-D11, HFFFPKSRIV; CLUS2-D12, RPHFFFPKSRIV) for 2 h at room temperature. Degradation products were analyzed via magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry (22, 24, 25), and ratios were calculated of the normalized ion intensities of the degradation product (“Degradable”) peaks over the normalized ion intensities of the corresponding reference (“Reference”) peptide peaks for each rung of the ladder (i.e. each of the individual peptides constituting the five-member nested set) (22, 25). Sequential amino acid truncations in the ladder are indicated. Replicate analyses (n = 6) were performed, and standard deviations are displayed as error bars.
Fig. 2.
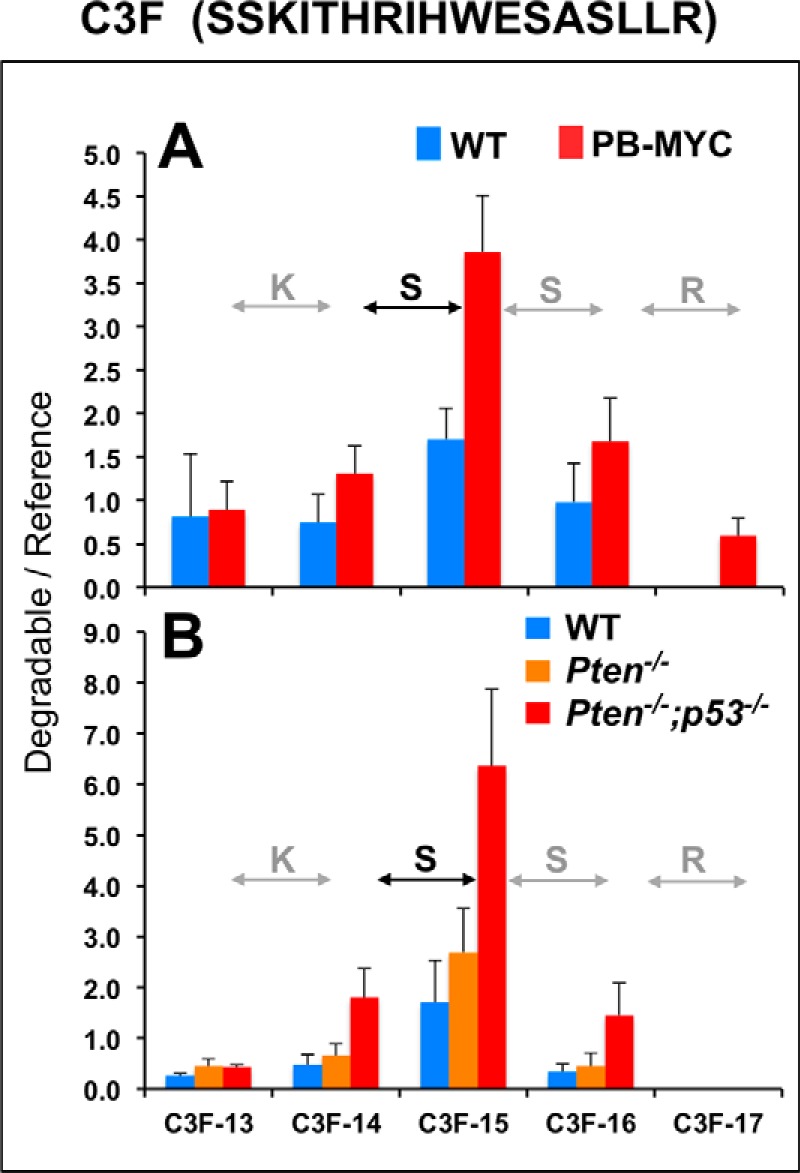
SSEAT assays of sera from mice with prostate cancer and corresponding wild-type strains using a complement C3 fragment as substrate. All mice are the same as described for Fig. 1. C3f is a complement C3 fragment (sequence listed at the top): NCBI Protein accession P01024 (residues 1304–1320). Pooled serum samples (5 μl each; as in Fig. 1) were incubated with substrate (100 pmol) and 2 pmol of each of the C3f-derived, non-degradable (all-D) reference peptides (supplemental Table S3: C3f-D13, ITHRIHWESASLL; C3f-D14, KITHRIHWESASLL; C3f-D15, SKITHRIHWESASLL; C3f-D16, SSKITHRIHWESASLL; C3f-D17, SSKITHRIHWESASLLR) for 1 h at room temperature. Degradation products were analyzed via magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry, and ratios were calculated of the normalized ion intensities of the degradation product (“Degradable”) peaks over the normalized ion intensities of the corresponding reference (“Reference”) peptide peaks for each rung of the ladder. Sequential amino acid truncations in the ladder are indicated (note that C-terminal Arg is removed first, followed by sequential S-S-K removal at the N terminus). Replicate analyses (n = 6) were performed, and standard deviations are displayed as error bars.
To verify that the observed differences in peptide degradation patterns were the result of PCa-induced changes in serum exopeptidase activities and not from a possible unrelated effect of the transgene chromosomal insertion or chance c-Myc-deregulated expression of proteases, we decided to analyze a second, independent mouse model for prostate cancer with a different molecular lesion or lesions. The PTEN and p53 tumor suppressors are among the most commonly inactivated or mutated genes in human prostate cancer. Prostate-specific, conditional inactivation of mouse Pten and p53 has been obtained using the Cre/loxP technique to generate transgenic mice expressing Cre after 7 weeks of age in prostatic epithelium (28). We obtained PtenloxP/loxP;Pb-Cre4 and p53loxP/loxP;Pb-Cre4 mice, hereinafter referred to as Pten−/− and p53−/−, as well as mice that had a combined Pten/p53 inactivation (referred to as Pten−/−;p53−/−) (28). Pten−/− mice develop an indolent invasive form of prostate cancer that progresses slowly, whereas Pten−/−;p53−/− mice develop rapidly progressive, invasive, high-volume prostate cancer.
Serum samples were prepared from both types of genetically altered mice (Pten−/− and Pten−/−;p53−/−) at 6 months of age, as well as from WT mouse controls that displayed normal prostate histology, to monitor the degradation of labeled CLUS2 and C3F peptides. Pronounced quantitative differences between cancer (red bars, progressive invasive; orange bars, indolent invasive) and controls (blue bars) were again noted for fragments CLUS-12 (Fig. 1B) and C3F-15 (Fig. 2B). Strikingly, in an invasive type-dependent manner, these two particular peptide fragments accumulated the most, by far, after incubation of the substrates in sera from mice with progressive invasive prostate cancer. As increased accumulation must be the result of a reduced rate of Arg-Pro dipeptide (CLUS2) and Ser (C3F) release from the N-terminal side of the peptide substrates, it can be attributed to equally reduced activities of aminopeptidases. Thus, our results suggest that changes in serum aminopeptidase activities are associated with prostate tumor development in mice and, secondly, that these changes are not unique to the molecular pathway responsible for the initiation of prostate tumorigenesis.
Freeze-frame Analysis of Serum Aminopeptidase Activities Cleaving Specific Amino Acids or Dipeptides
In previous studies, when analyzing large numbers of patient serum samples, we carried out multivariate statistical analysis of the full array of fragments (e.g. Ref. 24) generated after incubation of three different substrate peptides in each sample and followed that analysis with accurate quantitation of each fragment, to create a functional biomarker that allowed excellent class predictions using machine learning methods (8). Unfortunately, we did not have access to such large sample numbers in the mouse studies described here, which precluded a similar statistical analysis, as it would have been badly underpowered. Instead, we decided to focus on characterizing and quantifying individual steps of the serum aminopeptidase-dependent degradation of peptides, in particular the step(s) that resulted in differential amounts of certain fragments between cancer and control groups as already determined via steady-state quantitation (see above). This cannot be easily done using the assays we have described previously (8, 22) and above, as peptide degradation in serum is a continuous, highly time-dependent process that must be stopped at the site of a particular residue in order to monitor the precise kinetics and quantify the removal of that particular residue by one or more of the aminopeptidases in blood. We refer to such an assay as “freeze-frame analysis” of aminopeptidase-effected peptide degradation and have developed novel peptide substrates for this purpose, as described below.
Our initial results using two mouse models of prostate cancer with different molecular lesions indicated slower rates of N-terminal release of an Arg-Pro (RP)-dipeptide (CLUS2 peptide) and Ser (C3F) when incubated in sera of the tumor-bearing animals. We therefore synthesized novel substrate peptides that consisted of the following parts: (i) a non-degradable tail consisting of five d-arginines and five d-serines to create a hydrophilic, highly positively charged peptide that yielded a strong ion signal in MALDI-TOF-MS (yellow bar in the substrate diagram depicted in Fig. 3A and the residues in blue in the sequences listed in supplemental Table S4); (ii) an N-terminal cleavable moiety (e.g. Arg-Pro, Ser, or any other amino acid or selected dipeptides) indicated in bold red between brackets in supplemental Table S4; (iii) an identification tag consisting of one or two d-amino acids (in green in supplemental Table S4) to allow multiplexing of assays (i.e. to distinguish among the resulting cleavage products of the various assays); and (iv) a Gly residue that connects the N-t cleavable moiety with the entire peptide consisting of d-amino acids. Because Gly is achiral, the bond with the N-t residue or dipeptide is proteolytically cleavable, but the bond with the d-sequence is not. When a peptide substrate configured this way is added to serum or any other biological fluid containing aminopeptidases (and certain other proteases), the substrate (S) is converted into a stable product (P) by cleavage of just a single peptide bond in a time-dependent manner. Because both substrate and product have virtually identical MALDI-TOF ionization characteristics, conversion at any time point can be calculated using the following equation: P/P+S (Fig. 3A), where P and S are the intensities of, respectively, the product and substrate ion peak in the mass spectrum.
Fig. 3.

[Arg-Pro]-specific dipeptidyl aminopeptidase activity is reduced in sera from mice with aggressive prostate cancer. A, the peptide substrate (RP-G-SSRSRSRSRSR) designed for assaying the activity of [Arg-Pro]-specific aminopeptidases consists of (i) a non-degradable tail consisting of five d-Arg and six d-Ser; (ii) an N-terminal cleavable dipeptide (l-Arg and l-Pro); and (iii) a glycine residue that connects the N-t cleavable moiety with the all-d-peptide. Substrate (RPGSSRSRSRSRSR; highlighted in supplemental Table S4) and product (GSSRSRSRSRSR; shown as a yellow bar in the diagram) had virtually identical MALDI-TOF ionization characteristics. The percent substrate conversion was calculated using the following equation: P/P+S, where P and S are the intensities of the product and substrate ion peaks in the spectrum. B, MALDI-TOF mass spectra illustrating peptide substrate conversion in sera from mice with cancer and controls. PB-MYC, 12-month-old mice expressing human c-Myc in the prostate under control of a composite probasin promotor; WT, wild-type strain with the same genetic background as the PB-MYC transgenic strain. Sera were pooled from 18 mice each (PB-MYC or WT) and 5-μl samples incubated for 30 min at room temperature with the substrate (100 pmol) and 2 pmol of CLUS2-D8 (all-D) reference peptide (supplemental Table S3) as an internal standard. Reaction products were analyzed via magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry (22, 23). C, median substrate conversion (P/P+S) was ∼15% lower in sera from 18 individual mice with aggressive prostate cancer than in 18 individual healthy littermates. Boxplots are shown, and analysis of variation (Wilcoxon rank-sum test) was carried out. Each data point is averaged from triplicate analyses.
To optimize reaction conditions, we synthesized a non-degradable product with d-Ser as a tag and a non-degradable substrate, with the same tag and d-Arg-d-Pro at the N-t (see the bottom of supplemental Table S4), that can be spiked into serum in any amount or product-to-substrate ratio to determine the lower level of detection and the linear range of these assays. First, the lower level of detection was determined as 200 fmol of substrate or product spiked into 5 μl of serum and retrieved after 30 to 120 min using our standard reversed phase, particle-assisted, automated platform (23) for the MALDI-TOF-MS read-out (data not shown). We estimated that 10- to 20-fold of that amount (2 to 4 pmol per 5 μl of serum) would provide us with a stable quantitative readout, and we thus spiked 5 pmol of product together with 95 pmol of substrate into serum to mimic an assay result that would yield a 5% conversion rate. The calculated P/p + S value (from ion intensities) was 5.4% (left-hand panel in supplemental Fig. S2). We then carried out similar mixed analyses to mimic conversion rates of 10%, 25%, 50%, and 80% and obtained calculated values of 9.8%, 25.9%, 47.9%, and 81.7%, respectively (supplemental Fig. S2), with very good linear fit in the range of 5–100 pmol (R2 = 0.998 for the substrate and R2 = 0.991 for the product).
Reduced DPP4 Activity in Sera from Mice with Tumors of the Prostate
A peptide substrate with an N-t Arg-Pro dipeptide cleavable moiety ([RP]-pep), as described above, shown in Fig. 3A, and highlighted in yellow in supplemental Table S4, was incubated with serum pools from tumor-bearing PB-MYC mice or from healthy littermates. The MALDI-TOF mass spectra shown in Fig. 3B clearly illustrate that the substrate-to-product conversion rate was significantly slower in serum from the animals with cancer. Similarly, a peptide with a cleavable N-t serine (S-pep; supplemental Table S4) was used as a substrate and incubated in mouse sera from cancer and control groups, but no degradation was observed in either case after a 1-h reaction (supplemental Fig. S3). We therefore decided to only continue investigating the serum aminopeptidase activity or activities responsible for cleaving N-terminal Arg-Pro. The boxplots in Fig. 3C show that median conversion rates of the RP-pep in sera from 18 PB-MYC mice and 18 healthy controls were 0.64 and 0.75, respectively, a decrease of ∼15% with significant overall variation between the two groups (t test; p = 0.0003). Of note, in the course of all these analyses, we never detected a degradation product of the [RP]-peptide that had only the Arg removed (i.e. resulting in a “P-pep” product), which is in keeping with our earlier observations on the degradation patterns of the CLUS2 peptide in which a fragment with an N-t Pro was also never observed. These findings point toward the action of X-Pro-dipeptidyl peptidase(s) (29–31) in serum that removes N-t Arg-Pro from CLUS2 and RP-pep and suggest that this activity is lower in mice with prostate tumors.
DPP4 and DPP7 are two members of the XP-dipeptidyl peptidase family of proteases previously detected in serum and plasma, with DPP4 likely the more prominent of the two (29, 32). We obtained DPP4 double knockout (DPP4−/−) and WT control mice (33, 34) and determined [RP]-pep conversion rates in sera for both in the presence and absence of sitagliptin, a highly specific inhibitor of DPP4 (but not of DPPs 7, 8, and 9) that is commonly known under the trade name Januvia® and is used as an oral antihyperglycemic agent for the treatment of type 2 diabetes (30, 35). The results in Fig. 4 show that both a DPP4 double knockout and a specific inhibitor reduced the conversion rates from 62% to 2% to 3%, thus unequivocally identifying the protease in mouse serum that exhibits reduced activity in animals with advanced prostate cancer as DPP4. The remaining conversion activity likely results from fairly low levels of DPP7 or other dipeptidyl peptidases in blood.
Fig. 4.
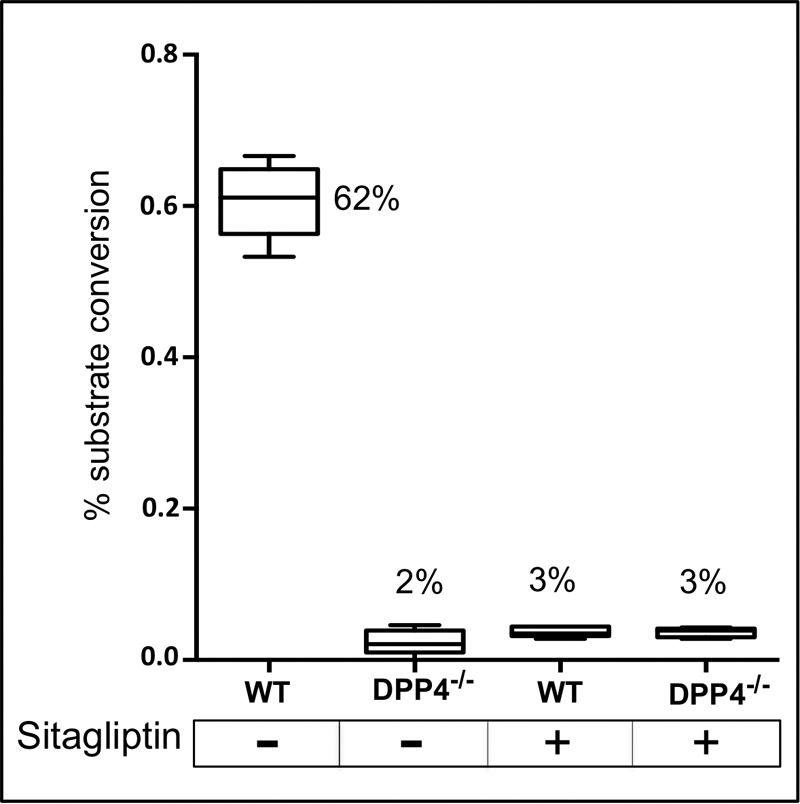
[Arg-Pro]-specific dipeptidyl aminopeptidase activity in mouse serum identified as DPP4. Peptide substrate (RP-G-SSRSRSRSRSR) designed for assaying the activity of [Arg-Pro]-specific aminopeptidases, assay conditions, and calculation of percent substrate conversion are described in the text and in Fig. 3. DPP4−/−, dipeptidyl aminopeptidase 4 null mice; WT, wild-type strain with the same genetic background as the DPP4 knockout strain. Serum samples were pooled from six animals each and analyzed in triplicate in the presence (+) or absence (−) of 10 μm sitagliptin, a highly specific DPP4 inhibitor (35) from Merck & Co (under the trade name JANUVIA®); boxplots and median percent conversion values are shown.
Activities but Not Amounts of DPP4 Are Reduced in Sera from Patients with Metastatic Prostate Cancer
To test whether the observed changes in DPP4 activities are relevant to human PCa, we analyzed three different sample cohorts: (i) one from 48 patients with localized disease (i.e. with primary tumors) collected less than a week prior to prostatectomy; (ii) one from 48 patients with mCRPC collected prior to the initiation of chemotherapy; and (iii) one from 48 age-matched, healthy controls (supplemental Tables S2 and S5). Comparison of [RP]-pep conversion rates in pooled sera from each cohort indicated appreciably less DPP4 activity in mCRPC patients than in both other groups; patients with localized PCa and healthy controls could not be distinguished on the basis of this assay (Fig. 5, upper panel on the left, shown on a pastel green field). Conversions could be almost totally blocked in all three groups by the addition of sitagliptin (supplemental Fig. S4), providing further evidence that DPP4 activity was being modulated.
Fig. 5.
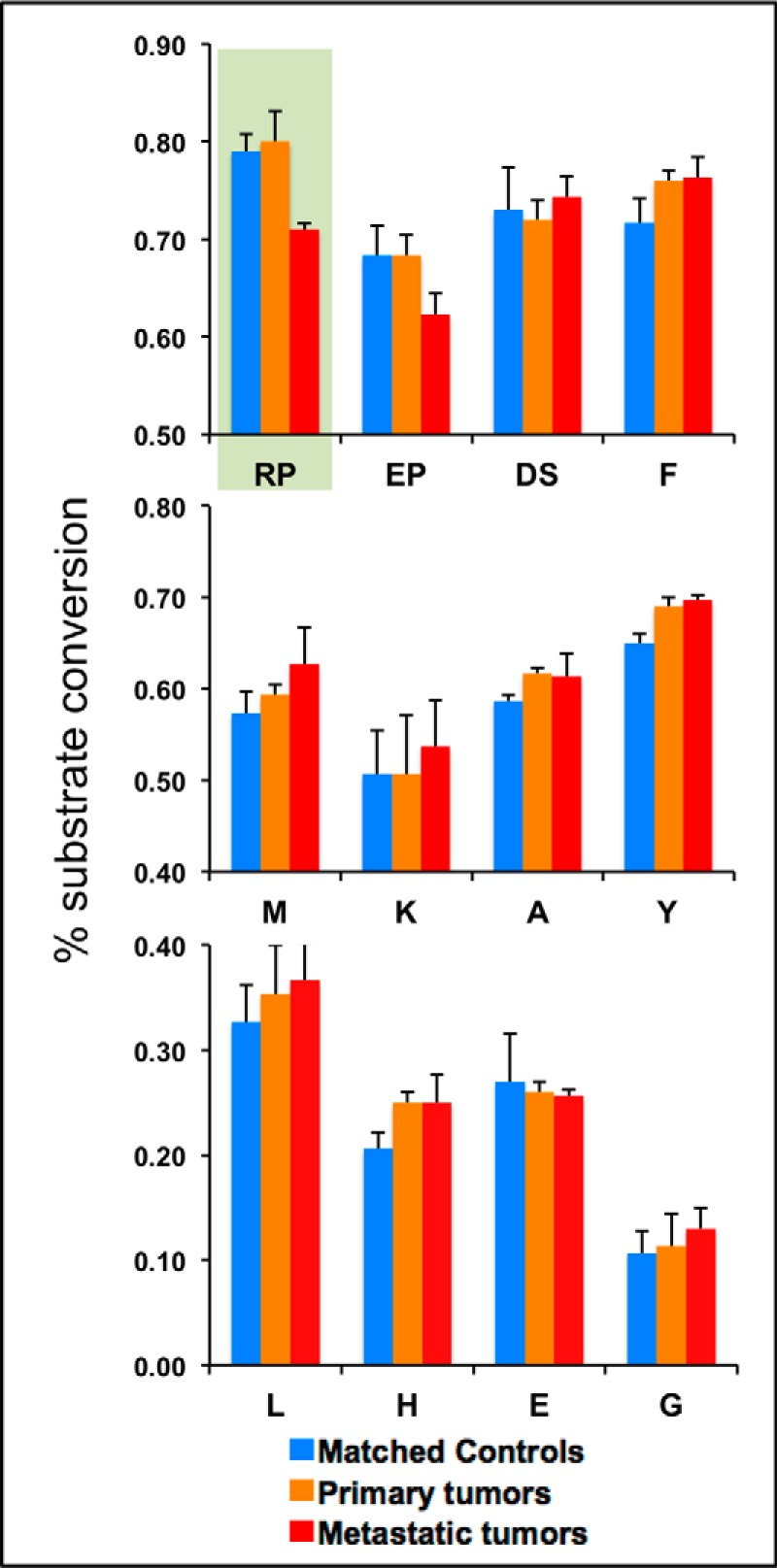
Selected aminopeptidase activities in sera from prostate cancer patients and matched controls. Peptide substrates designed for assaying the activity of specific aminopeptidases consist of a non-degradable tail and an N-terminal cleavable amino acid (F, M, K, A, Y, L, H, E, or G) or dipeptide (RP, EP, or DS) (see text, Fig. 3A, and supplemental Table S4); assay conditions and calculation of percent substrate conversion were as in Fig. 3, with modifications as described. Serum pools were prepared from 48 individual patients with localized prostate cancer (“primary tumors”; color coded in orange), 48 patients with metastatic, castrate-resistant prostate cancer (“metastatic tumors”; red), or 48 age-matched men without evidence of disease (“matched controls”; blue), and 5-μl samples were incubated with multiplexed substrates under the following conditions. Group 1, 100 pmol [RP]-peptide and 200 pmol [EP]-peptide, incubated for 30 min; group 2, 100 pmol each of [A]-, [F]-, [L]-, [M]-, [R]-, and [Y]-peptides, incubated for 30 min; and group 3, 100 pmol each of [E]-, [K]-, and [DS]-peptides, incubated for 3 h; 100 pmol [G]-peptide was incubated separately for 4 h. Replicate analyses (n = 3) were performed, and standard deviations are displayed as error bars.
We then addressed the question of whether the reduced activity of DPP4 in the serum of mCRPC patients is unique or more generally shared among all blood-based aminopeptidases. Because we did not have a good estimate of what aminopeptidases are present in blood and because of the fairly broad and overlapping specificities of several of these proteases (31), we decided to generate a small library of synthetic peptide substrates using the same design concept that was used for [RP]-pep and [S]-pep during the mouse studies. A total of 14 additional peptides were synthesized: 11 carrying a single amino acid as the cleavable moiety at the N-t, and 3 with cleavable dipeptides (supplemental Table S4). Conditions such as serum volume, substrate amounts, and reaction times were empirically optimized to yield MALDI-TOF-MS peaks with ion intensities similar to those of the [RP]-pep substrate and product (supplemental Figs. S5A–S5D). Individual assays were then multiplexed in serum samples based on optimal reaction times and resolution of all substrate and product peaks (m/z values); examples are shown in supplemental Figs. S6A–S6C. Conversion rates in pooled sera of the same three cohorts described above were then established for 11 different substrates (Fig. 5). Unlike DPP4-dependent [RP]-pep converting activity that is suppressed in patients with metastatic disease (Met), all but one of the newly tested substrates had similar or slightly elevated conversion rates in the Met group. Only the N-t Glu-Pro dipeptide was cleaved less efficiently in the Met pool than in the control pool, undoubtedly also the result of reduced DPP4 activity, as this protease is known to cleave Glu-Pro as well (30), and the conversions could be blocked by sitagliptin (data not shown).
Analysis of DPP4 activity in all 144 patient and control samples was then performed; the results (supplemental Table S5) are shown as dot plots in Fig. 6A. Conversion rates of the controls (Ctrl), patients with localized disease (Pri), and mCRPC patients (Met) had median values of 0.834, 0.801, and 0.719 (a 17% decrease versus controls), respectively, entirely in line with the changes associated with prostate tumor development in mouse models. The observed differences when comparing controls and Met patients or when comparing Pri and Met patients were statistically significant, having p values of 1.02E-03 and 1.1E-05. The simplest explanation for this observation would be reduced DPP4 levels in blood serum of patients with metastatic disease. Surprisingly, serum DPP4 levels in the same three groups, as determined by ELISA, were nearly identical, with median values of 555, 535, and 561 ng/ml, respectively (Fig. 6B), suggestive of (i) either post-translational modifications (PTMs) or allosteric changes that adversely affect DPP4 activity and/or (ii) the presence of a DPP4 inhibitor in a disease-stage-specific manner.
Fig. 6.

DPP4 activities but not amounts are reduced in sera from patients with metastatic prostate cancer. A, the peptide substrate (RP-G-SSRSRSRSRSR) designed for assaying the activity of DPP4 consists of a non-degradable tail and an N-terminal cleavable dipeptide (see text, Fig. 3A, and supplemental Table S4); assay conditions and calculation of percent substrate conversion were as in Fig. 3. Median percent substrate conversion (P/P+S) was ∼15% lower in sera from 48 individual patients with metastatic castrate-resistant prostate cancer (“Met”) than in 48 patients with primary, localized disease (“Pri”) and 48 age-matched men with no evidence of disease (“Ctrl”). Analysis of variation (Wilcoxon rank-sum test) was carried out. Each data point is averaged from triplicate analyses. B, serum DPP4 levels (ng/ml) were determined via ELISA, as described under “Experimental Procedures.” Each data point was averaged from duplicate analyses.
A Low-molecular-weight Inhibitor of DPP4 in Sera from Patients with Metastatic Prostate Cancer
To address the molecular basis or source of DPP4 inhibition in the serum of patients with advanced PCa, we carried out a series of biochemical analyses. First, we measured the activity of DPP4 that was affinity-captured from pooled sera using immobilized anti-DPP4 antibodies that did not block the active site. This on-bead assay is depicted in Fig. 7A. After the capture of DPP4 from each of the three serum pools, all other proteins and peptides were washed out. Subsequently, assay substrate ([RP]-pep) and PBS were added to the enzyme while it remained immobilized on magnetic particles, and the reaction product and uncleaved substrate were quantitated via MALDI-TOF-MS after 30 min of incubation. Whereas DPP4 activity can be measured quite effectively in this manner, we were unable to detect any differences between either of the two PCa groups and controls (Fig. 7A), indicating that PTMs or allosteric differences are unlikely to account for the reduced DPP4 activity in blood serum of mCRPC patients. In a second analysis, DPP4 was immuno-depleted from the serum pools by two rounds of capture on antibody-coated magnetic particles (see Fig. 7B), and 10 ng of rec DPP4 was then added per 20 μl of depleted serum, after which activity was measured in a standard assay (inset at right-hand side of bottom panel: rec DPP4). Depleted sera had very low levels of DPP4 activity (see inset: no rec DPP4). This procedure and assay were carried out for each of the two serum pools derived from the corresponding patient and control groups. The result shows a relative drop of rec DPP4 activity in depleted serum from patients with metastatic PCa (red bar), highly suggestive of the presence of a blood-based DPP4 inhibitor in sera from mCRPC patients. In a third set of analyses, using a 10-kDa cutoff membrane (see Fig. 7C), filtrates were prepared from serum pools that contained minimal to no DPP4 activity (see inset at right: no DPP4). Rec DPP4 was then added to filtrates of each of the three serum pools and assayed as usual. Again, we observed a relative loss of activity in the Met PCa pool.
Fig. 7.
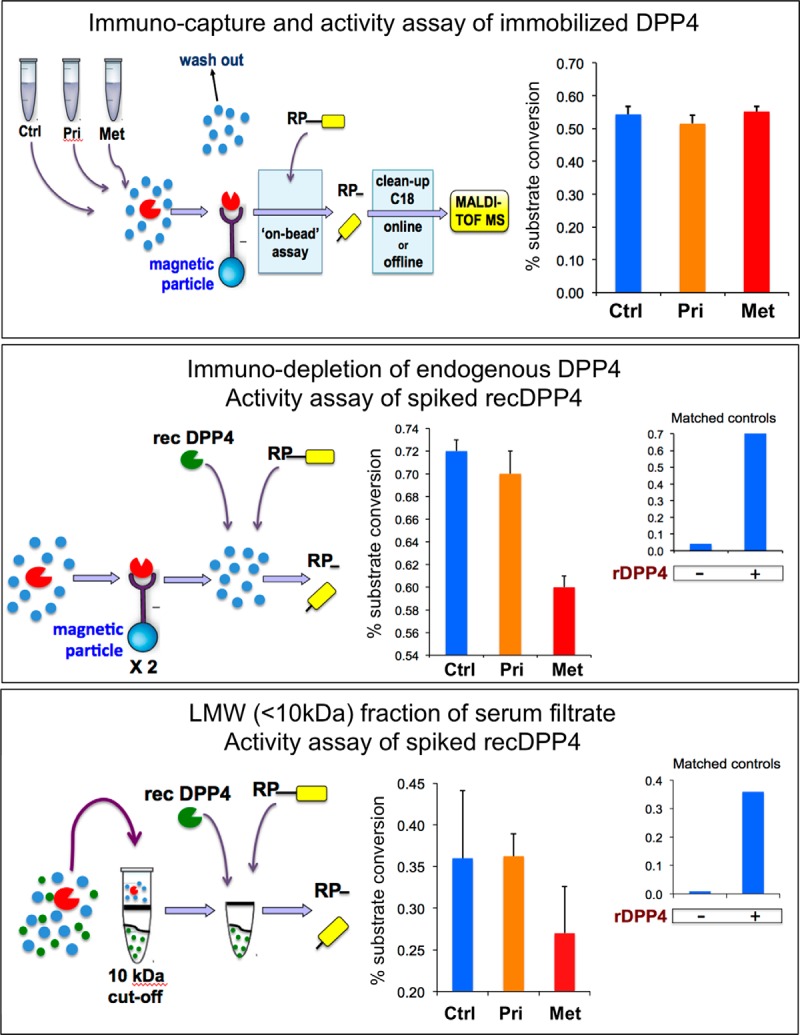
A low-molecular-weight inhibitor of DPP4 in sera from patients with metastatic prostate cancer. A, DPP4 was captured from 40-μl aliquots each of three separate pools of serum samples (Met, 48 patients with metastatic castrate-resistant prostate cancer; Pri, 48 patients with primary, localized disease; Ctrl, 48 age-matched men with no evidence of disease) using magnetic particles coated with anti-DPP4 antibodies. This was followed by extensive washing and on-bead assay of the enzymatic activity using a peptide substrate (RP-G-SSRSRSRSRSR) specifically designed for assaying the activity of DPP4 (see Fig. 3). Reaction products were analyzed via magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry, and the percent substrate conversion (P/P+S) was calculated. Replicate analyses (n = 3) were performed, and standard deviations are displayed as error bars in the graph on the right. B, DPP4 was removed from each of the three separate pools (40 μl each) of serum samples in two rounds of immuno-depletion, in a similar fashion as described above, which reduced endogenous serum DPP4 activity to less than 5% of the regular levels. Subsequent addition of 20 ng of recombinant (r) DPP4 completely restored activity in control samples (inset, right). Results of comparative activity assays of rDPP4, after spiking into depleted serum pools of each patient cohort, are shown in the center graph. C, filtrates (≤10-kDa cutoff) were prepared from each of the three separate pools (40 μl each) of serum, which virtually removed all endogenous serum DPP4 activity. Subsequent addition of 10 ng of rDPP4 to control serum filtrate resulted in substantial enzymatic activity as measured in our standard assay (inset, right). Results of comparative activity assays of rDPP4, after spiking into serum filtrates of each patient cohort, are shown in the center graph.
This last set of experiments was then repeated with filtrates reconstituted with recombinant DPP9, a close family member of DPP4, or with recombinant alanine aminopeptidase, a metalloprotease (supplemental Fig. S7). Whereas the activity of rec DPP9 was selectively inhibited in filtrates of serum from mCRPC patients, rec alanine aminopeptidase was not. Thus the results from the rec DPP4 and other reconstitution experiments are strongly suggestive of the presence of a low-molecular-weight inhibitor of DPP4, and potentially of other XP-dipeptidyl aminopeptidases, in blood serum of patients with metastatic PCa but not (or less so) in patients with localized disease or healthy controls. The inhibitor appears to have had little to no effect on metallo-aminopeptidases.
Potential Benefits of Adding DPP4 Activity to a Prediction Model with PSA
Having established reduced DPP4 activity (Fig. 6) and the likely presence of an inhibitor in serum of mCRPC patients, we wanted to investigate the potential benefit of adding DPP4 activity to a prediction model with PSA. Total PSA levels varied by cancer status as expected (supplemental Table S5); the median total PSA was 1.0 ng/ml for subjects in the healthy control group (interquartile range: 0.6–1.6), 6.7 ng/ml for primary prostate cancer subjects (interquartile range: 4.5–10.0), and 119 ng/ml for metastatic prostate cancer subjects (interquartile range: 67–220). DPP4 activity differed significantly with cancer status (Kruskal–Wallis, p value < 0.001). After we adjusted for total PSA, DPP4 activity remained a significant predictor of cancer status in comparisons of healthy controls to prostate cancer patients with either primary or metastatic prostate cancer (p value = 0.03) and of healthy controls and primary cancer patients to mCRPC patients (p value = 0.001). The interaction between DPP4 activity and total PSA was not significant in either comparison, indicating that the relationship between DPP4 activity and cancer status does not vary with total PSA for healthy controls versus primary or metastatic patients (p value = 0.6) or for healthy controls and primary patients versus mCRPC patients (p value = 0.9). Because age increases with cancer status, we also investigated these two logistic models after adjusting for age. DPP4 activity remained significant after we adjusted for age and total PSA for healthy controls versus primary and metastatic patients (p value = 0.012) and for healthy controls and primary patients versus mCRPC patients (p value = 0.006). It is therefore reasonable to assume that serum DPP4 activity could supplement or be a practical alternative for existing PCa biomarkers to classify cancer subtype and stage or mark a given clinical outcome of interest.
DISCUSSION
It has been our view that degradation patterns contained in the serum peptidome, when measured correctly, hold important information that might have direct clinical utility as a surrogate marker for the detection or classification of cancer (7). In recognition of the fact that endogenous exopeptidase activities contribute to cancer-type-specific information, we subsequently developed a sequence-specific exopeptidase activity test that allowed class prediction of cancer patients versus healthy controls with ≥90% sensitivity and specificity (8). We now describe the next generation of peptide-based freeze-frame reagents and related tests uniquely suited to probe a potentially altered balance of selected aminopeptidases, as opposed to the full array of exopeptidases, and/or their modulators in serum or plasma of patients. This is particularly relevant to cancer, as proteases are well-established components of tumor progression and invasiveness (4, 36, 37).
We also sought to exploit the merits of two well-characterized mouse models of prostate cancer, each with a distinct genetic lesion or lesions, to determine whether broad exopeptidase and targeted aminopeptidase activity tests can reveal proteolytic changes associated with tumor development and whether such changes are relevant to human PCa. One such proteolytic activity, resulting in the removal of an N-terminal Arg-Pro dipeptide from test peptides spiked in mouse sera, was notably impaired in mice with aggressive PCa relative to their cancer-free littermates. This occurred regardless of the molecular pathway responsible for the initiation of tumorigenesis. The results were fully reproducible when using a novel method and synthetic peptide substrate that enabled freeze-frame analysis of this specific, single-step cleavage. This in turn allowed us to quantify a 15% reduction in the activity of an aminopeptidase in blood sera of the PCa group versus a control group, with a significant overall variation (t test; p = 0.0003). By analyzing the same activities in selected knockout mice and evaluating the effect of a specific inhibitor, we subsequently identified the protease in question as DPP4.
Significant levels of DPP4 occur in body fluids, including serum, plasma, and urine (29, 32, 38, 39). Although DPP4 is expressed on numerous cell types, including kidney, liver epithelial cells, and endothelial cells of ventricular microvasculature (34, 38, 40), T cells are believed to provide the majority of its circulating form (also know as soluble CD26) by partially shedding surface CD26 into the blood stream following proteolytic cleavage (38, 41, 42). Known DPP4 substrates include growth factors, chemokines, and peptide hormones such as glucagon-like peptide 1, which helps control blood sugar levels (38, 43–45). In line with the results of our animal studies reported here, abnormal presence/absence, altered levels, and/or altered enzymatic activities of DPP4/CD26 have previously been associated with human disease, including cancer, prompting speculation about potential usefulness as a tumor biomarker (38, 46–53). The earliest observations came from measuring enzymatic activities in serum for a lack of sensitive nucleic acid probes or suitable ELISA reagents, making it difficult to speculate on the underlying reason(s) for the changing levels. Additionally, recent DPP4 mRNA and protein expression analyses in tumor tissues of various origins and disease states have seldom been linked to changes in activity in either tissue or blood. Thus, we set out to establish whether such correlations exist in the blood of PCa patients and controls.
Analysis of DPP4 activity, selected on the basis of reduced capacity in sera of tumor-bearing mice, indicated concordance in the blood of 48 patients with mCRPC relative to 48 patients with localized PCa and 48 age-matched healthy controls. The median value was 10% lower among the mCRPC patients relative to the localized PCa cohort and the age-matched healthy controls (Wilcoxon rank-sum test; p = 0.0001). However, no significant differences in DPP4 serum levels were found (Kruskal–Wallis; p = 0.8), with median values around 550 ng/ml in each group, which is in good agreement with previously reported values of ∼560 to 600 ng/ml in healthy donors (38, 51, 52, 54) and ∼580 ng/ml in prostate cancer patients (52), but in stark contrast to the much reduced levels in colorectal cancer (∼260 to 385 ng/ml), lung cancer (∼370 ng/ml), and pancreatic cancer (∼400 ng/ml) patients (51, 52). Our findings have two major implications. Firstly, reduced cell-surface expression or total absence of DPP4, as previously observed in many metastatic tumors of prostate origin but not in primary tumors or benign prostatic epithelial cells (50, 55), does not translate into noticeably reduced levels in blood. However, this may vary with solid tumors of different origin. Secondly, it must therefore be implied that the reduced DPP4 activity in blood of mCRPC patients is the result of impaired specific activity, most likely brought about by (i) PTMs or allosteric changes impeding enzyme function and/or (ii) the presence of a bona fide inhibitor.
Sialylation of DPP4 is enhanced in elderly individuals (56). Combined with the fact that hypersialylation may inhibit DPP4's proteolytic activity (57, 58), this might explain why serum DPP4 activity tends to decrease with age (29). However, our study showed that DPP4 activity remained a significant predictor of cancer status after adjusting for age (p = 0.006). Likewise, cell-surface sialylation of metastatic tumor cells is also known to occur (59) and might modify CD26 before shedding, even though there is currently no reported evidence of this. Still, we investigated the possibility that DPP4 activity could be inhibited via a PTM-based mechanism. To this end, we measured the activities of endogenous DPP4 isolated from sera of either mCRCP patients or healthy controls. Secondly, we also assayed rec DPP4 in immuno-depleted sera from the same two groups. Even though we cannot exclude the presence of PTMs on serum DPP4, our results strongly suggested that modification status had little to no effect on the specific activity, as it was found to be identical in both groups. We therefore postulated the existence of an inhibitor.
To date, the only known, naturally occurring DPP4 inhibitor in blood is a 115-kDa glycoprotein, glypican-3 (GPC3), which binds to and inhibits the activity of CD26 on the surface of certain cell types (60). Deletion of GPC3 causes Simpson–Golabi–Behmel overgrowth syndrome (61). The GPC3 serum concentration in healthy individuals is ∼4 ng/ml, but it has been found elevated to ∼100 ng/ml in hepatocellular carcinoma patients (62). However, GPC3 is rarely expressed, much less overexpressed, in prostate carcinoma tissues (63–65), making it unlikely that blood levels would increase as a result of PCa metastatic tumor growth.
We furthermore obtained experimental evidence, through reconstitution analyses of rec DPP4 in serum filtrates, of the existence and unique or increased presence in blood serum of mCRPC patients of a low-molecular-weight inhibitor of DPP4 and other dipeptidyl peptidases, but not of metallo-aminopeptidases such as alanine aminopeptidase. It is unlikely that either GPC3, if at all present at elevated concentrations, or most other protease inhibitors in blood would pass through the 10-kDa cutoff membranes used to prepare the filtrates containing this dipeptidyl-peptidase-inhibiting activity, unless a small fragment would retain such activity. We conclude that the dipeptidyl peptidase inhibitor is either a small to medium-sized polypeptide or a small molecule. The identity of this inhibitor is unknown at this point and will be the focus of future investigative efforts.
Another remaining question is whether the impaired activity of DPP4 in blood of mCRPC patients plays any role in tumor pathogenesis and progression. It has been previously noted that loss of DPP4 is associated with melanoma, colon, ovarian, lung, and prostate cancers and that restoring DPP4 expression can suppress tumor growth and enhance the survival of xenograft models (43, 66–69). In vitro studies also indicated that DPP4 inhibition increased the metastatic potential of some cancers in general and enhanced the invasion and metastasis of PCa cell lines in in vitro and in vivo metastasis assays in particular (68, 70–72). More specifically, it has been suggested that DPP4 inhibits the malignant phenotype of PCa cells by blocking the β-fibroblast growth factor signaling pathway and/or by degrading stromal derived growth factor-1, and that inhibition of DPP4 might be a trigger for PCa metastasis (71, 72). It should be noted that the experimental findings listed above pertain to tissue or cell-associated DPP4/CD26, not the circulating form that we have assayed and found to be the target of an inhibitor. Of course, if the inhibitor derives from metastatic tumor tissue, it may adversely affect cell-bound DPP4 activity just as well, perhaps contributing to cancer progression. Thus declining DPP4 activity in serum over time could potentially serve as an indicator of the aggressiveness of disease, an idea further substantiated by recent observations that increased plasma DPP4 is associated with better survival in several types of cancer (52).
In summary, we have described new peptide-based reagents uniquely suited to probe a potentially altered balance of selected aminopeptidases, such as DPP4, and/or their modulators in the serum or plasma of cancer patients. These tests offer the option of a targeted, proteolytic readout that may be either a supplement or a practical alternative for classical biomarker discovery and verification techniques. Future assays may also have diagnostic value, either alone or in combination with existing tests, for identifying cancer subtype and stage, or they may mark a given clinical outcome of interest. Further studies must focus on characterizing an inhibitor of DPP4 in sera of mCRPC patients. If such an inhibitor is released into the blood by metastatic PCa cells and could be identified, it might be possible to develop a noninvasive test for screening many more samples across a wider range of advanced disease stages and grades. This would validate our current findings and assist in developing a reliable measurement for classifying advanced malignancy and survival prediction.
Supplementary Material
Footnotes
Author contributions: A.N., J.V., A.J.V., B.S.C., H.L., and P.T. designed research; A.N., K.L., M.G., B.S.C., and H.L. performed research; S.Y., A.S., J.A.E., H.I.S., and B.S.C. contributed new reagents or analytic tools; A.N., K.L., J.P., M.G., J.V., M.A., A.J.V., H.L., and P.T. analyzed data; A.N. and P.T. wrote the paper; J.P. provided statistics and IT support; M.Y. provided oversight of protease assays; M.A. provided statistical analysis.
* This work was supported by U.S. National Institutes of Health Grant No. U24 CA126485 (to P.T.), which was part of the NCI-CPTAC Network; National Institutes of Health Grant No. P50 CA92629-SPORE (to H.I.S.); National Institutes of Health Grant No. R01 CA160816; the NIHR Oxford Biomedical Research Centre Program; and the Swedish Cancer Society (Project No. 11-0624) (to H.L.).
Disclosure statement: H.L. holds patents for free prostate-specific antigen (PSA), kallikrein-related peptidase 2, and intact PSA assays. H.L. and A.J.V. are named on a patent application for a statistical method to detect prostate cancer.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SSEAT
- sequence-specific exopeptidase activity test
- CD26
- cluster of differentiation 26
- DPP4
- dipeptidyl aminopeptidase 4
- GPC3
- glypican-3
- mCRPC
- metastatic castration-resistant prostate cancer
- Met
- metastatic disease
- PCa
- prostate cancer
- PSA
- prostate-specific antigen
- PTM
- post-translational modification
- rec
- recombinant
- WT
- wild type.
REFERENCES
- 1. Rifai N., Gillette M. A., Carr S. A. (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Ransohoff D. F. (2007) How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J. Clin. Epidemiol. 60, 1205–1219 [DOI] [PubMed] [Google Scholar]
- 3. Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 4. Matrisian L. M., Sledge G. W., Jr., Mohla S. (2003) Extracellular proteolysis and cancer: meeting summary and future directions. Cancer Res. 63, 6105–6109 [PubMed] [Google Scholar]
- 5. Lopez-Otin C., Matrisian L. M. (2007) Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808 [DOI] [PubMed] [Google Scholar]
- 6. Palermo C., Joyce J. A. (2008) Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 29, 22–28 [DOI] [PubMed] [Google Scholar]
- 7. Villanueva J., Shaffer D. R., Philip J., Chaparro C. A., Erdjument-Bromage H., Olshen A. B., Fleisher M., Lilja H., Brogi E., Boyd J., Sanchez-Carbayo M., Holland E. C., Cordon-Cardo C., Scher H. I., Tempst P. (2006) Differential exoprotease activities confer tumor-specific serum peptidome patterns. J. Clin. Invest. 116, 271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Villanueva J., Nazarian A., Lawlor K., Yi S. S., Robbins R. J., Tempst P. (2008) A sequence-specific exopeptidase activity test (SSEAT) for “functional” biomarker discovery. Mol. Cell. Proteomics 7, 509–518 [DOI] [PubMed] [Google Scholar]
- 9. Villanueva J., Philip J., Chaparro C. A., Li Y., Toledo-Crow R., DeNoyer L., Fleisher M., Robbins R. J., Tempst P. (2005) Correcting common errors in identifying cancer-specific serum peptide signatures. J. Proteome Res. 4, 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Check E. (2004) Proteomics and cancer: running before we can walk? Nature 429, 496–497 [DOI] [PubMed] [Google Scholar]
- 11. Diamandis E. P. (2004) Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: opportunities and potential limitations. Mol. Cell. Proteomics 3, 367–378 [DOI] [PubMed] [Google Scholar]
- 12. Jemal A., Siegel R., Ward E., Murray T., Xu J., Thun M. J. (2007) Cancer statistics, 2007. CA Cancer J. Clin. 57, 43–66 [DOI] [PubMed] [Google Scholar]
- 13. Albertsen P. C. (2010) Treatment of localized prostate cancer: when is active surveillance appropriate? Nat. Rev. Clin. Oncol. 7, 394–400 [DOI] [PubMed] [Google Scholar]
- 14. Rathkopf D., Scher H. I. (2013) Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 19, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shariat S. F., Semjonow A., Lilja H., Savage C., Vickers A. J., Bjartell A. (2011) Tumor markers in prostate cancer I: blood-based markers. Acta Oncol. 50 Suppl 1, 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lilja H., Cronin A. M., Dahlin A., Manjer J., Nilsson P. M., Eastham J. A., Bjartell A. S., Scardino P. T., Ulmert D., Vickers A. J. (2011) Prediction of significant prostate cancer diagnosed 20 to 30 years later with a single measure of prostate-specific antigen at or before age 50. Cancer 117, 1210–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vickers A. J., Roobol M. J., Lilja H. (2012) Screening for prostate cancer: early detection or overdetection? Annu. Rev. Med. 63, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fradet Y. (2009) Biomarkers in prostate cancer diagnosis and prognosis: beyond prostate-specific antigen. Curr. Opin. Urol. 19, 243–246 [DOI] [PubMed] [Google Scholar]
- 19. Scher H. I., Heller G. (2000) Clinical states in prostate cancer: toward a dynamic model of disease progression. Urology 55, 323–327 [DOI] [PubMed] [Google Scholar]
- 20. Smaletz O., Scher H. I., Small E. J., Verbel D. A., McMillan A., Regan K., Kelly W. K., Kattan M. W. (2002) Nomogram for overall survival of patients with progressive metastatic prostate cancer after castration. J. Clin. Oncol. 20, 3972–3982 [DOI] [PubMed] [Google Scholar]
- 21. Bianco F. J., Jr. (2008) Paradigms in androgen/castrate resistant states of prostate cancer in a biomarker era. Urol. Oncol. 26, 408–414 [DOI] [PubMed] [Google Scholar]
- 22. Villanueva J., Nazarian A., Lawlor K., Tempst P. (2009) Monitoring peptidase activities in complex proteomes by MALDI-TOF mass spectrometry. Nat. Protoc. 4, 1167–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Villanueva J., Philip J., Entenberg D., Chaparro C. A., Tanwar M. K., Holland E. C., Tempst P. (2004) Serum peptide profiling by magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry. Anal. Chem. 76, 1560–1570 [DOI] [PubMed] [Google Scholar]
- 24. Villanueva J., Lawlor K., Toledo-Crow R., Tempst P. (2006) Automated serum peptide profiling. Nat. Protoc. 1, 880–891 [DOI] [PubMed] [Google Scholar]
- 25. Villanueva J., Philip J., DeNoyer L., Tempst P. (2007) Data analysis of assorted serum peptidome profiles. Nat. Protoc. 2, 588–602 [DOI] [PubMed] [Google Scholar]
- 26. Mitrunen K., Pettersson K., Piironen T., Bjork T., Lilja H., Lovgren T. (1995) Dual-label one-step immunoassay for simultaneous measurement of free and total prostate-specific antigen concentrations and ratios in serum. Clin. Chem. 41, 1115–1120 [PubMed] [Google Scholar]
- 27. Ellwood-Yen K., Graeber T. G., Wongvipat J., Iruela-Arispe M. L., Zhang J., Matusik R., Thomas G. V., Sawyers C. L. (2003) Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4, 223–238 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z., Trotman L. C., Shaffer D., Lin H. K., Dotan Z. A., Niki M., Koutcher J. A., Scher H. I., Ludwig T., Gerald W., Cordon-Cardo C., Pandolfi P. P. (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durinx C., Neels H., Van der Auwera J. C., Naelaerts K., Scharpe S., De Meester I. (2001) Reference values for plasma dipeptidyl-peptidase IV activity and their association with other laboratory parameters. Clin. Chem. Lab. Med. 39, 155–159 [DOI] [PubMed] [Google Scholar]
- 30. Leiting B., Pryor K. D., Wu J. K., Marsilio F., Patel R. A., Craik C. S., Ellman J. A., Cummings R. T., Thornberry N. A. (2003) Catalytic properties and inhibition of proline-specific dipeptidyl peptidases II, IV and VII. Biochem. J. 371, 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barrett A. J., Rawlings N. D., Woessner J. F. (2004) Handbook of Proteolytic Enzymes, 2nd ed., Elsvier Academic Press, Amsterdam, The Netherlands [Google Scholar]
- 32. Farrah T., Deutsch E. W., Omenn G. S., Campbell D. S., Sun Z., Bletz J. A., Mallick P., Katz J. E., Malmstrom J., Ossola R., Watts J. D., Lin B., Zhang H., Moritz R. L., Aebersold R. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10, M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marguet D., Baggio L., Kobayashi T., Bernard A. M., Pierres M., Nielsen P. F., Ribel U., Watanabe T., Drucker D. J., Wagtmann N. (2000) Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc. Natl. Acad. Sci. U.S.A. 97, 6874–6879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tagore D. M., Nolte W. M., Neveu J. M., Rangel R., Guzman-Rojas L., Pasqualini R., Arap W., Lane W. S., Saghatelian A. (2009) Peptidase substrates via global peptide profiling. Nat. Chem. Biol. 5, 23–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thornberry N. A., Weber A. E. (2007) Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Curr. Top. Med. Chem. 7, 557–568 [DOI] [PubMed] [Google Scholar]
- 36. DeClerck Y. A., Mercurio A. M., Stack M. S., Chapman H. A., Zutter M. M., Muschel R. J., Raz A., Matrisian L. M., Sloane B. F., Noel A., Hendrix M. J., Coussens L., Padarathsingh M. (2004) Proteases, extracellular matrix, and cancer: a workshop of the path B study section. Am. J. Pathol. 164, 1131–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Puente X. S., Sanchez L. M., Overall C. M., Lopez-Otin C. (2003) Human and mouse proteases: a comparative genomic approach. Nat. Rev. Genet. 4, 544–558 [DOI] [PubMed] [Google Scholar]
- 38. Cordero O. J., Salgado F. J., Nogueira M. (2009) On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol. Immunother. 58, 1723–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Meester I., Scharpe S., Lambeir A. M. (2009) Dipeptidyl peptidases and related proteins: multifaceted markers and therapeutic targets. Clin. Chem. Lab. Med. 47, 245–247 [DOI] [PubMed] [Google Scholar]
- 40. Matheeussen V., Baerts L., De Meyer G., De Keulenaer G., Van der Veken P., Augustyns K., Dubois V., Scharpe S., De Meester I. (2011) Expression and spatial heterogeneity of dipeptidyl peptidases in endothelial cells of conduct vessels and capillaries. Biol. Chem. 392, 189–198 [DOI] [PubMed] [Google Scholar]
- 41. De Meester I., Vanhoof G., Hendriks D., Demuth H. U., Yaron A., Scharpe S. (1992) Characterization of dipeptidyl peptidase IV (CD26) from human lymphocytes. Clin. Chim. Acta 210, 23–34 [DOI] [PubMed] [Google Scholar]
- 42. Durinx C., Lambeir A. M., Bosmans E., Falmagne J. B., Berghmans R., Haemers A., Scharpe S., De Meester I. (2000) Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur. J. Biochem. 267, 5608–5613 [DOI] [PubMed] [Google Scholar]
- 43. Pro B., Dang N. H. (2004) CD26/dipeptidyl peptidase IV and its role in cancer. Histol. Histopathol. 19, 1345–1351 [DOI] [PubMed] [Google Scholar]
- 44. Brandt I., Lambeir A. M., Maes M. B., Scharpe S., De Meester I. (2006) Peptide substrates of dipeptidyl peptidases. Adv. Exp. Med. Biol. 575, 3–18 [DOI] [PubMed] [Google Scholar]
- 45. Lambeir A. M., Scharpe S., De Meester I. (2008) DPP4 inhibitors for diabetes—what next? Biochem. Pharmacol. 76, 1637–1643 [DOI] [PubMed] [Google Scholar]
- 46. Hino M., Nagatsu T., Kakumu S., Okuyama S., Yoshii Y., Nagatsu I. (1975) Glycylprolyl beta-naphthylamidase activity in human serum. Clin. Chim. Acta 62, 5–11 [DOI] [PubMed] [Google Scholar]
- 47. Haacke W., Kullertz G., Barth A. (1986) [Diagnostic value of the enzyme dipeptidyl peptidase IV (DP IV) in abdominal cancers]. Arch. Geschwulstforsch. 56, 145–153 [PubMed] [Google Scholar]
- 48. Kojima J., Ueno Y., Kasugai H., Okuda S., Akedo H. (1987) Glycylproline dipeptidyl aminopeptidase and gamma-glutamyl transpeptidase in human hepatic cancer and embryonal tissues. Clin. Chim. Acta 167, 285–291 [DOI] [PubMed] [Google Scholar]
- 49. Kojima K., Mihara R., Sakai T., Togari A., Matsui T., Shinpo K., Fujita K., Fukasawa K., Harada M., Nagatsu T. (1987) Serum activities of dipeptidyl-aminopeptidase II and dipeptidyl-aminopeptidase IV in tumor-bearing animals and in cancer patients. Biochem. Med. Metab. Biol. 37, 35–41 [DOI] [PubMed] [Google Scholar]
- 50. Bogenrieder T., Finstad C. L., Freeman R. H., Papandreou C. N., Scher H. I., Albino A. P., Reuter V. E., Nanus D. M. (1997) Expression and localization of aminopeptidase A, aminopeptidase N, and dipeptidyl peptidase IV in benign and malignant human prostate tissue. Prostate 33, 225–232 [DOI] [PubMed] [Google Scholar]
- 51. Cordero O. J., Ayude D., Nogueira M., Rodriguez-Berrocal F. J., de la Cadena M. P. (2000) Preoperative serum CD26 levels: diagnostic efficiency and predictive value for colorectal cancer. Br. J. Cancer 83, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Javidroozi M., Zucker S., Chen W. T. (2012) Plasma seprase and DPP4 levels as markers of disease and prognosis in cancer. Dis. Markers 32, 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kotackova L., Balaziova E., Sedo A. (2009) Expression pattern of dipeptidyl peptidase IV activity and/or structure homologues in cancer. Folia Biol. 55, 77–84 [PubMed] [Google Scholar]
- 54. Sedo A., Stremenova J., Busek P., Duke-Cohan J. S. (2008) Dipeptidyl peptidase-IV and related molecules: markers of malignancy? Expert Opin. Med. Diagn. 2, 677–689 [DOI] [PubMed] [Google Scholar]
- 55. Dinjens W. N., Ten Kate J., Kirch J. A., Tanke H. J., Van der Linden E. P., Van den Ingh H. F., Van Steenbrugge G. J., Meera Khan P., Bosman F. T. (1990) Adenosine deaminase complexing protein (ADCP) expression and metastatic potential in prostatic adenocarcinomas. J. Pathol. 160, 195–201 [DOI] [PubMed] [Google Scholar]
- 56. Smith R. E., Talhouk J. W., Brown E. E., Edgar S. E. (1998) The significance of hypersialylation of dipeptidyl peptidase IV (CD26) in the inhibition of its activity by Tat and other cationic peptides. CD26: a subverted adhesion molecule for HIV peptide binding. AIDS Res. Hum. Retroviruses 14, 851–868 [DOI] [PubMed] [Google Scholar]
- 57. Cuchacovich M., Gatica H., Pizzo S. V., Gonzalez-Gronow M. (2001) Characterization of human serum dipeptidyl peptidase IV (CD26) and analysis of its autoantibodies in patients with rheumatoid arthritis and other autoimmune diseases. Clin. Exp. Rheumatol. 19, 673–680 [PubMed] [Google Scholar]
- 58. Christopherson K. W., 2nd, Hangoc G., Mantel C. R., Broxmeyer H. E. (2004) Modulation of hematopoietic stem cell homing and engraftment by CD26. Science 305, 1000–1003 [DOI] [PubMed] [Google Scholar]
- 59. Passaniti A., Hart G. W. (1988) Cell surface sialylation and tumor metastasis. Metastatic potential of B16 melanoma variants correlates with their relative numbers of specific penultimate oligosaccharide structures. J. Biol. Chem. 263, 7591–7603 [PubMed] [Google Scholar]
- 60. Davoodi J., Kelly J., Gendron N. H., MacKenzie A. E. (2007) The Simpson-Golabi-Behmel syndrome causative glypican-3, binds to and inhibits the dipeptidyl peptidase activity of CD26. Proteomics 7, 2300–2310 [DOI] [PubMed] [Google Scholar]
- 61. Pilia G., Hughes-Benzie R. M., MacKenzie A., Baybayan P., Chen E. Y., Huber R., Neri G., Cao A., Forabosco A., Schlessinger D. (1996) Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 12, 241–247 [DOI] [PubMed] [Google Scholar]
- 62. Chen M., Li G., Yan J., Lu X., Cui J., Ni Z., Cheng W., Qian G., Zhang J., Tu H. (2013) Reevaluation of glypican-3 as a serological marker for hepatocellular carcinoma. Clin. Chim. Acta 423, 105–111 [DOI] [PubMed] [Google Scholar]
- 63. Zhang L., Liu H., Sun L., Li N., Ding H., Zheng J. (2012) Glypican-3 as a potential differential diagnosis marker for hepatocellular carcinoma: a tissue microarray-based study. Acta Histochem. 114, 547–552 [DOI] [PubMed] [Google Scholar]
- 64. Taylor B. S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B. S., Arora V. K., Kaushik P., Cerami E., Reva B., Antipin Y., Mitsiades N., Landers T., Dolgalev I., Major J. E., Wilson M., Socci N. D., Lash A. E., Heguy A., Eastham J. A., Scher H. I., Reuter V. E., Scardino P. T., Sander C., Sawyers C. L., Gerald W. L. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wesley U. V., Albino A. P., Tiwari S., Houghton A. N. (1999) A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J. Exp. Med. 190, 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wesley U. V., Tiwari S., Houghton A. N. (2004) Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int. J. Cancer 109, 855–866 [DOI] [PubMed] [Google Scholar]
- 68. Kikkawa F., Kajiyama H., Shibata K., Ino K., Nomura S., Mizutani S. (2005) Dipeptidyl peptidase IV in tumor progression. Biochim. Biophys. Acta 1751, 45–51 [DOI] [PubMed] [Google Scholar]
- 69. Havre P. A., Abe M., Urasaki Y., Ohnuma K., Morimoto C., Dang N. H. (2008) The role of CD26/dipeptidyl peptidase IV in cancer. Front. Biosci. 13, 1634–1645 [DOI] [PubMed] [Google Scholar]
- 70. Masur K., Schwartz F., Entschladen F., Niggemann B., Zaenker K. S. (2006) DPPIV inhibitors extend GLP-2 mediated tumour promoting effects on intestinal cancer cells. Regul. Pept. 137, 147–155 [DOI] [PubMed] [Google Scholar]
- 71. Wesley U. V., McGroarty M., Homoyouni A. (2005) Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res. 65, 1325–1334 [DOI] [PubMed] [Google Scholar]
- 72. Sun Y. X., Pedersen E. A., Shiozawa Y., Havens A. M., Jung Y., Wang J., Pienta K. J., Taichman R. S. (2008) CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF-1/CXCL12. Clin. Exp. Metastasis 25, 765–776 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.