

Summary
Because of their immunomodulatory properties, human bone marrow stromal cells (hBMSCs) represent promising stem cells for treatment of immune disorders. hBMSCs expansion precedes their clinical use, so the possibility that hBMSCs undergo spontaneous transformation upon long-term culture should be addressed. Whether hBMSCs retain immunosuppressive and anti-inflammatory properties upon oncogenic transformation remains unknown. Using sequentially mutated hBMSCs and spontaneously transformed hBMSCs, we report that, upon oncogenic transformation, hBMSCs lose immunosuppressive and anti-inflammatory properties in vitro and in vivo. Transcriptome profiling and functional assays reveal immune effectors underlying the loss of immunomodulation in transformed hBMSCs. They display a proinflammatory transcriptomic signature, with deregulation of immune and inflammatory modulators and regulators of the prostaglandin synthesis. Transformed hBMSCs lose their capacity to secrete the immunosuppressive prostacyclins prostaglandin E2 (PGE2) and PGI2 but produce proinflammatory thromboxanes. Together, the immunoregulatory profile adopted by hBMSCs largely depends on intrinsic genetic-molecular determinants triggered by genomic instability/oncogenic transformation.
Highlights
-
•
Oncogenic hBMSCs display robustly impaired immune properties
-
•
Transformed hBMSCs display a proinflammatory transcriptomic signature
-
•
Transformed hBMSCs lose capacity to secrete immunosuppressive prostacyclins
-
•
Transformed hBMSCs gain the capacity to produce proinflammatory thromboxanes
In this study, Menendez, Delgado, and colleagues demonstrate that human bone marrow stromal/mesenchymal stem cells lose their immune properties in vitro and in vivo upon oncogenic transformation by skewing its anti-inflammatory phenotype toward proinflammatory with a deregulation of γ interferon, tumor necrosis factor α, interleukin 2 (IL2), PGE2, PGI1, thromboxanes, IL10, IL8, and C-X-C motif chemokine 10. These data have implications not only in ex vivo expansion of hBMSC but also in microenvironment tumor biology.
Introduction
Human bone marrow stromal cells (hBMSCs) are a rare subset of bone marrow cells and constitute a promising source of multipotent progenitors for mesodermal tissues (Pittenger et al., 1999), being used worldwide in many clinical applications including tissue repair, treatment of graft-versus-host disease, and autoimmune diseases (García-Castro et al., 2008). The clinical potential of hBMSCs relies on key properties such as (1) multipotent differentiation, (2) long-term ex vivo expansion, (3) homing ability to damaged tissues, and (4) robust immunomodulatory properties (Bernardo and Fibbe, 2012, 2013; García-Castro et al., 2008). The mechanisms through which hBMSCs display reparative effects include the capacity to home to sites of damage, the ability to release anti-inflammatory factors, and the capacity to modulate immune responses (Bernardo and Fibbe, 2012; Marigo and Dazzi, 2011). hBMSCs secrete immunosuppressive factors including prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), transforming growth factor (TGF)-β, and nitric oxide (NO), thus modulating immune responses by inhibiting T cell activation and natural killer cell activity and inducing type II macrophage and dendritic cell differentiation and regulatory T cell (Bernardo and Fibbe, 2013; English, 2013; Herrero and Pérez-Simón, 2010; Ma et al., 2014; Yagi et al., 2010). However, it has been demonstrated that hBMSCs are not intrinsically immunoprivileged (Nauta et al., 2006), but they acquire immunosuppressive properties after exposure to an inflammatory environment (Prockop and Oh, 2012).
The immunosuppressive properties of allogeneic hBMSCs might be a double-edged sword. On one hand, they constitute the rationale for hBMSCs-based potential therapeutic approaches. On the other hand, they might enhance the ability of tumors to evade immune surveillance (Lazennec and Jorgensen, 2008; Momin et al., 2010). hBMSCs have been reported to inhibit or promote tumor growth, depending on yet undefined conditions (Momin et al., 2010; Stagg, 2008). Likewise, the experimental transformation of hBMSCs by different mechanisms gives rise to sarcoma formation in vivo, hence placing stromal mesenchymal stem cells as the cell of origin for certain sarcomas (Mohseny and Hogendoorn, 2011; Rodriguez et al., 2012). Practically, ex vivo expansion of stromal mesenchymal stem cells is a prerequisite for their clinical use (Barkholt et al., 2013) so that, when considering the use of ex vivo expanded hBMSCs, the possibility that they undergo senescence, genomic instability, and spontaneous transformation after long-term culture should be addressed (Barkholt et al., 2013; Estrada et al., 2013; Pan et al., 2014; Wang et al., 2005). Although in vitro spontaneous transformation seems rare, no information exists about the homeostasis of long-term cultured hBMSCs regarding the donor age, underlying disease, and source of stromal mesenchymal stem cells. Furthermore, although hBMSC-based clinical trials should represent the optimal source of evidence on the potential in vivo tumorigenic capacity of hBMSCs, current trials rarely focused on parameters relevant for assessing the transformation potential of allogeneic hBMSCs because they rarely evaluate long-term safety and efficacy of mesenchymal stem cells (MSCs) (Mishra et al., 2009; Momin et al., 2010). Additionally, stromal mesenchymal stem cells exposed to the tumor milieu could differentiate into carcinoma-associated fibroblasts, enhancing tumor growth (Mishra et al., 2009; Momin et al., 2010). Together, although it is an important concern for realizing the full clinical expectative of hBMSC, the oncogenic potential of hBMSCs remains poorly explored. Consequently, whether hBMSCs retain differentiation and immunosuppressive and anti-inflammatory properties upon oncogenic transformation remains unknown. Here, we take advantage of a collection of sequentially mutated hBMSCs ranging from wild-type to fully transformed hBMSCs (targeted with up to six oncogenic mutations; Funes et al., 2007; Rodriguez et al., 2013) to address whether hBMSCs at different stages of a well-characterized oncogenic process (normal, immortalized, and transformed; Funes et al., 2007; Rodriguez et al., 2013) retain immunomodulatory properties in vitro and in vivo. We describe an oncogenic-transformation-associated loss of the immunosuppressive and anti-inflammatory properties by hBMSCs and identify candidate immune effectors underlying this loss of immunomodulation capacity. These data have enormous implications not only in ex vivo expansion of hBMSCs but also in microenvironment tumor biology.
Results
Impaired In Vitro Homeostasis of Transformed hBMSC
We have very recently developed and characterized sarcoma models using several sequentially mutated hBMSCs (Funes et al., 2007; Rodriguez et al., 2013). This collection of hBMSCs ranges from wild-type (WT) (hBMSC-0H) to fully transformed hBMSC (Figure 1A; Funes et al., 2007; Rodriguez et al., 2013). The combination of oncogenic hits include p53 inactivation (hBMSC-1H), hBMSC-1H plus Rb inactivation and hTERT overexpression (hBMSC-3H), hBMSC-3H plus c-myc stabilization (hBMSC-4H), and hBMSC-4H plus H-RASv-12 (hBMSC-5H). In addition, the fusion oncogene FUS-CHOP was ectopically expressed in all the hBMSC genotypes (Funes et al., 2007; Rodriguez et al., 2013). Figure 1A summarizes the main features and tumorogenic potential of all the different hBMSCs. Briefly, hBMSCs harboring less than three oncogenic hits are nonimmortalized; hBMSC-3H-GFP, hBMSC-3H-FUS-CHOP (FC), and hBMSC-4H-GFP are immortalized, but not transformed; and hBMSC-4H-FC, hBMSC-5H-GFP, and hBMSC-5H-FC are transformed and originate sarcomas in vivo (Funes et al., 2007; Rodriguez et al., 2013). All hBMSC types, regardless of the number/nature of oncogenic hits, display a typical hBMSC phenotype (Menendez et al., 2009; Table S1 and Figure S1 available online). As expected, immortalized and transformed hBMSCs grow in vitro much faster than nonimmortalized hBMSCs (∼22, ∼28, and ∼4 doubling populations in 30 days, respectively; Figure 1B). Additionally, sequential acquisition of oncogenic hits in hBMSCs impairs their adipogenic differentiation ability but does not compromise osteogenic potential (Figures S2A and S2B). The accumulation of oncogenic events in transformed hBMSC-5H cells induce striking alterations in the expression of many genes involved in adipogenic differentiation, and consequently, these cells fail to activate the master regulators of adipogenic differentiation peroxisome proliferator-activated receptor γ and CCAAT/enhancer-binding protein alpha (Rodriguez et al., 2013). Thus, transformation of primary hBMSC is coupled to a differentiation impairment and proliferation advantage, hallmark properties of oncogenesis.
Figure 1.
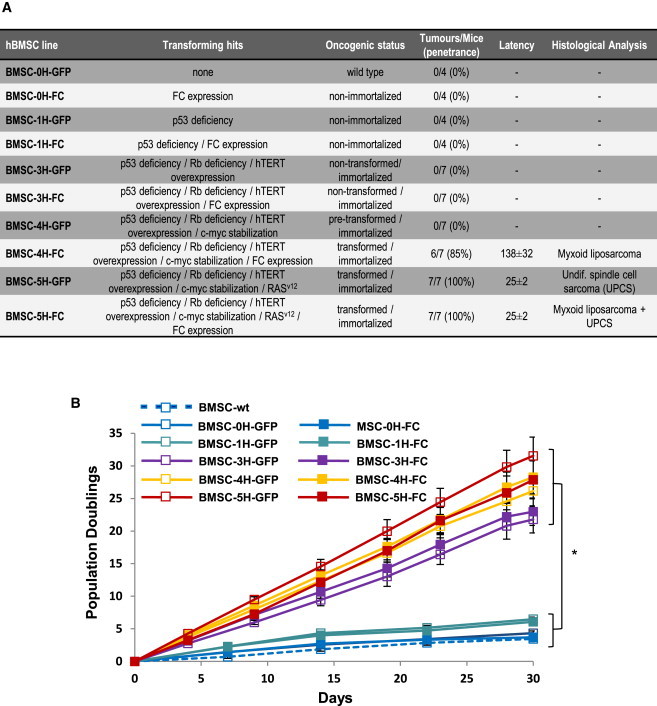
Transforming Mutations, Tumorogenic Potential, and In Vitro Growth Kinetics of the Indicated hBMSC Cultures
(A) Summary of the main features of the sequentially mutated collection of hBMSCs. Data on the in vivo tumorogenic potential (tumor penetrance, latency, and histological analysis) have been previously reported (Rodriguez et al., 2013). The latency represents the number of days until 1 cm3 tumor is observed.
(B) Proliferation was measured as cumulative population doublings in three independent triplicates. ∗p value immortalized/transformed hBMSCs versus nonimmortalized hBMSCs at day 30 < 0.01.
Transformed hBMSCs Lack Immunosuppressive and Anti-inflammatory Properties In Vitro and In Vivo
It remains elusive whether transformed hBMSCs retain immunomodulatory properties. We investigated the ability of immortalized and transformed hBMSCs to inactivate T cell responses and to inhibit inflammatory responses. The potential immunomodulatory activity of immortalized (hBMSC-3H) and transformed (hBMSC-4H and hBMSC-5H) hBMSCs was compared with WT-hBMSC (hBMSC-0H) and nonimmortalized hBMSC (hBMSC-1H). The addition of hBMSC-0H, hBMSC-1H, and hBMSC-3H hBMSCs to mixed lymphocyte culture (MLC) of peripheral blood mononuclear cells (PBMCs) from different donors significantly reduced the number of total cells in the culture and specifically decreased the number of cycling CD4 T cells (Figure 2A), suggesting they were efficient inhibiting the proliferative response of activated T cells. Because they lack class II major histocompatibility complex (MHC) and CD80 and CD40 costimulatory molecules, hBMSC-0H/ hBMSC-1H/ hBMSC-3H did not stimulate the proliferation of allogeneic PBMCs, supporting their “immune-privilege” status. On the other hand, hBMSC-4H and hBMSC-5H failed to inhibit cell proliferation in the MLC assays (Figure 2A). Moreover, hBMSC-0H/hBMSC-1H/hBMSC-3H significantly inhibited the production of the Th1-cytokines interleukin 2 (IL-2), interferon γ (IFNγ), and tumor necrosis factor α (TNF-α) on the allogeneic MLCs (Figure 2B). The immunosuppressive effects of hBMSC-0H/ hBMSC-1H/ hBMSC-3H were observed on allogeneic, but not on syngeneic, reactions (Figures 2A and 2B), indicating the requirement of activated immune responses. In contrast, hBMSC-4H and hBMSC-5H failed to decrease the production of Th1 cytokines by activated lymphocytes (Figure 2B). The observed effects were not a consequence of induced cell mortality, because the hBMSCs did not affect the apoptosis/survival of PBMCs on the MLCs.
Figure 2.
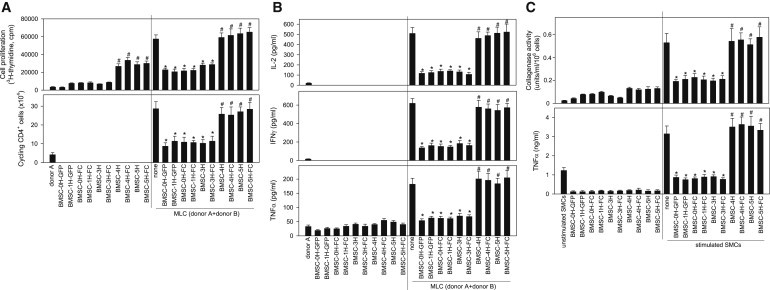
Transformed hBMSCs Lose Immunosuppressive and Anti-inflammatory Properties In Vitro
(A) MLCs established by coculturing responder PBMCs from donor A and stimulator PBMCs from donor B were treated with the indicated hBMSCs (right side of each panel). Cultures of PBMCs from donor A (syngeneic) alone and cultures of hBMSCs alone were used as basal controls (left side of each panel). Upper panel: proliferation was determined by measuring [3H]thymidine incorporation after 96 hr. Lower panel: responder PBMCs from donor B were CFSE labeled before being added to culture, and the number of cycling (CFSEmild/low) CD4 cells was determined by flow cytometry after 96 hr culture. ∗p value < 0.001 versus MLC without hBMSCs; #p value < 0.001 versus hBMSC-0H-treated cultures (n = 3 independent experiments; two-way ANOVA).
(B) Transformed hBMSCs fail to inhibit the production of Th1 cytokines by activated lymphocytes. MLCs were established and treated as described in (A). Cytokine contents were determined by ELISA after 48 hr culture. ∗p value < 0.001 versus MLC without hBMSCs; #p value < 0.001 versus hBMSC-0H-treated cultures (n = 3 independent experiments; two-way ANOVA).
(C) Transformed hBMSCs do not inhibit the inflammatory response in SMCs from RA patients. SMCs isolated from two RA patients were stimulated with lipopolysaccharide (for cytokine determination) or TNF-α (for collagenase activity assay) in the absence or presence of the indicated hBMSCs. Culture supernatants were assayed for collagenase activity (after 24 hr) or TNF-α contents (after 48 hr). ∗p value < 0.001 versus SMCs alone; #p value < 0.01 versus BMSC-0H-treated cultures.
We next investigated the capacity of the hBMSCs to regulate the inflammatory response of resident cells of the synovial membrane in patients with active rheumatoid arthritis (RA). Synovial-membrane-derived cells (SMCs) isolated from RA patients were cultured in the presence of hBMSCs and assayed for the production of the inflammatory mediator TNF-α and matrix-degrading enzymes (collagenase activity). Again, hBMSC-0H, hBMSC-1H, and hBMSC-3H treatment of RA SMC cultures decreased the secretion of TNF-α after stimulation with lipopolysaccharide (LPS) and reduced the collagenase activity after stimulation with TNF-α (Figure 2C). However, hBMSC-4H and hBMSC-5H did not inhibit the inflammatory response in RA SMCs (Figure 2C). Together, these data reveal that hBMSCs lose immunosuppressive and anti-inflammatory properties upon oncogenic transformation.
The lack of in vitro immunosuppressive and anti-inflammatory properties of transformed hBMSCs prompted us to analyze their in vivo properties in a 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced mouse model of inflammatory bowel disease that displays clinical, histopathological, and immunological features of human Crohn’s disease (Bouma and Strober, 2003; González et al., 2009; Sánchez et al., 2011). In both Crohn’s disease and our mouse model, activated Th1 and Th17 cells promote an exaggerated macrophage and neutrophil infiltration and activation, inducing a prolonged severe transmural inflamed intestinal mucosa, characterized by uncontrolled production of inflammatory cytokines and chemokines (Bouma and Strober, 2003; González et al., 2009; Sánchez et al., 2011). Inflammatory mediators such as cytokines and free radicals, produced by infiltrating cells and resident macrophages, play a critical role in colonic tissue destruction. TNBS-treated mice developed a severe illness characterized by bloody diarrhea, rectal prolapse, pancolitis accompanied by extensive wasting syndrome, and profound and sustained weight loss resulting in 50% mortality (Figure 3A). Macroscopic examination of colons showed striking hyperemia, inflammation, and necrosis (Figures 3B and 3C). However, mice treated with hBMSC-0H, hBMSC-1H, or hBMSC-3H displayed an increased survival rate, rapidly recovered body weight loss, improved the wasting disease, regained a healthy appearance, and only showed slight signs of colon inflammation, similar to control mice treated with vehicle (Figures 3A–3C). Histological examination of the colons showed that hBMSC-0H/hBMSC-1H/hBMSC-3H-treatment reduced the TNBS-induced transmural inflammation, depletion of mucin-producing goblet and epithelial cells, disseminated fibrosis, focal loss of crypts, and infiltration of inflammatory cells (Figure 3D). However, in line with the in vitro observations, treatment with transformed hBMSCs (hBMSC-4H and hBMSC-5H) failed to protect against experimental colitis in vivo (Figure 3), demonstrating that transformed hBMSCs lose their immunosuppressive and anti-inflammatory properties both in vitro and in vivo.
Figure 3.
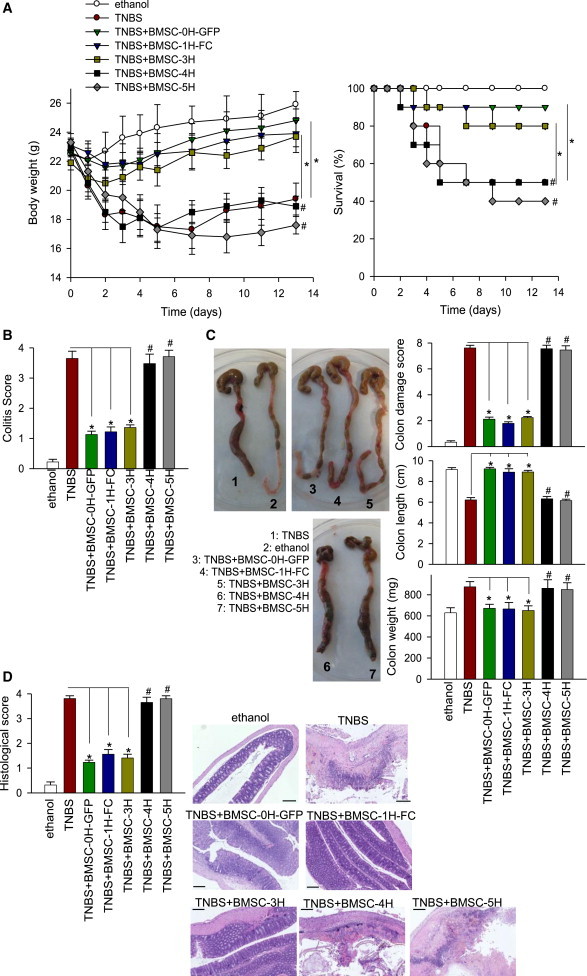
Treatment with Transformed hBMSCs Does Not Protect against Experimental Colitis In Vivo
Colitis was induced by intracolonic administration of TNBS (3 mg/mouse dissolved in 50% ethanol). Mice (n = 10 per group) were treated i.p. with the indicated hBMSCs (106), 8 hr after TNBS injection. Control mice received 50% ethanol.
(A and B) Clinical evolution was monitored by body weight changes and survival (A) as well as measuring the colitis score (B).
(C) Colon length and weight and macroscopic colonic damage score were evaluated at day 4.
(D) Histopathology was determined 4 days after cell infusion (four mice per group). No peritoneal tumors derived from transformed hBMSCs were observed. The scale bar represents 200 μm. ∗p value < 0.001 versus TNBS colitic mice (two-way ANOVA); #p value < 0.001 versus BMSC-0H-treated mice (paired Student’s t test).
Additionally, we investigated whether hBMSCs that acquired spontaneous transformation (Pan et al., 2014) also lose their immunomodulatory capacities. We observed that, similarly to the experimentally generated hBMSC-5H, three different lines of spontaneously transformed hBMSCs (Sp-T-BMSC-1-3) failed to suppress allogeneic T cell responses in vitro (Figure 4A) and the progression of experimental colitis in vivo (Figure 4B). These data with “clinical” hBMSCs that underwent spontaneous transformation validate the data on experimentally induced transformed hBMSCs, indicating that the loss of immune properties by hBMSCs upon oncogenic transformation may be a hallmark of transformed hBMSCs.
Figure 4.
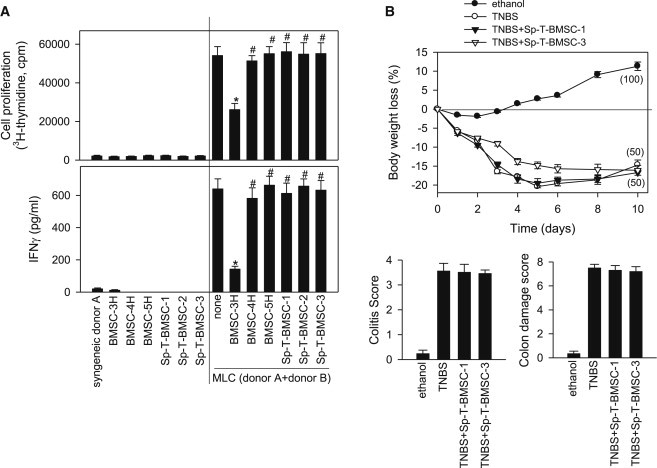
Spontaneously Transformed hBMSCs Lose Their Immunosuppressive Properties In Vitro and In Vivo
(A) MLCs were established by coculturing responder PBMCs from donor A and stimulator PBMCs from donor B. The indicated mitomycin-C-treated hBMSCs (BMSC-3H, BMSC-4H, and BMSC-5H) or mitomycin-C-treated Sp-T-BMSCs were added to the MLCs (right side of each panel). Cultures of PBMCs from donor A (syngeneic) and of mitomycin-C-treated hBMSCs and Sp-T-BMSCs alone were used as basal controls (left side of each panel). Upper panel: proliferation was determined by measuring [3H]thymidine incorporation after 96 hr. Lower panel: IFNγ contents were determined by ELISA after 48 hr culture. ∗p value < 0.001 versus MLC without BMSCs; #p value < 0.001 versus BMSC-3H-treated cultures (n = 3 independent experiments; two-way ANOVA).
(B) Colitis was induced by intracolonic administration of TNBS (3 mg/mouse dissolved in 50% ethanol). Mice (n = 10 per group) were treated i.p. with medium (TNBS group) or with two different lines of Sp-T-BMSCs (106) 12 hr after TNBS injection. Control mice (n = 5) received 50% ethanol. Clinical evolution was monitored by measuring the daily body weight loss (numbers in parentheses correspond to survival rates for each group), the colitis score (at day 4), and the macroscopic colonic damage score (at day 10).
Identification of Candidate Immune Effectors Underlying the Loss of Immunomodulation in Transformed hBMSCs
hBMSCs secrete immunosuppressive factors such as PGE-2, IDO-derived products, TGF-β, and NO, which modulate adaptive and innate immune responses (Bernardo and Fibbe, 2013; English, 2013; Herrero and Pérez-Simón, 2010; Ma et al., 2014; Yagi et al., 2010). To identify patterns of gene expression that could explain molecularly the lack of immunomodulatory properties by transformed hBMSCs, we performed a gene-expression profiling (GEP) comparing normal and transformed hBMSCs. Ingenuity Pathway Analysis (IPA) of genes differentially expressed between hBMSC-5H-GFP and hBMSC-0H-GFP revealed that many immunological pathways were significantly altered in hBMSC-5H-GFP cells (Figure 5A). Pathways such as the complement system, IL-6 and IL-10 signaling, dendritic cell maturation, or helper T cell differentiation are involved in the activation of the hBMSCs immunosuppressive properties (Chen et al., 2011; English, 2013; Yagi et al., 1998). Similarly, several disease-associated immunological pathways, mainly related to the proinflammatory cytokine IL-17, were altered in transformed hBMSC-5H-GFP cells (Figure 5A).
Figure 5.
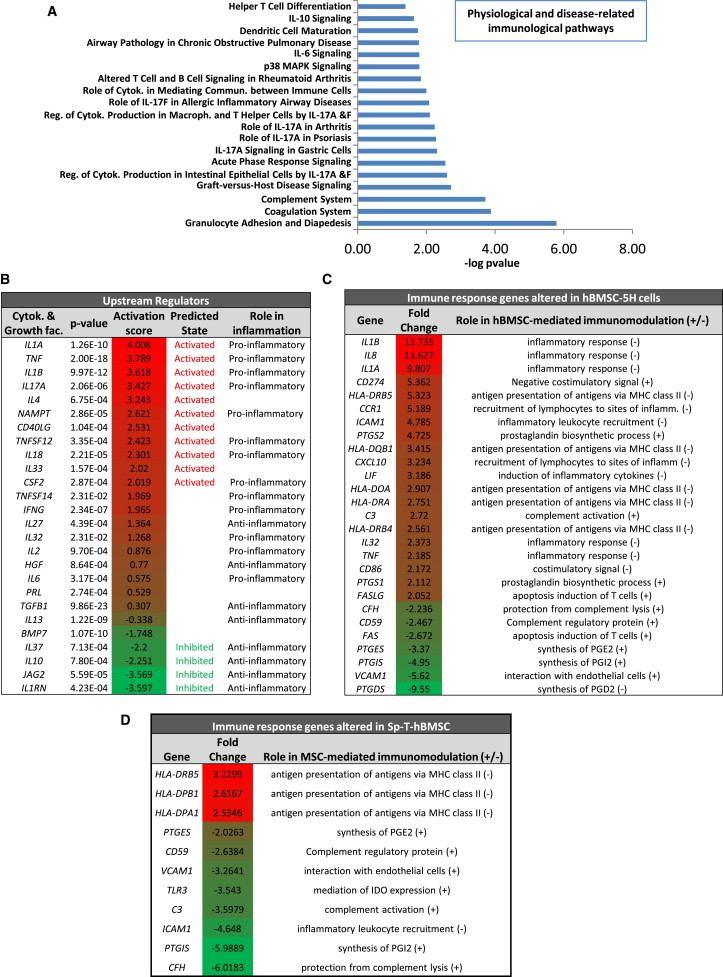
Gene-Expression Profiling Revealed an Impaired Immune Response Signaling in Transformed hBMSCs
Genes differentially expressed (p value < 0.05; regulation ≥ 2-fold) in hBMSC-5H versus hBMSC-0H were analyzed using the IPA software.
(A) Representation of the most significantly altered immunological pathways in hBMSC-5H.
(B) List of the cytokine and growth-factor-signaling pathways most significantly altered in hBMSC-5H cells. The corresponding activation Z scores are indicated and highlighted in red (activation)/green (inhibition) color scale. A regulator-signaling pathway is predicted to be active or inhibited if their Z score is higher than 2 or lower than −2, respectively. Whether each regulator functions as pro- or anti-inflammatory factor is indicated.
(C) List of genes involved in the induction (+) or inhibition (−) of immunosuppression altered in MSC-5H cells. The fold change expression is highlighted using red (activation)/green (inhibition) color scale.
(D) List of genes involved in the induction (+) or inhibition (−) of immunosuppression altered in Sp-T-hBMSC-1 as compared to syngeneic nontransformed hBMSCs. The fold change expression is highlighted using red (activation)/green (inhibition) color scale.
To identify regulators that could explain the gene-expression pattern observed in transformed hBMSC-5H-GFP, we looked for upstream regulators using the IPA software. We found a robust tendency of transformed hBMSC-5H-GFP cells toward activation of proinflammatory cytokines/growth-factors-mediated signaling pathways and inhibition of anti-inflammatory-molecules-driven signaling (Figure 5B). In addition, many regulators of hBMSCs immune properties were significantly altered in hBMSC-5H-GFP (Figure 5C). These include regulators of the prostaglandin synthesis (upregulation of prostaglandin synthase 1 [PTGS1] and PTGS2 and downregulation of prostaglandin E synthase [PTGES], prostaglandin I2 synthase [PTGIS], and prostaglandin D2 synthase [PTGDS]), inflammatory cytokines (upregulation of IL1B, IL8, IL1A, leukemia inhibitory factor [LIF], IL32, and TNF), complement system (upregulation of C3 and downregulation of CFH and CD59), MHC class II and costimulatory factors (upregulation of several MHC class II genes, CD274, and CD86), and other genes controlling the recruitment and proliferation of lymphocytes (upregulation of C-C chemokine receptor type 1 [CCR1], intracellular adhesion molecule 1 [ICAM1], C-X-C motif chemokine 10 [CXCL10], and LIF; Abdi et al., 2008; Bernardo and Fibbe, 2013; Chen et al., 2011; Dorris and Peebles, 2012; English, 2013; Herrero and Pérez-Simón, 2010; Iyer and Rojas, 2008; Kim et al., 2005; Ma et al., 2014; Nasef et al., 2008; Sandig et al., 2007; Yagi et al., 2010). Similarly, many positive regulators of immunomodulation like PTGES and PTGIS; the complement system factors C3, CFH, and CD59; the cell-adhesion molecule vascular cell adhesion protein 1, or the Toll-like receptor 3 (English, 2013), which mediate signaling leading to IDO activation, were downregulated in Sp-T-hBMSCs as compared to their parental nontransformed hBMSCs (Figure 5D). Also, Sp-T-hBMSCs displayed a downregulation of the signaling pathways controlled by TGF-β1 or IL-10 (data not shown) whereas MHC class II molecules were upregulated in Sp-T-hBMSCs (Figure 5D). Together, all these gene-expression data confirm that transformed hBMSCs display a proinflammatory rather than an anti-inflammatory transcriptomic signature and both experimentally and spontaneously transformed hBMSCs show a deregulation of key immune and inflammatory modulators.
We finally determined whether the transcriptional signature of transformed hBMSCs correlates with a different secretory profile of key immunoregulatory mediators. As expected, hBMSC-5H-GFP consistently produced more proinflammatory chemokines (CXCL10 and IL-8) than hBMSC-0H-GFP (Figure 6). Interestingly, transformation of hBMSCs did not affect the release of NO (Figure 6). As previously described (Gonzalez-Rey et al., 2010), WT-hBMSCs gained the capacity to secrete IL-10 following their stimulation with PBMCs; however, transformed hBMSC-5H-GFP failed to produce this anti-inflammatory cytokine under any condition (Figure 6). Similarly, hBMSC-5H-GFP lost their capacity to secrete immunosuppressive prostanoids, such as PGE2 and 6-keto-PGF1a, whereas they gained the capacity to produce proinflammatory thromboxanes (thromboxanes B2 and A2; Figure 6).
Figure 6.
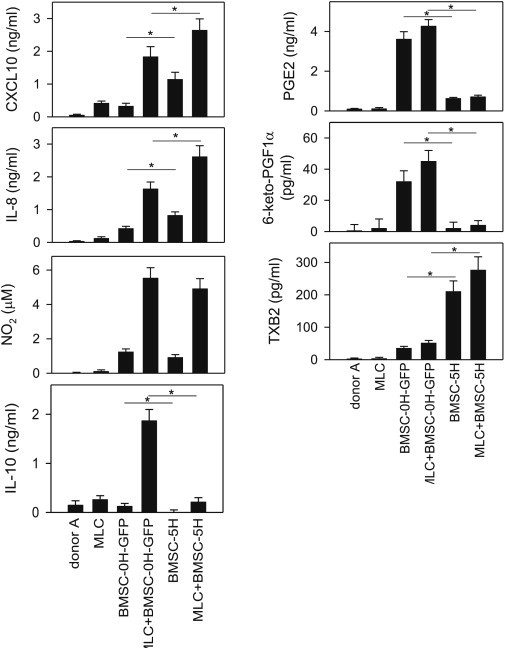
hBMSC Production of Candidate Immune Effectors Underlying the Loss of Immunomodulation in Transformed BMSCs
MLCs were established by coculturing PBMCs isolated from two mismatched donors. Cultures of PBMCs from donor A were used as basal syngeneic controls. hBMSC-0H and hBMSC-5H were added to the MLCs, and the production of CXCL10, IL-8, PGE2, IL-10, 6-keto-PGF1α, and TXB2 was determined in supernatants by ELISA after 48 hr culture. Levels of nitrite were determined using Griess reagent to measure indirectly the secretion of NO in culture supernatants. ∗p value < 0.001 (n = 6 independent experiments; paired t test student).
Discussion
Because of their robust immunomodulatory properties, hBMSCs represent a promising source of adult stem cells for tissue repair and treatment of many immune disorders (Bernardo and Fibbe, 2012, 2013; García-Castro et al., 2008). hBMSCs have the capacity to home to sites of damage and release anti-inflammatory factors modulating immune responses (Bernardo and Fibbe, 2012; Marigo and Dazzi, 2011). Ex vivo expansion of hBMSCs is a prerequisite for their clinical use (Barkholt et al., 2013), so the possibility that hBMSCs undergo senescence, genomic instability, and spontaneous transformation after long-term culture should be addressed when considering the use of ex vivo expanded hBMSCs (Barkholt et al., 2013; Estrada et al., 2013; Pan et al., 2014; Wang et al., 2005). We have recently reported how senescence induces phenotypic changes in hBMSCs and abrogates their protective activity in murine models of sepsis and graft-versus-host disease (Sepúlveda et al., 2014). However, the transformation potential of hBMSCs remains poorly explored and whether hBMSCs retain differentiation and immunomodulatory properties upon oncogenic transformation remains unknown. Here, we have harnessed a collection of sequentially mutated hBMSCs (Rodriguez et al., 2013) to report that, upon oncogenic transformation, hBMSCs lose their immunosuppressive and anti-inflammatory properties. This was observed in hBMSCs carrying oncogenic events of distinct nature, suggesting that the loss of immunomodulatory properties seems independent of “specific” oncogenic drivers. The reduced anti-inflammatory and immunomodulatory capacity of transformed hBMSCs does not seem to be a consequence of their cell division history, as hBMSC-4H require much-longer latency (138 days) than highly proliferative hBMSC-5H (25 days) to originate in vivo tumors, while displaying similar degree of immune deficiency. More importantly, these studies were validated in primary hBMSCs spontaneously transformed after long-term culture (Pan et al., 2014), which also failed to suppress allogeneic T cell responses in vitro and the progression of experimental colitis in vivo. These data indicate that the loss of immune properties by hBMSCs upon oncogenic transformation may be a hallmark of transformed hBMSCs.
It remains elusive how the complex transcriptional machinery triggered by transformation causes the loss in the immunoregulatory activity of hBMSCs. GEP identified various immune genes differentially expressed in transformed hBMSCs. Transformation of hBMSCs caused upregulation of proinflammatory cytokines and chemokines, such as TNF-α, IL1, IL8, IL-32, CXCL10, and LIF, and proinflammatory receptors/adhesion molecules such as ICAM and CCR1. In contrast, transformed hBMSCs failed to produce IL-10, an anti-inflammatory cytokine critically involved in the immunosuppressive action of hBMSC (Anderson et al., 2013; Gonzalez-Rey et al., 2010). Moreover, transformed hBMSCs upregulate the expression of human leukocyte antigen (HLA) class II and costimulatory (CD86) molecules. It is generally accepted that absence of MHC class II and costimulatory molecules is a characteristic phenotype of MSCs and one of the mechanisms underlying their immunosuppressive activities (Uccelli et al., 2008). Thus, transformed hBMSCs expressing MHC class II molecules and CD86 and secreting inflammatory cytokines and chemokines will favor the activation of T lymphocytes. Importantly, transformation of hBMSCs also induced important alterations in the synthesis pathway of prostanoids. Whereas transformed hBMSCs showed increased expression in COX2 (PTGS2) and COX1 (PTGS1), early acting enzymes in the prostanoids synthesis pathway, they displayed a reduced expression of PTGES, PTGIS, and PTGDS, enzymes downstream COX involved in the synthesis of PGE2, PGI2, and PGD2, respectively (Nicolaou et al., 2014). As a result, transformed hBMSCs produced very low levels of PGE2 and PGI2, as compared to normal hBMSCs. PGE2 is a critical mediator of the immunoregulatory activity of hBMSCs (Bernardo and Fibbe, 2013; Uccelli et al., 2008). Our study reports the production of PGI2 by normal hBMSCs. This finding is functionally relevant because prostacyclins are widely recognized as potent immunosuppressors (Nicolaou et al., 2014). As a consequence of the inhibition of the PTGES, PTGIS, and PTGDS, the activation of COX in transformed hBMSCs is shifted to the production of TXB2, which exerts potent inflammatory effects in several pathologies (Nicolaou et al., 2014).
Our data indicate that common oncogenic mutations sequentially induce the loss of the immunomodulatory capacity of hBMSCs by preventing the production of immunoregulatory prostacyclins and inducing a proinflammatory profile. This implies an important functional consequence. It is recognized that tissue resident hBMSCs function as sentinels of the microenvironment signals (inflammatory mediators or regulatory macrophages) and exert pro- or anti-inflammatory actions accordingly (Bernardo and Fibbe, 2013). We propose that, beside these extrinsic environmental signals, the immunoregulatory profile adopted by hBMSCs would largely depend on intrinsic genetic-molecular determinants triggered by senescence (Sepúlveda et al., 2014) and oncogenic transformation. Additionally, our data shed light onto the concept of hBMSC-mediated surveillance of immune response in the tumor because, upon oncogeneic transformation, hBMSCs lose their capacity to impair T cell and inflammatory responses. Overall, these data have enormous implications not only in ex vivo expansion of hBMSCs but also in microenvironment tumor biology.
Experimental Procedures
Generation and Culture of Mutated and Spontaneously Transformed hBMSCs
Wild-type hBMSCs (hBMSC-WT or hBMSC-0H) were obtained from Inbiobank. MSCs depleted of p53 (hBMSC-1H) were generated by lentiviral transduction with a p53-small hairpin RNA expression vector (pLVUH-shp53) as described (Rodriguez et al., 2011, 2013). The hBMSCs carrying three, four, or five different oncogenic hits (hBMSC-3H, hBMSC-4H, and hBMSC-5H) were developed and characterized elsewhere (Funes et al., 2007; Rodriguez et al., 2013). Briefly, hBMSCs were sequentially infected with retroviral particles carrying the following expression vectors: pBABE-puro-EST2 (hTERT expression) and pLXSN-neo-E6E7 (inactivation of p53 and Rb mediated by E6 and E7 antigens of human papillomavirus 16) to generate MSC-3H cells, pBABE-zeo-ST (introduction of the SV40 small T antigen to inactivate the protein phosphatase 2 phosphatase leading to c-myc stabilization) to generate MSC-4H cells, and pWZL-hygro-RasV12 (expression of oncogenic H-RASv-12) to generate MSC-5H cells. Following serial retroviral infections, drug selection with puromycin (1 μg/ml), neomycin (300 μg/ml), Zeocin (50 μg/ml), and hygromycin (100 μg/ml), respectively, was used to purify cell populations. Figure 1A summarizes the transforming hits and tumorogenic potential of each hBMSC genotype used in this study (Rodriguez et al., 2013). To overexpress FC, a hallmark fusion oncogene associated to human mixoid liposarcoma, each type of hBMSC (hBMSC-0H to -5H) was infected with either pRRL-EF1α-PGK-GFP (empty vector; GFP) or pRRL-EF1α-FUS-CHOP-PGK-GFP (FC-expressing vector; FC), as previously reported (Rodriguez et al., 2011, 2013). When transduction efficiency was <80%, the transduced GFP+ fraction was fluorescence-activated cell sorting enriched using a FACSAria cell sorter. The resulting hBMSC-GFP and hBMSC-FC were cultured in Advanced Dulbecco’s modified Eagles’ medium (DMEM) plus 10% fetal bovine serum (FBS) (Rodriguez et al., 2011, 2013). Spontaneously transformed hBMSCs (Sp-T-hBMSCs) were obtained from a previous study (Pan et al., 2014). Briefly, 46 different batches of hBMSCs were long-term cultured in DMEM supplemented with 10% FBS. Two out of five hBMSC cultures and two out of 41 liver-derived MSC cultures underwent a spontaneous genomic and functional oncogenic transformation. Transformed hBMSCs can form tumor in immunodeficient mice. Both primary transformed hBMSCs and hBMSCs retrieved from tumors formed in the mice were established as stable lines, which are used in this study (Pan et al., 2014).
In Vitro Growth Kinetics and Differentiation Assays
Growth kinetics were measured as cumulative population doublings (Rodriguez et al., 2011, 2013). An equal number of hBMSCs (2 × 105) between p6 and p12 were initially plated for each genotype, and cells were counted every 5 days and replated at 3 × 103 cells/cm2. For osteogenic and adipogenic differentiation assays (n = 3), 1 × 104 hBMSCs/cm2 were expanded in Advanced DMEM with 10% fetal calf serum. At confluence, medium was replaced with specific differentiation inductive medium. For adipogenic differentiation, cells were cultured in Adipogenic Differentiation Bullet Kit (Lonza) for 2 weeks. Differentiated cell cultures were stained with Oil Red O (Sigma). For osteogenic differentiation, cells were cultured in Osteogenic Differentiation Bullet Kit (Lonza) for 2 weeks. Differentiated cultures were stained with Alizarin Red S (Sigma; Rodriguez et al., 2011, 2013; Rubio et al., 2010, 2013; Sánchez et al., 2011).
Flow Cytometry Analysis
The immunophenotype of cultured hBMSCs was determined by flow cytometry using fluorochrome-conjugated monoclonal antibodies anti-CD90, CD73, CD105, CD44, CD166, CD106, CD45, CD34, HLA-DR, CD19, and CD14 (Miltenyi Biotec) as detailed elsewhere (Menendez et al., 2009; Sánchez et al., 2011).
T-Lymphocyte Proliferation and Cytokine Production
PBMCs were isolated from buffy coats from healthy volunteers by Ficoll-Hypaque gradients. MLCs were performed in 96-well, round-bottom plates by stimulating 105 responder PBMCs from donor A with 105 allogeneic HLA-mismatched mitomycin-C-treated stimulator PBMCs from donor B in complete medium (DMEM supplemented with 10% FBS, 20 mM L-glutamine, and 1% penicillin/streptomycin) in the absence or presence of the different genotypes of hBMSCs (2 × 104). In some experiments, hBMSCs and Sp-T-BMSCs were pretreated with mitomycin C (50 μg/ml; 20 min; 37°C) before being added to MLCs. Cells were pulsed with 5 μCi/well [3H]thymidine for the last 12 hr of a 96 hr culture and harvested onto membranes, and proliferation was determined by measuring [3H]thymidine uptake in a liquid scintillation counter. In similar experiments, responder PBMCs were labeled with 2.5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Molecular Probes) prior to setting up cocultures. After culture, cells were labeled with PerCP-conjugated anti-CD4 and fixed with 1% paraformaldehyde, and proliferating cells were determined by CFSE dilution in the CD4+ population. The number of cycling cells was calculated as the percent of CFSEmild/low cells that had divided by the total number of cells (Gonzalez-Rey et al., 2009; Sánchez et al., 2011). To determine cytokine production, MLCs were established in 24-well plates (in 1 ml) by stimulating 5 × 105 responder PBMCs from donor A with 5 × 105 allogeneic HLA-mismatched stimulator PBMCs from donor B in complete medium in the absence or presence of the different genotypes of hBMSCs (105). After 48 hr, levels of IL-2, CXCL10, IL-8, TNF-α, IL-10, and IFN-γ in the supernatants were determined by ELISA using capture/biotinylated detection antibodies from BD PharMingen and PrepoTech (Gonzalez-Rey et al., 2009; Sánchez et al., 2011). Levels of PGE2, thromboxane B2 (indirect determination of thromboxane A2), and 6-keto-PGF1a (indirect determination of prostacyclin PGI2) in supernatants (collected at 48 hr) were determined by ELISA kits purchased from Cayman Chemicals. The levels of NO in culture supernatants (at 48 hr) were determined indirectly by measuring the concentration of nitrite using the Griess reagent (Anderson et al., 2013).
Anti-inflammatory Studies
SMC cultures were established in 10% FBS/DMEM from synovial tissue obtained from two unrelated patients with active rheumatoid arthritis (RA). SMC cultures were conducted in RPMI supplemented with 8% heat-inactivated human serum, L-glutamine (20 mM), sodium pyruvate (1%), nonessential amino acids (1%), and penicillin/streptomycin (1%). We stimulated 2 × 105 SMCs with either LPS (1 μg/ml) or TNF-α (20 ng/ml) in the absence or presence of the different genotypes of hBMSCs (105), and after 24–48 hr, culture supernatants were assayed for TNF-α content and collagenase activity, respectively (Gonzalez-Rey et al., 2009; Sánchez et al., 2011). Collagenase activity in cell-free supernatants was determined using the EnzChek gelatinase/collagenase assay kit (Molecular Probes; Sánchez et al., 2011).
Induction of Experimental In Vivo Colitis
To induce in vivo colitis, 3 mg of TNBS (Sigma) in 50% ethanol (100 μl) was administered intrarectally in 7-week-old Bagg Albino/c male mice. Control mice received 50% ethanol alone. Animals were treated intraperitoneally (i.p.) with medium, with 106 BMSCs, or with 106 Sp-T-BMSCs 12 hr after TNBS instillation. Animals were monitored for the appearance of diarrhea, body weight loss, and survival. Colons were removed from the caecum to the anus, and colon length and weight were measured as markers of inflammation. Colons were evaluated for macroscopic damage (graded on a scale 0–10) based on criteria reflecting inflammation (hyperemia, bowel thickening, and ulceration extent). Scores for stool consistency and rectal bleeding were assessed as described (González et al., 2009). For histopathology analysis, a colon specimen was fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin-eosin (Sánchez et al., 2011). Inflammation was graded from 0 to 4 in a blinded fashion as described (González et al., 2009). The animal care committee of the University of Granada approved all mice protocols.
GEP
Exponentially growing wild-type (hBMSC-0H-GFP) and transformed (hBMSC-5H-GFP) hBMSCs were collected and stabilized in RNA later (Ambion) until RNA extraction. RNA was isolated using the Agilent Total RNA Isolation Kit and its quality checked in the Agilent 2100 Bioanalyzer. Total RNA samples were labeled with Cy3 using the Low-Input Quick Amp Labeling kit (Agilent; Barroso-delJesus et al., 2011). Samples were hybridized to Whole Human Genome 8x60K Microarray (G4851A), and arrays were scanned using an Agilent G2505B scanner. Each sample was labeled and hybridized as independent duplicates. Primary data were examined using GeneSpringGx 11.5 software (Silicon Genetics). Gene expression in the control and experimental groups was compared. Only genes satisfying the threshold of p value < 0.05 and a fold change expression >2 were included and assigned as significant. Analysis of pathways significantly altered in the experimental groups was performed using the Ingenuity Pathway software 8.0 (IPA; Ingenuity Systems; Bueno et al., 2013).
Statistical Analysis
All data are expressed as mean ± SEM. Statistical comparisons between experimental groups were performed with either a paired Student’s t test or Duncan’s multiple range test after two-way ANOVA. Statistical significance was defined as a p value < 0.05.
Author Contributions
P.M., M.D., and R.R. conceived the study, designed and performed experiments, analyzed data, and wrote the manuscript. A.H., M.R.-M., E.G.-R., and M.A.-B. performed and analyzed experiments. Q.P. provided key biological material/reagents and microarray data.
Acknowledgments
We thank Dr. Juan Funes and Prof. C. Boshoff (Cancer Research UK, London) for providing hBMSC-3H, -4H, and -5H cells. We thank Mar Roldán (GENyO) and Marta Caro (IPBLN-CSIC) for their technical assistance. This work was supported by the ISCIII/FEDER (PI10/00449 to P.M., CP11/00024—Miguel Servet—to R.R. and RTICC [RD12/0036/0015]), Excellence Grants from Junta de Andalucia (to M.D.), MINECO (SAF2013-43065 to P.M. and SAF2013-42946-R to R.R.), The Spanish Association Against Cancer and Fundación Sandra Ibarra (to P.M.), Health Canada (to P.M. and M.R.-M.), Grupo Español de Investigación en Sarcomas (J.M. Buesa-2012; to R.R.), and Obra Social Cajastur-IUOPA and Gobierno de Asturias (COF13-007; to R.R.). Q.P. was supported by the Netherlands Organization for Scientific Research (NWO/ZonMw) for a VENI grant (no. 916-13-032). P.M. also acknowledges the financial support from the Obra Social La Caixa-Fundaciò Josep Carreras.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
Mario Delgado, Email: mdelgado@ipb.csic.es.
Pablo Menendez, Email: pmenendez@carrerasresearch.org.
Accession Numbers
Microarray data have been deposited at Gene Expression Omnibus (GSE55108).
Supplemental Information
References
- Abdi R., Fiorina P., Adra C.N., Atkinson M., Sayegh M.H. Immunomodulation by mesenchymal stem cells: a potential therapeutic strategy for type 1 diabetes. Diabetes. 2008;57:1759–1767. doi: 10.2337/db08-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P., Souza-Moreira L., Morell M., Caro M., O’Valle F., Gonzalez-Rey E., Delgado M. Adipose-derived mesenchymal stromal cells induce immunomodulatory macrophages which protect from experimental colitis and sepsis. Gut. 2013;62:1131–1141. doi: 10.1136/gutjnl-2012-302152. [DOI] [PubMed] [Google Scholar]
- Barkholt L., Flory E., Jekerle V., Lucas-Samuel S., Ahnert P., Bisset L., Büscher D., Fibbe W., Foussat A., Kwa M. Risk of tumorigenicity in mesenchymal stromal cell-based therapies—bridging scientific observations and regulatory viewpoints. Cytotherapy. 2013;15:753–759. doi: 10.1016/j.jcyt.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Barroso-delJesus A., Lucena-Aguilar G., Sanchez L., Ligero G., Gutierrez-Aranda I., Menendez P. The Nodal inhibitor Lefty is negatively modulated by the microRNA miR-302 in human embryonic stem cells. FASEB J. 2011;25:1497–1508. doi: 10.1096/fj.10-172221. [DOI] [PubMed] [Google Scholar]
- Bernardo M.E., Fibbe W.E. Safety and efficacy of mesenchymal stromal cell therapy in autoimmune disorders. Ann. N Y Acad. Sci. 2012;1266:107–117. doi: 10.1111/j.1749-6632.2012.06667.x. [DOI] [PubMed] [Google Scholar]
- Bernardo M.E., Fibbe W.E. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Bouma G., Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- Bueno C., Ayllón V., Montes R., Navarro-Montero O., Ramos-Mejia V., Real P.J., Romero-Moya D., Araúzo-Bravo M.J., Menendez P. FLT3 activation cooperates with MLL-AF4 fusion protein to abrogate the hematopoietic specification of human ESCs. Blood. 2013;121:3867–3878. doi: 10.1182/blood-2012-11-470146. S1–S3. [DOI] [PubMed] [Google Scholar]
- Chen P.M., Yen M.L., Liu K.J., Sytwu H.K., Yen B.L. Immunomodulatory properties of human adult and fetal multipotent mesenchymal stem cells. J. Biomed. Sci. 2011;18:49. doi: 10.1186/1423-0127-18-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorris S.L., Peebles R.S., Jr. PGI2 as a regulator of inflammatory diseases. Mediators Inflamm. 2012;2012:926968. doi: 10.1155/2012/926968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol. Cell Biol. 2013;91:19–26. doi: 10.1038/icb.2012.56. [DOI] [PubMed] [Google Scholar]
- Estrada J.C., Torres Y., Benguría A., Dopazo A., Roche E., Carrera-Quintanar L., Pérez R.A., Enríquez J.A., Torres R., Ramírez J.C. Human mesenchymal stem cell-replicative senescence and oxidative stress are closely linked to aneuploidy. Cell Death Dis. 2013;4:e691. doi: 10.1038/cddis.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funes J.M., Quintero M., Henderson S., Martinez D., Qureshi U., Westwood C., Clements M.O., Bourboulia D., Pedley R.B., Moncada S., Boshoff C. Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc. Natl. Acad. Sci. USA. 2007;104:6223–6228. doi: 10.1073/pnas.0700690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Castro J., Trigueros C., Madrenas J., Pérez-Simón J.A., Rodriguez R., Menendez P. Mesenchymal stem cells and their use as cell replacement therapy and disease modelling tool. J. Cell. Mol. Med. 2008;12(6B):2552–2565. doi: 10.1111/j.1582-4934.2008.00516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González M.A., Gonzalez-Rey E., Rico L., Büscher D., Delgado M. Adipose-derived mesenchymal stem cells alleviate experimental colitis by inhibiting inflammatory and autoimmune responses. Gastroenterology. 2009;136:978–989. doi: 10.1053/j.gastro.2008.11.041. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rey E., Anderson P., González M.A., Rico L., Büscher D., Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut. 2009;58:929–939. doi: 10.1136/gut.2008.168534. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rey E., Gonzalez M.A., Varela N., O’Valle F., Hernandez-Cortes P., Rico L., Büscher D., Delgado M. Human adipose-derived mesenchymal stem cells reduce inflammatory and T cell responses and induce regulatory T cells in vitro in rheumatoid arthritis. Ann. Rheum. Dis. 2010;69:241–248. doi: 10.1136/ard.2008.101881. [DOI] [PubMed] [Google Scholar]
- Herrero C., Pérez-Simón J.A. Immunomodulatory effect of mesenchymal stem cells. Braz. J. Med. Biol. Res. 2010;43:425–430. doi: 10.1590/s0100-879x2010007500033. [DOI] [PubMed] [Google Scholar]
- Iyer S.S., Rojas M. Anti-inflammatory effects of mesenchymal stem cells: novel concept for future therapies. Expert Opin. Biol. Ther. 2008;8:569–581. doi: 10.1517/14712598.8.5.569. [DOI] [PubMed] [Google Scholar]
- Kim S.H., Han S.Y., Azam T., Yoon D.Y., Dinarello C.A. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22:131–142. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Lazennec G., Jorgensen C. Concise review: adult multipotent stromal cells and cancer: risk or benefit? Stem Cells. 2008;26:1387–1394. doi: 10.1634/stemcells.2007-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Xie N., Li W., Yuan B., Shi Y., Wang Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014;21:216–225. doi: 10.1038/cdd.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo I., Dazzi F. The immunomodulatory properties of mesenchymal stem cells. Semin. Immunopathol. 2011;33:593–602. doi: 10.1007/s00281-011-0267-7. [DOI] [PubMed] [Google Scholar]
- Menendez P., Catalina P., Rodríguez R., Melen G.J., Bueno C., Arriero M., García-Sánchez F., Lassaletta A., García-Sanz R., García-Castro J. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J. Exp. Med. 2009;206:3131–3141. doi: 10.1084/jem.20091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P.J., Mishra P.J., Glod J.W., Banerjee D. Mesenchymal stem cells: flip side of the coin. Cancer Res. 2009;69:1255–1258. doi: 10.1158/0008-5472.CAN-08-3562. [DOI] [PubMed] [Google Scholar]
- Mohseny A.B., Hogendoorn P.C. Concise review: mesenchymal tumors: when stem cells go mad. Stem Cells. 2011;29:397–403. doi: 10.1002/stem.596. [DOI] [PubMed] [Google Scholar]
- Momin E.N., Vela G., Zaidi H.A., Quiñones-Hinojosa A. The Oncogenic Potential of Mesenchymal Stem Cells in the Treatment of Cancer: Directions for Future Research. Curr. Immunol. Rev. 2010;6:137–148. doi: 10.2174/157339510791111718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasef A., Mazurier C., Bouchet S., François S., Chapel A., Thierry D., Gorin N.C., Fouillard L. Leukemia inhibitory factor: Role in human mesenchymal stem cells mediated immunosuppression. Cell. Immunol. 2008;253:16–22. doi: 10.1016/j.cellimm.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Nauta A.J., Westerhuis G., Kruisselbrink A.B., Lurvink E.G., Willemze R., Fibbe W.E. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood. 2006;108:2114–2120. doi: 10.1182/blood-2005-11-011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou A., Mauro C., Urquhart P., Marelli-Berg F. Polyunsaturated Fatty Acid-derived lipid mediators and T cell function. Front. Immunol. 2014;5:75. doi: 10.3389/fimmu.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q., Fouraschen S.M., de Ruiter P.E., Dinjens W.N., Kwekkeboom J., Tilanus H.W., van der Laan L.J. Detection of spontaneous tumorigenic transformation during culture expansion of human mesenchymal stromal cells. Exp. Biol. Med. (Maywood) 2014;239:105–115. doi: 10.1177/1535370213506802. [DOI] [PubMed] [Google Scholar]
- Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D., Moorman M.A., Simonetti D.W., Craig S., Marshak D.R. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Prockop D.J., Oh J.Y. Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol. Ther. 2012;20:14–20. doi: 10.1038/mt.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R., Rubio R., Gutierrez-Aranda I., Melen G.J., Elosua C., García-Castro J., Menendez P. FUS-CHOP fusion protein expression coupled to p53 deficiency induces liposarcoma in mouse but not in human adipose-derived mesenchymal stem/stromal cells. Stem Cells. 2011;29:179–192. doi: 10.1002/stem.571. [DOI] [PubMed] [Google Scholar]
- Rodriguez R., Rubio R., Menendez P. Modeling sarcomagenesis using multipotent mesenchymal stem cells. Cell Res. 2012;22:62–77. doi: 10.1038/cr.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R., Tornin J., Suarez C., Astudillo A., Rubio R., Yauk C., Williams A., Rosu-Myles M., Funes J.M., Boshoff C., Menendez P. Expression of FUS-CHOP fusion protein in immortalized/transformed human mesenchymal stem cells drives mixoid liposarcoma formation. Stem Cells. 2013;31:2061–2072. doi: 10.1002/stem.1472. [DOI] [PubMed] [Google Scholar]
- Rubio R., García-Castro J., Gutiérrez-Aranda I., Paramio J., Santos M., Catalina P., Leone P.E., Menendez P., Rodríguez R. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer Res. 2010;70:4185–4194. doi: 10.1158/0008-5472.CAN-09-4640. [DOI] [PubMed] [Google Scholar]
- Rubio R., Gutierrez-Aranda I., Sáez-Castillo A.I., Labarga A., Rosu-Myles M., Gonzalez-Garcia S., Toribio M.L., Menendez P., Rodriguez R. The differentiation stage of p53-Rb-deficient bone marrow mesenchymal stem cells imposes the phenotype of in vivo sarcoma development. Oncogene. 2013;32:4970–4980. doi: 10.1038/onc.2012.507. [DOI] [PubMed] [Google Scholar]
- Sánchez L., Gutierrez-Aranda I., Ligero G., Rubio R., Muñoz-López M., García-Pérez J.L., Ramos V., Real P.J., Bueno C., Rodríguez R. Enrichment of human ESC-derived multipotent mesenchymal stem cells with immunosuppressive and anti-inflammatory properties capable to protect against experimental inflammatory bowel disease. Stem Cells. 2011;29:251–262. doi: 10.1002/stem.569. [DOI] [PubMed] [Google Scholar]
- Sandig H., Pease J.E., Sabroe I. Contrary prostaglandins: the opposing roles of PGD2 and its metabolites in leukocyte function. J. Leukoc. Biol. 2007;81:372–382. doi: 10.1189/jlb.0706424. [DOI] [PubMed] [Google Scholar]
- Sepúlveda J.C., Tomé M., Fernández M.E., Delgado M., Campisi J., Bernad A., González M.A. Cell senescence abrogates the therapeutic potential of human mesenchymal stem cells in the lethal endotoxemia model. Stem Cells. 2014;32:1865–1877. doi: 10.1002/stem.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J. Mesenchymal stem cells in cancer. Stem Cell Rev. 2008;4:119–124. doi: 10.1007/s12015-008-9030-4. [DOI] [PubMed] [Google Scholar]
- Uccelli A., Moretta L., Pistoia V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- Wang Y., Huso D.L., Harrington J., Kellner J., Jeong D.K., Turney J., McNiece I.K. Outgrowth of a transformed cell population derived from normal human BM mesenchymal stem cell culture. Cytotherapy. 2005;7:509–519. doi: 10.1080/14653240500363216. [DOI] [PubMed] [Google Scholar]
- Yagi H., Deguchi K., Aono A., Tani Y., Kishimoto T., Komori T. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 1998;92:108–117. [PubMed] [Google Scholar]
- Yagi H., Soto-Gutierrez A., Parekkadan B., Kitagawa Y., Tompkins R.G., Kobayashi N., Yarmush M.L. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010;19:667–679. doi: 10.3727/096368910X508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.