

Abstract
The mechanistic target of rapamycin (mTOR) signaling pathway is a crucial cellular signaling hub that, like the nervous system itself, integrates internal and external cues to elicit critical outputs including growth control, protein synthesis, gene expression, and metabolic balance. The importance of mTOR signaling to brain function is underscored by the myriad disorders in which mTOR pathway dysfunction is implicated, such as autism, epilepsy, and neurodegenerative disorders. Pharmacological manipulation of mTOR signaling holds therapeutic promise and has entered clinical trials for several disorders. Here, we review the functions of mTOR signaling in the normal and pathological brain, highlighting ongoing efforts to translate our understanding of cellular physiology into direct medical benefit for neurological disorders.
Introduction
The mTOR signaling pathway acts as a molecular systems integrator to support organismal and cellular interactions with the environment. The mTOR pathway regulates homeostasis by directly influencing protein synthesis, transcription, autophagy, metabolism, and organelle biogenesis and maintenance. It is not surprising then that mTOR signaling is implicated in the entire hierarchy of brain function including the proliferation of neural stem cells, the assembly and maintenance of circuits, experience-dependent plasticity and regulation of complex behaviors like feeding, sleep and circadian rhythms. mTOR dysfunction is the root cause of several monogenetic disorders and is implicated in both neurodegenerative and neuropsychiatric diseases. Pharmacological manipulation of the mTOR pathway is proving to be a promising branch of neurotherapeutics. To provide a framework for considering the potential of these new therapeutic opportunities, in this review we will illustrate the inter-relatedness of neurological disorders through the lens of this multifaceted and ubiquitous molecular pathway.
The mTOR Signaling Network
The mTOR Complexes
From a scratch of soil culled from the ground of Easter Island (Rapa Nui in Polynesian), the soil bacteria Streptomyces hygroscopicus yielded the anti-fungal macrolide eponymously dubbed ‘rapamycin’ leading to the discovery of the mechanistic target of rapamycin (mTOR)(Brown et al., 1994; Sabatini et al., 1994). mTOR is a large (259kDa), highly conserved, serine/threonine kinase that is an atypical member of the phosphoinositide 3-kinase-related kinase family and is ubiquitously expressed in eukaryotic cell types, including neural cells(Sabatini et al., 1999)(Figure 1A).
Figure 1. Domain structure of the mTOR kinase and components of its protein complexes.
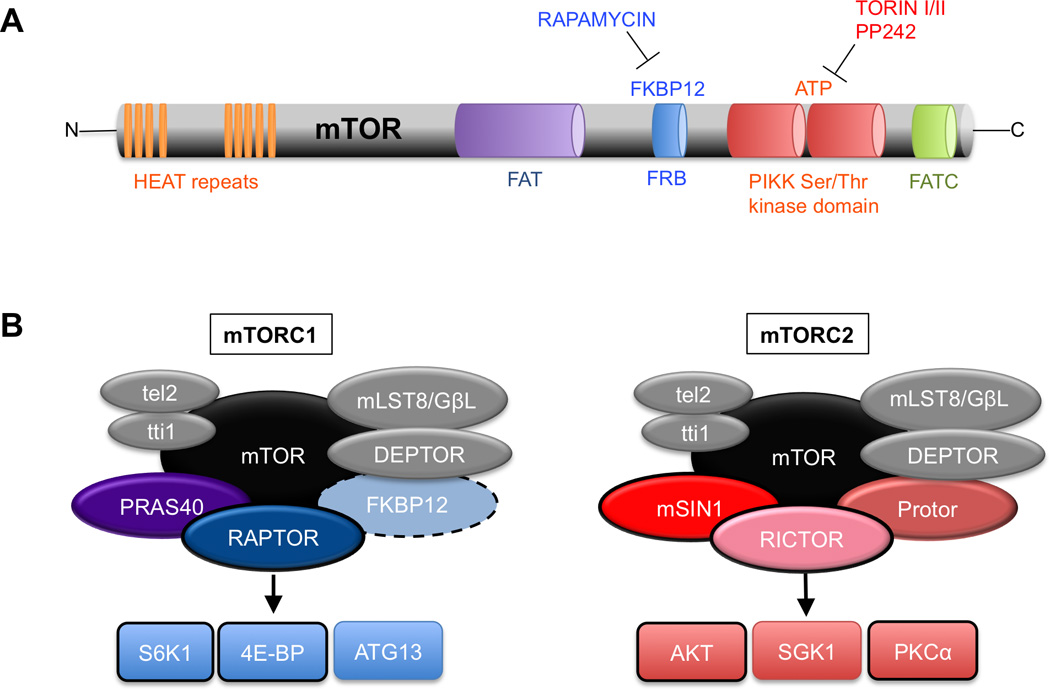
A. Domain organization of the mTOR kinase. HEAT (huntingtin, elongation factor 3, a subunit of phosphatase 2A and TOR1) repeats mediate protein interactions with Raptor, Rictor, and other proteins; FKBP12-rapamycin binding domain (FRB) is the site of rapamycin-mediated inhibition of mTORC1; The PIKK kinase domain contains the Ser/Thr catalytic activity and is the site of inhibition of kinase-site inhibitors such as Torin1 and Torin II, which inhibit both mTORC1 and mTORC2 activity; FATC = FRAP-ATM-TTRAP domain.
B. The components of mTORC1 and mTORC2. mTOR = mechanistic target of rapamycin; Raptor= scaffolding protein essential to mTORC1 activity and rapamycin sensitivity; PRAS40 = an inhibitor of mTORC1; DEPTOR = an inhibitor of mTORC1; mLST8/GβL = function unclear; Rictor = scaffold protein essential to mTORC2 function; mSIN1 = important for mTORC2 enzymatic activity toward AKT; Protor = mediates activity toward SGK. Black outlines indicate proteins that have been thoroughly examined in the nervous system. The dashed line around FKBP12 indicates that it is a non-obligate component of mTORC1.
mTOR function is mediated through two large biochemical complexes defined by their respective protein composition and have been extensively reviewed elsewhere(Dibble and Manning, 2013; Laplante and Sabatini, 2012)(Figure 1B). In brief, common to both mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) are: mTOR itself, mammalian lethal with sec13 protein 8 (mLST8; also known as GβL), and the inhibitory DEP domain containing mTOR-interacting protein (DEPTOR). Specific to mTORC1 is the regulator-associated protein of the mammalian target of rapamycin (Raptor) and proline-rich Akt substrate of 40 kDa (PRAS40)(Kim et al., 2002; Laplante and Sabatini, 2012). Raptor is essential to mTORC1 activity.
The mTORC2 complex includes the rapamycin insensitive companion of mTOR (Rictor), mammalian stress activated MAP kinase-interacting protein 1 (mSIN1), and proteins observed with rictor 1 and 2 (PROTOR 1 and 2)(Jacinto et al., 2006; Jacinto et al., 2004; Pearce et al., 2007; Sarbassov et al., 2004)(Figure 1B). Rictor and mSIN1 are both critical to mTORC2 function.
FKBP12 is a non-obligate mTOR-interacting protein that positively influences mTOR function and binds rapamycin only when incorporated into mTORC1 complex(Brown et al., 1994; Chen et al., 1995; Sabatini et al., 1994; Stan et al., 1994)(Figure 1B). The fact that rapamycin acts by blocking the interaction of FKBP12 with mTOR is a possible explanation for the pharmacological proclivity of rapamycin for mTORC1. Indeed, mTORC2 was originally considered rapamycin-insensitive; however, more recent studies have suggested that long-term rapamycin exposure also inhibits mTORC2(Jacinto et al., 2004; Loewith et al., 2002; Sarbassov et al., 2006). This may be partially explained by observations that high concentrations of rapamycin directly interact with mTOR kinase within the FKBP12-rapamycin binding domain (FRB) (Figure 1A;(Yang et al., 2013)).
Upstream Signaling
Extracellular activators of the mTOR pathway with relevance to the brain include brain-derived neurotrophic factor (BDNF), insulin, insulin-like growth factor 1 (IGF1), vascular endothelial growth factor (VEGF) and ciliary neurotrophic factor (CNTF), glutamate, and guidance molecules(Lenz and Avruch, 2005; Nie et al., 2010; Quevedo et al., 2002; Takei et al., 2004).
mTORC1 is potently activated by a small GTPase called Ras homolog enriched in brain (Rheb). The activity of Rheb is in turn suppressed by the Tuberous Sclerosis Complex (TSC), comprised of the tumor suppressors TSC1 (hamartin) and TSC2 (tuberin), and the recently defined TBC1D7, which act coordinately as a GTPase-activating protein (GAP) to convert active Rheb-GTP to inactive Rheb-GDP and suppress mTOR(Inoki et al., 2003; Li et al., 2004). Only TSC2 contains GAP function, however, both TSC1 and TSC2 are required for functionality of this heterodimer (Figure 2).
Figure 2. The mTOR Signaling Pathway.
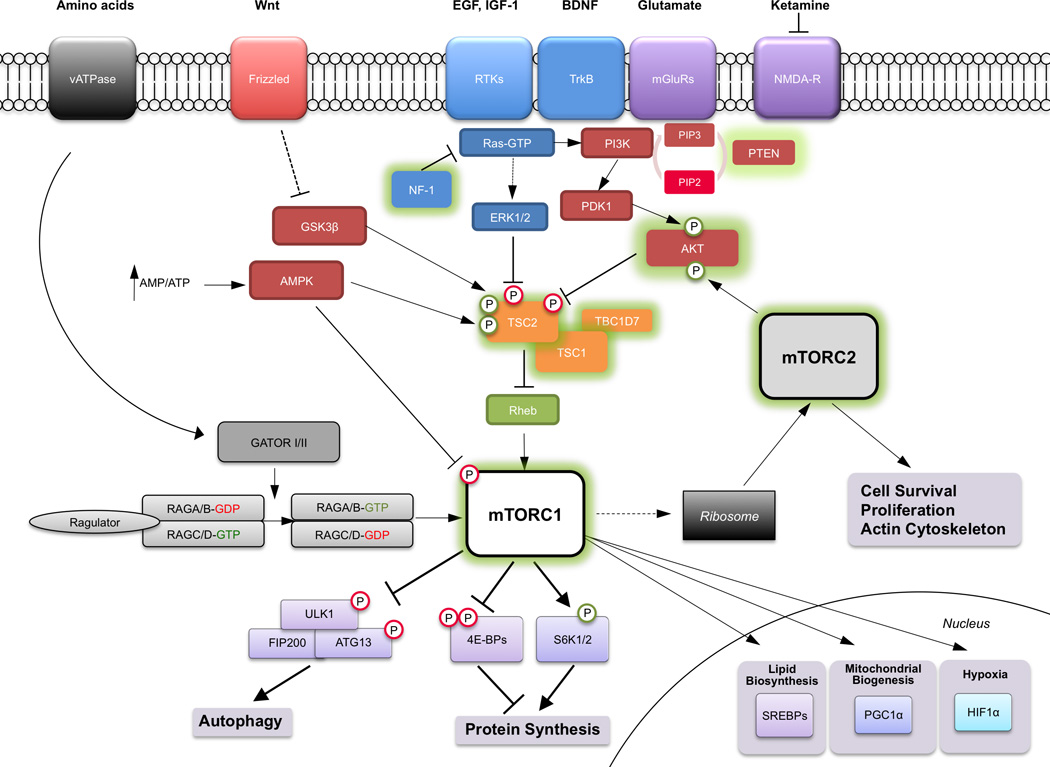
The mTOR complexes integrate signals from nutrients, growth factors, cytokines, and various intracellular influences to elicit a variety of crucial cellular responses. While there are thousands of mTOR substrates, those that have been best characterized in the regulation of crucial cellular processes such as protein synthesis and autophagy are depicted. Abbreviations not found in text include: RTKs = receptor tyrosine kinases; TrkB = tyrosine receptor kinase B, the receptor for BDNF (brain-derived neurotrophic factor); mGluRs = metabotropic glutamate receptors; NMDA-R = N-methyl-D-aspartate receptor; PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha).
The function of TSC is additionally under regulation by several kinase cascades. Inhibitory influences include extracellular signal related kinase (ERK), ribosomal S6 kinase (p90-RSK), glycogen synthase kinase 3β (GSK-3β) and AKT, which lead to an increase in mTOR activity through suppression of TSC2 activity. Independent of TSC2, AKT also stimulates mTOR activity directly by inhibiting PRAS40(Oshiro et al., 2007; Sancak et al., 2007; Vander Haar et al., 2007). Further upstream, phosphatase and tensin homolog deleted on chromosome 10 (PTEN) can inactivate PI3K and thus suppress signaling to mTOR (Figure 2).
AMP-dependent kinase (AMPK) regulates mTOR signaling via multiple cellular mechanisms. AMPK is activated by a high AMP/ATP ratio and thereby functions as a key sensor of relative energy status in the cell upstream of mTOR. First, AMPK phosphorylates and activates TSC2 function to suppress mTOR activity (Figure 2). Second, AMPK directly inhibits mTORC1 by phosphorylating Raptor(Gwinn et al., 2008). Thus, low energy states in which the AMP/ATP ratio is high, results in increased AMPK activity and suppression of mTOR-mediated growth pathways(Inoki et al., 2012).
An additional role for mTOR in cellular sensing of nutrients has been established, but not well-studied in neural systems (Figure 2). The mTOR pathway senses amino acids through the Rag family of GTPases and the TSC1/2 complex to promote the interaction of mTOR with Rheb(Menon et al., 2014; Zoncu et al., 2011a). The Rags are maintained inactive by a lysosome-bound complex called the ‘Ragulator’ and are activated by sequential action of two multiprotein complexes collectively termed GATOR1 and GATOR2, which demonstrate GAP activity to the Rags(Bar-Peled et al., 2012; Efeyan et al., 2013; Zoncu et al., 2011b).
Activation of mTORC2 in brain has not been widely studied. mTORC2 has an important role in cell survival and in the maintenance of the actin cytoskeleton and is implicated in the morphological regulation of actin-rich dendritic spines(Huang et al., 2013; Jacinto et al., 2004; Sarbassov et al., 2006; Thomanetz et al., 2013). In yeast and cancer cells, mTORC2 associates with the ribosome, which appears to be crucial to mTORC2 activation(Zinzalla et al., 2011). Since mTORC1 stimulates ribosome assembly, mTORC1 and mTORC2 are, in certain regards, both upstream and downstream of one another, making the dissection of their intertwined signaling properties complicated (Figure 2).
Downstream of mTOR
Protein Synthesis
mTOR regulates mRNA translation. The best-characterized substrates of mTORC1 are the p70 ribosomal S6 protein kinases 1 and 2 (S6K1/2) and the eukaryotic initiation factor 4E-binding proteins (4E-BPs) (Figure 2). S6K1/2 are partially redundant kinases encoded by separate genes. Both have unique and shared substrates. The ribosomal protein S6 (S6) is phosphorylated by both kinases(Fenton and Gout, 2011). S6K1 also phosphorylates eIF4B and eukaryotic elongation factor 2 kinase (eEF2K) further stimulating translation initiation and elongation of nascent peptide chains(Ma and Blenis, 2009).
The majority of regulated eukaryotic mRNAs contain a methylated guanosine repeat at their 5’-untranslated regions referred to as “the cap”. This structure is regulated by binding of the cap binding protein, eukaryotic initiation factor 4E (eIF4E). eIF4E is regulated by binding of 4E-BPs which inhibit the latter’s association with the mRNA cap and suppress translation initiation(Richter and Sonenberg, 2005). 4E-BPs compete with the scaffolding protein eIF4G for eIF4E binding. mTOR phosphorylation of 4E-BPs results in the release of eIF4E to the mRNA cap structure and subsequent recruitment of eIF4G and the RNA helicase eIF4A, among many other factors(Ma and Blenis, 2009; Sonenberg and Hinnebusch, 2007). Thus, translation is stimulated in direct response to mTORC1 activation.
Transcriptional Targets of mTOR
Although the role of mTORC1 in the control of protein synthesis has been widely studied, there is less known about how mTORC1 signaling affects gene expression. Among the best-studied transcription factors regulated by mTORC1 are the sterol-response binding proteins (SREBPs), which regulate lipogenesis(Peterson et al., 2011). The functions of the SREBPs in the brain are not fully understood; however, they have proposed roles in nutrient sensing, excitotoxicity, myelination, and neurodegenerative diseases(Barbero-Camps et al., 2013; Lebrun-Julien et al., 2014; Taghibiglou et al., 2009).
mTOR regulates the cellular response to hypoxia through regulation of the transcription and translation of the hypoxia inducible factor 1α (HIF1α)(Hudson et al., 2002). HIF1α is a transcription factor that shifts metabolism from oxidative phosphorylation toward glycolysis in response to low oxygen tension. HIF1α-mediated mechanisms are required for angiogenesis following hypoxia and have been implicated in stroke, neonatal hypoxic-ischemic injury, and neurodegenerative disease(Sharp and Bernaudin, 2004).
mTOR pathway activation stimulates the association of the YY1 transcription factor with the transcriptional co-activator PGC1α and results in the preferential activation of a mitochondrial gene program(Cunningham et al., 2007). A recent study demonstrated that rapamycin had beneficial effects on a mouse model of the neurodegenerative mitochondropathy Leigh’s Syndrome(Johnson et al., 2013b). Interestingly, rapamycin did not alter mitochondrial function or electron transport chain complex I assembly, yet it mitigated a wide range of metabolic derangements and improved overall survival.
Autophagy
Amino acids, macromolecules and damaged organelles are degraded and recycled by cells via the multistep process of autophagy. mTORC1 signaling regulates autophagy through inhibition of the unc-51-like kinase 1 (ULK1) complex (Figure 2). Inhibition of mTORC1 with rapamycin stimulates autophagy. In keeping with its central homeostatic and fundamental role in cellular physiology, autophagy has been linked to a wide spectrum of human pathology, most notably cancer, metabolic disease, and neurodegenerative disorders(Bové et al., 2011; Nixon, 2013).
The physiological function of autophagy in the brain remains unclear; however, genetic disruption of autophagy in the brain results in tremor, spasticity, axonal degeneration, widespread inclusion bodies with ubiquitinated protein, and early death(Hara et al., 2006; Komatsu et al., 2006; Komatsu et al., 2007). Significantly, these phenotypes resemble symptoms associated with neurodegenerative diseases characterized by accumulations of misfolded proteins (Figure 3). Recently, it was shown that autophagy regulates dendritic spine pruning during development that may contribute to mTOR pathway-dependent social behaviors (Tang et al., 2014). Importantly, autophagy is mechanistically distinct in neurons when compared to dividing cells. For example, unlike non-neuronal cells, Tsc2-deficient neurons display increased autolysosome accumulation and retain autophagic flux despite mTORC1 hyperactivation(Di Nardo et al., 2014). This is likely the result of a balance between inhibitory effects of mTORC1 with stimulatory effects of AMPK pathways on the ULK1 complex. More detailed characterization of the cellular mechanisms regulating autophagy in neurons will be crucial.
Figure 3. The mTOR Pathway in Neurodevelopmental and Neurodegenerative Diseases.
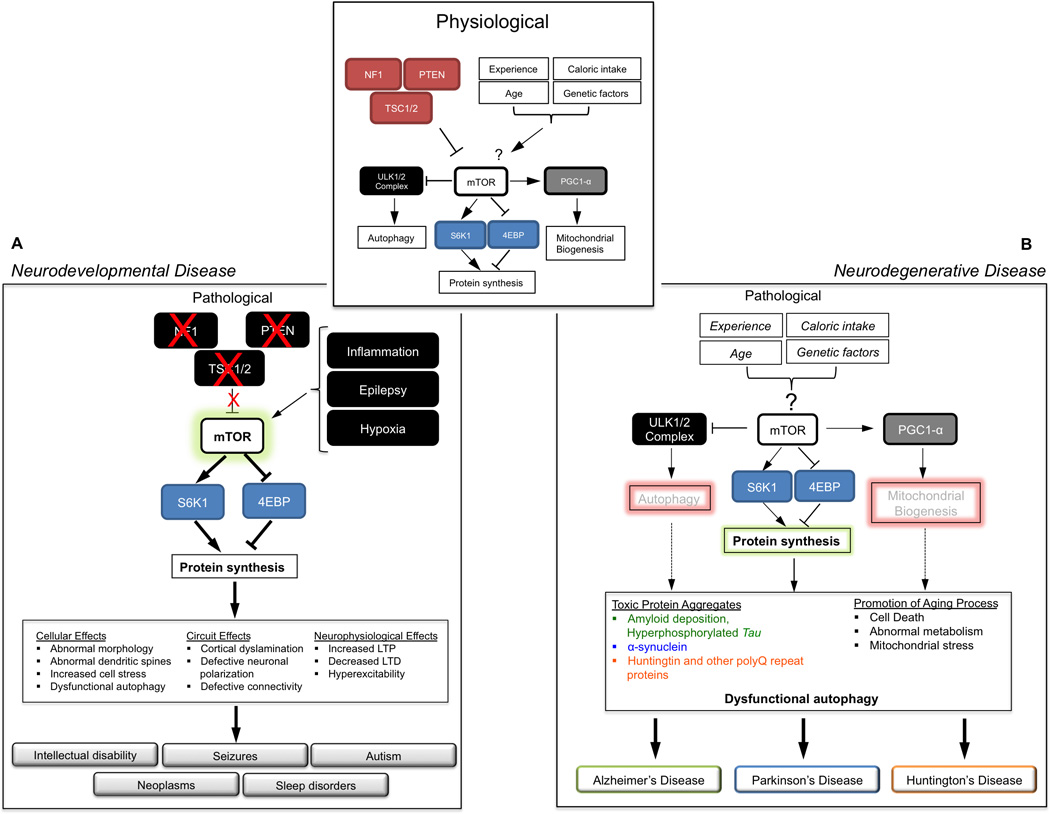
The small inset at the top depicts the physiological regulation of mTOR signaling by a variety of intrinsic and extrinsic factors. Downstream of mTOR, several cellular processes are regulated, including protein synthesis, mitochondrial biogenesis and autophagy.
A. Neurodevelopmental disease: The larger box depicts how loss of NF-1, TSC1/2, or PTEN, or environmental stimuli such as inflammation, epilepsy, or hypoxia may stimulate mTOR-dependent protein synthesis result in in a host of cellular, structural and physiological responses that culminate in clinical symptoms.
B. Neurodegenerative disease: The larger box models how changes in these factors may result in dysregulation of mTOR-dependent cellular processes, most notably autophagy, mitochondrial function and protein synthesis. Dysfunctional autophagy has been widely associated with neurodegenerative disease.
The Functions of mTOR in the Brain
Neural Development and Neural Stem Cells
The mouse mutant ‘flat-top’ carries a mutation in the mTOR gene and lacks a telencephalon, demonstrating a critical role for mTOR in early brain development(Hentges et al., 2001). While loss of mTOR early in development results in a catastrophic exhaustion of early progenitor cells necessary for brain development, over-activation may rapidly exhaust stem cell niches resulting in profound pathological consequences. A recent study expressed a mutant of mTOR that permitted activation of the kinase during specific developmental windows. Early embryonic activation of mTOR resulted in microcephaly with preserved hexalaminar cortical architecture whereas activating mTOR in post-mitotic neurons resulted in cortical hypertrophy and a disruption in cortical lamination, neurodegeneration with inclusion bodies containing ubiquitinated protein, and early death(Kassai et al., 2014).
Increased mTOR signaling due to loss of either TSC1/2 or PTEN results in profound changes in neuronal architecture and differentiation(Kwon et al., 2006; Tavazoie et al., 2005). TSC1/2-deficient neurons exhibit defects in cellular maturation as evidenced by the aberrant production of multiple axons(Choi et al., 2008). Loss of TSC1/2 function in hippocampal slice cultures results in an increased size of both neuronal somata and dendritic arbors but a decrease in dendritic spine number(Tavazoie et al., 2005). These findings were corroborated by observation of large neurons and dysplastic neural/glial cells in mouse models lacking Tsc1 in post-mitotic neurons or astrocytes(Meikle et al., 2007; Uhlmann et al., 2002).
To dissect the role of mTORC1 and mTORC2 in brain development several recent studies have utilized brain specific knockout of the Raptor and Rictor genes, respectively. A brain-specific Raptor knockout demonstrated microcephaly, a reduction in cell size, cell death and early post-natal lethality(Cloëtta et al., 2013). Interestingly, brain-specific Rictor knockouts also demonstrated a small brain, smaller neuronal somatic size and shorter dendritic processes(Thomanetz et al., 2013). The effects in Rictor knockouts appeared to be independent of mTORC1 activation, as phosphorylation of S6K1 and 4EBP1 did not significantly differ compared to controls. Despite many similarities in phenotype, the biochemical relationship between mTORC1 and mTORC2 in the brain remains largely unknown.
Oligodendrocyte-specific knockout of Raptor or Rictor were used to assess the relative contributions of mTORC1 and mTORC2 to myelination and oligodendrocyte maturation(Bercury et al., 2014; Wahl et al., 2014). Based on these studies mTORC1 appears to have a more prominent role than mTORC2 in promoting initiation of myelination, and myelin thickness in the spinal cord. It is hypothesized that mTORC1 plays a role in the control of lipogenesis and translation of myelin proteins(Lebrun-Julien et al., 2014). The most prominent effect on oligodendrocyte differentiation was observed in Raptor/Rictor double knockouts. Thus, although mTOR regulation of myelination is dominated by mTORC1, there is a synergistic effect of mTORC2 in development of these critical cells.
The interaction of mTOR signaling within and between diverse cell types in the brain is likely to be critical to the many functions of the pathway during development. This notion is demonstrated by comparing mouse models with a loss of Tsc1 in either neurons or astrocytes(Meikle et al., 2007; Uhlmann et al., 2002). Both mouse models exhibited seizures. Neuronal loss of Tsc1 demonstrated a significant effect on glial function as they demonstrate a loss of myelination in the cortex(Meikle et al., 2007). This findings echoes observations in the mTOR- or Raptor- ablated spinal cord(Bercury et al., 2014; Lebrun-Julien et al., 2014; Wahl et al., 2014). Reduction in white matter and impaired cortical connectivity is an important pathological feature of TSC(Peters et al., 2013). Regional variances in these changes may correlate with the incidence of neuropsychiatric phenotypes such as autism.
mTOR in Circuit Formation
Animal models with disrupted mTOR signaling provide evidence for a prominent role of the mTOR pathway in formation of functional neural circuits. For example, Tsc2+/− mice demonstrated aberrant retinotopic mapping. This process requires Eph receptor- and ERK-dependent regulation of mTORC1(Nie et al., 2010). This finding, taken together with other reports, demonstrates that axon guidance in response to extracellular guidance cues regulates mTOR-dependent pathways suggesting that regulation of local protein synthesis is a mechanism by which axons interact with their environment(Yoon et al., 2009). Identification of the axonal RNAs regulated by mTOR will likely provide insights into axonal biology and synaptogenesis.
Activity dependent local translation of protein at or near the synapse is a critical requirement for formation and maintenance of synapses (reviewed in(Jung et al., 2014)). The NMDA antagonist ketamine can increase mTOR pathway signaling. This increase correlated, at least temporarily, with an increase in synaptic and activity-dependent proteins such as Arc and Synapsin, and an increase in dendritic spines in vivo (Li et al., 2010). Another study demonstrated that in response to activity, mTOR signaling suppresses the translation of potassium channels in dendrites, potentially impacting neuronal membrane potential(Raab-Graham et al., 2006). These results suggest that the response to neuronal activity critically engages the mTOR signaling to produce long-lasting changes to neural circuits.
Aside from neuronal activity and growth factors, other types of cell-cell interactions also modulate mTOR signaling. One example is the recent identification of a role for MHC class I molecules in regulating insulin-mediated synapse formation(Dixon-Salazar et al., 2014). These results raise the possibility of connections between the immune system and mTOR activity that influence the development of neural circuitry.
Another emerging theme is that during injury to a neuronal process such as during spinal cord injury or denervation, there appears to be a recrudescence of developmentally critical pro-anabolic pathways such as mTOR signaling. In embryonic neurons, mTOR pathway components are enriched in developing axons and contribute to local protein synthesis(Choi et al., 2008; Nie et al., 2010). Axonal protein synthesis is also necessary for growth cone regeneration and mutation of TSC or PTEN stimulates process regeneration after injury(Lu et al., 2014; Park et al., 2008; Verma et al., 2005).
Synaptic Plasticity, Learning and Memory
The ability to make changes in synaptic strength is considered essential to the brain’s ability to store information. A role for mTOR signaling in synaptic plasticity was first gleaned when rapamycin blocked long-term synaptic facilitation in studies of Aplysia and crayfish(Beaumont et al., 2001; Casadio et al., 1999). Consistent with this finding, treatment of rat hippocampal slices with rapamycin strongly impaired protein synthesis-dependent late phase of long-term potentiation (L-LTP)(Tang et al., 2002). Tsc1+/− or Tsc2+/− mice that exhibit hyperactivation of mTOR also displayed a reduced threshold for L-LTP(Ehninger et al., 2008; von der Brelie et al., 2006). Similarly, knockout of 4E-BP2 (the dominant 4E-BP in the brain) lowered the threshold for induction of L-LTP by high-frequency stimulation(Banko et al., 2005). On the other hand, genetic ablation of neither the mTOR inhibitor (and rapamycin target) FKBP12 nor S6K1 completely phenocopied the effect of rapamycin. FKBP12 knockouts showed enhanced LTP, yet no defect in the threshold for its development(Hoeffer and Klann, 2010; Hoeffer et al., 2008). By contrast, S6K1 knockouts demonstrated impaired E-LTP but normal L-LTP. Thus, mTOR-mediated signaling may have complex roles in modulating synaptic plasticity through both S6K1 and 4EBP branches of the pathway.
mTORC1 has also been implicated in long-term depression (LTD), a model of weakened synaptic efficacy in the hippocampus and cerebellum. One form of LTD is dependent on metabotropic glutamate receptors (mGluRs). mGluR activation results in increased ERK-MAPK and PI3K-mTOR signaling and activation of protein synthesis near synapses. Consistently, inhibition of protein synthesis or mTOR signaling blocks mGluR-dependent LTD(Auerbach et al., 2011; Banko et al., 2006; Hou and Klann, 2004; Huber, 2000). This is thought to be secondary to local translation of mRNAs already transported to the dendrite(Lüscher and Huber, 2010). mGluR activity enhances the translational capacity of dendrites by activation of mTOR and S6K1 – yet mGluR-dependent LTD can still be elicited in S6K1/2 double knockout mice(Hou and Klann, 2004).
mTORC2 has also emerged as an important regulator of synaptic function. mTORC2 has been linked to control of the actin cytoskeleton in non-neuronal cells, raising the hypothesis that it could play a role in regulating neuronal processes(Jacinto et al., 2004). Disruption of Raptor or Rictor in cultured neurons impinged on process outgrowth(Urbanska et al., 2012). Forebrain-specific disruption of Rictor resulted in defects in the transition from E-LTP to L-LTP and a decrease in actin cytoskeleton turnover at hippocampal synapses(Huang et al., 2013). Since chronic rapamycin treatment reversed behavioral phenotypes of TSC1/2 mutant mice and can also impact mTORC2, these findings suggest that both mTORC1 and mTORC2 regulate synaptic physiology and behavior. Indeed, TSC1/2 can stimulate mTORC2 kinase activity independently of Rheb although this has not been confirmed in neural tissues(Huang et al., 2008).
In keeping with the abundant neurophysiological evidence linking mTOR signaling to synaptic plasticity, this pathway has important roles in cognitive behaviors. For example, Tsc2+/− mice – which show enhanced L-LTP and reduced LTD – demonstrate defects in hippocampus-dependent learning paradigms, which are reversible by a short course of rapamycin administration even in adulthood(Ehninger et al., 2008). S6K1 knockouts demonstrate abnormal fear conditioning and defective hippocampus-based learning while 4EBP2 knockouts demonstrate marked defects in spatial learning(Antion et al., 2008; Banko et al., 2007). Since S6K1 and 4EBP2 loss are expected to have at least partially opposing net downstream effects, these data suggest that any disruption in mTOR downstream signaling can affect cognition. These experiments highlight an emerging theme that an exquisite balance of mTOR signaling regulates synaptic function and behavior. Supporting this notion, Tsc1 forebrain-specific mutants demonstrate hyperexcitability and a disruption in excitatory/inhibitory (E/I) balance in hippocampal networks(Bateup et al., 2013).
Homeostatic Regulation
The maintenance of energy balance is a fundamental requirement of all living things, requiring the constant scaling of nutrient intake, metabolic demand, and motivational state. In mammals, the control of feeding is primarily integrated by opposing neurotransmitter and neuropeptide cell populations in the arcuate nucleus of the hypothalamus(Sternson, 2013; Woods et al., 2008). mTOR signaling appears to have a prominent, albeit complex, role in the molecular control of feeding behavior. Phosphorylation of S6K1 in the hypothalamus decreased with starvation, while overactivation of mTOR in catabolic pro-opiomelanocortin neurons reduced their activity and resulted in disinhibition of feeding and obesity(Cota et al., 2006; Mori et al., 2009; Yang et al., 2012). Treatment with the satiety signal leptin stimulated the phosphorylation of S6K1 and S6 in the hypothalamus in a rapamycin-dependent fashion suppressing feeding. Thus, leptin signals satiety and also conveys the signal to activate growth and anabolism. Consistently, rapamycin administration stimulated feeding but completely blocked the anorectic effects of leptin, consistent with the hypothesis that leptin signaling is mTOR-dependent(Cota et al., 2006). In aged mice, chronic rapamycin administration inhibited feeding, consistent with the pro-longevity effects of mTOR pathway suppression through mechanisms analogous to caloric restriction(Harrison et al., 2009; Johnson et al., 2013a; Kapahi and Zid, 2004). The neural mechanisms underlying this apparent discrepancy in age-dependent mTOR-related signaling in the hypothalamus remains unclear.
Circadian rhythms are 24-hour oscillations in behavior, gene expression, and physiology regulated by an autonomous transcriptional-translational-post-translational feedback loop(Takahashi et al., 2008). While light has been shown to activate phospho-S6 and phospho-S6K1 in the suprachiasmatic nucleus of the hypothalamus (SCN), the mechanisms by which activation of the mTOR pathway impacts circadian timing remains obscure(Cao et al., 2010). A recent study points to 4EBP-dependent translational regulation of vasoactive intestinal peptide in the SCN as a mechanisms that regulates phase shifts in response to light(Cao et al., 2013).
The circadian timekeeping system forms the foundation for rhythmic behaviors including sleep-wake oscillations(Saper et al., 2005). Genetic and microarray studies of sleep deprivation in mammals and flies have implicated the regulation of synaptic protein homeostasis as one of the major effects of sleep(Tononi and Cirelli, 2007). Interestingly, sleep mediates developmental cortical plasticity in an mTOR-dependent fashion(Seibt et al., 2012; Seibt and Frank, 2012).
Neuropathology of mTOR
Monogenic Disorders of the mTOR Pathway
Tuberous Sclerosis Complex
Tuberous Sclerosis Complex (TSC) is an autosomal dominant multisystem disorder caused by loss of either the TSC1 or TSC2 and is one of the paradigmatic monogenetic ‘mTORopathies’. TSC affects 1/6000 individuals worldwide(Crino et al., 2006). TSC manifests as tumor-hamartoma syndrome affecting multiple organs including the brain, skin eyes, kidneys, heart and lungs. TSC results in epilepsy (approximately 90% of patients), intellectual disability (approximately 50%), autism (approximately 50%) and other neuro-psychiatric morbidities including sleep disruption, ADHD and anxiety(Asato and Hardan, 2004; Bruni et al., 1995; Hunt and Stores, 1994; Husain et al., 2000) (Table 1). Missense mutations have been identified in over 700 locations for TSC2 and more than 200 in TSC1 without clear evidence for particular areas of vulnerability within either gene product(Dabora et al., 2001; Sancak et al., 2005). As a result, genotype-phenotype characterization has been difficult(van Slegtenhorst et al., 1999). TSC2 mutations seem to render a more severe phenotype, in keeping with its role as a functional GAP in the TS complex(Dabora et al., 2001) (Figure 3).
Table 1.
Neurological Disorders Associated with Dysfunctional mTOR Pathway Signaling
| Disease | Responsible Gene Product |
Neurological Manifestations |
Pathway Activity |
Reference |
|---|---|---|---|---|
| Neurodevelopmental | ||||
| Tuberous sclerosis complex | TSC1 or TSC2 | E, ASD, ID, Tubers, Hamartomas, multisystem benign tumors | Up | (Crino et al., 2006) |
| TBC1D7 Syndrome? | TBC1D7 | ID, macrocrania, neuropsychiatric | Up? | (Alfaiz et al., 2014) |
| Autism spectrum with macrocephaly | PTEN | ASD, E, MacroC | Up | (Zhou and Parada, 2012) |
| Cowden Syndrome | PTEN | MacroC, +/− ID, CA | Up | (Pilarski et al., 2013) |
| Bannayan-Riley-Rulvalcaba Syndrome | PTEN | MacroC, polyps, +/− ID, skin, WM cysts | Up | (Pilarski et al., 2013) |
| Lhermitte-Duclos Disease | PTEN | dysplastic gangliocytomas of the cerebellum | Up | (Pilarski et al., 2013) |
| Neurofibromatosis type I | NF1 | ID, E, OPG | Up | (Diggs-Andrews and Gutmann, 2013) |
| Non-syndromic autism | PTEN, mTOR, ?AKT, others… | ASD, E? | Up/Down | (O’Roak et al., 2012) |
| Epileptic encephalopathy | mTOR, others | E, ID | ? | (Allen et al., 2013) |
| Neurodegenerative | ||||
| Alzheimer’s Disease | APP, Presenilins, others | Dementia | Up? | (Caccamo et al., 2011; Spilman et al., 2010) |
| Parkinson’s Disease | a-synuclein, Parkin, PINK1, LRRK2, others | Movement disorder, dementia | Up? | (Bové et al., 2011) |
| Huntington’s Disease | Huntingtin | Movement disorder, neuropsychiatric | Up? | (Ravikumar et al., 2004) |
| Psychiatric | ||||
| Major Depressive Disorder | ? | Decreased mood, suicidality | Down? | (Autry et al., 2011; Li et al., 2011) |
| Schizophrenia | AKT1, DISC1, others | Hallucinations, delusions, thought disorder, depression | Up/Down? | (Emamian et al., 2004; Kim et al., 2009; Zhou et al., 2013) |
E = epilepsy; ID = intellectual disability; ASD = autism spectrum disorder; MacroC = macrocephaly; MicroC = microcephaly; OPG = optic pathway glioma; WM = white matter
The pathognomonic lesion in TSC brain is the cortical tuber and is present in over 80% of patients. Tubers are poorly understood, non-malignant, abnormalities of the developing cerebral cortex characterized histologically as a loss of normal cortical lamination, dysplastic neurons cells, large glial cells, and so-called “giant cells”. Cortical tuber burden was thought to correlate with the severity of neurologic symptoms. However, even patients without tubers can have very significant neurological symptoms including autism and intellectual disability. Still, seizure foci often localize to regions surrounding tubers suggesting that they are indeed epileptogenic(Mohamed et al., 2012).
Tumors in TSC are thought to arise from a loss-of-heterozygosity (LOH) resulting in an mTOR-hyperactivated state in homozygous-null cells. LOH has been convincingly demonstrated in most of the neoplasms associated with TSC including subependymal giant cell astrocytomas (SEGAs), angiomyolipomas, cardiac rhabdomyomas, and lymphangioleiomyomatosis (LAM). Biallelic inactivation has been detected, but in only a small number of cells within cortical tubers(Crino et al., 2010; Qin et al., 2010).
With regard to risk of neuropsychiatric symptoms, tuber volume may be a better marker than total tuber number(Jansen et al., 2008). With increasingly sophisticated modes of neuroimaging, more subtle and complex abnormalities in the TSC brain have been identified, including changes in white matter volume, myelination, and connectivity(Peters et al., 2013). These changes are being linked to specific phenotypes such as autism. For example, impaired connectivity of the arcuate fasciculus – the tract that connects Broca’s and Wernicke’s areas – is more common in patients with TSC that have autism compared to TSC patients without autism(Lewis et al., 2013). The cortex is not the only site of important CNS pathology in TSC. Emerging evidence has suggested that TSC patients have lesions in other brain areas including the cerebellum(Boer et al., 2008; Ertan et al., 2010; Reith et al., 2011).
While the TSC1/TSC2 complex interacts with more than 50 proteins, most remain poorly understood. One exception is the recently described TBC1D7, a TSC1-dependent component of the TSC1/2 complex that functions similarly to TSC1 and TSC2 on a cellular level(Dibble et al., 2012; Menon et al., 2014). Two recent studies have described humans carrying mutations in TBC1D7, which manifested clinically with intellectual disability, macrocrania (large skull), and anxiety. Since none of the cases had epilepsy, autism, tubers, or SENs, TBC1D7 mutations seem to diverge phenotypically from TSC1/2 mutations, at least in brain(Alfaiz et al., 2014; Capo-Chichi et al., 2013).
PTEN hamartoma Tumor Syndrome (PHTS)
PTEN is a dual specificity phosphatase that inhibits both the PI3K/AKT and MAPK pathways, and thus curbs mTOR signaling (reviewed in(Zhou and Parada, 2012)). PTEN is well understood to be an important human tumor-suppressor gene and its activity is a major regulator of growth mediated by the AKT/mTOR pathway. Germline mutations in PTEN are responsible for a group of rare disorders referred to as PTEN hamartoma tumor syndromes (PHTS)(Endersby and Baker, 2008; Pilarski et al., 2013). These include Cowden’s Syndrome (CS), Lhermitte-Duclos Disease (LDD) and Bannayan-Riley-Ruvacalba Syndrome (BRRS). CS is characterized by macrocephaly, benign hamartomas of the breast, thyroid, or endometrium, as well as malignant tumors. A minority of patients have co-morbid intellectual disability. LDD is characterized by dysplastic gangliocytomas of the cerebellum, which clinically cause ataxia, seizure, or increased intracranial pressure(Endersby and Baker, 2008). BRRS is the association of macrocephaly, developmental delay, neurocutaneous features, and/or intestinal polyps(Lynch et al., 2009). Mysteriously, the same mutations in PTEN can result in CS, LDD, or RBBS suggesting that many other factors cooperate to render specific phenotypes(Pilarski et al., 2013; Rodríguez-Escudero et al., 2011). In general, as in TSC, there is a poor genotype-phenotype correlation in PHTS (Table 1).
Interestingly, PTEN mutations that spare the lipid phosphatase domain are emerging as relatively common sporadic causes of autism, especially when associated with co-morbid macrocephaly (which is associated with autism in about 10–20% of cases)(Butler et al., 2005; Hobert et al., 2014; Klein et al., 2013; Varga et al., 2009; Zhou and Parada, 2012). As mentioned above, PTEN mutations have been estimated to occur in about 1% of sporadic autism, and PTEN gene testing is frequently recommended when macrocephaly is present with clinical features of autism(O'Roak et al., 2012). With striking similarity to human disease, mouse models with deletion of Pten in forebrain neurons results in macrocephaly, seizures and abnormal social interaction(Kwon et al., 2006). Pathologically, neurons demonstrate enlarged somata, increased length of both dendritic arbors and number of dendritic spines. Thus, the phenotype is similar but non-identical to that of TSC, possibly reflecting the many subtleties in the signaling pathway that await future discovery.
NF1
Neurofibromatosis is a common neurocutaneous disorder characterized by both benign and malignant tumors of the central and peripheral nervous system. NF1 is autosomally inherited in 1/2500-1/3000 live births. It has diverse manifestations in tissues that are primarily, although not exclusively, of neural crest origin. Clinical features of NF1 include the brain (glial tumors, macrocephaly), skin (café au lait spots, freckling, neurofibromas), bone (sphenoid wing dysplasia), kidney (renal artery stenosis), and endocrine systems. While epilepsy is seen in a relatively small percentage of cases (<10%), learning disabilities, ADHD, sleep disruption and anxiety are very common. Optic pathway gliomas occur in 15% of patients. Plexiform neurofibromas can undergo malignant transformation, and 3–5% of patients with NF1 develop malignant peripheral nerve sheath tumors. As in PTHS, macrocephaly is common (Table 1).
Mutations in the neurofibromin 1 (NF1) gene result in this disease. NF1 encodes a GTPase activating protein that suppresses the activity of the proto-oncogene Ras. In mice with a targeted disruption of the Nf1 gene, Schwann cells have increased levels of Ras activation and an increased growth rate. In its active state, Ras signals to several downstream effectors, including the ERK/MEPK and the PI3K/mTOR signaling pathways(Dasgupta and Gutmann, 2005; Dasgupta et al., 2005; Johannessen et al., 2005). While mTORC1 activity is essential for tumorigenesis downstream of NF1 loss(Johannessen et al., 2008), NF1 mediated regulation of mTORC1 appears independent of TSC/rheb(Banerjee et al., 2011). Clinical trials are in progress to test the efficacy of mTOR inhibitors and dual PI3K/mTOR inhibitors in NF1-related tumors(Endo et al., 2013).
Neurofibromin also plays a role in neurons. Nf1+/− mice also have learning and memory deficits. Lovastatin, which is an inhibitor of the beta-hydroxy-beta-methylglutary CoA reductase (HMG-CoA-reductase), and thus a blocker of Ras signaling, reversed spatial learning deficit and impairment of attention(Costa et al., 2001). Whether mTOR plays any role in neurons downstream of NF1 loss is unclear.
Other Neurodevelopmental Disorders
Fragile X Syndrome
The fragile X mental retardation protein 1 (FMRP) is deficient in a commonly inherited form of intellectual disability, Fragile X Syndrome(FXS). FMRP1 is an important translational repressor of hundreds of mRNAs in the brain(Darnell et al., 2011). In Fmr1−/y mutant mice, mTOR signaling is high, protein synthesis is elevated, and mGluR-dependent LTD is exaggerated(Auerbach et al., 2011; Huber et al., 2002; Sharma et al., 2010). Genetic deletion of S6K1 in Fmr1−/y mice reduced exaggerated protein synthesis and mitigated neurophysiological and behavioral defects(Bhattacharya et al., 2012). In addition, lovastatin rescued many of the phenotypes of Fmr1 mutants(Osterweil et al., 2013). While investigating the relationships between Tsc2+/− and Fmr1−/y mouse phenotypes, Bear and colleagues made the surprising finding that Tsc2+/− mice had reduced protein synthesis in the hippocampus. Fmr1−/y;Tsc2+/− double mutants demonstrated normalization in hippocampal mGluR-LTD, protein synthesis rates, and cognitive behaviors suggesting that FMRP and TSC1/2 balance one another(Auerbach et al., 2011). Whether this interaction occurs in other regions of the brain is not clear, and the biochemical events that might mediate such a relationship have not yet been determined.
Autism
Autism is a neurodevelopmental disorder defined by two cardinal features: 1) persistent social communication and social interaction deficit and 2) restricted and repetitive patterns of behavior. While the causes of autism appear heterogeneous, several genetic disorders that directly affect mTOR signaling are associated with autism, nominating this pathway as a possible etiological hub for the disorder. TSC patients display an autistic profile very similar to that of idiopathic autism(Jeste et al., 2008). Mutations in the PTEN gene have been estimated to cause 1–5% of autism(Zhou and Parada, 2012). High frequency of autism in monogenetic mTORopathies has suggested that regulation of mRNA translation may be a critical variable in the development of autism.
Studies in animal models have corroborated findings from human genetics. Tsc1+/− and Tsc2+/− mice and the spontaneously haploinsufficient Tsc2+/− Eker rat demonstrate abnormal social interaction reversible by rapamycin treatment(Ehninger et al., 2008; Goorden et al., 2007; Sato et al., 2012; Waltereit et al., 2011). Forebrain-specific deletion of Pten also resulted in abnormal social behavior(Kwon et al., 2006). Deletion of Tsc1 or Tsc2 in cerebellar Purkinje cells resulted in marked abnormalities in social behavior directly implicating mTOR signaling in the cerebellum as a mediator of social cognition(Reith et al., 2013; Tsai et al., 2012). 4E-BP2 knockout mice demonstrated autism-like features, thereby directly implicating mTOR-dependent protein synthesis in the control of social behavior(Gkogkas et al., 2013). Moreover, these mice demonstrated increased translation of the neuroligins, proteins which have been previously linked to ASD risk(Südhof, 2008). Remarkably, suppression of neuroligin 1 partially reversed the behavioral phenotypes in these mice(Gkogkas et al., 2013). In a parallel study, overexpression of eIF4E similarly resulted in defective social behavior and hippocampal physiology(Santini et al., 2013). These studies have illuminated translational regulation pathways as crucial mediators of higher order cognitive function and suggest that manipulation of these pathways may be pharmacological targets for treating autism.
Epilepsy
Epilepsy is a common neurological disorder affecting approximately 1% of population worldwide. Seizures are paroxysmal electrical events in the brain, often resulting in behavioral manifestations. The mainstay of therapy is anticonvulsant medications that address the symptoms but not the cause of the disorder. The mTOR pathway has received attention as a potential mediator of epilepsy since the connection between TSC and mTOR was made about a decade ago. It was initially recognized that mTOR is hyperactive in cells from cortical tubers and focal cortical dysplasias(Baybis et al., 2004; Miyata et al., 2004). More recently, several groups identified de novo somatic mutations of PI3K, AKT3, or mTOR genes in hemimegalencephaly, a condition associated with severe seizures(Lee et al., 2012; Poduri et al., 2012; Rivière et al., 2012). Human mutations in DEPDC5, a component of the GATOR complex, have been linked to both epilepsy with cerebral malformations and non-lesional epilepsy(Bar-Peled et al., 2013; Dibbens et al., 2013; Scheffer et al., 2014).
Animal models have further supported this association between mTOR and epilepsy. Essentially all neural mutant models of Tsc1 or Tsc2 homozygous loss lead to increased seizures(Carson et al., 2013; Goto et al., 2011; Meikle et al., 2007; Normand et al., 2013; Uhlmann et al., 2002; Zeng et al., 2011). Similarly, Pten knockout also leads to epilepsy in mice(Pun et al., 2012; Zhou et al., 2009). Importantly, epilepsy due to loss of Tsc1, Tsc2 or Pten all respond to rapamycin treatment, indicating the mTOR hyperactivation is necessary for this phenotype. Rapamycin seems effective in not only reducing seizures once they start but also in preventing seizures from ever developing indicating that mTOR may have an anti-epileptogenic effect in these genetic models(Meikle et al., 2008; Zeng et al., 2008). This has led to preclinical trials with mTOR inhibitors in other models of epileptogenesis, such as post-traumatic epilepsy(Berdichevsky et al., 2013; Guo et al., 2013; Huang et al., 2010). There are also some reports of mTOR inhibitors in temporal lobe epilepsy models but the level of efficacy seems to differ(Buckmaster and Lew, 2011; Zeng et al., 2009).
A recent genomics study of infantile spasms and Lennox-Gastaut Syndrome – forms of epileptic encephalopathy – identified a patient with de novo mutation in the mTOR gene itself, without accompanying brain malformations. The result nominates mTOR as a cause for these disorders and suggests that mTOR inhibitors represent possible treatments(Allen et al., 2013).
How mTOR mediates epileptogenesis is not yet clear. Given the many cellular and developmental functions of mTOR in neuronal differentiation, migration, axonal growth/sprouting and dendrite morphogenesis, it is likely that mTOR hyperactivation contributes to aberrant circuit formation. However, it is also likely that mTOR regulates neuronal excitability in already established neural circuits. Consistent with this model, Tsc1 inactivation in adult mice is rapidly followed by mTOR-dependent seizures(Abs et al., 2013).
One important concept to emphasize is that mTOR activity can be regulated by several non-genetic factors such as neuronal activation, hypoxia, and inflammatory responses. Thus, a combination of genetic “hits” (such as TSC1/2 or PTEN loss) and non-genetic “hits” (such as epilepsy, inflammatory response etc.) could synergistically hyperactivate mTOR and contribute to a common pathology (Figure 3B). Such combinatorial effects are just beginning to be explored and may provide one explanation for the variability of expression for genetic mTORopathies and may provide options for treatment as well(Ehninger et al., 2012; Waltereit et al., 2011).
Neurodegenerative Disease
Aging
One of the most compelling influences on extending lifespan across phyla is caloric intake. As a major gauge of nutrient signaling and energy balance, the mTOR pathway has emerged as a crucial hub in the regulation of overall lifespan (reviewed in(Johnson et al., 2013a)). There is an increasing body of pharmacologic and genetic data to support a role for mTOR in organismal longevity, particularly in females. Rapamycin extends the lifespan of various mouse strains; for reasons that remain unclear, the effect was greater in female mice(Harrison et al., 2009). Similarly, S6K1 knockout females demonstrated an extension of lifespan(Selman et al., 2009). In Drosophila, 4E-BP mediates lifespan extension in response to dietary restriction suggesting that protein synthesis is essential to the effects of caloric restriction on aging(Zid et al., 2009). How mTOR might mediate longevity in animals, whether mitochondrial function or autophagy are involved in this effect, and the role of female hormones such as estrogen that signal through mTOR are modifying these function remain unanswered.
Alzheimer’s Disease (AD)
AD is characterized clinically by progressive loss of short-term memory and cognitive dysfunction. The pathological hallmarks of this disorder are the accumulation of extracellular senile plaques and intracellular neurofibrillary tangles (NFT). The amyloid cascade hypothesis posits that abnormalities in the sequential cleavage of the amyloid precursor protein (APP) by β-secretase and then γ-secretase results in the generation of toxic oligomeric Aβ species, which initiate a cascade of cellular dysfunction resulting in synaptic and ultimately neuronal loss (reviewed in(Pei and Hugon, 2008; Wang et al., 2014))
Aβ accumulation correlates with an increase in mTOR signaling with increased levels of phosphorylated S6K1 accompanying the appearance of NFTs in post-mortem AD brain tissue(An et al., 2003). Accumulation of autophosphorylated mTOR at S2481 correlated with presence of pathological forms of tau and phospho-tau proteins(Li et al., 2005). Increased levels of AKT and its substrates were also identified in the temporal cortex of AD brains(Griffin et al., 2005). Pharmacologic or genetic manipulations that decrease the amount of Aβ in mouse brain are associated with corresponding decreases in mTOR pathway activity(Caccamo et al., 2011). Inhibition of mTOR signaling has a beneficial effect on both pathophysiological and behavioral outcomes in several mouse models of Aβ pathology(Caccamo et al., 2011; Spilman et al., 2010; Zhang et al., 2010). Because of the important role of autophagy in the clearance of misfolded protein aggregates, autophagy dysfunction has been implicated in AD pathogenesis, and the stimulation of autophagy by mTOR inhibition is being tested as a potential therapeutic(Nixon, 2013).
Parkinson’s Disease (PD)
PD is a common and debilitating neurodegenerative disorder characterized by resting tremor, bradykinesia, postural and autonomic instability, and rigidity. The pathological signature of PD is the loss of dopaminergic neurons in substantia nigra and the accumulation of α-synuclein-containing inclusion bodies (i.e. Lewy bodies). Increased numbers of autophagosomes have been reported in postmortem PD brains(Anglade et al., 1997). The accumulation of toxic proteins in PD brains suggests that in some cases, defects in autophagy may promote pathogenesis. An extension of this hypothesis is that the stimulation of autophagy could induce the appropriate handling of protein accumulations. Rapamycin induced autophagy and blocked the accumulation of ubiquitinated α-synuclein in α-synuclein-overexpressing cells and mice(Spencer et al., 2009; Webb et al., 2003). On the other hand, overexpression of constitutively activated AKT was also protective to both neuronal cell bodies and axons of the nigrostriatal projections in PD mouse models(Cheng et al., 2011; Ries et al., 2006). Thus, mTOR-pathway appears to participate in several ways in PD pathogenesis.
Huntington’s Disease (HD)
HD is an autosomal dominant genetic disorder characterized by progressive motor disorder, psychiatric dysfunction, and eventually, dementia. Disease-causing mutations in the huntingtin (HTT) gene result in expansion of polyglutamine repeats (CAG) in HTT and are thought to lead to the formation of pathogenic protein aggregates(reviewed in(Nixon, 2013; Ross et al., 2014)). When more than 35 repeats occur, disease ensues. It was hypothesized over a decade ago that stimulation of the clearance of HTT aggregates by stimulation of autophagy would benefit disease progression. Mutant HTT stimulates mTOR pathway activation and TOP-dependent translation in heterologous cell lines in cells expressing 74 CAG repeats (Q74) protein compared to those expressing the shorter, Q23 form suggesting that the suppression of autophagy may promote HD pathogenesis(Ravikumar et al., 2004). It remains unclear whether the activation of mTOR is essential to, or a compensation for, HD pathogenesis. In some studies, inhibition of mTOR-independent autophagy is protective(Tsvetkov et al., 2010). Others have shown that targeting the catalytic domain of mTOR is more effective than rapamycin analogs(Roscic et al., 2011).
Psychiatric Disease
Major depressive disorder is a debilitating, common, and costly worldwide illness. The NMDA receptor antagonist ketamine has been used with success for treatment-resistant depression(Berman et al., 2000; Zarate et al., 2006). Several reports over the past few years have directly implicated the mTOR pathway in mediating the rapid antidepressant effects of ketamine. Notably, ketamine administration to rats resulted in a rapid increase in phosphorylated mTOR, S6K1, and 4EBP. These biochemical changes correlated with increases in synaptic protein expression and synapse number at dendritic spines in the prefrontal cortex. Moreover, ketamine’s ability to ameliorate depressive symptoms was completely abrogated by pre-administration with rapamycin, suggesting that the effect of ketamine is mTOR-dependent(Li et al., 2010; Li et al., 2011). Ketamine has also been shown to disinhibit BDNF translation in a manner dependent on the S6K1 substrate eEF2K(Autry et al., 2011). An unresolved question is to what extent clinical depression results from acute or chronic dysfunction in mTOR signaling.
Schizophrenia is a common, chronic, and severe neuropsychiatric disorder characterized by delusions, hallucinations, disorganized thought and behavior, and affective flattening. Schizophrenia is estimated to affect 1% of the general population. A clear role for mTOR signaling in schizophrenia pathogenesis has not been established; however, inhibition of the disrupted in schizophrenia 1 (DISC1) gene product, a possible susceptibility locus for bipolar disorder and schizophrenia, resulted in increased phospho-AKT and phospho-S6 expression, neuronal hypertrophy, abnormal dendritic morphology and hyperexcitability accompanied by learning and memory deficits and depressive behavior(Kim et al., 2009; Zhou et al., 2013). Rapamycin was able to reverse the biochemical and behavioral effects of DISC1 knockdown, consistent with the hypothesis that one endogenous function of DISC1 is to curb mTOR signaling(Kim et al., 2009).
Studies from humans have shown a decrease in AKT1 signaling in the brain and peripheral lymphocytes of individuals with schizophrenia(Emamian et al., 2004). Along these lines, a brain-specific Rictor knockout showed a reduction in dopamine in the prefrontal cortex, elevated expression of norepinephrine transporter, and deficits in prepulse inhibition, a schizophrenia-associated endophenotype(Siuta et al., 2010). Thus, an increase in mTORC1 signaling secondary to suppression of DISC1 or a decrease in mTORC2 signaling secondary to loss of Rictor or AKT1 signaling, may contribute to schizophrenialike phenotypes in rodents.
Therapeutics
Since discovery of rapamycin in the 1960s, mTOR inhibitors have been used to prevent solid organ transplant rejection, to augment anti-cancer treatment regimens, and to prevent neovascularization of artificial cardiac stents. Once the relationship between TSC genes and the mTORC1 pathways was established, case series of TSC patients treated with rapamycin were reported(Franz et al., 2006). Subsequent clinical trials led to the approval of everolimus for SEGAs in TSC by the FDA in 2010 (US) and EMEA (Europe) in 2011, becoming the first ever approved therapy for patients with TSC(Krueger et al., 2010). Everolimus is FDA approved for treatment of SEGAs at any age and for renal angiomyolipomas over the age of 18. Subgroup analysis of everolimus-treated patients in the SEGA trial revealed improved fractional anisotrophy measures as a marker for white matter integrity on MR diffusion tensor imaging, consistent with effects seen in mouse models(Meikle et al., 2008; Tillema et al., 2012). These findings suggest that white matter integrity on diffusion tensor imaging may be a biomarker of TSC-associated neurocognitive deficits and respond to mTOR inhibitors(Peters et al., 2013). Recently, the first prospective phase I/II trial of 20 TSC patients showed that 12 weeks of everolimus treatment resulted in a reduction of seizure frequency and duration, increased quality of life and improved behaviors, as reported by parents(Krueger et al., 2013). Rapamycin has been reported to have efficacy to treat other manifestations of TSC associated with kidneys, lung and skin(Bissler et al., 2008; Davies et al., 2011; Haemel et al., 2010; McCormack et al., 2011). Additional multicenter randomized, placebo-controlled, double-blind phase II and phase III clinical trials are also underway to test the efficacy of everolimus separately for neurocognition and epilepsy (NCT01289912 and NCT01713946).
While rapalogs (allosteric inhibitors of mTOR) have been quite successful in clinical trials of TSC, their penetrance of blood-brain barrier is relatively poor. Furthermore, they preferentially inhibit mTORC1 rather than mTORC2 and have differential effects on S6K and 4E-BP1 arms of the downstream signaling pathways(Liu et al., 2006). Direct non-allosteric mTOR kinase inhibitors are currently in clinical development and display much stronger and complete suppression of mTORC1 kinase activity. Whether such compounds will enter the CNS and be effective and safe in diseases such as TSC is not yet known. Whether combination of mTORC1 inhibitors with extracellular factors (ephrins, diet etc) may have synergistic effects has not been yet explored.
Conclusions and Future Prospects
It is becoming clear that mTOR represents a point of convergence in brain disorders. While much of the attention has focused on the role of mRNA translation at the synapse, the identity of neuronal mRNAs regulated by the mTOR pathway still needs to been elucidated. Furthermore, mTOR regulates not only protein synthesis but also a plethora of metabolic processes and cytoskeletal dynamics, which are not well studied in neurons. The availability of rapalogues that can be utilized in proof of concept trials has accelerated research in this field. A deeper understanding of the fundamental roles of mTOR in neurodevelopment and degeneration may have implications not only for the prototypical mTORophathies such as TSC, but for a whole host of neurological disorders for which we desperately need therapies.
ACKNOWLDEGEMENTS
We wish to thank Robin Kleiman, Peter Tsai, Duyu Nie, Alessia Di Nardo, and Lara Boyle for critically reviewing the manuscript. Due to limited space we have not quoted all literature in the field, and we apologize to those whose articles are not referenced. J.L. is funded by NIH/NICHD K08HD071026-03. Research in Sahin lab is supported by the NIH (U01 NS082320, P20 NS080199, P30 HD018655), Department of Defense, Tuberous Sclerosis Alliance, Autism Speaks, Nancy Lurie Marks Family Foundation, Simons Foundation, Boston Children’s Hospital Translational Research Program, Novartis, Roche and Shire.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abs E, Goorden SM, Schreiber J, Overwater IE, Hoogeveen-Westerveld M, Bruinsma CF, Aganovic E, Borgesius NZ, Nellist M, Elgersma Y. TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol. 2013;74:569–579. doi: 10.1002/ana.23943. [DOI] [PubMed] [Google Scholar]
- Alfaiz AA, Micale L, Mandriani B, Augello B, Pellico MT, Chrast J, Xenarios I, Zelante L, Merla G, Reymond A. TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat. 2014;35:447–451. doi: 10.1002/humu.22529. [DOI] [PubMed] [Google Scholar]
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu Y-F, Madou MRZ, Marson AG, Mefford HC, Esmaeeli Nieh S, O'Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EPG, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W-L, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal I-G, Winblad B, Pei J-J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, Thomas G, Schuman EM, Rosenblum K, Klann E. Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn Mem. 2008;15:29–38. doi: 10.1101/lm.661908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asato MR, Hardan AY. Neuropsychiatric problems in tuberous sclerosis complex. J Child Neurol. 2004;19:241–249. doi: 10.1177/088307380401900401. [DOI] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P-f, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci U S A. 2011;108:15996–16001. doi: 10.1073/pnas.1019012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Merhav M, Stern E, Sonenberg N, Rosenblum K, Klann E. Behavioral alterations in mice lacking the translation repressor 4E-BP2. Neurobiol Learn Mem. 2007;87:248–256. doi: 10.1016/j.nlm.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero-Camps E, Fernández A, Martínez L, Fernández-Checa JC, Colell A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Aβ accumulation and tau pathology underlying Alzheimer's disease. Hum Mol Genet. 2013;22:3460–3476. doi: 10.1093/hmg/ddt201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. 2013;78:510–522. doi: 10.1016/j.neuron.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G, 2nd, Aronica E, Crino PB. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zhong N, Fletcher R, Froemke RC, Zucker RS. Phosphorylation and local presynaptic protein synthesis in calcium-and calcineurin-dependent induction of crayfish long-term facilitation. Neuron. 2001;32:489–501. doi: 10.1016/s0896-6273(01)00483-4. [DOI] [PubMed] [Google Scholar]
- Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL, Macklin WB. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J Neurosci. 2014;34:4466–4480. doi: 10.1523/JNEUROSCI.4314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky Y, Dryer AM, Saponjian Y, Mahoney MM, Pimentel CA, Lucini CA, Usenovic M, Staley KJ. PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy. J Neurosci. 2013;33:9056–9067. doi: 10.1523/JNEUROSCI.3870-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer K, Troost D, Jansen F, Nellist M, van den Ouweland AM, Geurts JJ, Spliet WG, Crino P, Aronica E. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology. 2008;28:577–590. doi: 10.1111/j.1440-1789.2008.00920.x. [DOI] [PubMed] [Google Scholar]
- Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Bruni O, Cortesi F, Giannotti F, Curatolo P. Sleep disorders in tuberous sclerosis: a polysomnographic study. Brain Dev. 1995;17:52–56. doi: 10.1016/0387-7604(94)00118-h. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–2347. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou X-P, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magrí A, Oddo S. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011;286:8924–8932. doi: 10.1074/jbc.M110.180638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Li A, Cho HY, Lee B, Obrietan K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30:6302–6314. doi: 10.1523/JNEUROSCI.5482-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Robinson B, Xu H, Gkogkas C, Khoutorsky A, Alain T, Yanagiya A, Nevarko T, Liu AC, Amir S, Sonenberg N. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron. 2013;79:712–724. doi: 10.1016/j.neuron.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capo-Chichi J-M, Tcherkezian J, Hamdan FF, Décarie JC, Dobrzeniecka S, Patry L, Nadon M-A, Mucha BE, Major P, Shevell M, Bencheikh BOA, Joober R, Samuels ME, Rouleau GA, Roux PP, Michaud JL. Disruption of TBC1D7, a subunit of the TSC1-TSC2 protein complex, in intellectual disability and megalencephaly. J Med Genet. 2013;50:740–744. doi: 10.1136/jmedgenet-2013-101680. [DOI] [PubMed] [Google Scholar]
- Carson RP, Fu C, Winzenburger P, Ess KC. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum Mol Genet. 2013;22:140–152. doi: 10.1093/hmg/dds414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey CH, Kandel ER. A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999;99:221–237. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- Chen J, Zheng XF, Brown EJ, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A. 1995;92:4947–4951. doi: 10.1073/pnas.92.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H-C, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M, Wang C, During M, Talloczy Z, Tanaka K, Komatsu M, Kobayashi K, Okano H, Kholodilov N, Burke RE. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci. 2011;31:2125–2135. doi: 10.1523/JNEUROSCI.5519-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloëtta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, Atanasoski S, Rüegg MA. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci. 2013;33:7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Yang T, Huynh DP, Pulst SM, Viskochil DH, Silva AJ, Brannan CI. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat Genet. 2001;27:399–405. doi: 10.1038/86898. [DOI] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- Crino PB, Aronica E, Baltuch G, Nathanson KL. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology. 2010;74:1716–1723. doi: 10.1212/WNL.0b013e3181e04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80. doi: 10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci. 2005;25:5584–5594. doi: 10.1523/JNEUROSCI.4693-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- Davies DM, de Vries PJ, Johnson SR, McCartney DL, Cox JA, Serra AL, Watson PC, Howe CJ, Doyle T, Pointon K, Cross JJ, Tattersfield AE, Kingswood JC, Sampson JR. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17:4071–4081. doi: 10.1158/1078-0432.CCR-11-0445. [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, Goto J, Dilsiz P, Talos DM, Clish CB, Kwiatkowski DJ, Sahin M. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet. 2014;23:3865–3874. doi: 10.1093/hmg/ddu101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, Crompton DE, Hughes JN, Bellows ST, Klein KM, Callenbach PMC, Corbett MA, Gardner AE, Kivity S, Iona X, Regan BM, Weller CM, Crimmins D, O'Brien TJ, Guerrero-López R, Mulley JC, Dubeau F, Licchetta L, Bisulli F, Cossette P, Thomas PQ, Gecz J, Serratosa J, Brouwer OF, Andermann F, Andermann E, van den Maagdenberg AMJM, Pandolfo M, Berkovic SF, Scheffer IE. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45:546–551. doi: 10.1038/ng.2599. [DOI] [PubMed] [Google Scholar]
- Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, Manning BD. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47:535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon-Salazar TJ, Fourgeaud L, Tyler CM, Poole JR, Park JJ, Boulanger LM. MHC class I limits hippocampal synapse density by inhibiting neuronal insulin receptor signaling. J Neurosci. 2014;34:11844–11856. doi: 10.1523/JNEUROSCI.4642-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013;493:679–683. doi: 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Sano Y, de Vries PJ, Dies K, Franz D, Geschwind DH, Kaur M, Lee YS, Li W, Lowe JK, Nakagawa JA, Sahin M, Smith K, Whittemore V, Silva AJ. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Molecular psychiatry. 2012;17:62–70. doi: 10.1038/mp.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–5430. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, Iida K-i, Matsumoto Y, Hakozaki M, Aoki M, Iwasaki H, Dobashi Y, Nishiyama K, Iwamoto Y, Oda Y. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19:450–461. doi: 10.1158/1078-0432.CCR-12-1067. [DOI] [PubMed] [Google Scholar]
- Ertan G, Arulrajah S, Tekes A, Jordan L, Huisman TAGM. Cerebellar abnormality in children and young adults with tuberous sclerosis complex: MR and diffusion weighted imaging findings. J Neuroradiol. 2010;37:231–238. doi: 10.1016/j.neurad.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille J-C, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- Goto J, Talos DM, Klein P, Qin W, Chekaluk YI, Anderl S, Malinowska IA, Di Nardo A, Bronson RT, Chan JA, Vinters HV, Kernie SG, Jensen FE, Sahin M, Kwiatkowski DJ. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci U S A. 2011;108:E1070–E1079. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PloS one. 2013;8:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemel AK, O'Brian AL, Teng JM. Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol. 2010;146:715–718. doi: 10.1001/archdermatol.2010.125. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges KE, Sirry B, Gingeras A-C, Sarbassov D, Sonenberg N, Sabatini D, Peterson AS. FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc Natl Acad Sci U S A. 2001;98:13796–13801. doi: 10.1073/pnas.241184198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert JA, Embacher R, Mester JL, Frazier TW, Eng C. Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur J Hum Genet. 2014;22:273–276. doi: 10.1038/ejhg.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjević K, Roman G, Costa-Mattioli M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci. 2013;16:441–448. doi: 10.1038/nn.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, Cao Z, Gruenthal M, Huang Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM. Role for Rapid Dendritic Protein Synthesis in Hippocampal mGluR-Dependent Long-Term Depression. Science. 2000;288:1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A, Stores G. Sleep disorder and epilepsy in children with tuberous sclerosis: a questionnaire-based study. Dev Med Child Neurol. 1994;36:108–115. doi: 10.1111/j.1469-8749.1994.tb11819.x. [DOI] [PubMed] [Google Scholar]
- Husain AM, Foley CM, Legido A, Chandler DA, Miles DK, Grover WD. Tuberous sclerosis complex and epilepsy: prognostic significance of electroencephalography and magnetic resonance imaging. J Child Neurol. 2000;15:81–83. doi: 10.1177/088307380001500203. [DOI] [PubMed] [Google Scholar]
- Inoki K, Kim J, Guan K-L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jansen FE, Vincken KL, Algra A, Anbeek P, Braams O, Nellist M, Zonnenberg BA, Jennekens-Schinkel A, van den Ouweland A, Halley D, van Huffelen AC, van Nieuwenhuizen O. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology. 2008;70:916–923. doi: 10.1212/01.wnl.0000280579.04974.c0. [DOI] [PubMed] [Google Scholar]
- Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. 2008;23:520–525. doi: 10.1177/0883073807309788. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, Rioth MJ, McClatchey A, Ryeom S, Cichowski K. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013a;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser E-B, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013b;342:1524–1528. doi: 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Gkogkas CG, Sonenberg N, Holt CE. Remote control of gene function by local translation. Cell. 2014;157:26–40. doi: 10.1016/j.cell.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid B. TOR pathway: linking nutrient sensing to life span. Sci Aging Knowledge Environ. 2004;2004:PE34. doi: 10.1126/sageke.2004.36.pe34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, Aiba A. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014;7:1626–1639. doi: 10.1016/j.celrep.2014.04.048. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim JY, Duan X, Liu CY, Jang M-H, Guo JU, Pow-anpongkul N, Kang E, Song H, Ming G-l. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Sharifi-Hannauer P, Martinez-Agosto JA. Macrocephaly as a clinical indicator of genetic subtypes in autism. Autism Res. 2013;6:51–56. doi: 10.1002/aur.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-i, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Iwata J-i, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, Mays M, Lopez CM, Kim MO, Franz DN. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–687. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- Kwon C-H, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Julien F, Bachmann L, Norrmén C, Trötzmüller M, Köfeler H, Rüegg MA, Hall MN, Suter U. Balanced mTORC1 Activity in Oligodendrocytes Is Required for Accurate CNS Myelination. J Neurosci. 2014;34:8432–8448. doi: 10.1523/JNEUROSCI.1105-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG. De novo somatic mutations in components of the PI3K–AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Avruch J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J Biol Chem. 2005;280:38121–38124. doi: 10.1074/jbc.C500363200. [DOI] [PubMed] [Google Scholar]
- Lewis WW, Sahin M, Scherrer B, Peters JM, Suarez RO, Vogel-Farley VK, Jeste SS, Gregas MC, Prabhu SP, Nelson CA, 3rd, Warfield SK. Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb Cortex. 2013;23:1526–1532. doi: 10.1093/cercor/bhs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, Li X-Y, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu R-J, Dwyer JM, Banasr M, Lee B, Son H, Li X-Y, Aghajanian G, Duman RS. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Alafuzoff I, Soininen H, Winblad B, Pei J-J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272:4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Inoki K, Guan K-L. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol. 2004;24:7965–7975. doi: 10.1128/MCB.24.18.7965-7975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Lu Y, Belin S, He Z. Signaling regulations of neuronal regenerative ability. Curr Opin Neurobiol. 2014;27C:135–142. doi: 10.1016/j.conb.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch NE, Lynch SA, McMenamin J, Webb D. Bannayan-Riley-Ruvalcaba syndrome: a cause of extreme macrocephaly and neurodevelopmental delay. Arch Dis Child. 2009;94:553–554. doi: 10.1136/adc.2008.155663. [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP, 3rd, Goldberg HJ, Young LR, Kinder BW, Downey GP, Sullivan EJ, Colby TV, McKay RT, Cohen MM, Korbee L, Taveira-DaSilva AM, Lee HS, Krischer JP, Trapnell BC. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–1606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell. 2014;156:771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H, Chiang ACY, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- Mohamed AR, Bailey CA, Freeman JL, Maixner W, Jackson GD, Harvey AS. Intrinsic epileptogenicity of cortical tubers revealed by intracranial EEG monitoring. Neurology. 2012;79:2249–2257. doi: 10.1212/WNL.0b013e3182768923. [DOI] [PubMed] [Google Scholar]
- Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG, Jr, Guan KL. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009;9:362–374. doi: 10.1016/j.cmet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, Sahin M. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci. 2010;13:163–172. doi: 10.1038/nn.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Normand EA, Crandall SR, Thorn CA, Murphy EM, Voelcker B, Browning C, Machan JT, Moore CI, Connors BW, Zervas M. Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron. 2013;78:895–909. doi: 10.1016/j.neuron.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O'Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro N, Takahashi R, Yoshino K-i, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem. 2007;282:20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang S-C, Chubykin AA, Sidorov M, Bianchi R, Wong RKS, Bear MF. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77:243–250. doi: 10.1016/j.neuron.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Huang X, Boudeau J, Pawłowski R, Wullschleger S, Deak M, Ibrahim AFM, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J-J, Hugon J. mTOR-dependent signalling in Alzheimer's disease. J Cell Mol Med. 2008;12:2525–2532. doi: 10.1111/j.1582-4934.2008.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Taquet M, Prohl AK, Scherrer B, van Eeghen AM, Prabhu SP, Sahin M, Warfield SK. Diffusion tensor imaging and related techniques in tuberous sclerosis complex: review and future directions. Future Neurol. 2013;8:583–597. doi: 10.2217/fnl.13.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105:1607–1616. doi: 10.1093/jnci/djt277. [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, Richards DA, Holland KD, Danzer SC. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. 2012;75:1022–1034. doi: 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, Taillon BE, Bouffard P, Kwiatkowski DJ. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. 2010;20:1096–1105. doi: 10.1111/j.1750-3639.2010.00416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo C, Salinas M, Alcázar A. Regulation of cap-dependent translation by insulin-like growth factor-1 in neuronal cells. Biochem Biophys Res Commun. 2002;291:560–566. doi: 10.1006/bbrc.2002.6479. [DOI] [PubMed] [Google Scholar]
- Raab-Graham KF, Haddick PCG, Jan YN, Jan LY. Activity-and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. 2006;314:144–148. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, Gambello MJ. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2013;51:93–103. doi: 10.1016/j.nbd.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Reith RM, Way S, McKenna J, Haines K, Gambello MJ. Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol Dis. 2011;43:113–122. doi: 10.1016/j.nbd.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci U S A. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière J-B, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GMS, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL, Majewski J, Bulman DE, O'Driscoll M, Shendure J, Graham JM, Boycott KM, Dobyns WB. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Escudero I, Oliver MD, Andrés-Pons A, Molina M, Cid VJ, Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet. 2011;20:4132–4142. doi: 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119:398–407. doi: 10.1111/j.1471-4159.2011.07435.x. [DOI] [PubMed] [Google Scholar]
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL, Tabrizi SJ. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–216. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Barrow RK, Blackshaw S, Burnett PE, Lai MM, Field ME, Bahr BA, Kirsch J, Betz H, Snyder SH. Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science. 1999;284:1161–1164. doi: 10.1126/science.284.5417.1161. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, Zonnenberg B, Verhoef S, Halley D, van den Ouweland A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13:731–741. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292. doi: 10.1038/ncomms2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, Licchetta L, Provini F, Bisulli F, Vadlamudi L, Gecz J, Connelly A, Tinuper P, Ricos MG, Berkovic SF, Dibbens LM. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–787. doi: 10.1002/ana.24126. [DOI] [PubMed] [Google Scholar]
- Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, Frank MG. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr Biol. 2012;22:676–682. doi: 10.1016/j.cub.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibt J, Frank MG. Translation regulation in sleep: Making experience last. Commun Integr Biol. 2012;5:491–495. doi: 10.4161/cib.21010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Tullet JMA, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson ICA, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- Siuta MA, Robertson SD, Kocalis H, Saunders C, Gresch PJ, Khatri V, Shiota C, Kennedy JP, Lindsley CW, Daws LC, Polley DB, Veenstra-Vanderweele J, Stanwood GD, Magnuson MA, Niswender KD, Galli A. Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol. 2010;8:e1000393. doi: 10.1371/journal.pbio.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell. 2007;28:721–729. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PloS one. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan R, McLaughlin MM, Cafferkey R, Johnson RK, Rosenberg M, Livi GP. Interaction between FKBP12-rapamycin and TOR involves a conserved serine residue. J Biol Chem. 1994;269:32027–32030. [PubMed] [Google Scholar]
- Sternson SM. Hypothalamic survival circuits: blueprints for purposive behaviors. Neuron. 2013;77:810–824. doi: 10.1016/j.neuron.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghibiglou C, Martin HGS, Lai TW, Cho T, Prasad S, Kojic L, Lu J, Liu Y, Lo E, Zhang S, Wu JZZ, Li YP, Wen YH, Imm J-H, Cynader MS, Wang YT. Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nat Med. 2009;15:1399–1406. doi: 10.1038/nm.2064. [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron. 2014 doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras A-C, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Thomanetz V, Angliker N, Cloëtta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Rüegg MA. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201:293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillema J-M, Leach JL, Krueger DA, Franz DN. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology. 2012;78:526–531. doi: 10.1212/WNL.0b013e318247ca8d. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Staying awake puts pressure on brain arousal systems. J Clin Invest. 2007;117:3648–3650. doi: 10.1172/JCI34250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–16987. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- Urbanska M, Gozdz A, Swiech LJ, Jaworski J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J Biol Chem. 2012;287:30240–30256. doi: 10.1074/jbc.M112.374405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst M, Verhoef S, Tempelaars A, Bakker L, Wang Q, Wessels M, Bakker R, Nellist M, Lindhout D, Halley D, van den Ouweland A. Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation. J Med Genet. 1999;36:285–289. [PMC free article] [PubMed] [Google Scholar]
- Vander Haar E, Lee S-I, Bandhakavi S, Griffin TJ, Kim D-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11:111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci. 2006;23:686–692. doi: 10.1111/j.1460-9568.2006.04594.x. [DOI] [PubMed] [Google Scholar]
- Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci. 2014;34:4453–4465. doi: 10.1523/JNEUROSCI.4311-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R, Japs B, Schneider M, de Vries PJ, Bartsch D. Epilepsy and Tsc2 haploinsufficiency lead to autistic-like social deficit behaviors in rats. Behav Genet. 2011;41:364–372. doi: 10.1007/s10519-010-9399-0. [DOI] [PubMed] [Google Scholar]
- Wang C, Yu J-T, Miao D, Wu Z-C, Tan M-S, Tan L. Targeting the mTOR signaling network for Alzheimer's disease therapy. Mol Neurobiol. 2014;49:120–135. doi: 10.1007/s12035-013-8505-8. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ, Cota D. Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu Rev Nutr. 2008;28:295–311. doi: 10.1146/annurev.nutr.28.061807.155505. [DOI] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron. 2012;75:425–436. doi: 10.1016/j.neuron.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BC, Zivraj KH, Holt CE. Local translation and mRNA trafficking in axon pathfinding. Results Probl Cell Differ. 2009;48:269–288. doi: 10.1007/400_2009_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zeng L-H, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Salemi J, Hou H, Zhu Y, Mori T, Giunta B, Obregon D, Tan J. Rapamycin promotes beta-amyloid production via ADAM-10 inhibition. Biochem Biophys Res Commun. 2010;398:337–341. doi: 10.1016/j.bbrc.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon C-H, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Parada LF. PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol. 2012;22:873–879. doi: 10.1016/j.conb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Zhou M, Li W, Huang S, Song J, Kim JY, Tian X, Kang E, Sano Y, Liu C, Balaji J, Wu S, Zhou Y, Zhou Y, Parivash SN, Ehninger D, He L, Song H, Ming G-L, Silva AJ. mTOR Inhibition ameliorates cognitive and affective deficits caused by Disc1 knockdown in adult-born dentate granule neurons. Neuron. 2013;77:647–654. doi: 10.1016/j.neuron.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011a;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011b;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]