

Abstract
Based on conventional data-dependent acquisition strategy of shotgun proteomics, we present a new workflow DeMix, which significantly increases the efficiency of peptide identification for in-depth shotgun analysis of complex proteomes. Capitalizing on the high resolution and mass accuracy of Orbitrap-based tandem mass spectrometry, we developed a simple deconvolution method of “cloning” chimeric tandem spectra for cofragmented peptides. Additional to a database search, a simple rescoring scheme utilizes mass accuracy and converts the unwanted cofragmenting events into a surprising advantage of multiplexing. With the combination of cloning and rescoring, we obtained on average nine peptide-spectrum matches per second on a Q-Exactive workbench, whereas the actual MS/MS acquisition rate was close to seven spectra per second. This efficiency boost to 1.24 identified peptides per MS/MS spectrum enabled analysis of over 5000 human proteins in single-dimensional LC-MS/MS shotgun experiments with an only two-hour gradient. These findings suggest a change in the dominant “one MS/MS spectrum - one peptide” paradigm for data acquisition and analysis in shotgun data-dependent proteomics. DeMix also demonstrated higher robustness than conventional approaches in terms of lower variation among the results of consecutive LC-MS/MS runs.
Shotgun proteomics analysis based on a combination of high performance liquid chromatography and tandem mass spectrometry (MS/MS) (1) has achieved remarkable speed and efficiency (2–7). In a single four-hour long high performance liquid chromatography-MS/MS run, over 40,000 peptides and 5000 proteins can be identified using a high-resolution Orbitrap mass spectrometer with data-dependent acquisition (DDA)1 (2, 3). However, in a typical LC-MS analysis of unfractionated human cell lysate, over 100,000 individual peptide isotopic patterns can be detected (4), which corresponds to simultaneous elution of hundreds of peptides. With this complexity, a mass spectrometer needs to achieve ≥25 Hz MS/MS acquisition rate to fully sample all the detectable peptides, and ≥17 Hz to cover reasonably abundant ones (4). Although this acquisition rate is reachable by modern time-of-flight (TOF) instruments, the reported DDA identification results do not encompass all expected peptides. Recently, the next-generation Orbitrap instrument, working at 20 Hz MS/MS acquisition rate, demonstrated nearly full profiling of yeast proteome using an 80 min gradient, which opened the way for comprehensive analysis of human proteome in a time efficient manner (5).
During the high performance liquid chromatography-MS/MS DDA analysis of complex samples, high density of co-eluting peptides results in a high probability for two or more peptides to overlap within an MS/MS isolation window. With the commonly used ±1.0–2.0 Th isolation windows, most MS/MS spectra are chimeric (4, 8–10), with cofragmenting precursors being naturally multiplexed. However, as has been discussed previously (9, 10), the cofragmentation events are currently ignored in most of the conventional analysis workflows. According to the prevailing assumption of “one MS/MS spectrum–one peptide,” chimeric MS/MS spectra are generally unwelcome in DDA, because the product ions from different precursors may interfere with the assignment of MS/MS fragment identities, increasing the rate of false discoveries in database search (8, 9). In some studies, the precursor isolation width was set as narrow as ±0.35 Th to prevent unwanted ions from being coselected, fragmented or detected (4, 5).
On the contrary, multiplexing by cofragmentation is considered to be one of the solid advantages in data-independent acquisition (DIA) (10–13). In several commonly used DIA methods, the precursor ion selection windows are set much wider than in DDA: from 25 Th as in SWATH (12), to extremely broad range as in AIF (13). In order to use the benefit of MS/MS multiplexing in DDA, several approaches have been proposed to deconvolute chimeric MS/MS spectra. In “alternative peptide identification” method implemented in Percolator (14), a machine learning algorithm reranks and rescores peptide-spectrum matches (PSMs) obtained from one or more MS/MS search engines. But the deconvolution in Percolator is limited to cofragmented peptides with masses differing from the target peptide by the tolerance of the database search, which can be as narrow as a few ppm. The “active demultiplexing” method proposed by Ledvina et al. (15) actively separates MS/MS data from several precursors using masses of complementary fragments. However, higher-energy collisional dissociation often produces MS/MS spectra with too few complementary pairs for reliable peptide identification. The “MixDB” method introduces a sophisticated new search engine, also with a machine learning algorithm (9). And the “second peptide identification” method implemented in Andromeda/MaxQuant workflow (16) submits the same dataset to the search engine several times based on the list of chromatographic peptide features, subtracting assigned MS/MS peaks after each identification round. This approach is similar to the ProbIDTree search engine that also performed iterative identification while removing assigned peaks after each round of identification (17).
One important factor for spectral deconvolution that has not been fully utilized in most conventional workflows is the excellent mass accuracy achievable with modern high-resolution mass spectrometry (18). An Orbitrap Fourier-transform mass spectrometer can provide mass accuracy in the range of hundreds of ppb (parts per billion) for mass peaks with high signal-to-noise (S/N) ratio (19). However, the mass error of peaks with lower S/N ratios can be significantly higher and exceed 1 ppm. Despite this dependence of the mass accuracy from the S/N level, most MS and MS/MS search engines only allow users to set hard cut-off values for the mass error tolerances. Moreover, some search engines do not provide the option of choosing a relative error tolerance for MS/MS fragments. Such negligent treatment of mass accuracy reduces the analytical power of high accuracy experiments (18).
Identification results coming from different MS/MS search engines are sometimes not consistent because of different statistical assumptions used in scoring PSMs. Introduction of tools integrating the results of different search engines (14, 20, 21) makes the data interpretation even more complex and opaque for the user. The opposite trend—simplification of MS/MS data interpretation—is therefore a welcome development. For example, an extremely straightforward algorithm recently proposed by Wenger et al. (22) demonstrated a surprisingly high performance in peptide identification, even though it is only marginally more complex than simply counting the number of matches of theoretical fragment peaks in high resolution MS/MS, without any a priori statistical assumption.
In order to take advantage of natural multiplexing of MS/MS spectra in DDA, as well as properly utilize high accuracy of Orbitrap-based mass spectrometry, we developed a simple and robust data analysis workflow DeMix. It is presented in Fig. 1 as an expansion of the conventional workflow. Principles of some of the processes used by the workflow are borrowed from other approaches, including the custom-made mass peak centroiding (20), chromatographic feature detection (19, 20), and two-pass database search with the first limited pass to provide a “software lock mass” for mass scale recalibration (23).
Fig. 1.

An overview of the DeMix workflow that expands the conventional workflow, shown by the dashed line. Processes are colored in purple for TOPP, red for search engine (Morpheus/Mascot/MS-GF+), and blue for in-house programs.
In DeMix workflow, the deconvolution of chimeric MS/MS spectra consists of simply “cloning” an MS/MS spectrum if a potential cofragmented peptide is detected. The list of candidate peptide precursors is generated from chromatographic feature detection, as in the MaxQuant/Andromeda workflow (16, 19), but using The OpenMS Proteomics Pipeline (TOPP) (20, 24). During the cloning, the precursor is replaced by the new candidate, but no changes in the MS/MS fragment list are made, and therefore the cloned MS/MS spectra remain chimeric. Processing such spectra requires a search engine tolerant to the presence of unassigned peaks, as such peaks are always expected when multiple precursors cofragment. Thus, we chose Morpheus (22) as a search engine. Based on the original search algorithm, we implement a reformed scoring scheme: Morpheus-AS (advanced scoring). It inherits all the basic principles from Morpheus but deeper utilizes the high mass accuracy of the data. This kind of database search removes the necessity of spectral processing for physical separation of MS/MS data into multiple subspectra (15), or consecutive subtraction of peaks (16, 17).
Despite the fact that DeMix workflow is largely a combination of known approaches, it provides remarkable improvement compared with the state-of-the-art. On our Orbitrap Q-Exactive workbench, testing on a benchmark dataset of two-hour single-dimension LC-MS/MS experiments from HeLa cell lysate, we identified on average 1.24 peptide per MS/MS spectrum, breaking the “one MS/MS spectrum–one peptide” paradigm on the level of whole data set. At 1% false discovery rate (FDR), we obtained on average nine PSMs per second (at the actual acquisition rate of ca. seven MS/MS spectra per second), and detected 40 human proteins per minute.
EXPERIMENTAL PROCEDURES
Sample Preparation
HeLa cell line was grown with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% antibiotics. Cells were collected and washed with PBS three times. Three separate cell pellets were lysed with ProteaseMAX™ Surfactant (0.1% w/v) in aqueous ammonium bicarbonate (50 mm) mixed with acetonitrile at a ratio of 9:1 (v:v) as previously described (2). Mixtures were incubated at 95 °C for 5 min and 15 min sonication (30% amplitude, 3:3 pulse) with a Branson sonicator. After that, protein extracts were reduced and alkylated via incubation with 10 mm of DTT and iodoacetamide, respectively. Proteins (80 μg) were digested with trypsin and incubated at 37 °C for 9 h. Trypsination was terminated by adding 5% acetic acid (vol.). Samples were incubated for 30 min at 45 °C in order to precipitate the detergent, and purified using spin filtration (Pall Nanosep® 10 kDa with Omega membrane).
LC-MS/MS Experiment
Three micrograms of each sample was injected to Thermo Scientific EASY-Spray columns (PepMap® RSLC, C18, 100 Å, 2 μm bead packed 50 cm column) connected to an Easy-nLC 1000 pump (Proxeon Biosystems, Odense, Denmark, now part of Thermo Fisher Scientific). Samples were loaded into the column with buffer A (99.9% water, 0.1% formic acid) and eluted in a 150 min LC-MS/MS experiment. The gradient was started from 2% and increased stepwise to 5% in 12 min, 19% in 88 min, and 30% buffer B (99.9% acetonitrile, 0.1% formic acid) in 15 min at a flow rate of 250 nL/min. It followed by a sharp increase to 98% buffer B in 15 min, then 8 min in 98% buffer B, followed by a sharp decrease to 2% buffer B in 2 min, and finally 10 min in 2% buffer B.
Data Acquisition
Mass spectra were acquired with an Orbitrap Q Exactive mass spectrometer (Thermo Scientific) in a data-dependent manner using a top-20 method. MS spectra were acquired at a resolution of 70,000 with maximum integration time of 250 ms and a target value of 3 × 106 ions. The m/z range was from 400 to 1200. Peptide fragmentation was performed via higher-energy collisional dissociation set at 25 V of normalized collisional energy. The MS/MS spectra were acquired at a resolution of 17,500, with a target value of 2 × 105 ions and a maximum integration time of 120 ms. For testing precursor selectivity, replicated experiments were performed with four different isolation widths: ±1.0, ±2.0, ±3.0, and ±4.0 Th as described in the main text. MS and MS/MS spectra in the profile mode were converted from Thermo .RAW files into mzML format using msconvert in ProteoWizard software package (25) (v. 3.0.5047 downloaded from http://proteowizard.sourceforge.net), with zero intensity sampling being ignored (filter: zeroSamples removeExtra 1–2). The OpenMS Proteomics Pipeline (TOPP) (24) (v. 1.11, downloaded from (http://open-ms.sourceforge.net) was used for calculating centroids of peaks from profile mass spectra, with signal-to-noise filter disabled in the PeakPickerHiRes. It was found important to use PeakPickerHiRes rather than the “Peak Picking” function in msconvert, because the former algorithm is more accurate.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (26) with the dataset identifier PXD000999.
Chromatographic Feature Detection
The FeatureFinderCentroided tool in TOPP was used for detecting and quantifying peptide chromatographic features from the centroided MS spectra, with the following parameters: m/z_tolerance = 0.01 Da, charge_low = 2, charge_high = 7, and feature_min_score = 0.5. The resultant features were exported from the FeatureXML format to CSV tables, containing for each feature the RT range, monoisotopic m/z, charge state, isotope distribution, and integrated ion intensity.
MS/MS Spectra Processing
Centroided MS/MS spectra were processed by msconvert with a filter “MS2Deisotope true 0.005Da.” MS/MS data (m/z and z of the precursor as well as RT of the MS/MS event) were matched with the feature list. Normally, at least one feature matched the nominal precursor, including RT, z, and m/z (the latter within 10 ppm). When no feature from the list matched, the original MS/MS spectrum was retained “as is.” If the ±2.0 Th isolation window overlapped with another detected feature's m/z, and MS/MS RT - with feature's RT range, the MS/MS spectrum was cloned, with the precursor m/z and z of the cloned MS/MS entry being copied from the matched feature. The resultant MS/MS spectra were written in the MGF format by a home-written Python script utilizing pymzml (27), and converted to mzML format by msconvert.
Search Engine
We used Morpheus (rev. 45) that has been designed by Wenger et al.(22) specifically for high resolution mass spectrometry. All options for spectral processing were unchecked. Parameters were set as: Protease = trypsin (no proline rule); Maximum Missed Cleavages = 3; Fixed Modifications = (carbamidomethylation of C); Variable Modifications = [oxidation of M, acetylation of protein N terminus]. UniProt Human Complete Proteome database (v. 2012_04, 89,601 human protein entries) was used as a source of target sequences. As a decoy, reversed protein sequences were generated on-the-fly and concatenated to the target database.
The first pass of search was done on the original MS/MS spectra with a large mass tolerance (±20 ppm) for both precursor and product monoisotopic peaks. The masses of reliably identified (in terms of FDR) peptides were used as “software lock mass” for MS scale recalibration. A Python script written in house and utilizing pyteomics package (28) was used for theoretical mass calculation. The RT-dependent calibration curve was calculated by the method of multivariate adaptive regression splines (MARS) (29) based on the RT and precursor m/z of unique peptide identifications (FDR < 1%). Python script was written using py-earth (https://github.com/jcrudy/py-earth) for nonlinear regression in mass error recalibration. All precursor masses in the MS/MS spectra as well as feature lists were then recalculated. The second pass database search was performed on deconvoluted MS/MS data with a narrower MS tolerance (10 ppm; the tolerance for MS/MS was still 20 ppm); it generated PSMs that were then rescored.
PSM Rescoring
The scoring scheme from the original Morpheus algorithm was reformulated. In the new scheme, the PSM score S is a summation of three subscores: S1, S2, and S3, reflecting three parameters: mass errors in MS and MS/MS, and relative fragment ion intensities, respectively. Three subscores played different roles in discriminating target and decoy peptide-spectrum matches.
Firstly, for all PSMs with q-value < 5 (equivalent of <5.0% FDR), precursor mass errors (deviations from the theoretical mass) were calculated, and the resultant mass error distribution (Fig. 2A) was fitted with normal distribution, giving the standard deviation of mass errors. Using the survival function of this normal distribution, for a given precursor mass error, a p value was calculated. The absolute p value ranging from 0.0001 to 1.0 was added to the final score of the precursor as S1. It also played the role as a soft mass tolerance. PSMs with precursor p value lower than 0.0001 were rejected as too improbable.
Fig. 2.

Scoring metrics of Morpheus-AS, A, precursor mass error distribution in MS; B, fragment mass error distribution in MS/MS; C, distribution of relative fragment ion abundances (log-transformed intensity fraction); D, score distribution after rescoring all PSMs; blue: target peptides; red: decoy peptide.
Furthermore, for each peptide sequence in the list, theoretical MS/MS product masses were calculated for only b- and y-series singly charged ions. Other types of ions were excluded as less probable products. By matching spectra with theoretical peaks for all PSMs in one set of data, an overall fragment mass error distribution was estimated the same way as estimating precursor mass errors (Fig. 2B). Then the σ (standard deviation) of mass error in MS/MS was calculated based on the distribution. In one PSM, each matching peak deviating from its theoretical value by less than 2σ (95.4% confidence interval) added 1.0 point to the score S2, to the maximum of (2n-2), where n is the number of residues in the peptide sequence. Additional to the original algorithm, if a complementary (b-y) pair of product peaks was found, an extra score of 1.0 was added to S2 for rewarding the confirmation of the precursor mass. As in the original Morpheus algorithm, this score of counting matched products is the major part of final score, which plays the most important role of identifying peptides. The scoring scheme was reformulated for the consideration of complementary peaks and postcalculated mass tolerance. Thus, S2 ranged from 0 to 3n-3. Comparing to the original Morpheus score, S2 provides larger space for discriminating between the target and decoy hits, and has less bias against short peptides, as the latter tend to produce more complementary fragments per unit length than longer peptides.
Because counting of matched fragments produces a discrete value, it is not optimal for a later estimation of the cutoff score in a continuous space. In order to smoothen the score distribution at the edge of the trimming threshold (e.g. at 1% FDR), the original Morpheus algorithm adds to the final score an absolute value of the matched fraction of ion intensity (ranges from 0 to 1.0), assuming that a true match will explain a larger fraction of ion peaks. Although that is generally true when comparing two alternative sequence assignments, in the case of analyzing chimeric spectra, the absolute value of ion matching varies significantly depending upon whether the assigned peptide is a primary or a secondary match. To better exploit the differences in the relative abundances of target and decoy fragments for chimeric spectra, distributions of log-transformed values of the matched fraction of ion intensity for target and decoy hits were investigated (Fig. 2C). We used the normal probability density function P(x, N) to calculate the different probabilities of being a target or decoy hit for a given fraction of intensity (ln I). The relative ratio of the probability densities of the target distribution (N3+) and decoy distribution (N3−) was calculated as
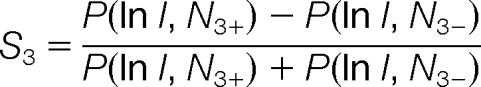 |
Which ranged from −1.0 to 1.0. As in the original Morpheus algorithm, the sub score S3 was used here to smoothen the score distribution at the edge of an arbitrary cutoff (1% FDR).
FDR Trimming
Duplications in the PSM list were removed, with the highest-scoring PSM retained, to ensure that one feature corresponded to only one peptide, and every deconvoluted spectrum was present only once. The filtered PSM list was sorted by score S in a descending order. The distribution of S for target and decoy peptides is shown in Fig. 2D. The FDR level was calculated as in MaxQuant/Andromeda, that is, as the ratio of decoy hits (FP, reversed sequences) and target hits (TP, forward sequences): FDR = FP/TP. The peptide list was trimmed to contain less than the chosen fraction of false identifications. As in some other search engines, sorting and trimming was done hierarchically three times for three peptide subpopulations, which possess different statistical properties in terms of FDR. First-pass: “high-risk” peptides with N-terminal proline—these peptides are rare because of trypsin specificity (no cleavage of prolyl bonds); second-pass: “medium-risk” peptides with the number of basic residues (Arg, Lys, and His) equal or larger than the charge z—these peptides are more stable in collisional dissociation (30); and third-pass: other peptides. Factors such as peptide length and charge were not considered as risks of causing false discoveries. The final PSM list was converted to OMSSA .csv format, which was used by Protein Herder from COMPASS package (31) (v. 1.0.4.5, downloaded from http://www.chem.wisc.edu/∼coon/software.php) to assign peptide sequences to protein groups using the principle of maximum parsimony.
Comparison with State-of-the-Art
The same set of three .RAW files was processed using Mascot MS/MS search engine (v. 2.3.02, Matrix Science), MaxQuant software (v. 1.4.1.2, downloaded from http://maxquant.org) with Andromeda search engine (16), and MS-GF+ (v. 9979) with Percolator (v. 2.07) (32). Precursor mass tolerance was set as 6 ppm. Products mass tolerance was set as 20 mmu in Mascot, 20 ppm in MaxQuant. MS-GF+ does not allow user-defined mass tolerance for MS/MS, thus we chose the default setting for Q-Exactive (parameter: -inst 3). PSMs from Mascot were filtered by E<0.05 for comparing the trimming by significance to FDR trimming. PSMs from Andromeda were filtered by 1.0% FDR. MS-GF+ resultant .mzid files were converted, rescored and filtered by Percorlator to 1.0% peptide FDR.
RESULTS
We used the HeLa cell lysate to test the performance of the DeMix workflow. The proteome was supposed to contain more than 10,000 expressed proteins (33), and had been comprehensively analyzed by Guo et al. (34) on a similar workbench. In a triplicate experiment, we obtained in each run with a 2 h chromatographic separation and the top-20 DDA strategy roughly 12,000 MS and 54,000 MS/MS spectra. At least 98% of MS/MS spectra were found simultaneously containing peaks of both protonated lysine (m/z 147.11280) and arginine (m/z 175.11895), indicating multiplexing of at least two different peptides (Table I).
Table I. Statistics of the triplicate analysis of unfractionated HeLa digest using 2 h-gradient LC-MS/MS.
| Experiment 1 | Experiment 2 | Experiment 3 | |
|---|---|---|---|
| Full MS (Survey) Scans | 11769 | 12136 | 12080 |
| Chromatographic Features | 76603 | 79372 | 81953 |
| Overlappinga | 70420 (91.9%) | 72933 (91.9%) | 75327 (91.9%) |
| Identified (-)d | 26948 (35.2%) | 28539 (36.0%) | 27742 (33.9%) |
| Identified (+)e | 34337 (44.8%) | 35937 (45.3%) | 34861 (42.5%) |
| MS/MS Scans | 53343 | 53977 | 53940 |
| LysH+ and ArgH+b | 52297 (98.0%) | 53080 (98.3%) | 53017 (98.3%) |
| MS/MS Spectral Clones | 134031 | 136143 | 136396 |
| Multiplexing Rate (x) | 2.51 | 2.52 | 2.53 |
| Peptide-spectrum Matchesc | 66042 | 68734 | 65420 |
| Success Rate | 49% | 50% | 48% |
| Identification Efficiency | 124% | 128% | 121% |
| Unique Peptide Sequences | 33066 | 34261 | 32412 |
| Protein Groups | 4726 | 4759 | 4682 |
a Overlapping was estimated in the RT-m/z space, where RT corresponds to feature's chromatographic trace (eluting peak width), and m/z equals to the MS/MS isolation width (±2.0 Th around the monoisotopic peak).
b Protonated lysine and arginine occurrences are calculated based on the co-existence of both peaks at m/z 147.11280 and 175.11895 in the MS/MS peak list, with 10 ppm tolerance.
c PSM list of unique peptide sequences was trimmed to 1.0% FDR.
d (-) Identified features without MS/MS cloning.
e (+) Identified features with MS/MS cloning allowed.
Chromatographic Feature Detection and MS/MS Deconvolution
We formed a set of possible precursors from a map of peptide-like chromatographic features in a three-dimensional space of monoisotopic m/z, retention time (RT), and ion intensity. We used The OpenMS Proteomics Pipeline (TOPP) to create this feature map, assembling full MS spectra into peptide features based on isotopic modeling and chromatographic tracing (20, 24). Thus obtained feature list was practically free from noise spikes, nonmonoisotopic assignments or nonpeptide artifacts. We detected around 80,000 peptide-like features in each of the three replicate experiments, in broad agreement with a previous report (4). In accordance with the estimation from MS/MS, we found that 92% of the listed features overlap with at least one other feature. This value was estimated by counting the number of co-eluting (RT-overlapping) features that have the distance between their monoisotopic masses smaller than the isolation width (± 2.0 Th) (Table I). Such features have a high chance of cofragmentation in MS/MS.
The deconvolution of chimeric MS/MS spectra was done by creating several spectral “clones” from a single original MS/MS spectrum. Alternative precursors from the feature map were assigned to individual clones when their chromatographic trace overlapped in the isolation window of the original MS/MS spectrum. Unlike in other deconvolution or demultiplexing methods, the cloning of MS/MS spectra did not result in any changes in the fragment list: in each spectral clone, the precursor was replaced by a new candidate, but the list of MS/MS fragments was kept the same as in the original MS/MS spectrum. Using this cloning method, we generated on average 2.5 MS/MS spectra out of one original MS/MS spectrum (Table I and supplemental Table S1).
Peptide-spectrum Matching and Rescoring
For each conventional PSM, a new score S was calculated reflecting three parameters: peptide molecular mass errors (Fig. 2A), mass errors of matched fragment peaks (Fig. 2B), and the relative abundances of these peaks (Fig. 2C). In this scoring scheme, mass tolerances were calculated a posteriori, separately for MS and MS/MS spectra, depending on the corresponding overall mass error distributions in the current dataset. Precursor mass errors were used to smoothly increase the score proportionally to the proximity to the theoretical values. Furthermore, theoretical MS/MS products were only calculated as singly charged b- and y-series ions, as the most probable types of fragments. No additional statistical assumption was made, for example, the prevalence of y-ions was not presumed. These omissions arguably leave space for further improvements, for example weighing fragment scores differently based on ion charge and type frequency and ion relative intensity. To widen the search space, we allowed up to three missed tryptic cleavages for candidate peptides from the sequence database.
Directly applying this rescoring method to the benchmark dataset, we obtained over 40,000 PSMs in each experiment with a 2 h gradient, which equals to >75% success rate of MS/MS identification for unique peptides at 1.0% FDR (Table II).
Table II. Comparison between the DeMix and MaxQuant workflow, with four database searching methods, based on the combined HeLa triplicate dataset.
| Search engine | MaxQuant |
DeMix |
||||||
|---|---|---|---|---|---|---|---|---|
| Andromeda | Mascot | MS-GF+ Percolator | Morpheus-AS | |||||
| Deconvolutiona | - | + | - | + | - | + | - | + |
| Peptide-spectrum matchesb | 115135 | 131597 | 78063 | 80096 | 118715 | 193450 | 122208 | 200292 |
| PSM per MS/MS | 0.714 | 0.816 | 0.484 | 0.497 | 0.736 | 1.199 | 0.758 | 1.242 |
| Unique peptides | 32646 | 38112 | 23133 | 23095 | 35468 | 45022 | 33712 | 41628 |
| PSM per peptide | 3.53 | 3.45 | 3.37 | 3.47 | 3.35 | 4.30 | 3.63 | 4.81 |
| Confetti supportedc | 86.4% | 84.6% | 89.3% | 89.4% | 85.4% | 83.0% | 86.3% | 84.6% |
| Protein groups | 4409 | 4642 | 3800 | 3801 | 4831 | 5443 | 4707 | 5167 |
| Strongly supportedd | 3597 | 3985 | 2865 | 2868 | 3776 | 4446 | 3691 | 4222 |
| Mean coverage | 24.4% | 25.1% | 22.9% | 22.9% | 24.7% | 25.7% | 24.6% | 25.6% |
a Deconvolution: secondary peptides in MaxQuant or spectra cloning in DeMix are allowed (+) or not (-).
b PSM lists were trimmed to E-value < 0.05 for Mascot, or to FDR < 0.01 for Andromeda, MS-GF+ Percolator and Morpheus-AS.
c Proportion of unique peptide sequences found in the Confetti database (34).
d Protein groups supported by two or more unique peptides are considered to be strongly supported; and mean coverage was calculated for these strongly supported proteins.
To relate the new score S with the “classical” scoring systems, we compared scores of the peptides identifications with those of other search engines (supplemental Fig. S1). The inhomogeneities in the new score distribution indicate that the new scoring system has a potential for further improvement.
One MS/MS Spectrum - More Peptide Identifications
The combination of cloning and rescoring yielded a total identification of 41,628 unique peptides at 1.0% FDR, which were grouped into 5167 human proteins covering around half of the expressed proteome (Table II and supplemental Table S2). Remarkably, the number of PSMs was 1.24 times the total number of the original MS/MS scans, breaking the “one MS/MS spectrum - one peptide” paradigm at the level of the whole data set for the first time. This identification efficiency was much higher than that of MaxQuant/Andromeda (0.82), which also enabled spectral deconvolution; it was 2.5 times of that of Mascot (0.50), when using E-value instead of FDR to trim the PSM list. The improvement obtained using the DeMix workflow was not because of the search engine; when Morpheus-AS was replaced with another modern search engine MS-GF+ Percolator (32), a similar efficiency was obtained (Table II).
The effective multiplicity distribution reflecting the number of identified peptides per MS/MS spectrum is shown in Fig. 3. In our workflow, less than 17.4% of all MS/MS spectra failed to produce a single hit in the database search, which was likely because of nontryptic, post-translationally modified or poorly fragmented peptides. At the same time, 50.7% of all MS/MS spectra produced a single hit. Over 30% were confidently assigned to two or more peptides, with a maximum of seven peptides identified from a single original MS/MS spectrum. In contrast, MaxQuant/Andromeda that also employs MS/MS spectra deconvolution, assigned less than 8% of all MS/MS spectra to ≥2 peptides. We were puzzled by such a difference in performance, but then found that the “second peptide” method implemented in MaxQuant was not efficient when the “first peptide” failed to be identified.
Fig. 3.
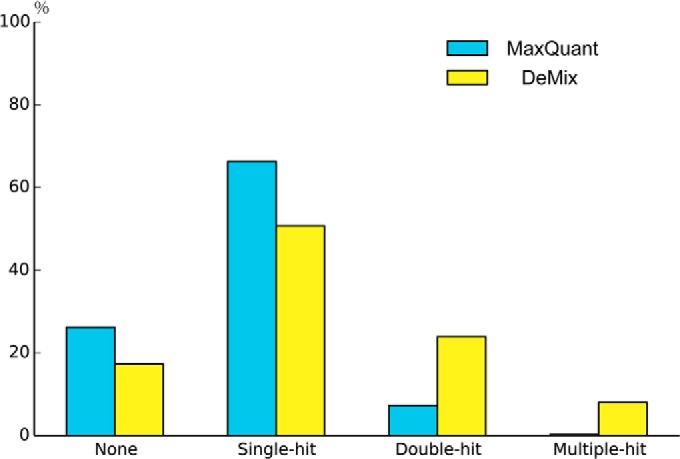
Deconvolution comparison: the distribution of peptide identifications per MS/MS spectrum for MaxQuant and the DeMix workflow.
Fig. 4B presents a typical MS/MS spectrum, which failed to produce a single identification in Andromeda search, but was assigned to four peptides from the feature map (Fig. 4A) and successfully identified in DeMix workflow. One-by-one annotation for the four peptides is shown in supplemental Fig. S2. For the 11.5% MS/MS spectra that were only assigned by DeMix workflow but not Andromeda, we found that 64% of these spectra produced identifications for alternative precursors, and 40% produced identifications only for alternative, but not primary, precursors.
Fig. 4.

Deconvolution example: a) a local feature map showing four peptides (boxes) being co-selected by the isolation window (red dash line) of a DDA acquisition (red X); square spots on the map are color-coded MS1 peaks in the RT-m/z space; b) the corresponding MS/MS spectrum (HELA3 #36982) was assigned to four peptide precursors in DeMix workflow, but failed in Andromeda search. The spectrum is annotated by “Expert System” with 20ppm mass tolerance (47). Only b-/y-type fragments are presented. 1) [HGDGTTLDIMLK]2+, m/z = 650.8311, Score=21.76, sp|P55786|PSA HUMAN: 754–765; 2) [ALVGGAVGGLAGAASK]2+, m/z =649.8724, Score=16.19, sp|Q96RL7|VP13A HUMAN: 2908–2923; 3) [FVAFSGEGQSLR]2+, m/z =649.3308, Score=13.65, sp|Q92890–1|UFD1 HUMAN: 326–337; 4) [QMCICADFEK]2+, m/z =651.2697 (original precursor), Score=11.19, sp|P14868|SYDC HUMAN: 255–264.
Comparing the average number of unique peptides in a protein group, we found that our workflow gave almost the same number, roughly eight peptides per protein, as Andromeda, with mean coverage of >25% for proteins having more than one unique peptides identified (Table II).
On average, one unique peptide sequence was supported by 4.8 PSMs by DeMix, compared with roughly 3.5 PSMs/peptide by MaxQuant. Therefore, DeMix peptides were re-identified more than one time by spectral clones. A majority (68.4%) of the spectral clones identified with alternative precursors provided extra support to identifications from the primary precursors in some other MS/MS scans; the remaining 31.6% supported new sequences. Such large fraction of “known” sequences identified using alternative precursors testifies to the validity of identification via MS/MS cloning.
Shown in supplemental Fig. S3, the distribution of the retention time differences between the primary spectra and secondary (clone) MS/MS spectra is very small, mostly less than 30 s, consistent with the typical width of the chromatographic peaks of only 15 s. If secondary peptides would give a broad distribution of RT differences with the primary peptides, that might indicate spurious identifications. Even so, there is a possibility that one peptide may have two distant chromatographic features. One possible cause of chromatographic splitting is isomerization of proline-containing peptides (35). Indeed, we found one example of this phenomenon. Peptide GVLLYGPPGTGK (P62195–2|PRS8_HUMAN: 177 - 188) was eluted separately at 88 min and 92.5 min (4.5 min difference). supplemental Fig. S4 shows the comparison of a pair of MS/MS spectra where the peptide was identified as a primary precursor and as a distant secondary precursor. A great deal of overlap was found between MS/MS fragments: the primary spectrum shared 12 out of 13 b- and y-ions with the secondary spectrum (supplemental Table S3). The correlation of ion intensities from the same fragments in the two MS/MS spectra is also very high: R2 = 0.895 (supplemental Fig. S5).
To additionally test the validity of peptide identifications, we made three sets of artificial spectra by shifting all original precursor masses in MS/MS spectra by +30 ppm (within the same isolation window), +3.0 Th (out of the isolation window), or by randomization of the whole precursor mass list. All three categories of these spectra yielded less than 1% identifications compared with the unmodified dataset (supplemental Table S4). The +30 ppm set had a higher number of identifications than the other two artificial sets, because of a small but nonzero probability that a real cofragmented peptide with that shifted mass is present in the same isolation window. Because <1% hit rate is exactly what would be expected at <1% FDR (the same level as the decoy hits), these results support both the validity of peptide identifications as well as the fair accuracy of our FDR estimates.
To further confirm that newly identified peptides really belong to the HeLa proteome, we mapped the identified sequences to the Confetti database (34), which hosts over 400,000 unique peptide sequences covering over 40% of more than 8000 detected proteins from HeLa cells. Without spectral cloning, 29,109 peptides identified by DeMix mapped on the Confetti with 4656 remaining unmapped (86.2% success rate). Enabling spectral cloning added 7931 peptides, of which 6088 peptides mapped and 1843 peptides did not (success rate of 76.7%). These figures agree well with the MaxQuant results where the mapping rates were around 85%. The somewhat lower success rate of the additional identifications produced by DeMix is understandable, because they were low-abundance peptides that have a lower a priori probability to be included in the Confetti dataset.
The above results indicate that extra identifications from spectral clones are mostly untargeted peptides during DDA. They can probably be targeted and identified in experiments with a longer LC gradient, enhanced digestion, or with prefractionation. Interestingly, replacing Morpheus-AS with MS-GF+Percolator yielded 8% more unique peptides without increasing the number of PSMs. But our investigation showed that these extra peptides had a lower rate (66%) of mapping to Confetti database. This could be because of the risk of model over-fitting when discriminating between the target and decoy hits by Percolator's machine learning.
One reason for a peptide not being targeted in DDA could be its low abundance. Investigating the abundance distribution of identified chromatographic features confirmed our expectation that the majority of newly identified peptides have relatively low abundances. As shown in Fig. 5 and Table I, the primary peptides identified without spectral cloning covered 35% of all features, which explained 71% of the total precursor ion current (sum of the peptides' absolute abundances) on the feature map. The average abundance of the identified primary peptide features was 8.64 (±0.56) on the log10-scale. By enabling spectral cloning, extra peptide identifications from deconvolution extended the coverage up to 45% of all features, and to 73% of the total precursor ion current. The average abundance of these additional peptide features was 8.23 (±0.42), nearly 2.5 times lower than the average abundance of the primary ones. Thus, deconvolution by spectral cloning provided an expansion of the dynamic range, which is currently considered to be one of the most important parameters in proteomics (36, 37). However, being technologically limited by the instrumental sensitivity, we found it extremely challenging to reliably identify any feature with an abundance below seven orders of magnitudes on the Q Exactive abundance scale.
Fig. 5.
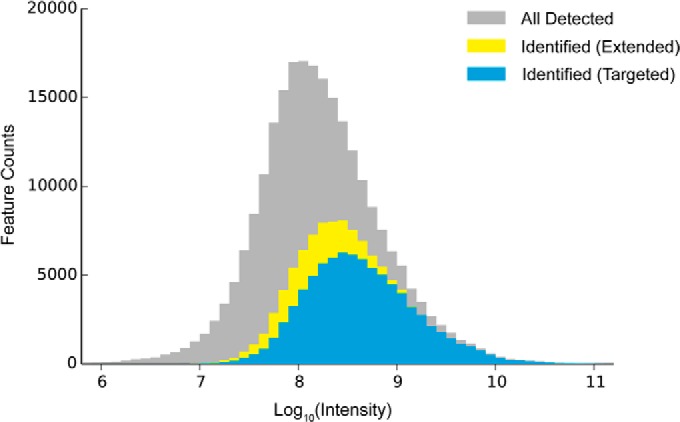
Abundance distributions of chromatographic features: gray: all peptide-like features detected in LC-MS; blue: identified peptides targeted in conventional DDA strategy; yellow: peptides additionally identified by feature-based deconvolution.
Multiplicity and Selectivity
To investigate the limit to the complexity of MS/MS data that can be efficiently deconvoluted by our method, we compared a new set of LC-MS/MS experiments with ascending widths of isolation, ±1.0, ±2.0, ±3.0, and ±4.0 Th. The results are presented in Table III.
Table III. Comparison of the results obtained with different MS/MS isolation window widths: ±1.0, ±2.0, ±3.0 and ±4.0 Th.
| MS/MS scan | Lys+ and Arg+ | Spectral clone | Multiplexing rate | PSM | Success rate | ID efficiency | |
|---|---|---|---|---|---|---|---|
| # | % | # | x | # | % | % | |
| ±1.0 | 45037 | 88.5 | 79739 | 1.77 | 42071 | 52.8 | 93.4 |
| ±2.0 | 44023 | 94.2 | 110671 | 2.51 | 46999 | 42.5 | 106.8 |
| ±3.0 | 46937 | 96.7 | 167529 | 3.57 | 57951 | 34.6 | 123.5 |
| ±4.0 | 48049 | 98.1 | 221313 | 4.61 | 63624 | 28.7 | 132.4 |
| Unique peptide | Effective peptide ID ratea | Protein group | Effective protein ID rateb | MaxQuant PSM | MaxQuant unique peptide | MaxQuant protein group | |
|---|---|---|---|---|---|---|---|
| ±1.0 | 27455 | 0.610 | 3995 | 0.0887 | 34733 | 24699 | 3676 |
| ±2.0 | 27777 | 0.631 | 4013 | 0.0912 | 33385 | 23706 | 3494 |
| ±3.0 | 28623 | 0.610 | 4112 | 0.0876 | 32268 | 22656 | 3445 |
| ±4.0 | 27716 | 0.577 | 4066 | 0.0846 | 29840 | 21187 | 3353 |
| Mean | 27893 | 4047 | 23062 | 3492 | |||
| S.D. | 506 | 53 | 1503 | 136 |
a Unique peptides per MS/MS scan.
b Protein groups per MS/MS scan.
The proportion of MS/MS spectra with co-existing protonated lysine and arginine well correlated with the size of the isolation window, varying from 88.5% for ±1.0 Th to 98.1% for ±4.0 Th window. The difference was much more considerable in comparing the multiplexing rates of spectral cloning: from 1.77x with the narrowest ±1.0 Th window, to 2.51x with a ±2.0 Th window, to 3.57x with a ±3.0 Th window, and to the maximum of 4.61× with the widest ±4.0 Th window. The multiplexing function of absolute window width is nicely (R2 = 0.994) described by a linear function:
The peptide identification efficiencies exceeded 100% for all windows except for the ±1.0 Th window. Widening the isolation window inevitably increased the spectral complexity during DDA. As a result, the success rate in peptide-spectrum matching declined with the window size. However, for a single LC-MS/MS run, DeMix yielded very similar numbers of unique peptides (∼28,000) and protein groups (∼4000) for different isolation windows, despite the huge differences in the numbers of deconvoluted spectra (MS/MS clones). This high stability in numbers disproved the suspicion that introducing a large amount of unidentifiable spectral clones may increase the number of random identifications from false discoveries. The maximum effective identification rate (number of unique peptides per MS/MS) was reached with a ±2.0 Th window, in accordance with previous studies (2, 4). Notably, DeMix workflow showed not only higher number of identifications, but also a three times smaller variation in this number across the four LC-MS/MS data sets, demonstrating higher robustness when varying precursor selectivity, compared with the state-of-the-art MaxQuant/Andromeda workflow (Table III).
Practical Software Solution
Our workflow shared some similarities with MaxQuant, which is a highly integrated all-in-one solution with very limited space for user-defined modifications. Thus, we had to reinvent some of the advanced features in MaxQuant workflow, such as the “software lock mass,” and to use open-sourced equivalent components, such as feature detection. Our workflow is not as integrated as MaxQuant, but it is more flexible: it allows much greater freedom for user-defined modifications. Users are only required to sequentially specify in different user-interfaces some parameters and file paths starting from the .RAW files and until the FDR-filtered lists of peptides and proteins are obtained. Because all the components in this pipeline are open-sourced, and mostly platform-independent (Windows/OSX/Linux), DeMix can be integrated, if needed, into a one-button solution resting on top of the TOPP platform; or ultimately, into an intelligent data-acquisition method at the instrument end. Source codes of DeMix workflow can be downloaded at https://github.com/userbz/DeMix
The computational performance of DeMix was reasonable, although not faster than MaxQuant. On our desktop computer (Intel quad-core i7 3.40 GHz with 16GB RAM), the complete analysis of the benchmark 2 h-gradient LC-MS/MS dataset was finished in roughly five hours, as opposed to three hours with MaxQuant. We found that the major bottlenecks are the feature detection (>10 GB memory and >3 h CPU time), and using in-house Python scripts in text (mzML) processing, for example,spectral cloning and PSM rescoring. Implementing multi-thread parallelization could increase the computational efficiency.
DISCUSSION
The DeMix workflow described in this study demonstrated a significant improvement in analyzing shotgun proteomics data with data-dependent acquisition (DDA). The observed improvements in MS/MS peptide identification were mainly attributed to two processes: spectral cloning and PSM rescoring (supplemental Table S4). At 1% FDR level, the number of peptide-spectrum matches significantly exceeded the total number of MS/MS spectra, breaking the “one MS/MS spectrum–one peptide” paradigm. Even though such a paradigm is attractive because of its simplicity, it is not realistic in analysis of complex proteomes without the significant narrowing of the precursor isolation window. Our estimates show that, in order to keep the degree of natural multiplexing below 10%, the isolation window has to be narrowed to 0.2 Th. This would present a challenge even for modern mass spectrometers, such as the Orbitrap Fusion (Thermo Scientific), which has the narrowest MS/MS window of 0.4 Th. Moreover, as the dynamic range of analysis increases, and new, low-abundant peptides become detectable, the paradigm is bound to break down again, demanding even further narrowing of the MS/MS window. Thus, natural multiplexing is a fact that the DDA practitioners have to accept and learn to deal with. Changing the paradigm to “one MS/MS spectrum–several peptides” will call for rethinking both data acquisition and data analysis in deep proteomics. The goal of modern instruments should be to record in multiplexed mode the MS/MS spectra of all precursors that are detected in MS spectra. Our data demonstrate that we at the moment are within a factor of two or less to this goal.
A respective adjustment in MS/MS search engines will also be needed. Direct matching of multiple precursors found in one MS/MS spectrum would be more elegant, and statistically more satisfactory, than processing of quasi-independent MS/MS clones. Furthermore, considering the simplicity of the scoring scheme, it is feasible to integrate the precursor deconvolution and peptide-spectrum matching into an intelligent data acquisition method.
It has become fashionable in recent times to compare unfavorably the DDA strategy to the alternative DIA, for its multiplexing nature and higher reproducibility (38). But objectively speaking, introducing into DDA extensive demultiplexing, as in our approach, brings DDA closer to DIA. In our study, the optimized isolation width ±2.0 Th was much narrower than that of most DIA methods, for example, the 25 Th wide window in SWATH (12). But the amount of evidence for peptide identification and thus the identification validity was also much higher in our approach, where each peptide was identified without a priori assumptions in the sequence database of the complete proteome, whereas SWATH (and some other DIA methods) only match a few fragment peaks to an MS/MS database containing only proteotypic peptides.
When widening the window above ±2.0 Th, we observed a decrease in spectral identification rate, which is closely related to spectral complexity and precursor intensity. Wider isolation window leads to more precursors being mixed together. Thus, mass conflict (two or more different fragments having an identical mass in one spectrum) will have a higher chance to happen. To avoid this conflict, Andromeda and some other search engine iteratively remove from the MS/MS spectra the peaks belonging from the previously identified peptide. Being too conservative, this method does not provide for the possibility that the same fragment mass belongs to different sequences, whereas such an event is statistically quite frequent for even-electron b- and y- fragments ions even in high resolution Orbitrap mass spectra (39). Thus, DeMix allows for multiple assignment of the same fragment peak. This increases the spectral identification rate but may also result in an increased rate of false matches. However, because these false matches would equally contain both direct and reversed (decoy) hits, trimming the output peptide list to <1% of reversed hits effectively checks this danger. This target-decoy approach may raise a concern of a bioinformatician because of the absence of a thorough statistical model for the scoring method, which precludes accurate calculation of p values for individual PSMs (40). However, our simple scoring method is unlikely to violate the basic presumptions of the target-decoy model, and thus the empirical estimation of FDR is satisfactory. Multiple evaluations performed in this study for a huge number of peptide identifications convinced us in the fair accuracy of the FDR estimates. In our opinion, using the FDR cutoff produces more reliable results than a probability (E-value) cutoff in analyzing chimeric spectra, because the FDR threshold will be automatically adjusted based on the actual decoy hit content, rather than on a presumed statistical model, as in the E-value cutoff.
Similarly, the phenomenon of decreased identification rate with an MS/MS window broadening has also been found in a recent study in metabolomics (41). The existence of the optimal window width for MS/MS seems to be a fundamental feature of tandem mass spectrometry, regardless of the method of analysis (DDA or DIA), or the analyte nature (peptides or metabolites). The width of the optimal window must however be analyte-dependent, as different analytes require different number of fragments to be uniquely identified. The mass accuracy in MS and MS/MS are two other important parameters. We are not aware of any theoretical study deducing the optimal window size by studying the effect of these parameters on tryptic peptide identification, but perhaps the time for such a study is ripe.
One of the fundamental problems faced by proteomics, especially in the DDA mode, is the insufficient overlap between the two consecutive LS-MS/MS runs in terms of identified peptides. One of the ways to alleviate this problem is to transfer peptide identifications through alignment of retention time or the order of elution (42, 43). Nevertheless, it is unlikely that perfect or nearly perfect overlap can be achieved without reaching the “bottom” of human proteome in a single LC-MS/MS analysis, as it is done for yeast proteome (7).
When the peptide abundance is very low (in our study, integrated intensity <107), it is nearly impossible to provide an informative MS/MS spectrum for reliable peptide identification (4). An increased sample load should help in detecting low abundant peptides, but in DIA the broadening of chromatographic peaks reduces the number of identifications, as the degree of multiplexing grows even further (38). In contrast, DDA with a much narrower MS/MS selection window should be more tolerant to chromatographic column overload. It appears that, because of a higher demand for chromatographic separation, the optimal on-column loading (300 ng) in DIA (38) is an order of magnitude lower than in the optimal DDA method (2). The same effect has recently been reported for the ion mobility platform (44). Thus DDA, by allowing larger sample loads, should provide more identifications in a single LC-MS/MS run than DIA. The reason why this has not been decisively demonstrated so far is that DDA could not properly handle the issue of natural multiplexing, which becomes more acute when the sample load increases. By employing demultiplexing, we have eliminated or at least dramatically reduced the impact of this problem on the number of identifications. As we mentioned before, the boundary between DDA and DIA are likely to be blurred in the future, and data analysis software can help a great deal in bridging the gap between the two approaches. But real breakthroughs in MS-based deep proteomics analysis can only come from hardware advances, such as higher acquisition rate (5), sensitivity (44), and mass accuracy (18), as well as better peptide separation (2), ionization (45), fragmentation (38), etc. Ultimately, reaching the “bottom” of the human proteome within a single analysis will require extension of the dynamic range of mass spectrometers by a few orders of magnitude (37).
Our final point reiterates the importance of high mass accuracy in proteomics, which can only be achieved by employing high resolution in both MS and MS/MS. For instance, in this study, the improvements came largely because of high mass accuracy intrinsically present in the Orbitrap data. We therefore remain strong advocates of employing high-resolution analyzers in proteomics, even though they are currently less sensitive than some low-resolution analyzers.
CONCLUSION
In this study, we presented a simple but robust workflow DeMix for analyzing complex proteomes with data-dependent LC-MS/MS acquisition. The workflow is able to deconvolute chimeric MS/MS spectra by spectral cloning for cofragmented precursors. Additionally, a reformulated scoring method based on Morpheus search engine was employed, increasing the peptide identification rate by utilizing the intrinsic high mass accuracy of the Orbitrap data. Tested on a benchmark dataset obtained from HeLa cell lysate, we achieved over 100% identification efficiency, and broke the “one MS/MS spectrum–one peptide ID” paradigm. With an instrument introduced to the market three years ago, we reached as high identification speed as using the next-generation instrument (5), and enabled rapid and deep analysis of human proteome twice as fast as in our previous workflow (2). We showed that integration of freely available, open-source and platform-independent software can immediately provide a practical and highly competitive solution for deep proteome profiling. Efforts to integrate this workflow with our accurate label-free quantification software (46) are under way to achieve a one-button operation.
Supplementary Material
Acknowledgments
We thank Craig Wenger from Coon group for helpful discussions.
Footnotes
Author contributions: B.Z. and R.A.Z. designed research; B.Z. and M.P. performed research; B.Z. contributed new reagents or analytic tools; B.Z. and A.C. analyzed data; B.Z. and R.A.Z. wrote the paper.
* This work was supported by the Knut and Alice Wallenberg Foundation as well as the Swedish Research council. Python scripts were developed and tested under Enthought® Canopy environment with a free academic license.
 This article contains supplemental Figs. S1 to S5 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S5 and Tables S1 to S4.
1 The abbreviations used are:
- DDA
- data-dependent acquisition
- AIF
- all-ion fragmentation
- DIA
- data-independent acquisition
- FDR
- false discovery rate
- LC-MS/MS
- liquid chromatography coupled to tandem mass spectrometry
- ppm (ppb)
- parts per million (billion)
- RT
- retention time
- Th
- Thomson, one unit of m/z
- PSM
- peptide-spectrum match
- SWATH
- sequential window acquisition of all theoretical fragment-ion spectra
- TOPP
- The OpenMS Proteomics Pipeline.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Pirmoradian M., Budamgunta H., Chingin K., Zhang B., Astorga-Wells J., Zubarev R. A. (2013) Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol. Cell. Proteomics 12, 3330–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thakur S. S., Geiger T., Chatterjee B., Bandilla P., Frohlich F., Cox J., Mann M. (2011) Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol. Cell. Proteomics 10, M110 003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Michalski A., Cox J., Mann M. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 10, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 5. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13(1):339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111 011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagaraj N., Kulak N. A., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. (2012) System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 11, M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Houel S., Abernathy R., Renganathan K., Meyer-Arendt K., Ahn N. G., Old W. M. (2010) Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J. Proteome Res, 9, 4152–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J., Bourne P. E., Bandeira N. (2011) Peptide identification by database search of mixture tandem mass spectra. Mol. Cell. Proteomics 10, M111 010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chapman J. D., Goodlett D. R., Masselon C. D. (2013) Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. [DOI] [PubMed] [Google Scholar]
- 11. Egertson J. D., Kuehn A., Merrihew G. E., Bateman N. W., MacLean B. X., Ting Y. S., Canterbury J. D., Marsh D. M., Kellmann M., Zabrouskov V., Wu C. C., MacCoss M. J. (2013) Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 10, 744–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gillet L. C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111 016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geiger T., Cox J., Mann M. (2010) Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteomics 9, 2252–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kall L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 15. Ledvina A. R., Savitski M. M., Zubarev A. R., Good D. M., Coon J. J., Zubarev R. A. (2011) Increased throughput of proteomics analysis by multiplexing high-resolution tandem mass spectra. Anal. Chem. 83, 7651–7656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 17. Zhang N., Li X. J., Ye M., Pan S., Schwikowski B., Aebersold R. (2005) ProbIDtree: an automated software program capable of identifying multiple peptides from a single collision-induced dissociation spectrum collected by a tandem mass spectrometer. Proteomics 5, 4096–4106 [DOI] [PubMed] [Google Scholar]
- 18. Zubarev R., Mann M. (2007) On the proper use of mass accuracy in proteomics. Mol. Cell. Proteomics 6, 377–381 [DOI] [PubMed] [Google Scholar]
- 19. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 20. Weisser H., Nahnsen S., Grossmann J., Nilse L., Quandt A., Brauer H., Sturm M., Kenar E., Kohlbacher O., Aebersold R., Malmstrom L. (2013) An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 12(4), 1628–1644 [DOI] [PubMed] [Google Scholar]
- 21. Nesvizhskii A. I. (2010) A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 73, 2092–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wenger C. D., Coon J. J. (2013) A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J. Proteome Res. 12, 1377–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox J., Michalski A., Mann M. (2011) Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass Spectr. 22, 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kohlbacher O., Reinert K., Gropl C., Lange E., Pfeifer N., Schulz-Trieglaff O., Sturm M. (2007) TOPP–the OpenMS proteomics pipeline. Bioinformatics 23, e191–197 [DOI] [PubMed] [Google Scholar]
- 25. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bald T., Barth J., Niehues A., Specht M., Hippler M., Fufezan C. (2012) pymzML–Python module for high-throughput bioinformatics on mass spectrometry data. Bioinformatics 28, 1052–1053 [DOI] [PubMed] [Google Scholar]
- 28. Goloborodko A. A., Levitsky L. I., Ivanov M. V., Gorshkov M. V. (2013) Pyteomics–a Python framework for exploratory data analysis and rapid software prototyping in proteomics. J. Am. Soc. Mass Spectr. 24, 301–304 [DOI] [PubMed] [Google Scholar]
- 29. Friedman J. H. (1991) Multivariate adaptive regression splines. Ann. Stat. 19, 1–67 [DOI] [PubMed] [Google Scholar]
- 30. Paizs B., Suhai S. (2005) Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 [DOI] [PubMed] [Google Scholar]
- 31. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., Coon J. J. (2011) COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Granholm V., Kim S., Navarro J. C., Sjolund E., Smith R. D., Kall L. (2014) Fast and accurate database searches with MS-GF+Percolator. J. Proteome Res. 13, 890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo X., Trudgian D. C., Lemoff A., Yadavalli S., Mirzaei H. (2014) Confetti: A Multi-protease Map of the HeLa Proteome for Comprehensive Proteomics. Mol. Cell. Proteomics 13(6):1573–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gesquiere J. C., Diesis E., Cung M. T., Tartar A. (1989) Slow isomerization of some proline-containing peptides inducing peak splitting during reversed-phase high-performance liquid chromatography. J. Chromatogr. A 478, 121–129 [Google Scholar]
- 36. Domon B., Aebersold R. (2006) Mass spectrometry and protein analysis. Science 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 37. Zubarev R. A. (2013) The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics 13, 723–726 [DOI] [PubMed] [Google Scholar]
- 38. Distler U., Kuharev J., Navarro P., Levin Y., Schild H., Tenzer S. (2014) Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 11(2), 167–170 [DOI] [PubMed] [Google Scholar]
- 39. Hubler S. L., Jue A., Keith J., McAlister G. C., Craciun G., Coon J. J. (2008) Valence parity renders z(*)-type ions chemically distinct. J. Am. Chem. Soc. 130, 6388–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gupta N., Bandeira N., Keich U., Pevzner P. A. (2011) Target-decoy approach and false discovery rate: when things may go wrong. J. Am. Soc. Mass Spectrom. 22, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu X., Chen Y., Subramanian R. (2014) Comparison of information-dependent acquisition, SWATH, and MS techniques in metabolite identification study employing ultrahigh-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Anal. Chem. 86(2), 1202–1209 [DOI] [PubMed] [Google Scholar]
- 42. Bateman N. W., Goulding S. P., Shulman N. J., Gadok A. K., Szumlinski K. K., Maccoss M. J., Wu C. C. (2014) Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell. Proteomics 13, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vincent C. E., Potts G. K., Ulbrich A., Westphall M. S., Atwood J. A., 3rd, Coon J. J., Weatherly D. B. (2013) Segmentation of precursor mass range using “tiling” approach increases peptide identifications for MS1-based label-free quantification. Anal. Chem. 85, 2825–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baker E. S., Burnum-Johnson K. E., Jacobs J. M., Diamond D. L., Brown R. N., Ibrahim Y. M., Orton D. J., Piehowski P. D., Purdy D. E., Moore R. J., Danielson W. F., 3rd, Monroe M. E., Crowell K. L., Slysz G. W., Gritsenko M. A., Sandoval J. D., Lamarche B. L., Matzke M. M., Webb-Robertson B. J., Simmons B. C., McMahon B. J., Bhattacharya R., Perkins J. D., Carithers R. L., Jr., Strom S., Self S. G., Katze M. G., Anderson G. A., Smith R. D. (2014) Advancing the high throughput identification of disease specific protein signatures using multiplexed ion mobility spectrometry. Mol. Cell. Proteomics 3(4), 1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hahne H., Pachl F., Ruprecht B., Maier S. K., Klaeger S., Helm D., Medard G., Wilm M., Lemeer S., Kuster B. (2013) DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat, Methods 10, 989–991 [DOI] [PubMed] [Google Scholar]
- 46. Lyutvinskiy Y., Yang H., Rutishauser D., Zubarev R. A. (2013) In silico instrumental response correction improves precision of label-free proteomics and accuracy of proteomics-based predictive models. Mol. Cell. Proteomics 12, 2324–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Neuhauser N., Michalski A., Cox J., Mann M. (2012) Expert system for computer-assisted annotation of MS/MS spectra. Mol. Cell. Proteomics 11, 1500–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.