

Abstract
Kainate receptors (KARs) are ionotropic glutamate receptors that also activate noncanonical G-protein-coupled signaling pathways to depress the slow afterhyperpolarization (sAHP). Here we show that long-term depression of KAR-mediated synaptic transmission (KAR LTD) at rat hippocampal mossy fiber synapses relieves inhibition of the sAHP by synaptic transmission. KAR LTD is induced by high-frequency mossy fiber stimulation and natural spike patterns and requires activation of adenosine A2A receptors. Natural spike patterns also cause long-term potentiation of NMDA receptor-mediated synaptic transmission that overrides the effects of KAR LTD on the cellular response to low-frequency synaptic input. However, KAR LTD is dominant at higher frequency synaptic stimulation where it decreases the cellular response by relieving inhibition of the sAHP. Thus we describe a form of glutamate receptor plasticity induced by natural spike patterns whose primary physiological function is to regulate cellular excitability.
Introduction
Kainate receptors (KARs) are unique among glutamate receptors in exhibiting both canonical ionotropic and noncanonical metabotropic functions. At postsynaptic densities they mediate a component of ionotropic synaptic responses (Castillo et al., 1997; Vignes and Collingridge, 1997) but also inhibit both the medium and slow components of the afterhyperpolarization (mAHP and sAHP) via their metabotropic action (Cherubini et al., 1990; Melyan et al., 2002, 2004; Fisahn et al., 2005; Ruiz et al., 2005; Fernandes et al., 2009; Segerstrale et al., 2010). Therefore, although classically considered ionotropic receptors, the increasing importance of the metabotropic actions of KARs is forcing a reassessment of their physiological function.
Mossy fiber synapses between dentate granule cells and CA3 pyramidal cells in the hippocampus exhibit NMDA receptor (NMDAR)-independent long-term potentiation (LTP) in response to high-frequency stimulation (HFS). This form of mossy fiber LTP is expressed presynaptically as an increase in the probability of glutamate release from the presynaptic terminal resulting in an increase in AMPA receptor (AMPAR)-mediated synaptic transmission (Nicoll and Schmitz, 2005). Mossy fiber synapses also exhibit other forms of synaptic plasticity including postsynaptically expressed LTP of NMDARs (Kwon and Castillo, 2008a; Rebola et al., 2008), and LTP and LTD of AMPARs (Lei et al., 2003; Rebola et al., 2011).
At mossy fiber synapses, KARs are located on both presynaptic boutons and postsynaptic spines (Darstein et al., 2003). Activation of presynaptic KARs facilitates subsequent glutamate release which in turn facilitates the induction of presynaptically expressed mossy fiber LTP (Bortolotto et al., 1999; Contractor et al., 2001; Schmitz et al., 2001; Kamiya et al., 2002; Pinheiro and Mulle, 2008) (but see Kwon and Castillo, 2008b). Activation of postsynaptic KARs results in a small, slow decaying synaptic response that is a minor component of mossy fiber postsynaptic potentials (EPSPs) (Castillo et al., 1997; Vignes and Collingridge, 1997; Mulle et al., 1998) but contributes to EPSP summation (Frerking and Ohliger-Frerking, 2002; Rebola et al., 2007; Sachidhanandam et al., 2009; Straub et al., 2011). Long-term depression of postsynaptic KARs (KAR LTD) is induced at mossy fiber synapses in response to low-frequency stimulation (LFS) or postsynaptic depolarization (Selak et al., 2009; Chamberlain et al., 2012). Furthermore, it is not clear what role KAR LTD plays given that KAR EPSPs are considerably smaller than either AMPAR or NMDAR EPSPs.
We hypothesized that synaptic plasticity of KARs alters the inhibition of the AHP in CA3 pyramidal neurons by synaptic KAR activation thereby providing a mechanism for synaptic plasticity to regulate cellular excitability. Here we provide evidence for depression of the sAHP by synaptic KARs and relief of this inhibition by KAR LTD induced by HFS or naturally occurring spike patterns recorded from dentate granule cells. We show that KAR LTD and NMDAR LTP can be induced concurrently by naturally occurring spike patterns and investigate the summative effect of these two forms of synaptic plasticity on cellular response to synaptic stimulation. Finally, we demonstrate the pre-eminent functional consequence of KAR LTD is the relief of sAHP inhibition.
Materials and Methods
Slice preparation.
Transverse hippocampal slices were prepared from male juvenile (P13–P15) Wistar rats. Brains were immediately removed after cervical dislocation and immersed in ice-cold aCSF containing the following (in mm): 119 NaCl, 10 glucose, 26 NaHCO3, 2.5 KCl, 1 NaH2PO4, 1 CaCl2, and 5 MgSO4. Individual hippocampi were mounted on agar and 400–500 μm slices were cut using a DSK 1000 microslicer (DSK). After dissection, slices were transferred to aCSF containing the following (in mm): 119 NaCl, 10 glucose, 26 NaHCO3, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, and 2.5 CaCl2, maintained at 35°C for 30 min, and then stored at room temperature. After dissection, slices were left for a minimum of 1 h before recordings were made. All solutions were saturated with 95% O2 and 5% CO2. All experiments were performed in accordance with Home Office guidelines as directed by the Home Office Licensing Team at the University of Bristol.
Electrophysiological recordings.
Slices were placed in a submerged recording chamber perfused with aCSF (as above) at 35°C. CA3 pyramidal cells were visualized using infrared-differential interference contrast optics on an Olympus BX-51WI microscope. Patch electrodes with a resistance of 4–5 MΩ were pulled from borosilicate filamented glass capillaries (Harvard Apparatus) using a vertical puller (PC-10; Narishige). For voltage-clamp recordings pipettes were filled with intracellular solution containing the following (in mm): 117 CsMeSO3, 8 NaCl, 10 HEPES, 5 QX-314, 4 Mg-ATP, 0.3 Na-GTP, 0.2 EGTA, 0.1 bestatin, and 0.1 leupeptin, set to pH 7.4, 280–285 mOsm. For current-clamp recordings pipettes were filled with a potassium-based intracellular solution containing the following in mm: 135 KGluconate, 8 NaCl, 10 HEPES, 0.2 EGTA, 2 MgATP, and 0.3 NaGTP.
Recordings from CA3 pyramidal neurons were made with a Multiclamp 700A amplifier (Molecular Devices), filtered at 4 kHz, and digitized at 10 kHz using a data acquisition board and Signal acquisition software (CED). For voltage-clamp experiments cells were clamped at −60 mV (without junction potential correction). Series resistance was monitored throughout the experiments and cells that showed >20% changes were discarded from subsequent analysis. Recordings were also rejected from analysis if the series resistance was >30 MΩ. AMPAR EPSCs were recorded in the presence of picrotoxin (50 μm; GABAAR antagonist) and L-689,560 (5 μm; NMDAR antagonist). When recording KAR EPSCs GYKI 53655 (40 μm) was also included to block AMPAR. To record NMDAR EPSCs, cells were clamped at −50 mV, the extracellular Mg2+ concentration was reduced to 0.3 mm, and NBQX (20 μm) was added to block AMPAR and KAR. Current-clamp recordings were done in the presence of picrotoxin (50 μm) and CGP 55845 (1 μm) to block GABAAR and GABABR, respectively. Bridge balance was used for all current-clamp recordings.
Synaptic responses were evoked with 100 μs square voltage steps applied at 0.05 Hz through a bipolar stimulating electrode located in the granule cell layer to stimulate mossy fiber axons. In all experiments with AMPAR and NMDAR EPSCs and some experiments on KAR EPSCs the purity of mossy fiber responses was confirmed by 5 min application of the group II mGluR agonist DCG-IV (1 μm) (Kamiya et al., 1996). Any responses not inhibited by >80% were discarded from subsequent analysis. The peak amplitude of evoked synaptic responses was measured to calculate response amplitude. Control test pulses were given every 20 s. In the paired-pulse stimulation protocol, two pulses were delivered with 40 ms (AMPAR and KAR) or 100 ms (NMDAR) intervals. Natural stimulus patterns were presented while maintaining cells in voltage-clamp at −60 mV.
IsAHPs were recorded in CA3 neurons voltage-clamped at −50 mV in the presence of picrotoxin (50 μm; GABAAR antagonist), L-689,560 (5 μm; NMDAR antagonist), GYKI 53655 (40 μm; AMPAR antagonist), and CGP 55845 (1 μm; GABABR antagonist) and induced by a depolarizing voltage step to 0 mV for 200 ms applied every 20 s. Recording pipettes were filled with a potassium based intracellular solution containing the following in mm: 135 KGluconate, 8 NaCl, 10 HEPES, 0.2 EGTA, 2 MgATP, and 0.3 NaGTP. Measurement of IsAHP amplitude was made at the peak of the response ∼300 ms after returning the membrane potential to −50 mV and was clearly distinguishable from ImAHP. The data were filtered at 1 kHz and sampled at 2 kHz. Synaptic stimulation was applied to mossy fibers every 20 and 10 s before IsAHP.
Data analysis.
Data were analyzed using Signal acquisition software (CED). Three consecutive traces were averaged together to produce a mean response every minute. EPSC amplitude measurements were taken from the mean traces and normalized to the mean baseline EPSC amplitude. Quantification of plasticity was calculated by averaging the amplitude of control EPSCs measured for 10 min before induction and comparing it with the average amplitude of EPSCs between 30 and 35 min following induction. 1/CV2 was calculated from EPSC amplitude measurements made during 10 min of baseline recording and 20–30 min after plasticity induction. Values were then normalized to the baseline for each experiment. Spike delay was calculated as the average time taken to spike from the first of the 10 stimuli, spike number as the total number of spikes and depolarization envelope was calculated as the area under the curve created by joining points in the voltage trace just before each stimulation. Spike delay, number, and depolarization were all averaged for 10 min before natural stimulus pattern (NSP) and 15–20 min after. Data are plotted as the mean ±SEM. Statistical tests were performed with paired or unpaired Student's t tests or ANOVA as appropriate. *, **, and *** denote p < 0.05, 0.01, and 0.001 respectively. Synaptic plasticity was assessed by comparing the mean normalized EPSC amplitudes 30–35 min after induction.
Drugs.
UCL2077, L-689,560, DCG-IV, CGP 55845, and ZM 241385 were purchased from Tocris Bioscience. Picrotoxin and PKC 19–36 were purchased from Sigma-Aldrich. NBQX and GYKI 53655 (custom synthesized) were purchased from Ascent Scientific.
Results
KAR inhibition of sAHP
We first characterized the effects of KAR-mediated transmission at hippocampal mossy fiber synapses on the sAHP in CA3 pyramidal neurons (Melyan et al., 2004; Ruiz et al., 2005). To test whether sAHP was depressed by KAR activation, whole-cell voltage-clamp recordings were made from CA3 pyramidal neurons in acutely prepared hippocampal slices and sAHP currents (IsAHP) elicited by depolarizing the membrane potential to 0 mV for 200 ms in the presence of GYKI 53655, picrotoxin, CGP 55845, and L-689,560 to inhibit AMPARs, GABAARs, GABABRs, and NMDARs respectively. This protocol produced robust, stable IsAHP of average amplitude 71.0 ± 7.6 pA and duration 4.3 ± 0.6 s. In control experiments IsAHP amplitude showed only a small nonsignificant depression over 40 min recordings (84 ± 12%; n = 6; p > 0.05). We occasionally saw a small mAHP current but lack of consistency when recording in the whole-cell configuration (Buchanan et al., 2010) prevented further analyses of these currents. Stimulation of the mossy fiber pathway with 10 stimuli at 25 Hz every 20 s produced KAR-mediated postsynaptic currents (KAR EPSCs) and led to a fully reversible decrease in the amplitude of IsAHP (Fig. 1A) (55.6 ± 11.5%; n = 6; p < 0.01) without any change in input resistance (194.6 ± 21.0 MΩ vs 225.1 ± 22.5 MΩ; n = 6; p > 0.05) or holding current (48.4 ± 26.1 pA vs 39.9 ± 28.3 pA; n = 6; p > 0.05). The depression of IsAHP was also reversed by the addition of the AMPAR/KAR antagonist NBQX (10 μm) (Fig. 1B) (from 56.2 ± 9.5% of baseline to 90.6 ± 5.3%; n = 5; p < 0.01) or the mGluR group II agonist DCG-IV (1 μm) that selectively inhibits neurotransmitter release from mossy fiber terminals (Kamiya et al., 1996) (Fig. 1C) (from 54.9 ± 9.1% of baseline to 85.6 ± 10.6%; n = 6; p < 0.01) demonstrating that the depression of IsAHP was mediated by activation of synaptic KARs at mossy fiber synapses.
Figure 1.
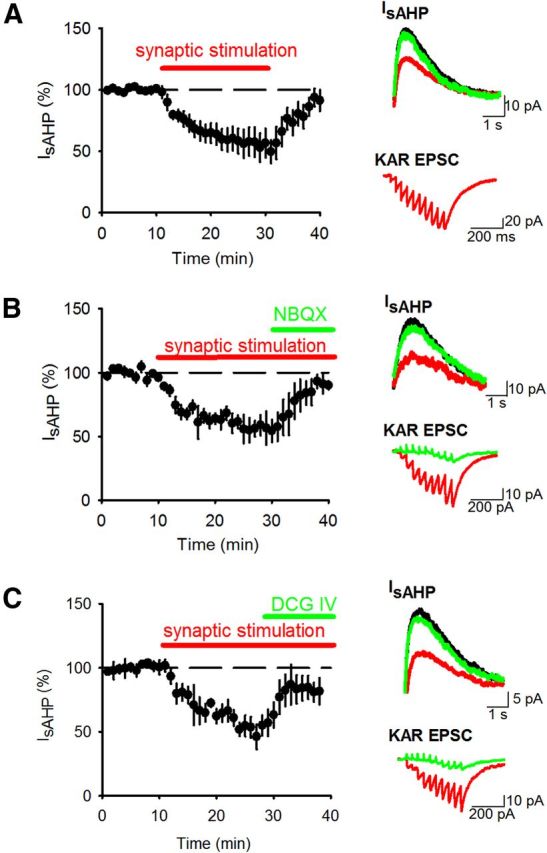
Synaptic KAR stimulation depresses IsAHP. A, IsAHPs were stimulated with 200 ms depolarizations to 0 mV from a holding potential of −60 mV. Synaptic stimulation of 10 stimuli at 25 Hz produced a depression in IsAHP. Example traces show IsAHPs (top) and synaptic responses (bottom) during baseline (black), synaptic stimulation (red) and after recovery (green). B, Application of NBQX (10 μm) to block KARs reversed the depression of IsAHP by synaptic stimulation. C, Application of DCG-IV (1 μm) to inhibit glutamate release from mossy fiber terminals reversed the depression of IsAHP by synaptic stimulation.
KAR LTD induced by HFS or natural stimulus patterns
KAR LTD can be induced by prolonged LFS or postsynaptic depolarization (Selak et al., 2009; Chamberlain et al., 2012); however, other forms of plasticity at this synapse are induced by HFS therefore we tested whether KAR LTD can be induced by HFS by characterizing the effects of HFS on KAR-mediated transmission at hippocampal mossy fiber synapses. AMPAR EPSCs were recorded in the presence of L-689,560 and picrotoxin and KAR EPSCs were pharmacologically isolated with additional GYKI53655. One-hundred or more stimuli presented at 25 Hz typically induces presynaptic LTP associated with decreases in paired pulse ratio (PPR) and EPSC amplitude variance (Nicoll and Schmitz, 2005). However, it has been shown that lower numbers of stimuli can induce postsynaptically expressed NMDAR LTP without engaging presynaptic LTP (Kwon and Castillo, 2008a; Rebola et al., 2008). We tested a HFS protocol of 50 stimuli at 25 Hz and found that this induced a robust LTD of KAR EPSCs (Fig. 2A) (54.4 ± 5.2%; n = 6; p < 0.01) but no presynaptic LTP measured with AMPAR EPSCs (Fig. 2B) (110.2 ± 32.4%; n = 7; p > 0.05). Analysis of PPR and EPSC amplitude variance using 1/CV2 revealed no change for either KAR EPSCs (Fig. 2A) (PPR 1.9 ± 0.1 to 2.0 ± 0.1; p > 0.05; norm 1/CV2 1.51 ± 0.59; p > 0.05) or AMPAR EPSCs (Fig. 2B) (PPR 2.9 ± 0.3 to 2.8 ± 0.2; p > 0.05; norm 1/CV2 0.83 ± 0.19; p > 0.05). In agreement with previously published data PPR for KAR EPSCs was lower compared with AMPAR EPSCs (Ito et al., 2004; Perrais et al., 2009) and similar to our previous studies (Martin et al., 2007; Chamberlain et al., 2012), mossy fiber KAR EPSCs recorded at 34°C were stable before and after HFS demonstrating no underlying rundown in response amplitude over the course of the experiments. Therefore, at the mossy fiber synapse, KAR LTD can be induced by HFS in the absence of presynaptic LTP.
Figure 2.
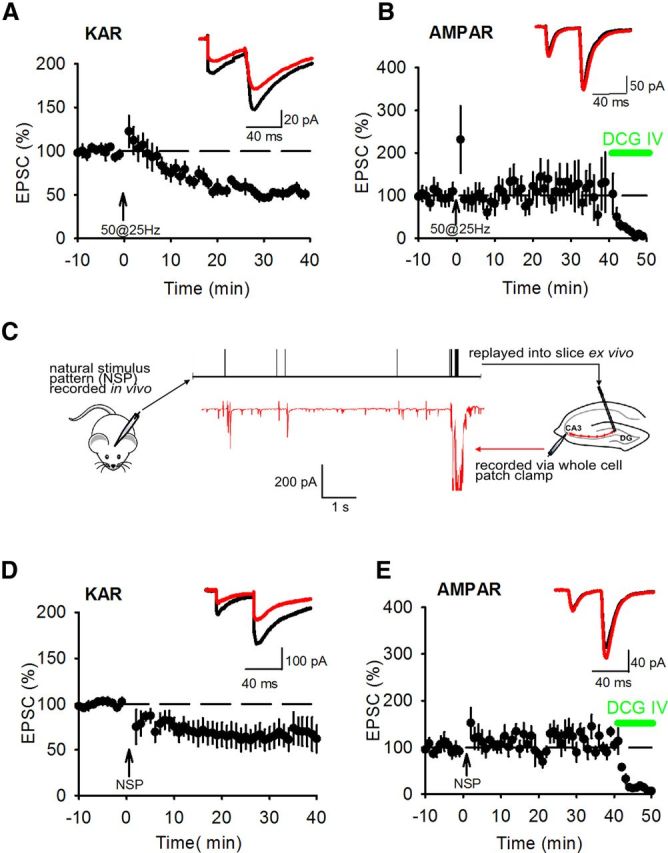
HFS induces LTD of KAR EPSCs. A, 50 stimuli at 25 Hz (arrow) was sufficient to induce LTD of KAR EPSCs. Example traces show EPSCs during baseline (black) and after HFS (red). PPR was unchanged by KAR LTD induction. B, Fifty stimuli at 25 Hz was insufficient to induce LTP of AMPAR EPSCs or change PPR. DCG-IV (1 μm) was applied at the end of each experiment to confirm mossy fiber input. C, Illustration of NSP. A granule cell was recorded in vivo and a spike pattern with high density of spikes (black) replayed into the stimulation electrode of a slice experiment while recording the synaptic response in a CA3 pyramidal neuron (red). The NSP was sufficient to induce KAR LTD (D) but insufficient to induce presynaptic LTP measured by AMPAR EPSCs (E).
The different plasticity thresholds for KAR LTD and presynaptic LTP suggest that patterns of activity that occur at mossy fiber synapses in vivo may engage KAR LTD more readily than presynaptic LTP. It has been shown that some patterns of high-frequency granule cell activity recorded from awake rats are able to induce presynaptic LTP at mossy fiber synapses when replayed into slices (Gundlfinger et al., 2010; Mistry et al., 2011). We investigated whether a NSP of duration 10 s and containing 56 stimuli which incorporated a burst of 50 stimuli in 2 s (Fig. 2C) (chosen as an epoch of activity with a high density of granule cell spiking) (Wiebe and Staubli, 1999, 2001; Mistry et al., 2011) could induce KAR LTD or presynaptic LTP.
Similar to HFS of 50 stimuli at 25 Hz, the NSP induced KAR LTD (Fig. 2D) (59.6 ± 11.6%; n = 7, p < 0.05) with no change in PPR (Fig. 2D) (1.5 ± 0.2 vs 1.7 ± 0.2; p > 0.05) or EPSC amplitude variance (norm 1/CV2 0.89 ± 0.31; p > 0.05) but interestingly the NSP did not induce presynaptic LTP measured with AMPAR EPSCs (Fig. 2E) (100.8 ± 12.9%; n = 7; p > 0.05) and there was no change in PPR (Fig. 2E) (3.5 ± 0.5 vs 3.6 ± 0.3; p > 0.05) or EPSC amplitude variance (norm 1/CV2 0.89± 0.20; p > 0.05). Indeed, three consecutive repetitions of the NSP separated by 20 s were insufficient to induce reliable presynaptic LTP (176.8 ± 55.5%; n = 8; p > 0.05; data not shown). We attribute the lack of presynaptic LTP to the high threshold for LTP induction in slices from rats compared with mice (Schmitz et al., 2003; Kwon and Castillo, 2008a; Rebola et al., 2008; Mistry et al., 2011). We conclude that the NSP contains sufficient numbers of stimuli at a high enough frequency to engage KAR LTD but not presynaptic LTP.
KAR LTD relieves the inhibition of IsAHP
We hypothesized that KAR LTD alters the inhibition of the sAHP by synaptic KAR activation thereby providing a mechanism for synaptic plasticity to regulate cellular excitability. To test whether IsAHP was depressed by KAR activation and then relieved of this depression following KAR LTD, we recorded IsAHP and stimulated mossy fiber synapses with 10 stimuli at 25 Hz every 20 s. After IsAHP stabilized we then stimulated mossy fiber synapses with the NSP which relieved ∼50% of IsAHP inhibition induced by 25 Hz synaptic stimulation (Fig. 3A) (from 40.4 ± 7.9% to 65.2 ± 8.2; n = 5; p < 0.01 compared with synaptic stimulation). The NSP also reduced the KAR EPSC without a change in PPR (Fig. 3A) (39.5 ± 9.1%; p < 0.01; PPR-2.0 ± 0.3 to 1.8 ± 0.4; p > 0.05; n = 5). Similarly, induction of KAR LTD with 50 stimuli at 25 Hz also led to a reversal of the synaptic stimulation-induced depression of IsAHP, a robust LTD and no change in PPR (Fig. 3B) (IsAHP from 58.4 ± 2.5% to 85.0 ± 4.6%; p < 0.01; KAR EPSC to 47.5 ± 4.6% p < 0.01; PPR from 1.7 ± 0.5 to 1.8 ± 0.5; p > 0.05; n = 5). To confirm the role of KARs in the relief of IsAHP inhibition by the NSP (Fig. 3A), we repeated the experiments with the addition of CNQX (10 μm) which prevented the depression of IsAHP by synaptic stimulation (Fig. 3C) (92.1 ± 9.4%; p > 0.05; n = 5) and the effect of the NSP on IsAHP (Fig. 3C) (98.5 ± 13.2%; p > 0.05; n = 5). These results demonstrate that KAR LTD partially relieves the inhibition of IsAHP by synaptic KAR activation.
Figure 3.
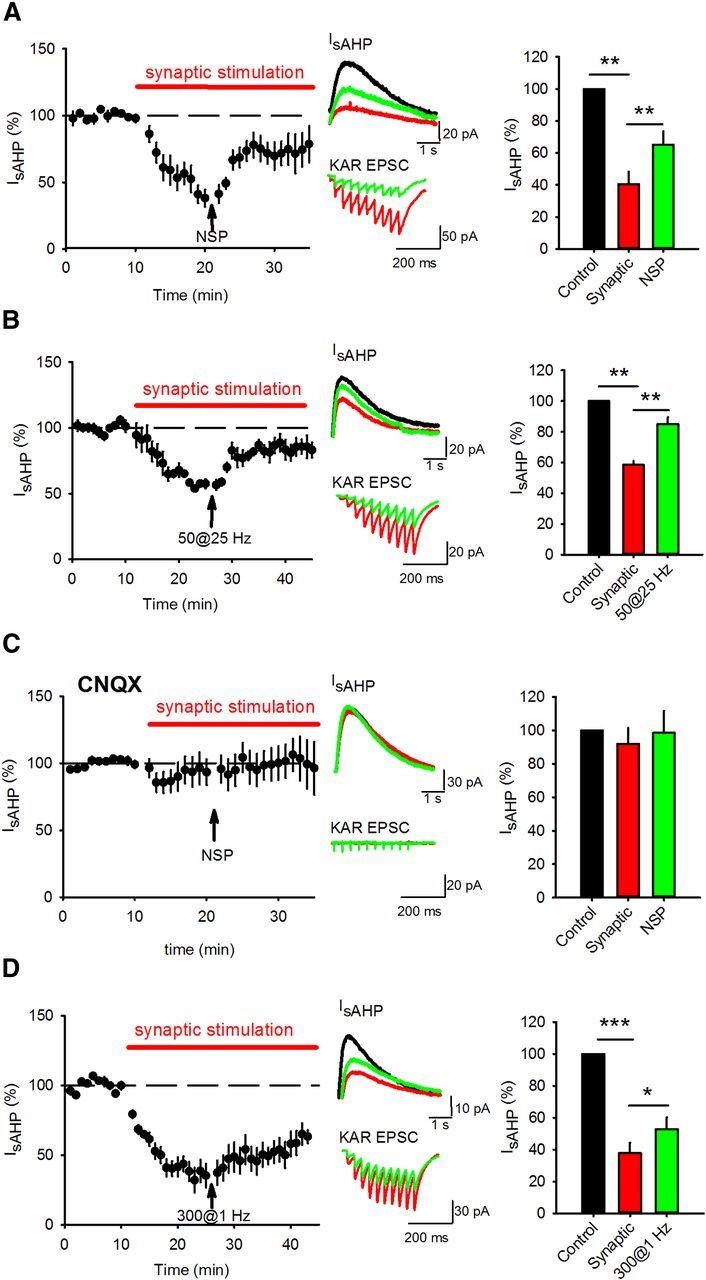
KAR LTD relieves the KAR-mediated inhibition of sAHP. A, IsAHP amplitude was depressed by synaptic stimulation of 10 stimuli at 25 Hz but the depression was partially relieved by the NSP. Example traces show IsAHPs (top) and synaptic responses (bottom) during baseline (black), after synaptic stimulation (red) and after the NSP (green). Bar graph shows quantification of IsAHP amplitude depression and subsequent relief. B, Depression of IsAHP by synaptic stimulation was also relieved by HFS of 50 stimuli at 25 Hz. C, In the presence of CNQX (10 μm) to block KARs, synaptic stimulation did not depress IsAHP and the NSP had no effect on IsAHP amplitude. D, Depression of IsAHP by synaptic stimulation was relieved by LFS of 300 stimuli at 1 Hz but with a slower time course.
KAR LTD can also be induced using LFS of 300 stimuli at 1 Hz (Selak et al., 2009; Chamberlain et al., 2012) so we next tested whether this protocol could also relieve the inhibition of IsAHP by synaptic KAR activation. Stimulation of mossy fiber synapses with 300 stimuli at 1 Hz also induced KAR LTD and relieved the inhibition of IsAHP by synaptic KAR activation but with a slower time course than NSP or 50 stimuli at 25 Hz induced KAR LTD (Fig. 3D) (IsAHP from 37.7 ± 6.7% to 52.7 ± 7.4%, p < 0.05; KAR EPSC to 59.9 ± 12.5%, p < 0.05; PPR from 2.0 ± 0.2 to 1.9 ± 0.2; p > 0.05; n = 6).
Induction of NMDAR LTP
We have so far considered two independent forms of synaptic plasticity at the mossy fiber synapse, presynaptic LTP expressed as an increase in the probability of glutamate release and postsynaptic KAR LTD. Both are induced by HFS. A third form of synaptic plasticity, NMDAR LTP, also exists at the mossy fiber synapse but its induction requires fewer stimuli at high-frequency than presynaptic LTP (Kwon and Castillo, 2008a; Rebola et al., 2008). We therefore investigated whether HFS with low numbers of stimuli or the NSP could induce NMDAR LTP. Stable NMDAR EPSCs were recorded at −50 mV in the presence of 0.3 mm Mg2+ and 20 μm NBQX. HFS of 24 stimuli at 25 Hz (Kwon and Castillo, 2008a) induced robust LTP of NMDAR EPSCs (Fig. 4A) (315.7 ± 56.5%; n = 6; p < 0.05) with no corresponding change in PPR (Fig. 4A) (2.5 ± 0.1 vs 2.3 ± 0.2; p > 0.05) or EPSC amplitude variance (norm 1/CV2 2.59 ± 0.98; p > 0.05). NMDAR LTP was also induced by the NSP (Fig. 4B) (192.7 ± 28.9%; n = 7; p < 0.05) with no corresponding change in PPR (Fig. 4B) (2.7 ± 0.3 vs 2.5 ± 0.4; p > 0.05) or EPSC amplitude variance (norm 1/CV2 2.09 ± 1.56; p > 0.05). Interestingly, we were unable to induce KAR LTD with HFS of 24 stimuli at 25 Hz (Fig. 4C) (100.7 ± 26.9%; n = 6; p > 0.05) again without a change in either PPR (1.68 ± 0.23 vs 1.65 ± 3.4; p > 0.05) or EPSC amplitude variance (norm 1/CV2 2.67 ± 1.57; p > 0.05). The higher threshold for KAR LTD versus NMDAR LTP could be because NMDARs are blocked during measurement of KAR EPSCs. However, removal of the NMDAR antagonist during HFS of 24 stimuli at 25 Hz did not result in KAR LTD (Fig. 4D) (120.3 ± 10.0%, n = 5, p > 0.05) indicating that activation of NMDARs did not lower the threshold for KAR LTD to that seen for NMDAR LTP. In addition, HFS of 10 stimuli at 25 Hz was insufficient to induce NMDAR LTP (Fig. 4E) (146.8 ± 40.0%, n = 7, p > 0.05). Thus we describe 3 different forms of synaptic plasticity at the mossy fiber synapse that have different thresholds for induction. NMDAR LTP is induced by the fewest number of stimuli, then KAR LTD and finally presynaptic LTP with both NMDAR LTP and KAR LTD induced by the NSP.
Figure 4.
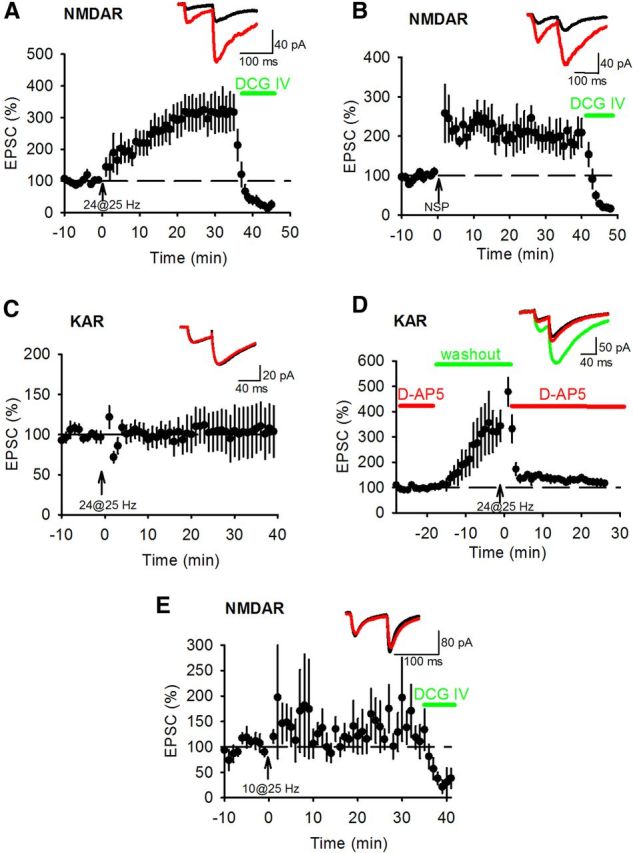
HFS induces LTP of NMDAR EPSCs. (A) Twenty-four stimuli at 25 Hz (arrow) was sufficient to induce NMDAR LTP. Example traces show EPSCs during baseline (black) and after HFS (red). PPR was unchanged by NMDAR LTP induction. DCG-IV (1 μm) was applied at the end of each experiment to confirm mossy fiber input. B, The NSP was sufficient to induce NMDAR LTP but did not change PPR. C, Twenty-four stimuli at 25 Hz was insufficient to induce KAR LTD or change PPR. D, Twenty-four stimuli at 25 Hz was insufficient to induce KAR LTD with NMDAR unblocked. D-AP5 (50 μm) was washed out for 15 min before presentation of 24 stimuli at 25 Hz and then immediately reapplied. Example traces show EPSCs during baseline (black), after D-AP5 washout (red) and after HFS and reapplication of D-AP5 (green). E, Ten stimuli at 25 Hz was insufficient to induce NMDAR LTP or change PPR.
KAR LTD requires adenosine A2A receptor activation
KAR LTD at mossy fiber synapses induced by LFS or depolarization (15 min at +30 mV) is dependent on activation of mGluR5 and PKC (Selak et al., 2009; Chamberlain et al., 2012), both of which are also required for NMDAR LTP. An additional requirement for NMDAR LTP is the activation of adenosine A2A receptors (A2ARs) (Rebola et al., 2008). Given the similarity in induction protocols and mechanisms between NMDAR LTP and KAR LTD, we tested the requirement for A2ARs in KAR LTD with the A2AR antagonist ZM 241385 (50 nm). Inhibition of A2ARs prevented KAR LTD induced by HFS with 50 stimuli at 25 Hz (Fig. 5A) (97.3 ± 7.8%, p > 0.05; PPR from 1.6 ± 0.2 to 1.7 ± 0.2; p > 0.05; n = 10). Therefore, our data reveal similar mechanisms underlie NMDAR LTP and KAR LTD at mossy fiber synapses.
Figure 5.
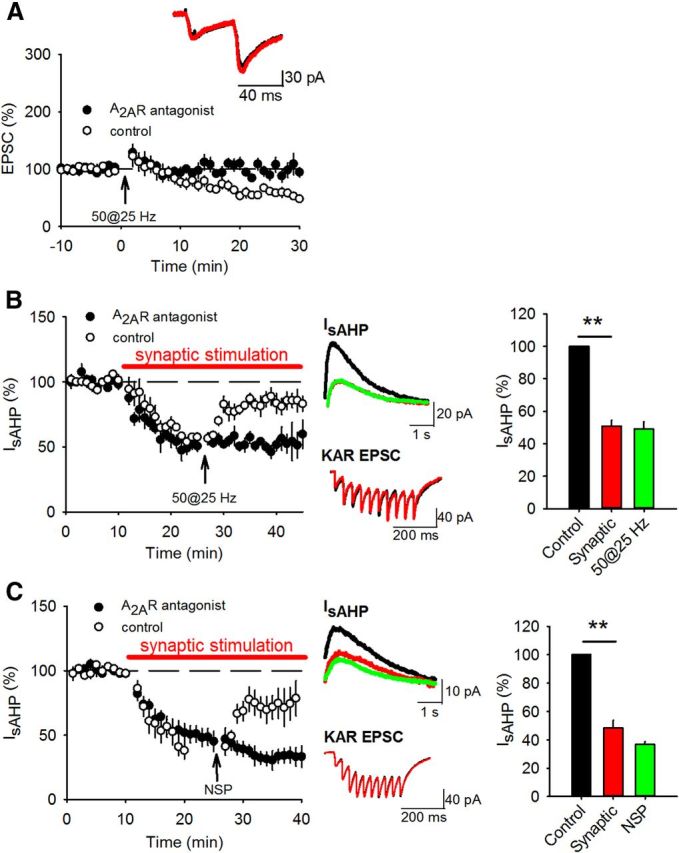
KAR LTD and the relief of IsAHP inhibition require adenosine A2A activation. A, Application of the A2AR antagonist ZM 241385 (50 nm) blocked KAR LTD induced by 50 stimuli at25 Hz. Control data from Figure 2A are included for comparison. PPR was unchanged. Example traces show EPSCs during baseline (black) and after HFS (red). B, C, IsAHP amplitude was depressed by synaptic stimulation of 10 stimuli at 25 Hz in the presence of the A2AR antagonist but the relief of depression by 50 stimuli at 25 Hz (B) or the NSP (C) was blocked. Control data from Figures 3B (B) and 3A (C) are included for comparison. Example traces show IsAHPs (top) and synaptic responses (bottom) during baseline (black), after synaptic stimulation (red) and after the 50 stimuli at 25 Hz or NSP (green). Bar graph shows quantification of IsAHP amplitude depression.
Inhibition of KAR LTD is predicted to also inhibit the relief of IsAHP inhibition by synaptic KAR activation. In the presence of the A2AR antagonist, activation of synaptic KARs depressed IsAHP in a similar fashion to that observed in control conditions. However, stimulation of mossy fiber synapses with 50 stimuli at 25 Hz did not result in KAR LTD or relief of IsAHP depression (Fig. 5B) (from 50.9 ± 3.7% to 49.1 ± 4.6%, p > 0.05; n = 5). Similarly, in the presence of the A2AR antagonist stimulation of mossy fiber synapses with the NSP did not induce KAR LTD or relieve the inhibition of IsAHP by synaptic KAR activation (Fig. 5C) (from 48.4 ± 5.5% to 38.0 ± 2.0%, p > 0.05; n = 5). Thus, the canonical and noncanonical effects of KAR LTD are inhibited by an adenosine A2AR antagonist supporting a mechanism that mirrors that of NMDAR LTP.
KAR LTD decreases cellular excitability by relieving inhibition of the sAHP
KAR and NMDAR kinetics are relatively slow and the amplitude of synaptic responses relatively small compared with AMPARs at mossy fiber synapses (Castillo et al., 1997; Vignes and Collingridge, 1997; Mulle et al., 1998; Rebola et al., 2011). Therefore, the major impact of postsynaptic NMDAR and KAR is the duration of EPSPs rather than amplitude (Sachidhanandam et al., 2009). However, it is difficult to predict what effect the combination of NMDAR LTP and KAR LTD may have on the duration of EPSPs and their temporal summation. Furthermore, the impact of KAR LTD on sAHP will also affect EPSP temporal summation reducing cellular excitability.
To explore the consequences of concurrent changes in postsynaptic NMDAR and KAR we examined the cellular response to 10 synaptic stimuli presented at 3, 10, or 20 Hz before and after induction of NMDAR LTP and KAR LTD by the NSP. Ten stimuli at 3, 10, or 20 Hz was insufficient to induce any form of long-term synaptic plasticity at mossy fiber synapses (Figs. 2, 4). EPSP amplitude was set so that the first action potential occurred on EPSP 3–6 in control conditions. EPSP amplitudes varied across experiments (range 1.2–11.8 mV) but there was no correlation between initial EPSP amplitude and frequency of synaptic stimulation or result of NSP stimulation (data not shown). In agreement with the observation that presynaptic LTP is not induced by the NSP and that NMDAR and KAR have little influence on peak EPSP amplitude, the NSP produced no change in the amplitude of the first EPSP at any frequency of stimulation (3 Hz: 8.0 ± 1.2 mV vs 10.2 ± 1.2 mV; 10 Hz: 4.9 ± 1.3 mV vs 5.0 ± 1.5 mV; 20 Hz: 6.3 ± 1.3 mV vs 5.7 ± 1.1 mV). However, during 3 Hz synaptic stimulation the first action potential occurred earlier following the NSP (Fig. 6A) (delay to spike 50.1 ± 4.3%; n = 5; p < 0.001), whereas at 10 or 20 Hz synaptic stimulation there was no change (Fig. 6A) (delay to spike 97.5 ± 18.4%, n = 7, p > 0.05 and 91.9 ± 11.9%, n = 7, p > 0.05 for 10 and 20 Hz, respectively). In addition, we analyzed the number of spikes during synaptic stimulation at each frequency before and after the NSP. At 3 Hz there was an increase in the number of spikes (Fig. 6A) (number of spikes 147.8 ± 17.9%; n = 5; p < 0.05), whereas at 10 and 20 Hz there was a decrease after the NSP (Fig. 6A) (number of spikes 68.1 ± 6.3; n = 7; p < 0.01 and 70.5 ± 10.6%; n = 8; p < 0.05 for 10 and 20 Hz, respectively). We also examined the sustained depolarization resulting from temporal summation of synaptic responses (depolarization envelope, see experimental procedures) as another measure of NMDAR and KAR activation. During synaptic stimulation at 3 Hz there was an increase in the size of the depolarization envelope after the NSP (Fig. 6A) (depolarization envelope 340.0 ± 15.8%; n = 5; p < 0.001), whereas at 10 and 20 Hz there was a decrease (Fig. 6A) (depolarization envelope 65.7 ±14.0%, n = 7, p < 0.05 and 75.9 ± 6.5%, n = 7, p < 0.01 for 10 and 20 Hz, respectively).
Figure 6.
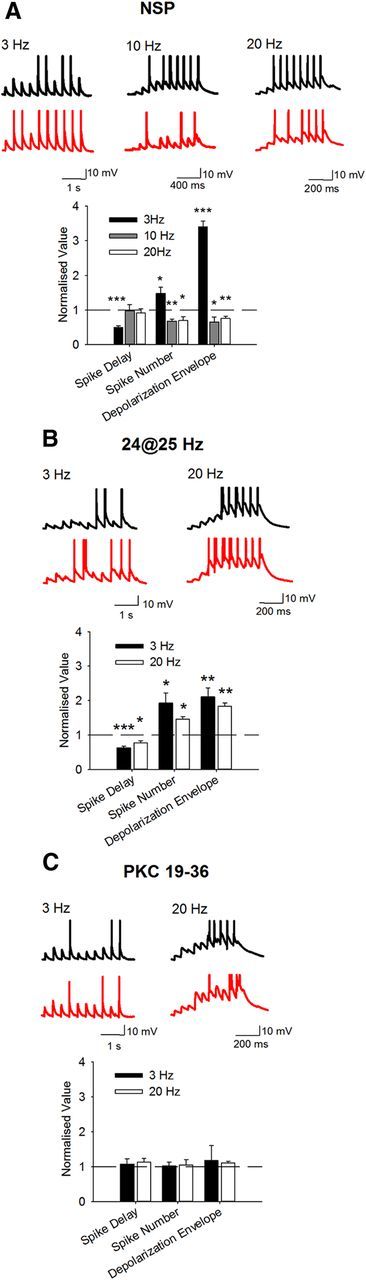
The NSP induces frequency-dependent effects on EPSP summation and cellular excitability. A, 10 synaptic stimuli at 3, 10, and 20 Hz produced action potentials on approximately the fourth stimulus. The NSP increased cellular excitability in response to 3 Hz stimulation measured by a reduction in delay to first spike, an increase in the number of spikes and an increase in underlying depolarization envelope. The NSP decreased cellular excitability in response to 10 or 20 Hz stimulation measured by a decrease in number of spikes and underlying depolarization envelope but caused no change in delay to first spike for 10 synaptic stimuli. Example traces show response to 10 stimuli during baseline (black) and after the NSP (red). B, Induction of NMDAR LTP but not KAR LTD by 24 stimuli at 25 Hz increased cellular excitability in response to 3 and 20 Hz stimulation measured by a reduction in delay to first spike, an increase in the number of spikes and an increase in underlying depolarization envelope. C, Introduction of the PKC inhibitory peptide PKC 19–36 (4.8 μm) into the patch pipette prevented the NSP causing any change in delay to first spike, the total number of spikes or the underlying depolarization envelope for 10 synaptic stimuli at 3 or 20 Hz.
NMDAR LTP is predicted to reduce spike delay and increase the number of spikes and depolarization envelope, whereas the reverse is predicted for KAR LTD. Therefore, these results suggest that NMDAR LTP is the dominant form of plasticity in the cellular response to 3 Hz synaptic stimulation and KAR LTD is dominant in the response to 10 and 20 Hz stimulation. To test this we used a stimulation protocol of 24 stimuli at 25 Hz previously shown to induce NMDAR LTP but not KAR LTD (Fig. 4). Using 3 Hz synaptic stimulation, 24 stimuli at 25 Hz produced changes in response to synaptic stimulation that were similar to those obtained with the NSP. The first action potential occurred earlier and both the number of spikes and depolarization envelope increased (Fig. 6B) (delay to spike 62.8 ± 5.0%, p < 0.001; spike number 192.5 ± 29.6%, p < 0.05; depolarization envelope 210.3 ± 26.2%, p < 0.01; n = 8). However, using 20 Hz synaptic stimulation, 24 stimuli at 25 Hz produced the opposite changes in response to synaptic stimulation to the NSP. Similar to the effects using 3 Hz, the first action potential occurred earlier and both the number of spikes and depolarization envelope increased (Fig. 6B) (delay to spike 76.7 ± 6.8%, p < 0.05; spike number 146.1 ± 7.0%, p < 0.05; depolarization envelope 183.0 ± 9.8%, p < 0.01; n = 6).
Both NMDAR LTP and KAR LTD induced by the NSP can be blocked by inhibition of PKC (Kwon and Castillo, 2008a; Selak et al., 2009; Chamberlain et al., 2012). Therefore, to test the dependence of the observed changes in excitability on these forms of postsynaptically expressed synaptic plasticity we repeated the experiments including the PKC inhibitory peptide PKC19–36 in the pipette solution. With PKC activity blocked there was no change in spike delay, number of spikes or depolarization envelope after the NSP in response to trains of 3 or 20 Hz synaptic stimulation (Fig. 6C) (3 Hz-delay to spike 107.2 ± 14.6%, p > 0.05; spike number 101.4 ± 11.4%, p > 0.05; depolarization envelope 117.0 ± 42.8%, p > 0.05; n = 8; 20 Hz delay to spike 112.5 ± 10.8%, p > 0.05; spike number 104.0 ± 15.7%, p > 0.05; depolarization envelope 110.2 ± 4.5%, p > 0.05; n = 6).
These results reveal that NMDAR LTP and KAR LTD induced by the NSP produce a frequency-dependent effect on cellular excitability. Three Hertz synaptic stimulation showed an increase in depolarization envelope and reduction in spike delay, whereas 10 and 20 Hz stimulation showed a decrease in depolarization envelope and the total number of spikes but no change in spike delay. NMDAR LTP reduces spike delay and increases the number of spikes and depolarization envelope, whereas the reverse is predicted for KAR LTD. Therefore, these results demonstrate that NMDAR LTP is the dominant form of plasticity in the cellular response to 3 Hz synaptic stimulation and predict that KAR LTD is dominant in the response to 10 and 20 Hz stimulation.
The effect of KAR LTD on EPSP temporal summation could be a result of a reduction in either the canonical or noncanonical signaling pathways or both. To test this we investigated the effect of KAR LTD and NMDAR LTP on EPSP summation in the absence of the sAHP. We first used bath application of 10 μm UCL2077 (Zaitsev and Anwyl, 2012) which rapidly blocked IsAHP (Fig. 7A) (to 8.2 ± 17.3%, p < 0.01; n = 3). Inhibition of sAHP had little effect on the change in cellular response to synaptic stimulation at 3 Hz caused by the NSP. Delay to spike was still decreased and number of spikes and depolarization envelope were still increased although the increase in number of spikes was enhanced (Fig. 7B) (delay to spike 67.4 ± 14.3%, p < 0.05; spike number 178.2 ± 35.1%, p < 0.05; depolarization envelope 347.7 ± 105.5%, p < 0.05; n = 7). In stark contrast, inhibition of sAHP with UCL2077 reversed the effects of the NSP on 10 and 20 Hz synaptic stimulation. Delay to spike was now decreased and the number of spikes and depolarization envelope were both increased (Fig. 7B) (10 Hz delay to spike 77.4 ± 5.7%, p < 0.05; number of spikes 135.9 ± 10.8%; p < 0.05; depolarization envelope 143.0 ± 16.4%; p < 0.05; n = 8; 20 Hz delay to spike 77.4 ± 7.8%, p < 0.05; number of spikes 127.7 ± 7.7%; p < 0.05; depolarization envelope 135.3 ± 11.9%; p < 0.05; n = 9). Similarly, inhibiting the sAHP by inclusion of 1 mm cAMP in the pipette solution (Pedarzani and Storm, 1993) also had little effect on the change in cellular response to synaptic stimulation at 3 Hz caused by the NSP. Delay to spike was still decreased and number of spikes and depolarization envelope were still increased although the increase in number of spikes was enhanced (Fig. 7C) (delay to spike 63.6 ± 12.8%, p < 0.05; number of spikes 208.3 ± 26.8%; p < 0.05; depolarization envelope 198.4 ± 11.2%; p < 0.05; n = 6). In addition, inhibition of sAHP with cAMP reversed the effects of the NSP on 20 Hz synaptic stimulation. Delay to spike was now decreased and the number of spikes and depolarization envelope were both increased (Fig. 7C) (delay to spike 63.4 ± 11.9%, p < 0.05; spike number 242.8 ± 93.6%, p < 0.05; depolarization envelope 125.7 ± 7.0%; p < 0.05; n = 6). Addition of cAMP to the pipette solution had no effect on NMDAR LTP or KAR LTD induced by the NSP (data not shown). These results indicate that in the absence of sAHP, NMDAR LTP induced by the NSP is dominant over KAR LTD suggesting that the most important effect of KAR LTD is via the noncanonical signaling pathway and the inhibition of the sAHP.
Figure 7.
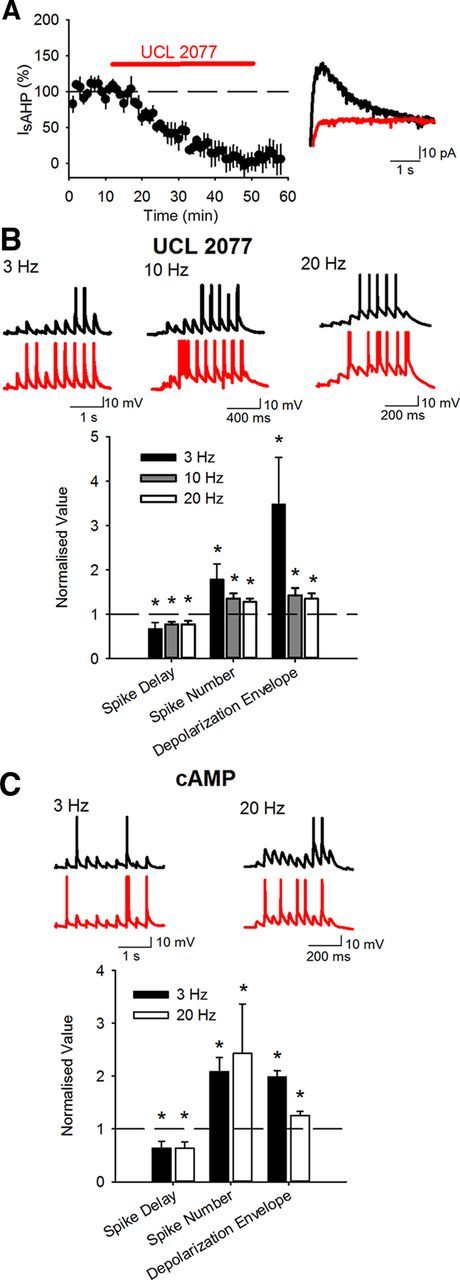
Blockade of sAHP prevented NSP-induced decreases in cellular excitability. A, Application of UCL2077 (10 μm) blocked IsAHP. Example traces show IsAHPs during baseline (black), after UCL2077 (red). B, After incubation in UCL2077 the NSP increased cellular excitability measured by a reduction in delay to first spike and increases in the number of spikes and the underlying depolarization envelope in response to 10 synaptic stimuli at 3, 10, and 20 Hz. Example traces show response to 10 stimuli during baseline (black) and after NSP (red). C, With IsAHP blocked by inclusion of cAMP (1 mm) in the pipette solution, the NSP increased cellular excitability measured by a reduction in delay to first spike and increases in the number of spikes and the underlying depolarization envelope in response to 10 synaptic stimuli at 3 and 20 Hz.
Discussion
The discovery of postsynaptic KARs at mossy fiber synapses that produce small amplitude KAR EPSCs compared with AMPAR or NMDAR EPSCs (Castillo et al., 1997; Vignes and Collingridge, 1997) suggests their physiological ionotropic function is relatively subtle (Sachidhanandam et al., 2009). Here, we describe a major role for synaptic KARs in regulating cellular excitability via inhibition of the sAHP. Furthermore, we show that the inhibition of sAHP can be modulated by synaptic plasticity of KARs.
KAR regulation of AHP function has previously been demonstrated by agonist application to CA1 or CA3 pyramidal neurons (Cherubini et al., 1990; Melyan et al., 2002; Fisahn et al., 2005; Ruiz et al., 2005; Fernandes et al., 2009) and for tonic KAR activation in hippocampal interneurons (Segerstrale et al., 2010). Synaptically activated KAR regulation of the AHP has been shown in CA1 and CA3 pyramidal neurons (Melyan et al., 2004; Ruiz et al., 2005). The specific identity of AHP currents regulated by KARs is unclear. mAHP currents mediated by SK channels are shown to be regulated in interneurons (Segerstrale et al., 2010), whereas the sAHP is regulated in CA1 pyramidal neurons (Cherubini et al., 1990; Melyan et al., 2002, 2004) and both mAHP and sAHP currents are regulated in CA3 pyramidal neurons (Fisahn et al., 2005; Ruiz et al., 2005; Fernandes et al., 2009). We found that synaptic KAR regulation of sAHP in CA3 pyramidal neurons is the dominant effect of KAR LTD on cellular excitability.
Interestingly, we observed the effect of the NSP on cellular excitability had a frequency-dependent component. The NSP was able to induce both KAR LTD and NMDAR LTP but not presynaptic LTP leading to the question of what the combined impact of NMDAR increase versus KAR decrease might be. Therefore we looked at the impact of the NSP on the response to 10 synaptic stimuli at various frequencies. We found a differential effect dependent on the frequency of synaptic stimulation. At 3 Hz cellular excitability changed as expected if NMDAR LTP is the dominant form of plasticity whereas at 10 or 20 Hz it appeared that KAR LTD is dominant (Fig. 6). Evidence suggests that at higher frequencies there is a larger contribution of the sAHP, as activation of the sAHP by suprathreshold EPSP trains is dependent on stimulation number and frequency (Wu et al., 2004), and the threshold for sAHP activation in CA3 pyramidal cells is minimally 3 Hz with half maximal sAHP achieved at 8 Hz (Podlogar and Dietrich, 2006). Therefore, at 3 Hz synaptic stimulation, NMDARs dominate temporal summation increasing the depolarization envelope and decreasing spike delay. At 10 or 20 Hz synaptic stimulation, sAHPs become more dominant because sAHPs shunt NMDAR EPSPs (Fernandez de Sevilla et al., 2007) and reduce the depolarization envelope during trains of synaptic stimulation (Wu et al., 2004). Thus, at 10 or 20 Hz synaptic stimulation the role of KARs in regulating the sAHP becomes the dominant factor. The ionotropic function of postsynaptic KARs also regulates cellular excitability and response to synaptic stimulation (Sachidhanandam et al., 2009) and therefore plasticity of KARs is likely to alter the ionotropic function of KARs. However, our data suggest that the concurrent plasticity of NMDARs and the metabotropic KAR function are dominant under our experimental conditions.
Postsynaptic KARs at mossy fiber synapses are composed primarily of GluK2 and GluK5 subunits (Mulle et al., 1998; Darstein et al., 2003; Ruiz et al., 2005; Fernandes et al., 2009) although it is not currently clear which subunit is critical for nonionotropic signaling (Ruiz et al., 2005; Fernandes et al., 2009). Metabotropic coupling is thought to occur via Gq/11 G-proteins that inhibit channels underlying both the sAHP and mAHP via activation of PLC and PKC (Ruiz et al., 2005) although it is also reported that pertussis toxin inhibits the coupling (Melyan et al., 2002). KAR LTD induced by LFS is dependent on the GluK5 subunit and its interactions with SNAP25 and PICK-1. mGluR5 and PKC activation promote GluK5 interaction with SNAP25 and allow KAR endocytosis (Selak et al., 2009). LFS-induced KAR LTD also requires PKC-mediated phosphorylation and SUMOylation of GluK2 (Chamberlain et al., 2012). Intriguingly, we also found KAR LTD induced by HFS was dependent on the activation of adenosine A2A receptors (Fig. 5), which was previously shown to be required for the induction of NMDAR LTP at mossy fiber synapses (Rebola et al., 2008). Indeed, the mechanism and induction protocols we describe for KAR LTD are remarkably similar to NMDAR LTP with the exception that KAR LTD does not require the activation of NMDARs (Kwon and Castillo, 2008a; Rebola et al., 2008). Interestingly, NMDARs can induce a short-term depression of KAR EPSCs at the mossy fiber synapse (Ghetti and Heinemann, 2000; Rebola et al., 2007). It is possible that even though NMDARs are not required for KAR LTD, they may contribute by lowering the induction threshold, although our experiments suggest this is not the case (Fig. 4).
The NSP we have used is taken from the activity of a single granule neuron in the rat hippocampus during performance of a delayed nonmatch to sample memory task (Wiebe and Staubli, 1999, 2001; Mistry et al., 2011). We have selected it as an epoch of activity that contained a high density and duration of high-frequency activity (Mistry et al., 2011). We found this NSP was sufficient to induce NMDAR LTP and KAR LTD but not presynaptic LTP in rat hippocampal slices because the threshold for induction of presynaptic LTP was higher than KAR LTD or NMDAR LTP. This suggests that NMDAR LTP and KAR LTD may be the most commonly occurring forms of plasticity at mossy fiber synapses in vivo. Our data show that the coincident induction of both forms of plasticity led to a shift in preferred synaptic input frequency from high (10–20 Hz) to low (<3 Hz). Response to higher frequency synaptic input is reduced because KAR LTD removes the inhibition of the sAHP that is dominant at higher frequencies. In contrast, response to lower frequency synaptic input is enhanced because the sAHP is not engaged and NMDAR LTP causes an increase in EPSP temporal summation. In addition, NMDAR LTP may render mossy fiber synapses capable of undergoing NMDAR-dependent AMPAR LTP (Rebola et al., 2011) so KAR LTD induction in tandem with NMDAR LTP may be necessary as a protective mechanism against increases in cell excitability. Our data provide evidence for previously undescribed roles for postsynaptic KARs and KAR plasticity at hippocampal mossy fiber synapses.
Footnotes
This work was funded by BBSRC (S.E.L.C., J.R.M.) and Wellcome Trust (L.A.A., J.R.M.), and by studentships awarded by MRC (J.H.L.P.S.) and BBSRC/Eli Lilly (L.M.T.G.R.). We thank N. Marrion, A. Randall, and J. Henley for helpful discussions.
References
- Bortolotto ZA, Clarke VR, Delany CM, Parry MC, Smolders I, Vignes M, Ho KH, Miu P, Brinton BT, Fantaske R, Ogden A, Gates M, Ornstein PL, Lodge D, Bleakman D, Collingridge GL. Kainate receptors are involved in synaptic plasticity. Nature. 1999;402:297–301. doi: 10.1038/46290. [DOI] [PubMed] [Google Scholar]
- Buchanan KA, Petrovic MM, Chamberlain SE, Marrion NV, Mellor JR. Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron. 2010;68:948–963. doi: 10.1016/j.neuron.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Chamberlain SE, González-Gonzalez IM, Wilkinson KA, Konopacki FA, Kantamneni S, Henley JM, Mellor JR. SUMOylation and phosphorylation of GluK2 regulate kainate receptor trafficking and synaptic plasticity. Nat Neurosci. 2012;15:845–852. doi: 10.1038/nn.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Rovira C, Ben-Ari Y, Nistri A. Effects of kainate on the excitability of rat hippocampal neurones. Epilepsy Res. 1990;5:18–27. doi: 10.1016/0920-1211(90)90062-Z. [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/S0896-6273(01)00191-X. [DOI] [PubMed] [Google Scholar]
- Darstein M, Petralia RS, Swanson GT, Wenthold RJ, Heinemann SF. Distribution of Kainate receptor subunits at hippocampal mossy fiber synapses. J Neurosci. 2003;23:8013–8019. doi: 10.1523/JNEUROSCI.23-22-08013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Catches JS, Petralia RS, Copits BA, Xu J, Russell TA, Swanson GT, Contractor A. High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron. 2009;63:818–829. doi: 10.1016/j.neuron.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Fuenzalida M, Porto Pazos AB, Buño W. Selective shunting of the NMDA EPSP component by the slow afterhyperpolarization in rat CA1 pyramidal neurons. J Neurophysiol. 2007;97:3242–3255. doi: 10.1152/jn.00422.2006. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Heinemann SF, McBain CJ. The Kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurones. J Physiol. 2005;562:199–203. doi: 10.1113/jphysiol.2004.077412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Ohliger-Frerking P. AMPA receptors and Kainate receptors encode different features of afferent activity. J Neurosci. 2002;22:7434–7443. doi: 10.1523/JNEUROSCI.22-17-07434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti A, Heinemann SF. NMDA-dependent modulation of hippocampal Kainate receptors by calcineurin and Ca(2+)/calmodulin-dependent protein kinase. J Neurosci. 2000;20:2766–2773. doi: 10.1523/JNEUROSCI.20-08-02766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundlfinger A, Breustedt J, Sullivan D, Schmitz D. Natural spike trains trigger short- and long-lasting dynamics at hippocampal mossy fiber synapses in rodents. PLoS ONE. 2010;5:e9961. doi: 10.1371/journal.pone.0009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Contractor A, Swanson GT. Attenuated plasticity of postsynaptic kainite receptors in hippocampal CA3 pyramidal neurons. J Neurosci. 2004;24:6228–6236. doi: 10.1523/JNEUROSCI.1302-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S, Manabe T. Kainate receptor-dependent short-term plasticity of presynaptic Ca2+ influx at the hippocampal mossy fiber synapses. J Neurosci. 2002;22:9237–9243. doi: 10.1523/JNEUROSCI.22-21-09237.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008a;57:108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Role of glutamate autoreceptors at hippocampal mossy fiber synapses. Neuron. 2008b;60:1082–1094. doi: 10.1016/j.neuron.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, Pelkey KA, Topolnik L, Congar P, Lacaille JC, McBain CJ. Depolarization-induced long-term depression at hippocampal mossy fiber-CA3 pyramidal neuron synapses. J Neurosci. 2003;23:9786–9795. doi: 10.1523/JNEUROSCI.23-30-09786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/S0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Lancaster B, Wheal HV. Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci. 2004;24:4530–4534. doi: 10.1523/JNEUROSCI.5356-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry R, Dennis S, Frerking M, Mellor JR. Dentate gyrus granule cell firing patterns can induce mossy fiber long-term potentiation in vitro. Hippocampus. 2011;21:1157–1168. doi: 10.1002/hipo.20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-E. [DOI] [PubMed] [Google Scholar]
- Perrais D, Pinheiro PS, Jane DE, Mulle C. Antagonism of recombinant and native GluK3-containing kainate receptors. Neuropharmacology. 2009;56:131–140. doi: 10.1016/j.neuropharm.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Podlogar M, Dietrich D. Firing pattern of rat hippocampal neurons: a perforated patch-clamp study. Brain Res. 2006;1085:95–101. doi: 10.1016/j.brainres.2006.02.050. [DOI] [PubMed] [Google Scholar]
- Rebola N, Sachidhanandam S, Perrais D, Cunha RA, Mulle C. Short-term plasticity of kainate receptor-mediated EPSCs induced by NMDA receptors at hippocampal mossy fiber synapses. J Neurosci. 2007;27:3987–3993. doi: 10.1523/JNEUROSCI.5182-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Rebola N, Carta M, Lanore F, Blanchet C, Mulle C. NMDA receptor-dependent metaplasticity at hippocampal mossy fiber synapses. Nat Neurosci. 2011;14:691–693. doi: 10.1038/nn.2809. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci. 2005;25:11710–11718. doi: 10.1523/JNEUROSCI.4041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidhanandam S, Blanchet C, Jeantet Y, Cho YH, Mulle C. Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J Neurosci. 2009;29:5000–5008. doi: 10.1523/JNEUROSCI.5807-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Nicoll RA. Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science. 2001;291:1972–1976. doi: 10.1126/science.1057105. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Breustedt J, Nicoll RA. Presynaptic kainate receptors impart an associative property to hippocampal mossy fiber long-term potentiation. Nat Neurosci. 2003;6:1058–1063. doi: 10.1038/nn1116. [DOI] [PubMed] [Google Scholar]
- Segerstråle M, Juuri J, Lanore F, Piepponen P, Lauri SE, Mulle C, Taira T. High firing rate of neonatal hippocampal interneurons is caused by attenuation of afterhyperpolarizing potassium currents by tonically active kainate receptors. J Neurosci. 2010;30:6507–6514. doi: 10.1523/JNEUROSCI.4856-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak S, Paternain AV, Aller IM, Picó E, Rivera R, Lerma J. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron. 2009;63:357–371. doi: 10.1016/j.neuron.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Straub C, Hunt DL, Yamasaki M, Kim KS, Watanabe M, Castillo PE, Tomita S. Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nat Neurosci. 2011;14:866–873. doi: 10.1038/nn.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Wiebe SP, Stäubli UV. Dynamic filtering of recognition memory codes in the hippocampus. J Neurosci. 1999;19:10562–10574. doi: 10.1523/JNEUROSCI.19-23-10562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebe SP, Staubli UV. Recognition memory correlates of hippocampal theta cells. J Neurosci. 2001;21:3955–3967. doi: 10.1523/JNEUROSCI.21-11-03955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Disterhoft JF. Slow afterhyperpolarization governs the development of NMDA receptor-dependent afterdepolarization in CA1 pyramidal neurons during synaptic stimulation. J Neurophysiol. 2004;92:2346–2356. doi: 10.1152/jn.00977.2003. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Anwyl R. Inhibition of the slow afterhyperpolarization restores the classical spike timing-dependent plasticity rule obeyed in layer 2/3 pyramidal cells of the prefrontal cortex. J Neurophysiol. 2012;107:205–215. doi: 10.1152/jn.00452.2011. [DOI] [PubMed] [Google Scholar]