

Abstract
Background
Prokaryotic CRISPR-Cas systems confer resistance to viral infection and thus mediate bacteria-phage interactions. However, the distribution and functional diversity of CRISPRs among environmental bacteria remains largely unknown. Here, comparative genomics of 75 Salinispora strains provided insight into the diversity and distribution of CRISPR-Cas systems in a cosmopolitan marine actinomycete genus.
Results
CRISPRs were found in all Salinispora strains, with the majority containing multiple loci and different Cas array subtypes. Of the six subtypes identified, three have not been previously described. A lower prophage frequency in S. arenicola was associated with a higher fraction of spacers matching Salinispora prophages compared to S. tropica, suggesting differing defensive capacities between Salinispora species. The occurrence of related prophages in strains from distant locations, as well as spacers matching those prophages inserted throughout spacer arrays, indicate recurring encounters with widely distributed phages over time. Linkages of CRISPR features with Salinispora microdiversity pointed to subclade-specific contacts with mobile genetic elements (MGEs). This included lineage-specific spacer deletions or insertions, which may reflect weak selective pressures to maintain immunity or distinct temporal interactions with MGEs, respectively. Biogeographic patterns in spacer and prophage distributions support the concept that Salinispora spp. encounter localized MGEs. Moreover, the presence of spacers matching housekeeping genes suggests that CRISPRs may have functions outside of viral defense.
Conclusions
This study provides a comprehensive examination of CRISPR-Cas systems in a broadly distributed group of environmental bacteria. The ubiquity and diversity of CRISPRs in Salinispora suggests that CRISPR-mediated interactions with MGEs represent a major force in the ecology and evolution of this cosmopolitan marine actinomycete genus.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-936) contains supplementary material, which is available to authorized users.
Keywords: Salinispora, CRISPR-Cas, Prophages, Mobile genetic elements, Immunity, Evolution
Background
CRISPRs (clustered regularly interspaced short palindromic repeats) have been detected in approximately 85% of archaeal and 50% of bacterial genomes [1]. They are considered a means of prokaryotic adaptive immunity against bacteriophages [2], which are major determinants of prokaryotic abundance, diversity and community structure [3]. CRISPRs consist of conserved repeats separated by variable spacers, the latter representing incorporated fragments of viral or plasmid DNA that specify immunity upon subsequent encounters [4]. Many CRISPRs are associated with Cas gene arrays, which can be classified into three major types and ten subtypes [5, 6] and are considered essential for CRISPR function. The activity of CRISPR-Cas systems proceeds in three stages: the acquisition of protospacer sequences from foreign genetic elements and their integration into the CRISPR array, constitutive transcription of the array, and target interference through transcribed crRNA [2]. In response, phages have developed mechanisms to evade CRISPR action [7–9], suggesting a co-evolutionary arms race between bacteria and phages.
Comparative genomics has given insight into CRISPRs from Actinobacteria [10, 11], Firmicutes [12, 13], Cyanobacteria [14, 15], enterobacteria [16], and Archaea [17]. In addition, mathematical modeling has presented important concepts about CRISPR dynamics during phage-bacteria interactions [18, 19]. Most of what is known about CRISPRs has been derived from pathogenic or industrially relevant bacteria such as Salmonella [20] and Streptococcus [12]. In the case of environmental bacteria, it has been shown that CRISPRs are widespread in Cyanobacteria except for the major marine lineages Prochlorococcus and Synechococcus [15]. In freshwater Cyanobacteria, CRISPRs were used to illustrate specific host-cyanophage interactions [14]. Furthermore, CRISPRs have been linked to host-phage co-evolution, community structuring and biogeographic patterns in microbial mats [21], acidophilic biofilms [22], and hot spring microbiota [23].
CRISPRs also control genetic exchange [24, 25] and intraspecies recombination [26], hence mediating evolutionary processes [27]. They may also regulate gene expression via crRNAs [28] and ‘self-targeting spacers’ that match elements in the host genome [29]. CRISPR activity has also been linked to DNA repair [30] and can affect various bacterial phenotypes including biofilm formation [31], swarming motility [32], and pathogenicity [33]. Despite the insights afforded by these studies, the distribution, diversity and functional roles of CRISPR-Cas systems among closely related environmental bacteria remain largely unknown.
In the present study, we analyzed CRISPR-Cas and prophage content in 75 Salinispora strains from seven global collection sites. This actinomycete genus has a pan-tropical distribution in marine sediments [34, 35] and is comprised of three closely related species; the cosmopolitan S. arenicola and the regionally confined sister taxa S. pacifica and S. tropica [36, 37]. The species have been further divided into 16S rRNA phylotypes (i.e. single nucleotide variants), with the highest diversity in S. pacifica and the lowest in S. tropica [35] . The genus is recognized for the production of diverse secondary metabolites [38], with the associated biosynthetic pathways showing evidence of extensive horizontal gene transfer [39, 40].
The diversity and distribution of CRISPR-Cas systems in Salinispora spp. was investigated to (i) assess the role of CRISPRs in phage defense, (ii) characterize past interactions with foreign genetic elements, (iii) elucidate linkages between CRISPR features and Salinispora microdiversity, and (iv) identify biogeographic signatures in CRISPR and prophage content. The detected diversity of CRISPR-Cas systems, including spacers that match foreign genetic elements, supports a role in host immunity. Spacer arrays illustrated recurring encounters with related phages as well as geographically confined MGEs. These findings suggest the presence of complex CRISPR-mediated interactions between Salinispora spp. and foreign genetic elements that may influence the ecology and evolution of this broadly distributed marine actinomycete genus.
Results and discussion
CRISPR content in 75 Salinisporastrains
Genome sequences from 75 Salinispora strains derived from seven global collection sites were analyzed for CRISPR-Cas content (Additional file 1). In total, 335 CRISPR loci were detected, with an average of 4.4 per strain (Table 1) but considerable among strain variability, ranging between 1 and 12 (Additional file 1). Unlike many genera for which multiple genome sequences are available, all 75 Salinispora strains harbored CRISPRs, suggesting they are an ecologically relevant feature of this genus. Salinispora CRISPR content exceeded the average reported for mesophilic bacteria [1] and marine bacterial metagenomes [41] and accounted for up to 0.3% of some genomes, which is approximately a third of the reported ‘prokaryotic maximum’ [2]. CRISPRs were concentrated in genomic islands, which represent the major regions of gene acquisition in Salinispora spp. [40]. Prior evidence of extensive horizontal gene transfer in Salinispora spp. [39, 40], coupled with the ubiquity of CRISPR-Cas systems detected in the present study, suggests an ongoing dynamic between CRISPR-mediated immunity and the acquisition of foreign genetic material.
Table 1.
Summary of CRISPR-Cas and prophage content in Salinispora spp.
| Species | Genomes analyzed | Total CRISPRs | Avg. loci/strain (per Mb) | Avg. locus size ± SD (bp) | Loci with Cas arrays (%) | Total spacers | Avg. spacers/ strain (±SD) | Avg. prophages/strain | Spacers matching Salinisporaprophages/known MGEs (%)* |
|---|---|---|---|---|---|---|---|---|---|
| S. arenicola | 37 | 169 | 4.5 (0.8) | 1243 ± 1087 | 78 (56) | 3033 | 82 ± 52 | 1.3 | 18.3/2.5 |
| S. pacifica | 31 | 136 | 4.4 (0.8) | 1110 ± 1087 | 54 (63) | 2153 | 69 ± 42 | 1.1 | 8.9/0.6 |
| S. tropica | 7 | 30 | 4.3 (0.8) | 1362 ± 1086 | 14 (63) | 551 | 79 ± 57 | 2.1 | 4.5/0.2 |
*only considering perfect matches (100% sequence identity/coverage).
The 335 CRISPR loci contained 5737 spacers, of which 68% were observed only once across all genomes. Extensive differences in spacer content were detected among strains isolated at the same time from the same site (Additional file 1), suggesting that spatiotemporal encounters with mobile genetic elements (MGEs) may be highly variable. On average, S. arenicola and S. tropica contained more spacers per strain than S. pacifica, however, the numbers varied greatly among strains within each species (Table 1).
Diversity and evolution of Cas array subtypes
The 75 Salinispora strains contained 146 Cas arrays (Table 1), all of which can be classified as type I based on the inclusion of a cas3 gene [6]. Cas arrays could be further grouped into six subtypes (Figure 1), of which five occurred in all three species and one (I-U_Sa) was only observed in S. arenicola. In total, 60% of the CRISPRs were associated with Cas arrays (Table 1), with up to five different array subtypes in some strains (Additional file 1). Three of these subtypes (I-E, I-C, I-B) have been previously characterized [6], with the most common (I-E) occurring in 49 strains. Almost two-thirds of the I-E arrays were associated with paired loci, i.e., two CRISPRs (one with inverted repeat sequences) flanking internalized cas genes, as often observed in Archaea [42]. Eleven strains contained two I-E or I-C arrays (Additional file 1). BLAST analysis of the associated cas3 genes indicated that the two arrays in a given strain were acquired as independent events from different sources based on sequence similarities to homologs in different actinomycetes (Verrucosispora vs. Streptomyces spp. for I-E arrays and Frankia vs. Stackebrandtia spp. for I-C arrays). In all three species, the GC content of I-C arrays was lower than the overall genomic GC content. This was especially apparent in S. pacifica (64.2% vs. 69.8% GC), suggesting that I-C arrays have been acquired from distantly related taxa. To the best of our knowledge, three of the Cas array subtypes detected (herein designated as I-U_csb3, I-U_csx17 and I-U_Sa) have not previously been described despite containing known cas genes (csb1, csb2, csb3, csx17). These subtypes were designated as I-U based on convention [6]. However, the Integrated Microbial Genomes (IMG) database [43] revealed that a variety of bacteria from different phylogenetic groups possess equivalent arrays, indicating these are not unique to Salinispora spp.
Figure 1.
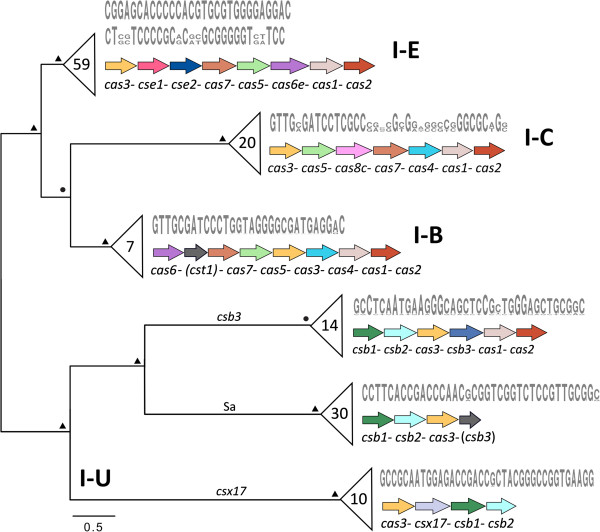
cas3 phylogeny and CRISPR repeat diversity. Condensed maximum likelihood phylogeny of cas3 nucleotide sequences reveals clades corresponding to Cas array subtype. The two major clades delineate known (I-E, I-C, I-B) and previously undescribed (I-U) subtypes. The order of genes for each subtype is displayed on the right. Gene annotations in parentheses designate hypothetical proteins with low identity to those indicated. In the I-B arrays, cas8b was replaced by a larger gene related to cst1. The total number of each array subtype among the 75 genomes is shown in the condensed nodes. Six arrays were missing several genes and therefore excluded from the analysis. Nodal support values (● above 80%, ▲ 100%) were obtained by 1000 bootstrap replicates (see Additional file 2 for the full tree including strain names and bootstrap values). Consensus repeat sequences in the associated CRISPR loci (indicated in gray) were specific to each array subtype and mostly showed considerable conservation.
cas3 is the signature gene of type I arrays [6]. A cas3 phylogeny revealed clades that corresponded to Cas array subtype as opposed to taxonomic relationships (Figure 1). The finding of cas3 sequence similarities across species boundaries supports the concept that Cas arrays evolve independent of their hosts [20, 44]. Furthermore, sequences within the array subtypes reveal evidence of recombination, as different Salinispora species shared virtually identical cas3 genes. The same patterns were observed with cas1 genes and corresponding protein sequences (Additional file 2), the most common phylogenetic marker for CRISPR-Cas systems. The delineation of the Cas array subtypes was supported by the repeat sequences, which frequently shared subtype-specific conservation (Figure 1) and averaged between 29 nt (subtypes I-E and I-B) and 37 nt (subtypes I-C and I-U).
Cas-associated CRISPRs contained significantly more spacers than Cas-devoid loci (p < 1 × 10-10), as might be expected given that cas genes are required for spacer integration [2]. Furthermore, subtypes I-E, I-C and I-B contained significantly more spacers (p < 0.0001) than the three I-U subtypes. Considering the latter, I-U_Sa and I-U_csx17 lack cas1 and are thus potentially unable to incorporate additional spacers, as cas1 is involved in spacer integration [2].
CRISPRs illustrate interactions with foreign genetic elements
We assessed defensive functions of Salinispora CRISPRs by analyzing for perfect matches between Salinispora spacers and mobile genetic elements (MGEs). These included 97 prophages that were identified in the 75 genomes (Additional file 3) as well as MGEs deposited in the Aclame database [45] (the latter referred to as ‘known MGEs’). On average, 11% of spacers matched Salinispora prophages (Table 1). Prophage-devoid strains had a higher fraction of matching spacers than prophage-harboring strains (p < 0.05), supporting a functional role of CRISPRs in phage immunity. In addition, 1.1% of spacers matched known MGEs, which was comparable to observations for marine bacterial metagenomes [41] and oral pathogens [26]. Some spacers matched homologous elements from different viral genomes, suggesting they may target multiple phage strains. CRISPRTarget [46] revealed that MGEs matched by Salinispora spacers are associated with various protospacer-associated motifs (PAMs), short sequences important for protospacer acquisition [2]. This suggests that Salinispora spp. can detect different PAMs and integrate a large diversity of spacers. When including lower-quality matches (100% identity over at least 18 nt) the majority (77%) of spacers matched plasmids, suggesting that a major role for Salinispora CRISPRs is to defend against plasmid integration. As no information about the plasmid content of the strains investigated is currently available, we focused on the role of CRISPRs in phage defense, while realizing this may not present a complete picture of CRISPR functionality in Salinispora spp.
CRISPRs indicate differing defensive capacities among Salinisporaspecies
S. arenicola had four-fold more spacers matching Salinispora prophages and twelve-fold more spacers matching known MGEs than S. tropica. This corresponded to the fact that only two-third of S. arenicola but all S. tropica strains harbored prophages, with 1.3 vs. 2.1 prophages per genome, respectively (Table 1). A substantial number of S. arenicola spacers that matched Salinispora prophages were located in the I-U_Sa Cas arrays, which are specific to S. arenicola. This additional array and spacer diversity may provide superior defensive capacities for S. arenicola, which potentially contributes to its broader geographic distribution [35]. S. pacifica had an intermediate fraction of spacers matching Salinispora prophages and known MGEs, with 1.1 prophages per genome (Table 1). There was a significantly lower frequency of prophages among phylotype C and F strains (p < 0.01). While these phylotypes also contained significantly more spacers (p < 0.01), the fraction of those spacers matching Salinispora prophages and known MGEs was similar to other phylotypes. The differing phage sensitivities between S. pacifica phylotypes are thus independent from or only partially related to CRISPRs.
In contrast, the total numbers of CRISPR loci or spacers were uncorrelated with prophage content in all three species (R2 < 0.01). For instance, strains CNS-051 and CNS-205 contained 11 and 8 CRISPRs with 119 and 140 spacers, respectively. Despite these similarities, these strains harbored 0 and 5 prophages, respectively (Additional file 1). The number and diversity of Cas arrays were also uncorrelated with prophage content (R2 < 0.001). For instance, S. pacifica strain DSM-45549 contained four Cas array subtypes and three prophages while the Cas-devoid S. pacifica strain CNS-103 only contained one prophage (Additional file 1). Thus, the number of CRISPR loci as well as the diversity of associated Cas arrays appear to be affected by factors other than phage exposure.
History of Salinisporainteractions with a common prophage
We focused on a common prophage that is related to the Streptomyces SV1 phage and was detected in 24 Salinispora strains from all three species (Figure 2A, Additional file 3). Six percent of Salinispora spacers matched SV1-related sequences, suggesting that this phage represents a major challenge to the genus. Strains without an integrated SV1 prophage had a larger fraction of spacers matching SV1 in Cas-associated loci (89%) compared to those with an integrated SV1 prophage (75%), supporting a specific targeting of this phage. The history of encounters with SV1-related phages was determined for six strains per species (three harboring and three lacking SV1) by analyzing the location of matching spacers within spacer arrays according to the concept that ancestral spacers are commonly located at the ‘trailer’ end and more recent spacers at the ‘leader’ end of a spacer array [4]. Matching spacers, the majority with unique sequences, were detected throughout the spacer arrays (Figure 2B) suggesting recurring encounters with SV1-related phages over time. Given that the SV1 phage represents a vector for genetic exchange [47], it is interesting to speculate that it may represent a source of beneficial genetic material in addition to a survival challenge.
Figure 2.
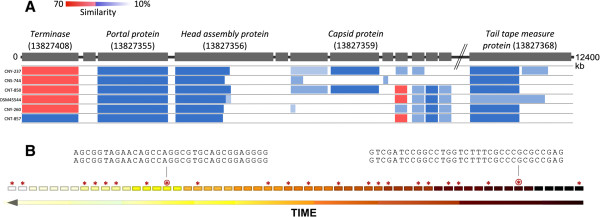
Streptomyces SV1-related prophage content in Salinispora spp. A) Comparison of genes from six intact, SV1-related prophages in Salinispora genomes with the SV1 type phage (gray; with GenBank Gene IDs in parentheses) by color-coding percent sequence similarities over a 12400 bp region within SV1. B) History of SV1 encounters in S. tropica CNR-699 as reflected in multiple spacer matches in a single array. Incomplete matches are indicated by asterisks, perfect matches by encircled asterisks with sequences shown above.
Linkages of CRISPR-Cas features with microdiversity
Salinispora microdiversity on the subspecies level has been defined based on 16S rRNA phylotypes (Additional file 1) and a multilocus phylogeny (Additional file 4). We detected several correlations between CRISPR-Cas features and microdiversity. For instance, one well-supported S. pacifica lineage contained the only strains (CNT-796 and CNT-851) with a modified I-C array lacking cas1/cas2, suggesting these genes have been lost in this lineage. Another S. pacifica lineage (containing strains CNQ-768 and CNS-103) was unique in being entirely devoid of cas genes. Also, certain clades were characterized by chromosomal relocations of CRISPR-Cas systems, as seen with I-E arrays in S. pacifica (strains CNT-796 and CNT-851) and S. tropica (strains CNS-197 and CNR-699).
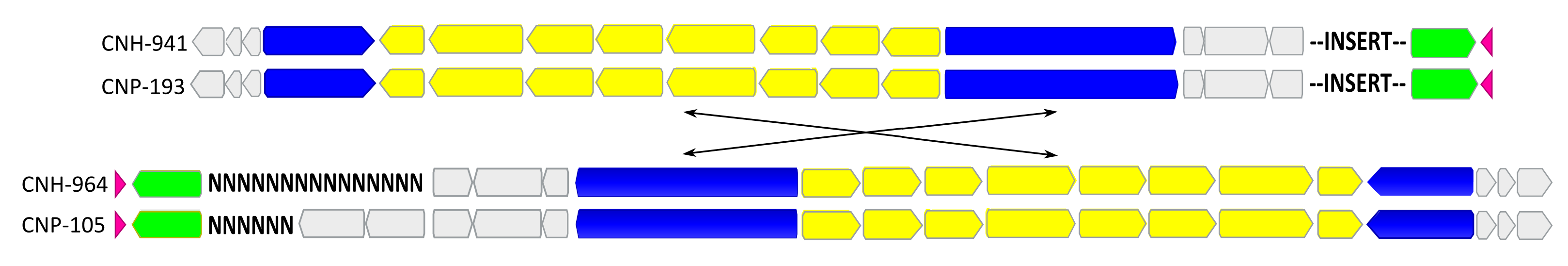
The most distinct linkages were observed among the four S. arenicola phylotype B strains, which contained significantly more CRISPRs and spacers than strains from S. arenicola phylotypes A and ST (p < 0.05). Many spacers were unique to phylotype B, underlining that spacer composition can reflect population structure and evolutionary relationships [48, 49]. CRISPR characteristics not only distinguished phylotype B from other phylotypes, but also the two subclades within phylotype B (strains CNH-941 and CNP-193 vs. CNH-964 and CNP-105; Additional file 4). For instance, a paired CRISPR locus and flanking genes were inverted in one of the subclades (Additional file 5). Furthermore, there were subclade-specific differences in spacer content. While multiple spacers were shared by all phylotype B strains, which is consistent with observations among other closely related bacteria [46], spacer array alignments revealed three sets of spacers that were specific to one of the subclades (Figure 3). This probably illustrates subclade-specific deletions or insertions of whole spacer groups [49]. Sixty-five percent of the group 1 spacers in CNH-941 and CNP-193 matched plasmids from Alphaproteobacteria, while the group 2 spacers in CNH-964 and CNP-105 equally matched phages and largely gammaproteobacterial plasmids. This may coincide with differing defensive capacities or varying modes of interaction with MGEs between the two subclades. While prophage content appeared independent of these observations (Additional file 2) MGEs are also involved in diversification [50, 51], niche adaptation [52], and microdiversity [53]. It is hence interesting to speculate that these differences may influence the evolutionary or ecological divergence within S. arenicola phylotype B.
Figure 3.
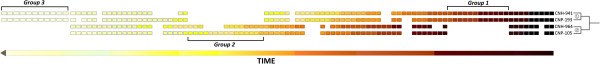
Subclade-specific spacer content in S. arenicola phylotype B. Alignment of spacers from a homologous CRISPR locus present in all four S. arenicola phylotype B strains. The phylogenetic relationships among the four strains are depicted on the right. Spacers are indicated by rectangles arranged from the oldest (trailer end; right) to the most recent spacers (leader end; left). Vertically aligned spacers are identical and separated by subclade-specific groups of spacers (1, 2, 3), which were likely deleted from the respective other lineage or selectively acquired.
Biogeographic patterns in CRISPR and prophage content
The strains analyzed in this study originate from seven global collection sites and were derived from independent sediment samples. While sampling efforts were not uniform across locations and may have affected the biogeographic patterns observed, it is interesting to note that 40% of the spacers observed in more than one strain were restricted to specific locations and/or biomes, the latter describing major oceanic regions distinguished by oceanographic factors such as nutrient concentrations and primary productivity [54]. Location-specific spacers provide evidence of exposure to local virus populations [41, 55], with the majority of localized spacers occurring in strains from the Sea of Cortez (Figure 4A). This is a highly productive sea [56] enclosed by a distinct geographical barrier and the only site classified as a Coastal biome [54]. While these results are preliminary, it is intriguing to speculate that spacer sequences can be used to trace location-specific interactions with distinct MGE pools, as also observed in other ecosystems [23, 57, 58]. A more nuanced biogeographic pattern was the detection of identical spacers with location-specific nucleotide substitutions, as found in strains from Hawaii, Fiji and Palau (Figure 4B). This may illustrate the presence of widespread MGEs that maintain location-specific genetic variants. Furthermore, SV1-related prophages could be resolved into geographically confined lineages (Figure 4C), supporting the concept that Salinispora strains are exposed to location-specific MGEs.
Figure 4.

Biogeographic patterns among Salinispora spacers and prophages. A) Distribution of spacers unique to certain locations (SC = Sea of Cortez, HW = Hawaii, FJ = Fiji, PY = Palmyra, BH = Bahamas, PA = Palau) and biomes; B) Location-specific spacer variants reflected in single nucleotide polymorphisms (marked in red) within a conserved 41 nt spacer in strains from Hawaii (3 strains with 3 variants), Fiji (4 strains with 1 variant) and Palau (1 strains with 1 variant); C) Maximum likelihood phylogeny (1000 bootstrap replicates) of SV1-related prophages in genomes from geographically distant Salinispora strains, showing location-specific SV1 lineages.
Self-targeting spacers
Several studies have reported the occurrence of ‘self-targeting spacers’ that match regions within the host genome [29]. While self-targeting spacers can be deleterious and strongly selected against [29, 59], they have also been suggested to function as regulatory elements [20, 60, 61] or to affect genome content [62]. Interestingly, a third of the 75 Salinispora strains harbored such spacers, with perfect matches to e.g. a cytochrome P450 within a terpenoid biosynthetic pathway [40] and two DNA-modifying genes (Table 2). However, experimental evidence would be required to determine potential regulatory roles. In addition, several self-targeting spacers matched resident prophages, suggesting that CRISPR interference may be ineffective in some cases. Alternatively, self-targeting may be prevented by selective self vs. non-self mechanisms, such as variations in spacer flanking sequences [63].
Table 2.
Chromosomal matches of select self-targeting spacers
| Strain | Spacer match (IMG Gene ID) | Spacer sequence (above) and matching chromosomal region with adjacent nucleotides (5′-3′; below) |
|---|---|---|
| S. arenicola CNH-964/S. arenicola CNP-105 | Adenylosuccinate lyase (2515702456/2518452715) | GCCCACCTTGCCGTGCCACCACGCCTCCCGCACCTCGTT |
| GTGCTGCTGCCCACCTTGCCGTGCCACCACGCCTCCCGCACCTCGTTGAACTCCG | ||
| 23S rRNA methyltransferase (2515702034/2518452486) | CCGAGCGGGTCGAGCTGACCGTCGGGGCGGTGGCCCCGGG | |
| GCGGAGGCCGAGCGGGTCGAGCTGACCGTCGGGGCGGTGGCCCCGGGCGGGCAC | ||
| S. arenicola CNX-481 | Cytochrome P450 (2518471737) | TACCGACGCAGCCATAACTCGTGCTAGGACGG |
| CTGATGGCTACCGACGCAGCCATAACTCGTGCTAGGACGGTGCCCGCG |
Conclusions
This study describes a comprehensive survey of CRISPR-Cas systems among a large collection of strains from a cosmopolitan marine actinomycete genus. The finding of ubiquitous and diverse CRISPR-Cas systems suggests that Salinispora maintains a robust mechanism to mediate interactions with MGEs, which may be of ecological and evolutionary relevance in virally rich marine sediments [3]. Future surveys of CRISPR-Cas systems will provide additional opportunities to assess the evolutionary history of MGE exposure, the effectiveness of these systems as mechanisms of adaptive defense, and how CRISPRs may be linked to the ecology and evolution of Salinispora.
Methods
Genome sequences and CRISPR-Cas classification
The 75 Salinispora genome sequences (Additional file 1) were downloaded from the IMG database (https://img.jgi.doe.gov). CRISPRs were predicted using CRISPRFinder [64] on pseudochromosomes generated from the genome sequences (i.e. contigs assembled using a closed reference genome) [39] and unmapped contigs. Only CRISPRs classified as ‘confirmed’ were considered. Predicted CRISPRs were manually checked and adjacent loci combined if separated by Ns and having the same repeat sequences. Annotated cas genes were verified by determining similarities to known cas genes using BLAST [65] and UniProt [66]. The naming of cas genes and their classification into Cas array subtypes was done following [6]. The IMG database was searched for equivalent Cas arrays in other sequenced bacterial genomes. CRISPRmap was used to classify repeats into motifs, families, and superclasses based on similarities to known repeat sequences [67]. Repeat consensus sequences were obtained using WebLogo [68].
Phylogenetic and structural analyses of Cas arrays
cas1 and cas3 nucleotide and corresponding Cas1 and Cas3 amino acid sequences were aligned using MAFFT v7.017 (L-INS-i algorithm, 100PAM/k = 2 scoring matrix, gap open penalty 1.53, offset value 0.123) [69] and manually curated. The best substitution models (cas1: TN93 + G + I; Cas1: WAG + G + F; cas3: T92 + G; Cas3: JTT + G) were determined using MEGA5 [70]. Maximum likelihood phylogenies were computed with MEGA5 (using the best model and 100 bootstrap replicates) and RAxML (with default settings and 1000 bootstrap replicates) implemented on the CIPRES Science Portal [71], always giving the same topology. Nucleotide sequences of cas1 [KM526976-KM527070] and cas3 [KJ677987-KJ678124] have been deposited at GenBank (Additional file 6). Architectures of selected loci and flanking regions were analyzed with progressiveMauve [72]. Spacer arrangement in S. arenicola phylotype B was evaluated by aligning concatenated spacer sequences (sorted from trailer to leader end) with MAFFT [69].
Prophage prediction and sequence comparison
Prophages were predicted using PHAST [73] on both the pseudochromosomes and unmapped contigs. Predicted intact prophages classified as being related to the Streptomyces SV1 phage were compared with the sequenced SV1 type phage (GenBank accession number NC_018848) using the CGView Comparison Tool [74]. Nucleotide sequences of SV1-related prophages were aligned using Mugsy [75] and the resulting alignment file converted to Fasta using the Galaxy web server [76]. The alignment was manually curated and the best substitution model (GTR + G) determined using MEGA5 [70]. A maximum likelihood phylogeny was computed using MEGA5 with 1000 bootstrap replicates (Additional file 7).
Analysis of spacers
Spacers were extracted from genome sequences and sorted by unique (only found once across all 75 genomes) and shared (found in ≥2 genomes). Spacers were searched against different databases (Aclame MGE_0.4, PHAST_virus, PHAST_prophage_virus, CRISPRFinder spacer) with the standard BLAST parameters for short query sequences (word size 7; match/mismatch scores 1,-3; gap costs 5,2) using Geneious Pro v5.5 (available from http://geneious.com). In addition, short-query BLAST was used to determine spacers matching Salinispora prophages as well as self-targeting spacers matching non-CRISPR regions. Furthermore, short-query BLAST against Salinispora prophages was done with spacers from five representative strains from each species that were sorted by Cas-associated, Cas-devoid, associated with known Cas array subtypes (I-E, I-C, I-B), and associated with herein designated Cas array subtypes (I-U). Only perfect matches with 100% identity over the entire spacer length were considered. A separate BLAST search against Aclame was performed which also considered incomplete hits (100% sequence identity over at least 18 nt), as this may still be indicative of the targeted MGE type. The 18 nt threshold corresponds to 2/3 of the average Salinispora repeat length, which has been suggested as the minimum for a functioning spacer [1]. Also, 100% coverage hits are possibly rare since the vast majority of phage diversity is likely still unknown [3].
Statistical evaluation
The number of CRISPR loci, prophages and MGE genes per strain were normalized by genome size and gene count, respectively. Values were compared by species, location, and biome (both between and within species) as well as phylotype (only within species) using the Kruskal-Wallis one-way analysis of variance implemented in R [77] to test for significant differences. In case of a significant result (p < 0.05) the Wilcoxon rank-sum test implemented in R [77] was used to test the specific sample pairs for significant differences (p < 0.05). The fraction of spacers matching Salinispora prophages in strains with and without prophages was compared using Student’s t-test. Correlations between the number of CRISPRs/spacers/Cas arrays and prophages were calculated using least squares regression.
Availability of supporting data
All supporting data are included within the article and its additional files.
Electronic supplementary material
Additional file 1: Overview of genome and CRISPR features of 75 Salinispora strains. Origin (location, biome, latitude/longitude, sampling date, depth) and general genome characteristics (genome size, gene count, 16S rRNA phylotype), CRISPR content (number of loci and spacers, Cas array diversity, CRISPRmap classification of repeats), number of prophages, and MGE content of 75 Salinispora strains analyzed in the present study. (XLSX 32 KB)
{kind=link}
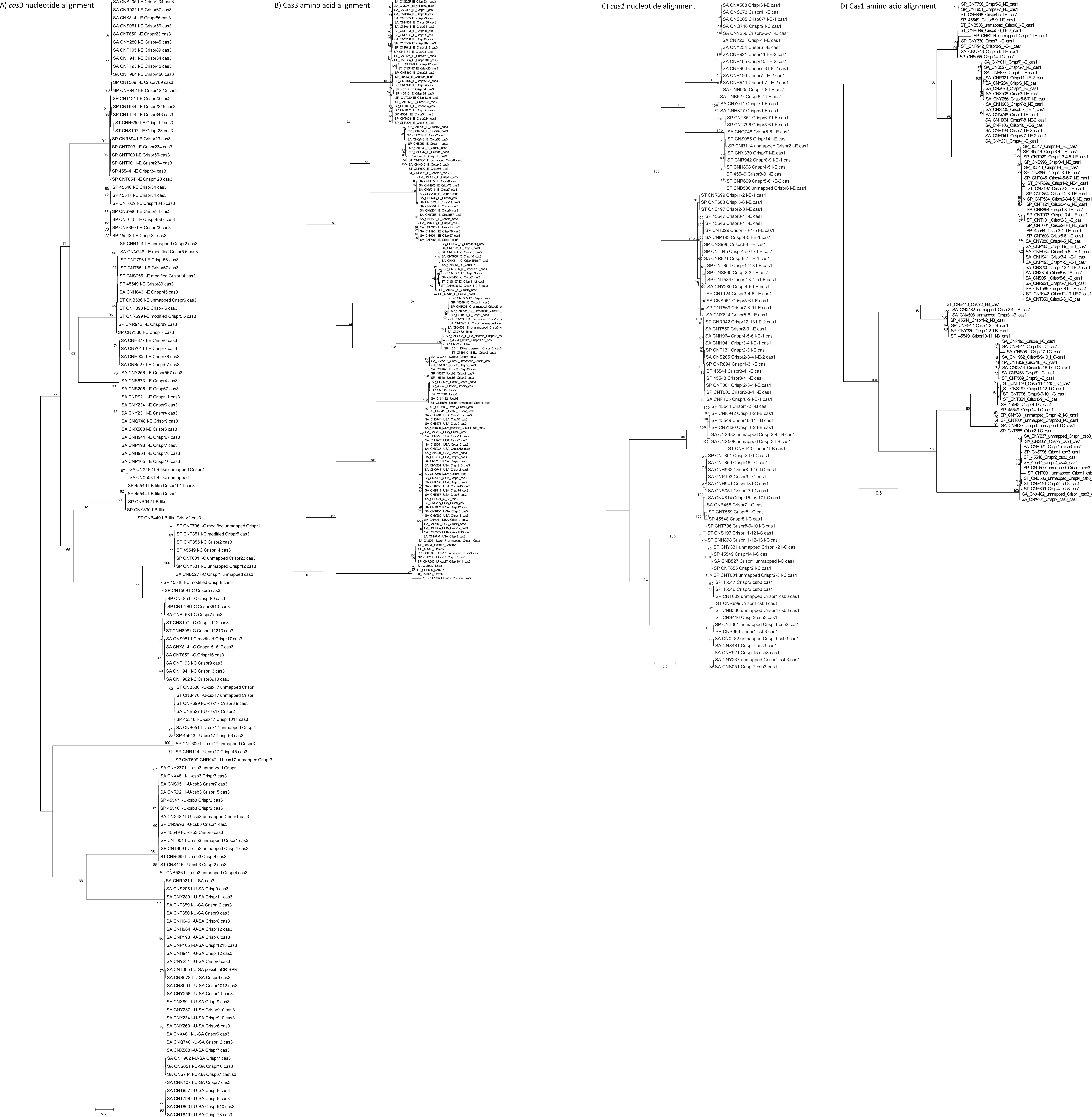
Additional file 2: Phylogeny of cas genes and Cas proteins. Maximum likelihood phylogenies of aligned cas1/cas3 nucleotide as well as Cas1/Cas3 amino acid sequences (1000 bootstrap replicates with only those >50 shown). Species names abbreviated (SA = S. arenicola, SP = S. pacifica, ST = S. tropica) followed by strain number, Cas array subtype, and internal CRISPR locus ID. (PNG 5 MB)
Additional file 3: Detected prophages. Prophages detected in the 75 Salinispora genomes, classified based on sequence similarities with known prophages [73], length in Kb, number of coding sequences (CDS) and GC content (% GC). (XLSX 21 KB)
{kind=link}
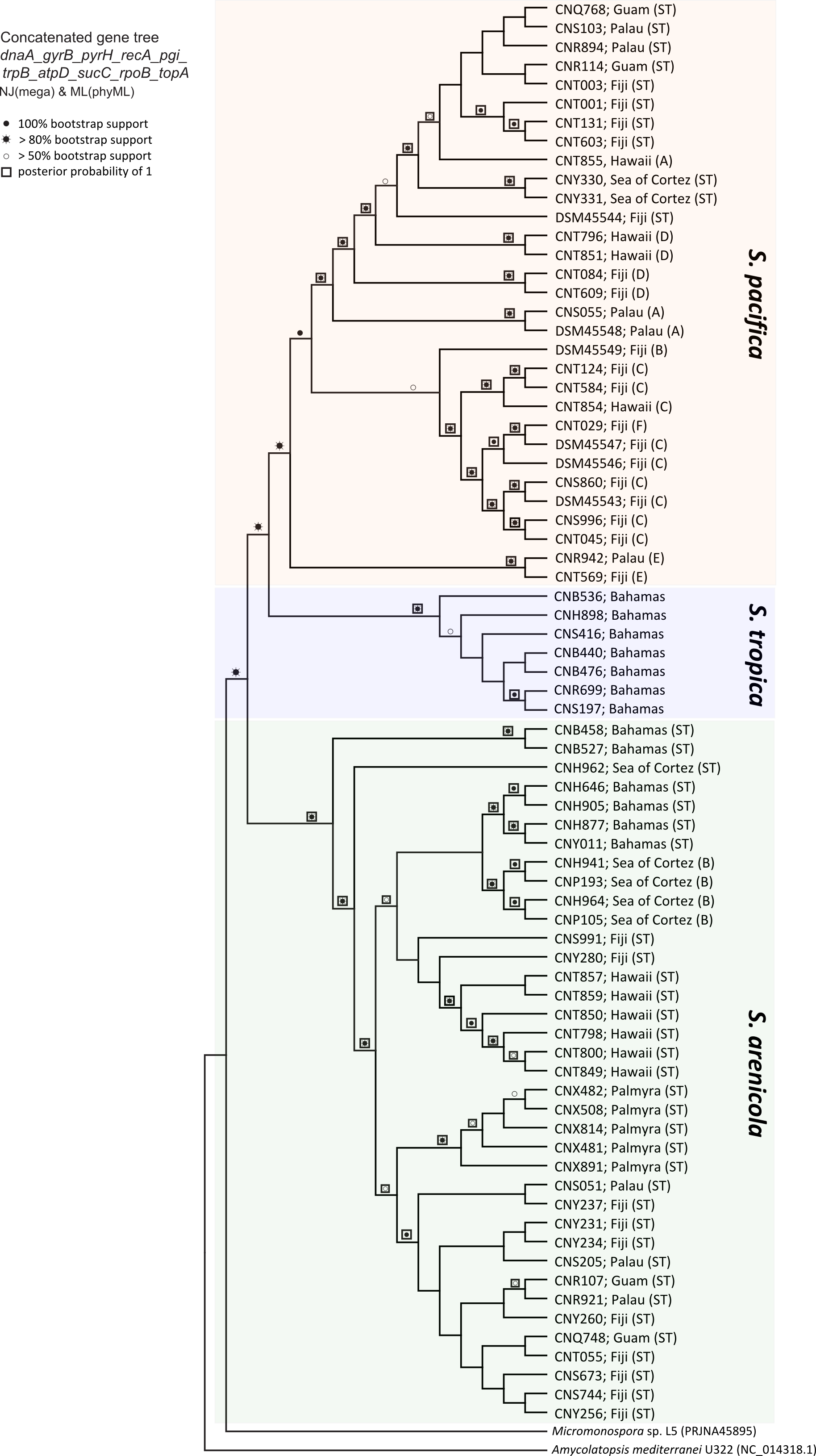
Additional file 4: Salinispora species phylogeny. Maximum likelihood phylogeny (1000 bootstrap replicates) of ten single-copy, concatenated housekeeping genes from 75 Salinispora genomes labeled with origin and phylotype (ST; A-F). A detailed description can be found in the original publication [39]. (PNG 715 KB)
{kind=link}
Additional file 5: Subclade-specific architectures of CRISPR loci and flanking genes. progressiveMauve alignment of paired CRISPR loci and flanking genes in S. arenicola phylotype B, showing that the arrays are inverted in subclade 1 (strains CNH-941 and CNP-193) compared to subclade 2 (strains CNH-964 and CNP-105). Blue: CRISPRs, yellow: cas genes, green: integrases; pink: tRNAs. (PNG 138 KB)
Additional file 6: Salinispora cas gene accession numbers. GenBank accession numbers of Salinispora cas1 and cas3 sequences used for phylogenetic analyses. (XLSX 16 KB)
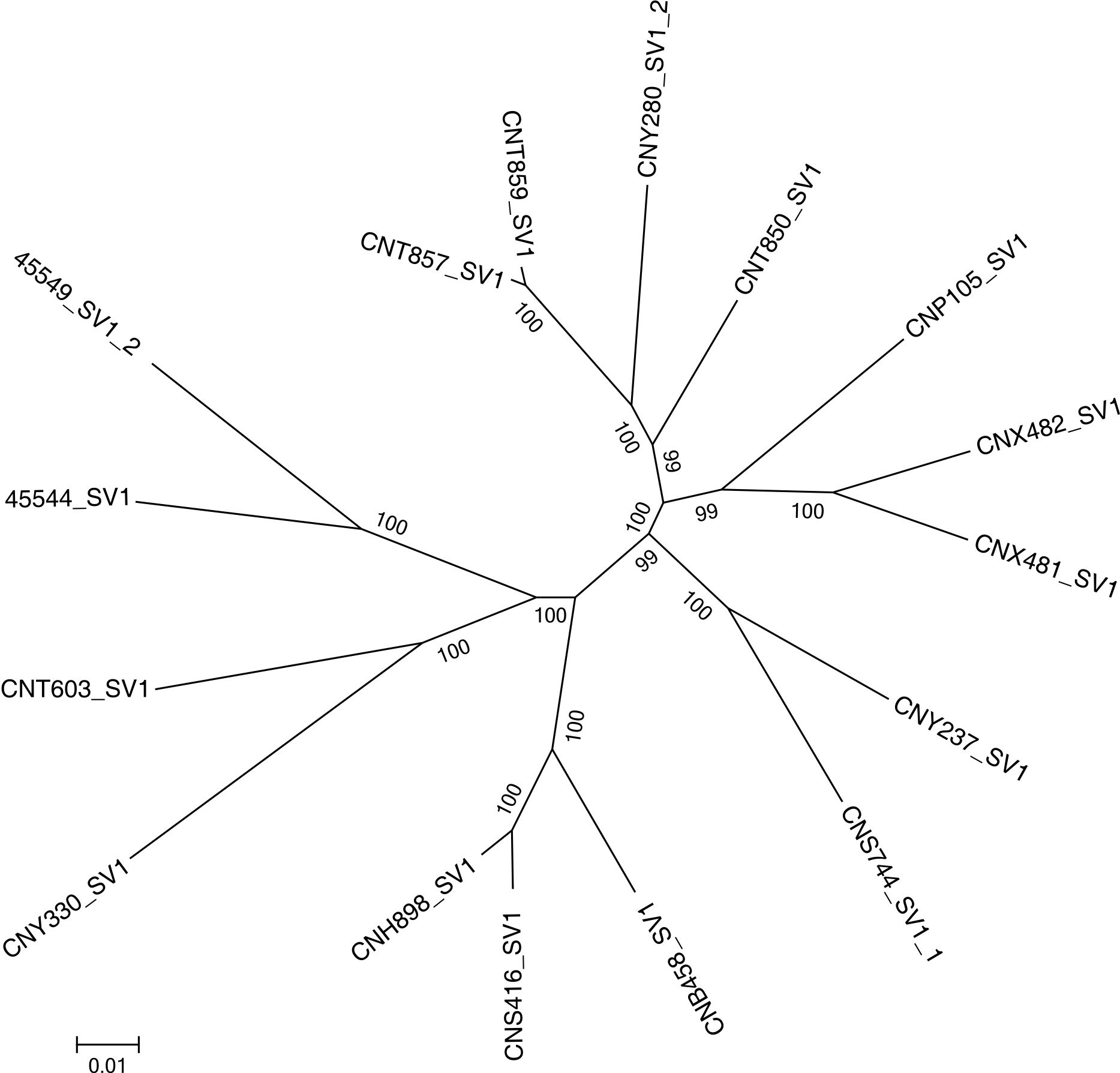{kind=link}
Additional file 7: SV1 prophage phylogeny. Maximum likelihood phylogeny (1000 bootstrap replicates) of conserved regions within SV1-related prophages in Salinispora genomes. (PNG 274 KB)
Acknowledgments
We thank Eduardo Santamaría-del-Ángel for statistical advice. Katherine Duncan and Juan Ugalde are thanked for valuable suggestions. MW was supported by a fellowship within the postdoc program of the German Academic Exchange Service (DAAD). NM-A acknowledges a graduate fellowship from Consejo Nacional de Ciencia y Tecnología (CONACyT-213497). PRJ acknowledges support from the National Science Foundation (OCE-1235142). Genome sequencing was conducted by the U.S. Department of Energy Joint Genome Institute and supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MW participated in study design, carried out CRISPR analyses, and wrote the manuscript. NM-A carried out sequence alignments and phylogenetic analyses. PRJ participated in study design and preparation of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Matthias Wietz, Email: matthias.wietz@uni-oldenburg.de.
Natalie Millán-Aguiñaga, Email: nmillana@ucsd.edu.
Paul R Jensen, Email: pjensen@ucsd.edu.
References
- 1.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorek R, Lawrence CM, Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem. 2013;82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- 3.Suttle CA. Marine viruses - major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 4.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. Evolution and classification of the CRISPR-cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skennerton CT, Angly FE, Breitbart M, Bragg L, He S, McMahon KD, Hugenholtz P, Tyson GW. Phage encoded H-NS: a potential achilles heel in the bacterial defence system. PLoS One. 2011;6:e20095. doi: 10.1371/journal.pone.0020095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seed KD, Lazinski DW, Calderwood SB, Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489–491. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, Fan X, Xie J. Comparative genomic structures of Mycobacterium CRISPR-cas. J Cell Biochem. 2012;113:2464–2473. doi: 10.1002/jcb.24121. [DOI] [PubMed] [Google Scholar]
- 11.Pleckaityte M, Zilnyte M, Zvirbliene A. Insights into the CRISPR/Cas system of Gardnerella vaginalis. BMC Microbiol. 2012;12:301. doi: 10.1186/1471-2180-12-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath P, Coûté-Monvoisin A-C, Romero DA, Boyaval P, Fremaux C, Barrangou R. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int J Food Microbiol. 2009;131:62–70. doi: 10.1016/j.ijfoodmicro.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 13.Palmer KL, Gilmore MS. Multidrug-resistant enterococci lack CRISPR-cas. MBio. 2010;1:e00227-10. doi: 10.1128/mBio.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuno S, Yoshida T, Kaneko T, Sako Y. Intricate interactions between the bloom-forming cyanobacterium Microcystis aeruginosa and foreign genetic elements, revealed by diversified clustered regularly interspaced short palindromic repeat (CRISPR) signatures. Appl Environ Microbiol. 2012;78:5353–5360. doi: 10.1128/AEM.00626-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai F, Axen SD, Kerfeld CA. Evidence for the widespread distribution of CRISPR-cas system in the Phylum Cyanobacteria. RNA Biol. 2013;10:1–7. doi: 10.4161/rna.24571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Díez-Villaseñor C, Almendros C, García-Martínez J, Mojica FJM. Diversity of CRISPR loci in Escherichia coli. Microbiology. 2010;156:1351–1361. doi: 10.1099/mic.0.036046-0. [DOI] [PubMed] [Google Scholar]
- 17.Garrett RA, Vestergaard G, Shah SA. Archaeal CRISPR-based immune systems: exchangeable functional modules. Trends Microbiol. 2011;19:549–556. doi: 10.1016/j.tim.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Weinberger AD, Sun CL, Pluciński MM, Denef VJ, Thomas BC, Horvath P, Barrangou R, Gilmore MS, Getz WM, Banfield JF. Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput Biol. 2012;8:e1002475. doi: 10.1371/journal.pcbi.1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levin BR. Nasty viruses, costly plasmids, population dynamics, and the conditions for establishing and maintaining CRISPR-mediated adaptive immunity in bacteria. PLoS Genet. 2010;6:e1001171. doi: 10.1371/journal.pgen.1001171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Touchon M, Rocha EPC. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS One. 2010;5:e11126. doi: 10.1371/journal.pone.0011126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heidelberg JF, Nelson WC, Schoenfeld T, Bhaya D. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One. 2009;4:e4169. doi: 10.1371/journal.pone.0004169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–1050. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- 23.Held NL, Whitaker RJ. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ Microbiol. 2009;11:457–466. doi: 10.1111/j.1462-2920.2008.01784.x. [DOI] [PubMed] [Google Scholar]
- 24.Brodt A, Lurie-Weinberger MN, Gophna U. CRISPR loci reveal networks of gene exchange in archaea. Biol Direct. 2011;6:65. doi: 10.1186/1745-6150-6-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe T, Nozawa T, Aikawa C, Amano A, Maruyama F, Nakagawa I. CRISPR regulation of intraspecies diversification by limiting IS transposition and intercellular recombination. Genome Biol Evol. 2013;5:1099–1114. doi: 10.1093/gbe/evt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fricke WF, Mammel MK, McDermott PF, Tartera C, White DG, Leclerc JE, Ravel J, Cebula TA. Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J Bacteriol. 2011;193:3556–3568. doi: 10.1128/JB.00297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampson TR, Saroj SD, Llewellyn AC, Tzeng Y-L, Weiss DS. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013;497:254–257. doi: 10.1038/nature12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 2010;26:335–340. doi: 10.1016/j.tig.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Babu M, Beloglazova N, Flick R, Graham C, Skarina T, Nocek B, Gagarinova A, Pogoutse O, Brown G, Binkowski A, Phanse S, Joachimiak A, Koonin EV, Savchenko A, Emili A, Greenblatt J, Edwards AM, Yakunin AF. A dual function of the CRISPR-cas system in bacterial antivirus immunity and DNA repair. Mol Microbiol. 2011;79:484–502. doi: 10.1111/j.1365-2958.2010.07465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cady KC, O’Toole GA. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J Bacteriol. 2011;193:3433–3445. doi: 10.1128/JB.01411-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zegans ME, Wagner JC, Cady KC, Murphy DM, Hammond JH, O’Toole GA. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J Bacteriol. 2009;191:210–219. doi: 10.1128/JB.00797-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunderson FF, Cianciotto NP. The CRISPR-associated gene cas2 of Legionella pneumophila is required for intracellular infection of amoebae. MBio. 2013;4:e00074-13. doi: 10.1128/mBio.00074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen PR, Mafnas C. Biogeography of the marine actinomycete Salinispora. Environ Microbiol. 2006;8:1881–1888. doi: 10.1111/j.1462-2920.2006.01093.x. [DOI] [PubMed] [Google Scholar]
- 35.Freel KC, Edlund A, Jensen PR. Microdiversity and evidence for high dispersal rates in the marine actinomycete ‘Salinispora pacifica’. Environ Microbiol. 2012;14:480–493. doi: 10.1111/j.1462-2920.2011.02641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maldonado LA, Stach JEM, Pathom-aree W, Ward AC, Bull AT, Goodfellow M. Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie Van Leeuwenhoek. 2005;87:11–18. doi: 10.1007/s10482-004-6525-0. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed L, Jensen PR, Freel KC, Brown R, Jones AL, Kim B-Y, Goodfellow M. Salinispora pacifica sp. nov., an actinomycete from marine sediments. Antonie Van Leeuwenhoek. 2013;103:1069–1078. doi: 10.1007/s10482-013-9886-4. [DOI] [PubMed] [Google Scholar]
- 38.Fenical W, Jensen PR. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 39.Ziemert N, Lechner A, Wietz M, Millán-Aguiñaga N, Chavarria K, Jensen PR. Diversity and evolution of secondary metabolism in the marine actinomycete Salinispora. Proc Natl Acad Sci U S A. 2014;111:E1130–E1139. doi: 10.1073/pnas.1324161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Penn K, Jenkins C, Nett M, Udwary DW, Gontang E, McGlinchey RP, Foster B, Lapidus A, Podell S, Allen EE, Moore BS, Jensen PR. Genomic islands link secondary metabolism to functional adaptation in marine actinobacteria. ISME J. 2009;3:1193–1203. doi: 10.1038/ismej.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorokin VA, Gelfand MS, Artamonova II. Evolutionary dynamics of clustered irregularly interspaced short palindromic repeat systems in the ocean metagenome. Appl Environ Microbiol. 2010;76:2136–2144. doi: 10.1128/AEM.01985-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lillestøl RK, Shah SA, Brügger K, Redder P, Phan H, Christiansen J, Garrett RA. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol. 2009;72:259–272. doi: 10.1111/j.1365-2958.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- 43.Markowitz VM, Mavromatis K, Ivanova NN, Chen I, Min A, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 44.Shah SA, Garrett RA. CRISPR/Cas and Cmr modules, mobility and evolution of adaptive immune systems. Res Microbiol. 2011;162:27–38. doi: 10.1016/j.resmic.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Leplae R, Hebrant A, Wodak SJ, Toussaint A. ACLAME: a classification of mobile genetic elements. Nucleic Acids Res. 2004;32:D45–D49. doi: 10.1093/nar/gkh084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, Brown CM. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stuttard C. Cotransduction of his and trp loci by phage SV1 in Streptomyces venezuelae. FEMS Microbiol Lett. 1983;20:467–470. doi: 10.1111/j.1574-6968.1983.tb00169.x. [DOI] [Google Scholar]
- 48.Lopez-Sanchez M-J, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol. 2012;85:1057–1071. doi: 10.1111/j.1365-2958.2012.08172.x. [DOI] [PubMed] [Google Scholar]
- 49.Yin S, Jensen MA, Bai J, Debroy C, Barrangou R, Dudley EG. The evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in clustered regularly interspaced short palindromic repeat (CRISPR) spacer composition. Appl Environ Microbiol. 2013;79:5710–5720. doi: 10.1128/AEM.00950-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brüssow H, Canchaya C, Hardt W-D. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paul JH. Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008;2:579–589. doi: 10.1038/ismej.2008.35. [DOI] [PubMed] [Google Scholar]
- 52.Maruyama F, Kobata M, Kurokawa K, Nishida K, Sakurai A, Nakano K, Nomura R, Kawabata S, Ooshima T, Nakai K, Hattori M, Hamada S, Nakagawa I. Comparative genomic analyses of Streptococcus mutans provide insights into chromosomal shuffling and species-specific content. BMC Genomics. 2009;10:358. doi: 10.1186/1471-2164-10-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Middelboe M, Holmfeldt K, Riemann L, Nybroe O, Haaber J. Bacteriophages drive strain diversification in a marine Flavobacterium: implications for phage resistance and physiological properties. Environ Microbiol. 2009;11:1971–1982. doi: 10.1111/j.1462-2920.2009.01920.x. [DOI] [PubMed] [Google Scholar]
- 54.Longhurst AR. Ecological Geography of the Sea. San Diego: Academic Press; 1998. [Google Scholar]
- 55.Cui Y, Li Y, Gorgé O, Platonov ME, Yan Y, Guo Z, Pourcel C, Dentovskaya SV, Balakhonov SV, Wang X, Song Y, Anisimov AP, Vergnaud G, Yang R. Insight into microevolution of Yersinia pestis by clustered regularly interspaced short palindromic repeats. PLoS One. 2008;3:e2652. doi: 10.1371/journal.pone.0002652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Álvarez-Borrego S. Phytoplankton biomass and production in the Gulf of California: a review. Bot Mar. 2012;55:119–128. [Google Scholar]
- 57.Flores CO, Valverde S, Weitz JS. Multi-scale structure and geographic drivers of cross-infection within marine bacteria and phages. ISME J. 2013;7:520–532. doi: 10.1038/ismej.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kunin V, He S, Warnecke F, Peterson SB, Garcia Martin H, Haynes M, Ivanova N, Blackall LL, Breitbart M, Rohwer F, McMahon KD, Hugenholtz P. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 2008;18:293–297. doi: 10.1101/gr.6835308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paez-Espino D, Morovic W, Sun CL, Thomas BC, Ueda K, Stahl B, Barrangou R, Banfield JF. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat Commun. 2013;4:1430. doi: 10.1038/ncomms2440. [DOI] [PubMed] [Google Scholar]
- 60.Aklujkar M, Lovley DR. Interference with histidyl-tRNA synthetase by a CRISPR spacer sequence as a factor in the evolution of Pelobacter carbinolicus. BMC Evol Biol. 2010;10:230. doi: 10.1186/1471-2148-10-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Touchon M, Charpentier S, Clermont O, Rocha EPC, Denamur E, Branger C. CRISPR distribution within the Escherichia coli species is not suggestive of immunity-associated diversifying selection. J Bacteriol. 2011;193:2460–2467. doi: 10.1128/JB.01307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS, Richter C, Przybilski R, Pitman AR, Fineran PC. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 2013;9:e1003454. doi: 10.1371/journal.pgen.1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 66.The UniProt Consortium Update on activities at the universal protein resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lange SJ, Alkhnbashi OS, Rose D, Will S, Backofen R. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 2013;41:8034–8044. doi: 10.1093/nar/gkt606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller M, Pfeiffer W, Schwartz T: Creating the CIPRES science gateway for inference of large phylogenetic trees.Proceedings of the Gateway Computing Environments Workshop: 14 Nov 20101–8.
- 72.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView comparison tool. BMC Genomics. 2012;13:202. doi: 10.1186/1471-2164-13-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics. 2011;27:334–342. doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blankenberg D, Taylor J, Nekrutenko A. Making whole genome multiple alignments usable for biologists. Bioinformatics. 2011;27:2426–2428. doi: 10.1093/bioinformatics/btr398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.R Core Team . R: A Language and Environment for Statistical Computing. Volume 1. Vienna: R Foundation for Statistical Computing; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Overview of genome and CRISPR features of 75 Salinispora strains. Origin (location, biome, latitude/longitude, sampling date, depth) and general genome characteristics (genome size, gene count, 16S rRNA phylotype), CRISPR content (number of loci and spacers, Cas array diversity, CRISPRmap classification of repeats), number of prophages, and MGE content of 75 Salinispora strains analyzed in the present study. (XLSX 32 KB)
Additional file 2: Phylogeny of cas genes and Cas proteins. Maximum likelihood phylogenies of aligned cas1/cas3 nucleotide as well as Cas1/Cas3 amino acid sequences (1000 bootstrap replicates with only those >50 shown). Species names abbreviated (SA = S. arenicola, SP = S. pacifica, ST = S. tropica) followed by strain number, Cas array subtype, and internal CRISPR locus ID. (PNG 5 MB)
Additional file 3: Detected prophages. Prophages detected in the 75 Salinispora genomes, classified based on sequence similarities with known prophages [73], length in Kb, number of coding sequences (CDS) and GC content (% GC). (XLSX 21 KB)
Additional file 4: Salinispora species phylogeny. Maximum likelihood phylogeny (1000 bootstrap replicates) of ten single-copy, concatenated housekeeping genes from 75 Salinispora genomes labeled with origin and phylotype (ST; A-F). A detailed description can be found in the original publication [39]. (PNG 715 KB)
Additional file 5: Subclade-specific architectures of CRISPR loci and flanking genes. progressiveMauve alignment of paired CRISPR loci and flanking genes in S. arenicola phylotype B, showing that the arrays are inverted in subclade 1 (strains CNH-941 and CNP-193) compared to subclade 2 (strains CNH-964 and CNP-105). Blue: CRISPRs, yellow: cas genes, green: integrases; pink: tRNAs. (PNG 138 KB)
Additional file 6: Salinispora cas gene accession numbers. GenBank accession numbers of Salinispora cas1 and cas3 sequences used for phylogenetic analyses. (XLSX 16 KB)
Additional file 7: SV1 prophage phylogeny. Maximum likelihood phylogeny (1000 bootstrap replicates) of conserved regions within SV1-related prophages in Salinispora genomes. (PNG 274 KB)