

Summary
High fat diets (HFD) lead to obesity and inflammation in the central nervous system. Estrogens and Estrogen Receptor alpha (ERα) protect premenopausal females from the metabolic complications of inflammation and obesity related disease. Here we demonstrate that hypothalamic PGC-1α regulates ERα and inflammation in vivo. HFD significantly increased palmitic acid (PA) and sphingolipids in the CNS of males when compared to female mice. PA, in vitro, and HFD, in vivo, reduced PGC-1α and ERα in hypothalamic neurons and astrocytes of male mice and promoted inflammation. PGC-1α depletion with ERα overexpression significantly inhibited PA-induced inflammation, confirming that ERα is a critical determinant of the anti-inflammatory response. Physiologic relevance of ERα-regulated inflammation was demonstrated by reduced myocardial function in male but not female mice following chronic HFD exposure. Our findings show for the first time that HFD/PA reduces PGC-1α and ERα, promoting inflammation and decrements in myocardial function in a sex-specific way.
Keywords: Obesity, sexual dimorphism, estrogen receptor alpha (ERα), peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), inflammation, astrocytes, neurons, hypothalamus
Introduction
In the last 50 years, obesity has become a global epidemic. The World Health Organization estimates that more than half a billion adults worldwide are obese (Gregor and Hotamisligil, 2011). Obesity is associated with chronic, low-grade inflammation in adipose tissues and in the central nervous system (CNS) (Hotamisligil, 2006; Thaler et al., 2011). CNS inflammation, in turn, leads to insulin and leptin resistance and facilitates the onset of cardiovascular disease; (Purkayastha et al., 2011; Thaler et al., 2011; Zhang et al., 2008) however, the mechanisms underlying CNS inflammation and accompanying co-morbidities remain unclear.
The saturated long-chain fatty acid palmitic acid (PA) induces inflammation in the hypothalamus, and the plasma concentration of PA is significantly increased in obesity (Opie and Walfish, 1963; Reaven et al., 1988). Furthermore, PA crosses the blood–brain barrier (Dhopeshwarkar and Mead, 1973; Smith and Nagura, 2001) and high-fat diets (HFD) promote CNS uptake of PA (Karmi et al., 2010; Wang et al., 1994). Intracerebroventricular infusion of PA activates pro-inflammatory responses in the hypothalamus and promotes insulin resistance (Posey et al., 2009). Thus, PA-induced hypothalamic inflammation could play a critical role in the development of obesity-related diseases such as cardiovascular disease.
Although obesity affects both males and females, there is a sexual dimorphism in the development of metabolic complications associated with obesity (Shi et al., 2009; Sugiyama and Agellon, 2012). Premenopausal women are protected from the adverse effects of obesity; however, the prevalence of metabolic disorders increases significantly after menopause (Ford, 2005). Interestingly, 17β-estradiol (E2) and its receptor, Estrogen Receptor α (ESR1/ERα), protect against obesity-related diseases (Musatov et al., 2007; Xu et al., 2011). Abnormal adiposity and glucose intolerance have been associated with polymorphisms and point mutations of the human ERα gene (Okura et al., 2003, 2013; Smith et al., 1994), and mice lacking ERα (ERKO) have increased adiposity and impaired glucose tolerance (Heine et al., 2000).
The peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) is a transcriptional co-activator involved in multiple metabolic pathways (Lin et al., 2005). PGC-1α is reduced in adipose tissue in genetically obese mice (Lepob/Lepob) as well as following HFD exposure (Crunkhorn et al., 2007). However, its role in the hypothalamus in the response to HFD has not been characterized. Two studies have demonstrated that PGC-1α transcriptionally regulates ERα in vitro (Bourdoncle et al., 2005; Tcherepanova et al., 2000); however, it is unknown if this regulation occurs in the hypothalamus in vivo.
In the present study, we evaluate the sexually dimorphic response to chronic HFD exposure. We demonstrate PA and sphingolipids are increased in the CNS of male when compared to female mice following consumption of HFD. We show that chronic HFD exposure promotes hypothalamic inflammation and reduces ERα in male but not female mice. Further, we demonstrate that hypothalamic inflammation depresses myocardial function in male but not female mice. Treatment of neuronal and astrocyte cell cultures with PA, to mimic the in vivo effects of HFD exposure, increases inflammation and reduces ERα. PA-induced inflammation is enhanced in the absence of ERα, and E2 pre-treatment protects against PA-induced inflammation only when ERα levels are restored. The mechanism by which fatty acids reduce ERα involves depletion of PGC-1α in neurons and astrocytes following PA-treatment or HFD-feeding in male but not female mice. Our data demonstrate a sexually dimorphic response to HFD or PA: HFD suppresses PGC-1α only in male mice, leading to down-regulation of ERα and increased CNS inflammation, which is associated with decrements in myocardial function.
Results
Long-term exposure to HFD promotes hypothalamic inflammation in male mice
Age-matched male and female mice were fed chow diet or HFD for 16 weeks. HFD males and females gained significantly more weight than controls (Fig. 1a). Weight gain was matched in male and female mice (Fig. 1b). HFD feeding significantly increased hypothalamic PA levels in males but not females (Fig. 1c). Consistent with previous findings (De Souza et al., 2005; Thaler et al., 2011), HFD feeding increased pro-inflammatory cytokines (Interleukin -IL1β, Tumor necrosis factor -TNFα Il6) and decreased the anti-inflammatory cytokine Il10 in the hypothalamus of male but not female mice (Fig. 1d and Fig. S1 a–c). Similar patterns of inflammation were found in the hippocampus and in the cortex (Fig. S1 d, e). No such changes were observed in females, despite similar weight gain following the HFD (Fig. 1d and Fig. S1 a–e).
Figure 1. Chronic exposure to a high-fat diet promotes inflammation.

(a) Body weight at time of sacrifice. Chow M, n=29; HFD M, n=30; chow F, n=30; HFD F, n=32. (b) Body-weight gain following HFD exposure. (c) Palmitic acid amount in total brain. n=4/group. (d) mRNA levels of inflammatory markers in hypothalamic tissue following diet exposure. Chow M and F, n=9; HFD M, n=10; HFD F, n=12. (e) Hypothalamic sphigolipids content. n=5/group. (f) Oral glucose tolerance test (OGTT) and (g) area under the OGTT curve of male and female mice fed chow or HFD for 14 weeks. Chow M and F, n=4; HFD M and F, n=5. (h) Percent fractional shortening (%FS) following diet exposure. n=7/group. (i) Representative echocardiograms. LVEDd, Left Ventricular End-Diastolic Diameter; LVEDs, Left Ventricular End-Systolic Diameter. Data are presented as mean ±SEM. *p < 0.05., **p < 0.01 and ***p < 0.001. See also Figure S1.
Sphingolipids are important signal transduction metabolites in immune-inflammatory responses and are antagonists of insulin signaling (Summers, 2006). Consumption of HFD increases sphingolipid levels in plasma of rodents and humans (Haus et al., 2009; Holland et al., 2013). We found greater accumulation of ceramides in the hypothalamus of male HFD mice when compared to females (Fig. 1e). Glucosylceramides were also significantly higher in HFD males than in females (Fig. 1e), whereas the levels of sphingomyelins were lower in the HFD females. Collectively these data suggest that decreased production of sphingolipids protects the hypothalamus of females from the pro-inflammatory effects of the HFD.
HFD impairs glucose tolerance more in male than in female mice
To determine the metabolic consequences of HFD-induced inflammation, an oral glucose tolerance test (OGTT) was performed on male and female mice fed chow or HFD. Previous studies suggest female humans and rodents have improved glucose tolerance compared to males (Macotela et al., 2009; Yki-Jarvinen, 1984). Consistent with this, chow females had lower glucose levels than males at every time point following the glucose gavage (Fig. 1f). Although glucose homeostasis was impaired in both HFD male and female mice, there was a significant increase in glucose levels in HFD males when compared to females on the same diet (Fig. 1f, g). Importantly, basal glucose levels and glucose clearance were similar between female mice exposed to the HFD and chow-fed males (neither of which had increased markers of inflammation in the hypothalamus); however, HFD exposure in males significantly impaired glucose homeostasis and this correlated with increased inflammation in the CNS as well as in the periphery (Fig. S1 f). Notably, female mice showed higher plasma β-Hydroxybutyrate levels (Fig. S1 g), consistent with reports that ketone bodies have anti-inflammatory effects (Gasior et al., 2006). Lastly, E2 is known for its anti-inflammatory role in the CNS (Barreto et al., 2009). E2 levels were significantly increased in females but unchanged in males following HFD exposure (Fig. S1 h), suggesting that E2, inhibits inflammation in HFD females.
Long-term exposure to HFD impairs myocardial function in male mice
Obesity is associated with comorbidities, including cardiovascular disease (Grundy et al., 2004), and the incidence of cardiovascular disease is sex dependent, with an increased risk in males when compared to premenopausal females (Czubryt et al., 2006; Ozbey et al., 2002). Further, work published by Purkayastha S. et al. demonstrates that hypothalamic inflammation induces hypertension (Purkayastha et al., 2011). To evaluate whether the sexual dimorphism in hypothalamic inflammation is associated with differences in cardiovascular function, we assessed myocardial function in male and female mice following HFD exposure. Consistent with previous reports (Battiprolu et al., 2012; Volkers et al., 2014), HFD feeding reduced myocardial function in male mice as demonstrated by decreased percent fractional shortening (%FS) (Fig. 1h, i), but not in female mice (Fig. 1h, i). This suggests that, independent of similar body weight gain following HFD exposure, females are protected from the adverse effects of HFD. Our results are supported by Louwe et al., who showed a sex-dependent effect of HFD on cardiovascular function (Louwe et al., 2012). These data provide the physiological relevance of sexual dimorphisms in hypothalamic inflammation.
Long-term exposure to HFD decreases hypothalamic ERα expression in male mice
To investigate why females are protected from the adverse effects of the HFD, we measured ERα in hypothalamic tissue (Fig. 2a–e) since it has previously been shown that ERα protects against obesity-related diseases (Heine et al., 2000; Musatov et al., 2007). We found that ERα expression was significantly lower in the arcuate (ARC) of male but not female mice following HFD exposure (Fig. 2a–e). Additionally, Erα and not Erβ (Fig. S2 f), levels were significantly decreased and inflammation increased in the hippocampus, cortex and adipose tissue of HFD male mice, confirming a correlation between decreased ERα and higher tissue inflammation (Fig. S2 c, d). Collectively, these results indicate there is a sexually dimorphic response to HFD exposure, with greater PA, sphingolipids, inflammation, and glucose levels in HFD males when compared to females. These findings suggest reductions in myocardial function in male mice are correlated with reductions in ERα expression in the CNS.
Figure 2. Chronic exposure to a high-fat diet decreases ERα.
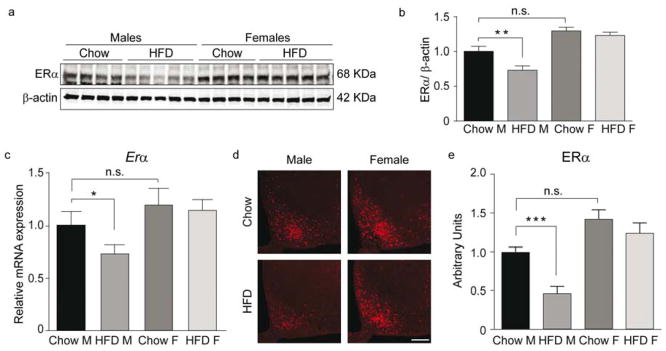
(a–b) Representative immunoblot (a) and quantification (b) of ERα in the hypothalamus. Chow M and F, n=9; HFD M and F, n=10. (c) mRNA levels of Erα in hypothalamic tissue. Chow M and F, n=9; HFD M, n=10; HFD F, n=12. (d–e) Representative confocal images showing ERα immunoreactivity in the ARC (d) and relative quantification (e). n=10/group. Scale bar: 125 μm. Data are presented as mean ±SEM. *p < 0.05, **p < 0.01 and ***p < 0.001. See also Figure S2.
E2 pretreatment protects against PA-induced inflammation in neurons through ERα
Given the increased PA in the CNS of HFD males and the associated reductions in ERα, we addressed whether PA influences ERα in neurons. We exposed N43 cells (a hypothalamic cell line expressing ERα (Fig. S3 a)) and primary hypothalamic neurons to PA, which significantly increased the expression of markers of inflammation (Fig. 3a, d) and decreased ERα expression (Fig. 3b, c, e, f), supporting the hypothesis that reductions in ERα are permissive for PA-induced inflammation. To validate the sexually dimorphic response to PA-induced inflammation, we generated primary male and female hypothalamic neuronal cultures. PA increased inflammation and reduced ERα in male but not female primary neuronal cultures (Fig. 3g). Importantly, only ERα was affected; PA treatment did not modulate Erβ expression (Fig. S3 b). Additionally, we determined that PA exposure significantly increased ceramide levels (Fig. 3h), consistent with our findings in vivo. To determine if these results were specific to PA, we tested the inflammatory response to stearic and linoleic acid. Consistent with the literature (Arruda et al., 2011), stearic acid induced inflammation, while linoleic acid did not (Fig. S3 c). Importantly, ERα was significantly decreased following stearic acid confirming that loss of ERα is permissive for increased inflammation (Fig. S3 c).
Figure 3. 17-β estradiol, through activation of ERα modulates PA-induced inflammation in hypothalamic neurons.

(a–c) N43 cells were pre-treated for 1 h with E2 and then treated for 8 h as indicated. (a) mRNA levels of Tnfα and Il6 in N43 cells. mRNA (b) and protein (c) levels of ERα in N43 cells following treatments. n=3. (d–f) Primary hypothalamic neurons were pre-treated for 1 h with E2 followed by 8 h of the indicated treatments. (d) mRNA levels of Tnfα and Il6 in primary hypothalamic neurons following the aforementioned treatments. mRNA of Erα (e) and ERα protein levels (f) in primary neurons. n=5. (g) mRNA levels of the indicated genes following 8 h treatment in primary male and female hypothalamic neurons. Males, n=5; females n=8. (h) Ceramides content in N43 cells following pre-treatment for 12 h with E2, where indicated, and treated for 8 h. n=3. (i) N43 cells were transfected with siRNAs, pretreated 48 h later for 1 h with E2, and cultured as indicated. mRNA levels of inflammatory markers following the treatments. The insert is a representative immunoblot of N43 cells transfected with control (UNR) siRNA or siRNA for ERα. n=3. (j) N43 cells were infected either with AdGFP (empty vector) or with AdGFP-ERα and then treated as indicated. Data represent mRNA levels of inflammatory markers following treatments. Insert shows representative immunoblots of N43 cells infected with the aforementioned viral constructs demonstrating ERα protein levels 48 h after the adenoviral exposure. n=3. Data are presented as mean ± SEM. *p< 0.05, **p< 0.01, ***p < 0.001. See also Figure S3.
E2 acts through ERα to produce an anti-inflammatory effect in astrocytes (Barreto et al., 2009), and E2 is increased in female mice following HFD exposure (Fig. S1 h). To determine whether E2/ERα mediates a similar anti-inflammatory role in neurons, we co-treated neuronal cells with E2 and PA. Co-treatment with E2 did not decrease the PA-induced inflammation (Fig. S3 d); however, ERα expression was significantly decreased (Fig. S3 e, f). Importantly, when we pre-treated the cells with E2 prior to PA treatment, ERα expression was not reduced (Fig. 3b, c, e, f) and the PA-induced inflammation and increased ceramide levels were significantly inhibited (Fig. 3a, d, h). To assess the role of E2/ERα in mediating these protective effects, we transfected N43 cells with an ERα-specific siRNA to downregulate ERα expression. Despite E2 pretreatment, lack of ERα abolished the E2-induced anti-inflammatory effect (Fig. 3j), demonstrating that E2 requires ERα to decrease PA-induced inflammation.
ERα overexpression significantly protects against PA-induced inflammation in neurons
To determine whether ERα is sufficient to inhibit PA-mediated inflammation, we infected N43 cells with an adenoviral construct designed to overexpress ERα and treated with PA. ERα overexpression significantly decreased the pro-inflammatory effect of PA, suggesting that ERα protects against PA-induced inflammation (Fig. 3k).
Absence of ERα in microglia is associated with inability of E2 to protect against the proinflammatory effects of PA
More than 50% of the brain is composed of non-neuronal cells; microglia and astrocytes represent the most abundant cell types. Since it has been established that HFD activates glial cells (Thaler et al., 2011), we evaluated the expression of ERα in microglia. To this end, we used CX3CR1GFP/GFP mice expressing the GFP construct in microglial cells and performed immunofluorescence with a previously validated ERα antibody (Fig. S2 b) to analyze the co-localization of ERα. Consistent with previously published reports (Saijo et al., 2011; Wu et al., 2013), analysis of the Pearson’s coefficient confirmed ERα did not co-localize with microglial cells in the hypothalamus (Fig. 4a, b). To determine whether the absence of ERα in microglia affects the ability of E2 to attenuate PA-induced inflammation, we treated BV2 cells, a microglial cell line lacking ERα, with E2 and PA. Despite E2 pretreatment, E2 anti-inflammatory activity was not observed (Fig. 4c). These data confirm that E2 requires ERα to mediate its anti-inflammatory effects.
Figure 4. E2 does not ameliorate PA-induced inflammation in microglia due to lack of ERα expression.

(a) Representative confocal images of ERα immunoreactivity in the ARC of CX3CR1 GFP/GFP male mice. Scale bar: 125 μm. (b) Colocalization analysis of ERα and microglia in the ARC. Pearson’s coefficient in dataset volume and in ROI volume: −0.0175. Scale bar: 30 μm. (c) BV2 cells were pre-cultured for 1 h with E2 and then treated for 8 h as indicated (c) Data represent mRNA levels of inflammatory markers in BV2 cells. Results are presented as mean ±SEM. *p < 0.05. n=3.
HFD exposure promotes astrogliosis in male but not female mice
Chronic HFD exposure significantly activates astrocytes in rodents and humans (Lee et al., 2013; Thaler et al., 2011). Analysis of astrogliosis based on expression and staining of glial fibrillary acidic protein (GFAP) revealed an increase in the hypothalamus of male but not female mice (Fig. 5a–c). These data are consistent with enhanced expression of markers of inflammation in male but not female mice exposed to HFD (Fig. 1d).
Figure 5. HFD exposure promotes astrogliosis in male mice.

(a) mRNA levels of Gfap in the hypothalamus. n=12/group. (b–c) Representative confocal images (b) and quantification (c) of GFAP immunoreactivity in the ARC. n=10/group. Scale bar: 125 μm. (d–e) mRNA levels of Gfap (d), Il6, Il1β (e) in primary astrocytes. Primary astrocytes were cultured for 1 h with E2 as indicated and then treated for 8 h. n=3–4. (f–g) Representative immunoblot (f) and quantification (g) of ERα protein levels in primary astrocytes treated as above. n=3/group. Data are presented as mean ±SEM. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4.
To further elucidate the sexually dimorphic response to HFD, we prepared primary hypothalamic astrocytes from male and female pups. Astrocyte activation was analyzed based on the expression of Gfap and markers of inflammation (Il6 and Il1β. Both male and female astrocytes responded to PA treatment by increasing Gfap, Il6 and Il1β (Fig. 5d, e). Interestingly, despite similar basal expression of Gfap and inflammatory genes in male and female astrocytes, there was significantly lower expression of these markers following PA in female astrocytes (Fig. 5d, e). Consistent with previous studies (Spence et al., 2013), ERα was expressed in these cells (Fig. S 4). Increased markers of inflammation correlated with ERα protein levels, with males showing significant reductions in ERα following exposure to PA (Fig. 5f, g). Pretreatment of astrocytes with E2 significantly decreased markers of inflammation in the presence of ERα (Fig. 5e). These data further confirm the sexually dimorphic response to PA, with greater inflammation in male astrocytes correlating with a reduction in ERα.
PGC-1α drives ERα loss in the hypothalamus of male mice
To address the mechanism by which HFD influences ERα within the ARC nucleus, we focused on PGC-1α, a key transcriptional co-activator involved in different metabolic pathways (Lin et al., 2005). PGC-1α transcriptionally regulates ERα in vitro (Bourdoncle et al., 2005; Tcherepanova et al., 2000); however, its role in vivo and in the hypothalamus is unclear.
Consistent with a putative interaction between PGC-1α and ERα, we first demonstrated that they are co-localized in the hypothalamus (Fig. 6a, b). To further demonstrate that PGC-1α transcriptionally regulates ERα we evaluated ERα transcripts in PGC-1α−/− mice. Consistent with the hypothesis that exposure to HFD reduces ERα expression in the CNS via effects on PGC-1α, we found that Erα mRNA levels were significantly reduced in PGC-1α−/− mice (Fig. 6c). Importantly, in ERKO mice, Pgc-1α expression was unaltered (Fig. 6d), suggesting that PGC-1α regulates ERα but ERα does not regulate PGC-1α. To confirm this finding, N43 cells were transfected with siRNA against PGC-1α ERα mRNA and protein levels were significantly reduced following PGC-1α downregulation (Fig. 6e–g). Collectively these results indicate that in vitro, as well as in vivo, PGC-1α transcriptionally regulates ERα.
Figure 6. Reductions in hypothalamic PGC-1α are associated with reductions in ERα.

(a–b) IMARIS imaging of co-localization between ERα (Alexa 488) and PGC-1α Alexa reconstructed using confocal images of the hypothalamus of a male mouse. Pearson’s coefficient in ROI volume = 0.3893; Pearson’s coefficient in colocalized volume = 0.1796. Scale bar: 250 μm. (b) IMARIS imaging of the co-localization (yellow areas) between ERα and PGC-1α. Pearson’s coefficient in ROI volume = 0.4551; Pearson’s coefficient in colocalized volume = 0.3167. Scale bar: 250 μm. (c) mRNA levels of Erα in cortex from PGC-1α−/− male mice. n=3 WT and n=5 PGC-1α −/−. (d) mRNA levels of Pgc-1α in hypothalamus from ERKO male mice. n=6 WT and n=5 ERKO. (e–g) N43 cells transfected with a control siRNA (UNR) or siRNA for PGC-1α 48 h later ERα levels were measured by qPCR (e) and Western blot (f). (f, g) Representative immunoblot (f) and quantification (g) of ERα and PGC-1α levels of N43 cells transfected with UNR or PGC-1α siRNA. Data are presented as mean ±SEM, and *p < 0.05.
HFD exposure decreases PGC-1α in the hypothalamus of male mice
To evaluate if HFD influences PGC-1α in vivo, we analyzed PGC-1α expression following HFD exposure. HFD reduced PGC-1 α expression in the hypothalamus, hippocampus, cortex and visceral adipose tissue of male but not female mice (Fig. 7c and Fig. S5 a–c). Western blot and immunofluorescence revealed reductions in PGC-1α in the hypothalamus of HFD male but not female mice (Fig. 7a, b, d, e). Treatment of N43 cells, primary neurons and primary astrocytes with PA significantly reduced PGC-1α expression (Fig. S5 d–f). To confirm that these effects are specific to PGC-1α and not other nuclear transcription factors, we analyzed the expression of other nuclear transcription factors in the hypothalamus and found they were not altered by sex or HFD (Fig. S5 g). Interestingly, PGC-1α is also decreased following stearic acid treatment, consistent with reductions in PGC-1α being associated with inflammation (Fig. S5 f). Thus, our findings demonstrate that HFD exposure and PA treatment reduce PGC-1α.
Figure 7. PGC-1α is decreased in the hypothalamus of male mice exposed to chronic HFD.

Representative immunoblot (a) and quantification (b) of PGC-1α protein levels in the hypothalamus. n=12/group. (c) mRNA levels of PGC-1α in hypothalamus. n=10/group. (d–e) Representative confocal images showing PGC-1α immunoreactivity in the ARC (d) and its relative quantification (e). n=10/group. Scale bar: 125 μm. (f) mRNA expression of inflammatory markers from N43 cells transfected with a control siRNA (UNR) or siRNA for PGC-1α, followed by infection with AdGFP or with AdGFP-ERα and treated as indicated. n=3. Data are presented as mean ±SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S5.
ERα overexpression is sufficient to significantly inhibit PA induced inflammation
To determine whether reductions in PGC-1α or changes in ERα are associated with PA-induced inflammation, we overexpressed ERα in cells transfected with siRNA targeting PGC-1α and treated with PA. PGC-1α downregulation enhanced the pro-inflammatory effect of PA (Fig. 7f); however, ERα overexpression significantly inhibited PA-induced inflammation both in the presence and absence of PGC-1α (Fig. 7f). These results demonstrate that ERα overexpression is sufficient to significantly inhibit PA-induced inflammation even in the absence of PGC-1α.
Discussion
We demonstrate for the first time a sexually dimorphic response to HFD exposure, with HFD males having higher levels of PA and sphingolipids in the CNS than females. Our findings further demonstrate that males and not females have CNS and peripheral inflammation despite comparable weight gain following HFD consumption. Additionally, we show that reductions in PGC-1α and ERα facilitate inflammation. Lastly, our data demonstrate a physiologic relevance of ERα-regulated inflammation to metabolic homeostasis by demonstrating only in males a reduction in myocardial function.
ERα functions in the CNS to regulate food intake, glucose homeostasis, and energy expenditure (Musatov et al., 2007; Xu et al., 2011). HFD exposure induces hypothalamic expression of inflammatory cytokines such as TNFα, IL1β and IL6 in male mice (De Souza et al., 2005; Thaler et al., 2011). CNS inflammation has been associated with hypertension in male mice (Purkayastha et al., 2011). Epidemiological studies have characterized a sexual dimorphism in obesity-related metabolic complications, such as cardiovascular disease, with a higher prevalence in men and postmenopausal women than in pre-menopausal women (Shi et al., 2009; Sugiyama and Agellon, 2012). Our study extends these associations by characterizing HFD-induced increases in PA, sphingolipids, and inflammation, and associated reductions in myocardial function in male but not female mice.
Our findings demonstrate that exposure of neurons and astrocytes to PA decreases ERα and leads to inflammation. These data are consistent with our previous study showing that ERα deletion induces inflammation in adipose tissues (Davis et al., 2013). Additionally, E2 pre-treatment protects against PA-induced inflammation, only if ERα is present, demonstrating E2, which is elevated in the HFD females, requires ERα to promote its anti-inflammatory effect. Importantly, these data support previous studies indicating that E2 suppresses pro-inflammatory cytokines and increases the production of anti-inflammatory cytokines in an ERα dependent manner in the CNS (Spence et al., 2013; Vegeto et al., 2003).
Our finding that, in the absence of ERα, E2 does not have protective effects against PA-induced inflammation is inconsistent with a previously proposed anti-inflammatory role for E2 acting through ERα in microglial cells (Arevalo et al., 2010; Barreto et al., 2009). This may be explained by the use of a microglial cell line lacking ERα in our study. Along with other studies we show that microglia do not express ERα in vivo (Saijo et al., 2011; Wu et al., 2013), therefore we suggest that E2 does not act through ERα in microglial cells. Saijo et al. show that ERβ in microglia has a protective role in modulating inflammation (Saijo et al., 2011). However, we did not see changes in Erβ expression in males or females following HFD exposure; nevertheless, our data do not rule out the possibility that ERβ, in other experimental settings, modulates inflammation in microglial cells.
Our data suggest that astrocytes and neurons are responsible for the sex-related differences that we see in vivo since markers of inflammation are significantly increased following PA exposure in male neurons and astrocytes compared to females. Importantly, ERα is also decreased in male neurons and astrocytes following PA exposure and is partially restored following E2 pretreatment. These data, along with other studies (Spence et al., 2013), suggest that ERα is required in astrocytes and neurons to modulate the fatty acid-induced inflammatory response.
Our finding that PGC-1α regulates ERα expression in the hypothalamus is the first demonstration that this transcriptional modulation occurs in vivo. These results extend previous reports showing that PGC-1α modulates ERα transcriptional activity in vitro (Bourdoncle et al., 2005; Tcherepanova et al., 2000). We hypothesize that PGC-1α and ERα constitute a hypothalamic signaling system involved in the response to HFD. This appears to be specific for PGC-1α–ERα since neither HFD nor sex affects other nuclear transcription factors.
Depletion of ERα in different hypothalamic neuronal populations affects food intake, energy expenditure and body weight (Xu et al., 2011). We demonstrate that, in the presence of fatty acids, hypothalamic Pgc-1α expression is reduced in a sexually dimorphic manner, implicating reduced hypothalamic PGC-1α in the development of obesity. This is consistent with reports of reduced PGC-1α expression in the adipose tissue of insulin-resistant and morbidly obese individuals (Wilson-Fritch et al., 2004), and that polymorphisms of the PGC-1α gene correlate with increased risk of type 2 diabetes (Ek et al., 2001), findings that further indicate a critical role for PCG-1α in modulating metabolic function. These data, however, contradict previously published studies of total body PGC-1α knockout mice, as well as neuronal inactivation of PGC-1α where reduced PGC-1α expression protected against diet-induced obesity (Lin et al., 2004; Ma et al., 2010). In those studies, PGC-1α deficiency led to degenerative lesions in the brain that worsened with age (Lin et al., 2004; Lucas et al., 2012; Ma et al., 2010). PGC-1α regulates a host of genes and in both of these published models PGC-1α was knocked down during development, likely provoking developmental compensations that could explain the lean phenotype. Importantly, our data are the first demonstration that HFD/PA reduces PGC-1α in the hypothalamus and that this reduction correlates with impaired myocardial function. Further research using inducible knockdown of PGC-1α in adult mice is required to evaluate the role of PGC-1α in response to HFD-induced hypothalamic inflammation independent of developmental compensatory effects.
Loss of PGC-1α and ERα increases inflammation in the periphery, as well as in conditions of nutrient excess and aging (Hsiao et al., 2013; Sczelecki et al., 2013). Here we demonstrate PGC-1α is a determinant of PA-induced hypothalamic inflammation. To confirm whether loss of PGC-1α or loss of ERα modulates the HFD/PA-induced inflammatory response, we knocked down PGC-1α in hypothalamic neurons and simultaneously overexpressed ERα following PA-induced inflammation. ERα ameliorated PA-induced inflammation, showing that it is a critical determinant of the anti-inflammatory pathway. Nevertheless, we are aware that ERα overexpression alone does not preclude the PA-induced inflammatory response. This suggests that other factors are necessary to completely inhibit the PA-induced inflammation.
In summary, HFD/PA-driven reductions in PGC-1α suppress ERα, and cause hypothalamic inflammation, which correlates with impaired myocardial function. Our data reveal a sexually dimorphic response to the HFD, with males having increased CNS PA and sphingolipids when compared to females and that this is associated with increased inflammation. The inflammatory response to fatty acid exposure implicates the PGC-1α-ERα pathway as the molecular basis for sexually dimorphic HFD-induced hypothalamic inflammation. These critical insights into the sexually dimorphic response to HFD and its impact on myocardial function may provide a basis for future development of sex-specific treatments for obesity and its associated metabolic disorders.
Experimental Procedures
Animals and body weight
Animal care and procedures were approved by the University of Texas Southwestern Medical Center. C57BL/6 mice purchased from the Jackson Laboratory (The Jackson Laboratory, Bar Harbor, MA, USA), ERKO mice (gift from P. Chambon) (Dupont et al., 2000), CX3CR1 GFP/GFP mice (Strain B6.129P-Cx3cr1tm1Litt/J, The Jackson Laboratory) and PGC-1α−/− mice (gift from B. Spiegelman) were housed in a temperature-controlled environment in groups of two to five at 22°C–24°C using a 12-hour light/dark cycle. Mice were fed standard phytoestrogen-free chow (#2916, Harlan-Teklad, Madison, WI) and exposed to 42% HFD (#88137, Harlan Teklad) at 8 weeks of age. Water was provided ad libitum.
Fatty Acids Analysis
Fatty acids were quantified using pentafluorobenzoyl bromide derivatization prior to ECNI-GC-MS detection, as described previously (Quehenberger et al., 2011).
Sphingolipids quantitation
Sphingolipid were quantified by liquid chromatography/electrospray ionization/tandem mass spectrometry with a TSQ Quantum Ultra Triple Stage Quadrupole Mass Spectrometer (Thermo Scientific, Waltham, MA) equipped with an electrospray ionization probe and interfaced with an Agilent 1100 HPLC (Agilent Technologies, Santa Clara, CA) (Holland et al., 2013).
Oral glucose tolerance test
Mice were fasted for 3 hours (starting at 7 a.m.). Glucose (2.5g/kg body weight) was gavaged; at the indicated time points blood samples from the tail vein were collected. Glucose concentrations were measured using glucose strips and a glucometer (Bayer Contour®, Whippany, NJ, USA). Mice did not have access to food throughout the experiment.
Echocardiography
Echocardiograms were performed on conscious mice using a Vevo 2100 system with a MS400C scanhead. Left Ventricular End-Diastolic Diameter (LVEDd) and Left Ventricular End-Systolic Diameter (LVEDs) were measured from M-mode recordings. Fractional shortening (FS) was calculated as (LVEDd-LVEDs)/LVEDd and expressed as a percentage. Measurements were made from 2D parasternal short axis views in diastole at the level of the papillary muscles (Battiprolu et al., 2012).
Cell culture
N43 cells (CELLutions Biosystem Inc., Cedarlane, Burlington, NC, USA) and BV2 cells (gift from C.K. Glass) were cultured in HyClone™ DMEM medium (Thermo Scientific), containing 10% charcoal:dextran stripped fetal bovine serum (Gemini, West Sacramento, CA) 100 units/mL penicillin G sodium and 100 μg/mL streptomycin sulfate and 100 mg/L sodium pyruvate. Cells were grown for 24 h before treatments with medium containing 2% charcoal:dextran stripped fetal bovine serum. Cells were pretreated for the indicated time with 10−8 M E2 (Sigma, St-Louis, MO) conjugated with fatty acid free BSA (MP Biomedicals, LLC, Solon, OH) and then treated for 8 h with 10−8 M E2 or 100 μM PA (Matreya, Pleasant Gap, PA) conjugated with BSA alone or in combination.
RNA interference in hypothalamic cell cultures
Cells were cultured in 6-well plates and transfected at 50% confluence with siRNAs targeting murine ERα (Thermo Scientific), murine PGC-1α (Thermo Scientific) or with an unrelated control siRNA (UNR, Thermo Scientific) by means of Lipofectamine® RNAiMAX transfection reagent (Life Technologies), according to the manufacturer’s instructions. 48 h after siRNA transfection, cells were treated as indicated or directly lysed for protein or RNA extraction.
Adenoviral constructs and infections
Adenoviruses expressing FLAG-ERα (AdERα) and GFP were constructed as previously described (Luo et al., 2007). N43 cells were infected with the indicated adenoviral constructs in 1 mL of serum-free medium. 4 h after virus exposure, 1 mL of completely supplemented medium was added. 48 h later, cells were treated as indicated.
qPCR
For analysis of gene expression, mice were anesthetized and decapitated. Tissues were stored in RNAlater® (Ambion, Life Technologies, Carlsbad, CA) at 4°C. 24 h later, tissues were homogenized in 1 mL of TRIzol® (Ambion, Life Technologies). For analysis of gene expression in cell cultures, cells were washed twice with cold PBS and lysed in 1 mL of TRIzol® (Ambion, Life Technologies).
RNA from tissue or cells was extracted using RNeasy Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Total RNA (1 μg) was reverse-transcribed using the SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. qPCR reactions were carried out on an ABI PRISM 7700 Sequence Detection System (Applied Biosystems). The ΔΔCT method was used for relative quantification analysis.
Western blot
Mouse hypothalamic tissue was dissected, immediately frozen in liquid nitrogen after extraction and homogenized. Protein concentration in the supernatants was evaluated using the bicinchoninic acid technique (Pierce Biotechnology, Rockford, IL).
N43 cells and primary neuronal cultures were collected, washed with cold PBS and lysed using RIPA buffer supplemented with protease and phosphatase inhibitors (Roche Applied Science). Cell lysates were quantified as previously described.
Proteins (30 μg) from tissues or cell lysates were separated on Criterion™ TGX™ precast gels (Biorad, Hercules, USA) and electrotransferred to membranes using the Trans-Blot® Turbo™ transfer system (Biorad). Nonspecific binding sites were saturated by incubating membranes for 1 h in TBS supplemented with 5% non-fat powdered milk (w:v in 20 mM TBS), followed by overnight incubation with primary antibodies. Labeling was revealed with appropriate IRDye® secondary antibodies (LiCor, Lincoln, NE) using the ODYSSEY® Quantitative Fluorescence Imaging Systems (LiCor).
Fluorescence microcoscopy
Mice were anesthetized and perfused with 10% formalin. Brains were dissected and post-fixed in 10% formalin for 24 h followed by 2 h treatment with 30% sucrose in PBS. Brain sections were cut at 30 μm using a Thermo Scientific HM 450 sliding microtome (Thermo Scientific). The sections were permeabilized in 0.01% Triton and blocked in 3% donkey serum (Jackson Immuno Research, West Grove, PA, USA) for 2 h. Brain sections were incubated in the primary antibodies overnight (ERα, Santa Cruz; GFAP, Abcam, Cambridge, England; PGC-1α Calbiochem) followed by the respective secondary antibodies (Alexa Fluor®, Life Technologies) for 2 h. Sections were then placed on gelatinized slides, mounted with VECTASHIELD anti-fading medium with DAPI (Vector Laboratories, Burlingame, CA) and coverslipped. Slides were analyzed using a TCS SP2 confocal fluorescence microscope (Leica Microsystems GmbH). Five pictures containing the ARC region of the hypothalamus were taken from at least 5 mice per group and staining was quantified using ImageJ software (http://rsb.info.nih.gov/ij/).
Statistical analysis
Data are presented as mean ±SEM. Statistical analyses were performed with GraphPad Prism software (GraphPad Software Inc.) and Microsoft Office 2010 Excel. Comparisons between 2 conditions were made using the unpaired 2-tailed Student t test. One-way ANOVA was used for comparison of more than 2 groups. Scheffe’s F test was employed for post-hoc analysis. p < 0.05 was considered to be statistically significant.
Supplementary Material
Highlights.
Male and female mice differ in their metabolic response to fatty acids
Hypothalamic inflammation correlates with reductions in myocardial function
HFD-induced downregulation of ERα is permissive for increased inflammation
Fatty acids decrease PGC-1α in male but not female mice
Acknowledgments
DJC is supported by NIH/NIDDK P01 088761-01 and the Klarman Foundation for Eating Disorders. MHT is supported (in part) by the Helmholtz Alliance ICEMED through the Initiative and Networking Fund of the Helmholtz Association, the DZD - German Center for Diabetes Research and Portfolio Grant - Metabolic Dysfunction.
Footnotes
The authors declare that there are no conflicts of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, Carvalheira JB, Velloso LA. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. 2011;152:1314–1326. doi: 10.1210/en.2010-0659. [DOI] [PubMed] [Google Scholar]
- Barreto G, Santos-Galindo M, Diz-Chaves Y, Pernia O, Carrero P, Azcoitia I, Garcia-Segura LM. Selective estrogen receptor modulators decrease reactive astrogliosis in the injured brain: effects of aging and prolonged depletion of ovarian hormones. Endocrinology. 2009;150:5010–5015. doi: 10.1210/en.2009-0352. [DOI] [PubMed] [Google Scholar]
- Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122:1109–1118. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg ML, Omran SF, Weir J, Meikle PJ, Watt MJ. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J Physiol. 2012;590:4377–4389. doi: 10.1113/jphysiol.2012.233288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdoncle A, Labesse G, Margueron R, Castet A, Cavailles V, Royer CA. The nuclear receptor coactivator PGC-1alpha exhibits modes of interaction with the estrogen receptor distinct from those of SRC-1. J Mol Biol. 2005;347:921–934. doi: 10.1016/j.jmb.2005.01.048. [DOI] [PubMed] [Google Scholar]
- Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:15439–15450. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, Espira L, Lamoureux L, Abrenica B. The role of sex in cardiac function and disease. Can J Physiol Pharmacol. 2006;84:93–109. doi: 10.1139/y05-151. [DOI] [PubMed] [Google Scholar]
- Davis KE, Neinast MD, Sun K, Skiles WM, Bills JD, Zehr JA, Zeve D, Hahner LD, Cox DW, Gent LM, et al. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol Metab. 2013;2:227–242. doi: 10.1016/j.molmet.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar GA, Mead JF. Uptake and transport of fatty acids into the brain and the role of the blood-brain barrier system. Adv Lipid Res. 1973;11:109–142. doi: 10.1016/b978-0-12-024911-4.50010-6. [DOI] [PubMed] [Google Scholar]
- Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- Ek J, Andersen G, Urhammer SA, Gaede PH, Drivsholm T, Borch-Johnsen K, Hansen T, Pedersen O. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia. 2001;44:2220–2226. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- Gasior M, Rogawski MA, Hartman AL. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. 2006;17:431–439. doi: 10.1097/00008877-200609000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual review of immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C American Heart, A., National Heart, L., and Blood, I. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, Kirwan JP. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes. 2009;58:337–343. doi: 10.2337/db08-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, et al. An FGF21-adiponectin-cERαmide axis controls energy expenditure and insulin action in mice. Cell metabolism. 2013;17:790–797. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum Mol Genet. 2013;22:1826–1842. doi: 10.1093/hmg/ddt036. [DOI] [PubMed] [Google Scholar]
- Karmi A, Iozzo P, Viljanen A, Hirvonen J, Fielding BA, Virtanen K, Oikonen V, Kemppainen J, Viljanen T, Guiducci L, et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59:2171–2177. doi: 10.2337/db09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeveld M, Aerts JM. Glycosphingolipids and insulin resistance. Prog Lipid Res. 2009;48:196–205. doi: 10.1016/j.plipres.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Lee D, Thaler JP, Berkseth KE, Melhorn SJ, Schwartz MW, Schur EA. Longer T(2) relaxation time is a marker of hypothalamic gliosis in mice with diet-induced obesity. Am J Physiol Endocrinol Metab. 2013;304:E1245–1250. doi: 10.1152/ajpendo.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell metabolism. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Louwe MC, van der Hoorn JW, van den Berg SA, Jukema JW, Romijn JA, van Dijk KW, Rensen PC, Smit JW, Steendijk P. Gender-dependent effects of high-fat lard diet on cardiac function in C57Bl/6J mice. Appl Physiol Nutr Metab. 2012;37:214–224. doi: 10.1139/h11-153. [DOI] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, Cowell RM. Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking pgc-1alpha. PloS one. 2012;7:e42878. doi: 10.1371/journal.pone.0042878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236–1247. doi: 10.1038/nprot.2007.135. [DOI] [PubMed] [Google Scholar]
- Ma D, Li S, Lucas EK, Cowell RM, Lin JD. Neuronal inactivation of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) protects mice from diet-induced obesity and leads to degenerative lesions. J Biol Chem. 2010;285:39087–39095. doi: 10.1074/jbc.M110.151688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009;58:803–812. doi: 10.2337/db08-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, Kaplitt MG, Ogawa S. Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:2501–2506. doi: 10.1073/pnas.0610787104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okura T, Koda M, Ando F, Niino N, Ohta S, Shimokata H. Association of polymorphisms in the estrogen receptor alpha gene with body fat distribution. Int J Obes Relat Metab Disord. 2003;27:1020–1027. doi: 10.1038/sj.ijo.0802378. [DOI] [PubMed] [Google Scholar]
- Opie LH, Walfish PG. Plasma free fatty acid concentrations in obesity. N Engl J Med. 1963;268:757–760. doi: 10.1056/NEJM196304042681404. [DOI] [PubMed] [Google Scholar]
- Ozbey N, Sencer E, Molvalilar S, Orhan Y. Body fat distribution and cardiovascular disease risk factors in pre- and postmenopausal obese women with similar BMI. Endocr J. 2002;49:503–509. doi: 10.1507/endocrj.49.503. [DOI] [PubMed] [Google Scholar]
- Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nature medicine. 2011;17:883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quehenberger O, Armando AM, Dennis EA. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. Biochim Biophys Acta. 2011;1811:648–656. doi: 10.1016/j.bbalip.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad F, Hester KD, Yang G, Hannun YA, Bielawski J. Altered adipose and plasma sphingolipid metabolism in obesity: a potential mechanism for cardiovascular and metabolic risk. Diabetes. 2006;55:2579–2587. doi: 10.2337/db06-0330. [DOI] [PubMed] [Google Scholar]
- Sczelecki S, Besse-Patin A, Abboud A, Kleiner S, Laznik-Bogoslavski D, Wrann CD, Ruas JL, Haibe-Kains B, Estall JL. Loss of Pgc-1alpha expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am J Physiol Endocrinol Metab. 2013 doi: 10.1152/ajpendo.00578.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Seeley RJ, Clegg DJ. Sexual differences in the control of energy homeostasis. Frontiers in neuroendocrinology. 2009;30:396–404. doi: 10.1016/j.yfrne.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- Smith QR, Nagura H. Fatty acid uptake and incorporation in brain: studies with the perfusion model. J Mol Neurosci. 2001;16:167–172. doi: 10.1385/JMN:16:2-3:167. discussion 215–121. [DOI] [PubMed] [Google Scholar]
- Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, Itoh N, Sofroniew MV, Voskuhl RR. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J Neurosci. 2013;33:10924–10933. doi: 10.1523/JNEUROSCI.0886-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama MG, Agellon LB. Sex differences in lipid metabolism and metabolic disease risk. Biochemistry and cell biology. 2012;90:124–141. doi: 10.1139/o11-067. [DOI] [PubMed] [Google Scholar]
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Tcherepanova I, Puigserver P, Norris JD, Spiegelman BM, McDonnell DP. Modulation of estrogen receptor-alpha transcriptional activity by the coactivator PGC-1. J Biol Chem. 2000;275:16302–16308. doi: 10.1074/jbc.M001364200. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2011;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambon P, et al. Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci USA. 2003;100:9614–9619. doi: 10.1073/pnas.1531957100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkers M, Doroudgar S, Nguyen N, Konstandin MH, Quijada P, Din S, Ornelas L, Thuerauf DJ, Gude N, Friedrich K, et al. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med. 2014;6:57–65. doi: 10.1002/emmm.201303183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SW, Wang M, Grossman BM, Martin RJ. Effects of dietary fat on food intake and brain uptake and oxidation of fatty acids. Physiol Behav. 1994;56:517–522. doi: 10.1016/0031-9384(94)90295-x. [DOI] [PubMed] [Google Scholar]
- Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JA. Targeting estrogen receptor beta in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2013;110:3543–3548. doi: 10.1073/pnas.1300313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, Zhang X, Zou F, Gent LM, Hahner LD, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell metabolism. 2011;14:453–465. doi: 10.1016/j.cmet.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yki-Jarvinen H. Sex and insulin sensitivity. Metabolism. 1984;33:1011–1015. doi: 10.1016/0026-0495(84)90229-4. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.