

Abstract
Background
Microdeletions in the NRXN1 gene have been associated with a range of neurodevelopmental disorders, including autism spectrum disorders, schizophrenia, intellectual disability, speech and language delay, epilepsy and hypotonia.
Results
In the present study we performed array CGH analysis on 10,397 individuals referred for diagnostic cytogenetic analysis, using a custom oligonucleotide array, which included 215 NRXN1 probes (median spacing 4.9 kb). We found 34 NRXN1 deletions (0.33% of referrals) ranging from 9 to 942 kb in size, of which 18 were exonic (0.17%). Three deletions affected exons also in the beta isoform of NRXN1. No duplications were found. Patients had a range of phenotypes including developmental delay, learning difficulties, attention deficit hyperactivity disorder (ADHD), autism, speech delay, social communication difficulties, epilepsy, behaviour problems and microcephaly. Five patients who had deletions in NRXN1 had a second CNV implicated in neurodevelopmental disorder: a CNTNAP2 and CSMD3 deletion in patients with exonic NRXN1 deletions, and a Williams-Beuren syndrome deletion and two 22q11.2 duplications in patients with intronic NRXN1 deletions.
Conclusions
Exonic deletions in the NRXN1 gene, predominantly affecting the alpha isoform, were found in patients with a range of neurodevelopmental disorders referred for diagnostic cytogenetic analysis. The targeting of dense oligonucleotide probes to the NRXN1 locus on array comparative hybridisation platforms provides detailed characterisation of deletions in this gene, and is likely to add to understanding of the importance of NRXN1 in neural development.
Keywords: Copy number variants, Autism spectrum disorders, NRXN1, Neurodevelopmental disorders, Epilepsy, Microcephaly, Neurexins
Background
Neurexin 1 (NRXN1; 2p16.3) is a member of a small family of proteins, which also includes neurexin 2 and neurexin 3, originally identified as synaptic transmembrane receptors for the black-widow spider toxin α-latrotoxin [1]. Neurexins play a role in synapse maturation by fine-tuning synaptic properties and regulating synaptic transmission through interaction with neuroligins [2] and mediate trans-synaptic interactions that help to shape the synapse [3]. Each neurexin gene (NRXN1-3) produces two major isoforms, α- and β-, with different extracellular but similar intracellular and transmembrane structures [4]. α-NRXNs, which act as synaptic organisers, have six LNS (Laminin G, NRXN, Sex-hormone-binding globulin) domains, with three intercalated epidermal growth factor (EGF)-like domains and have been shown to interact with neurexophilins [5] and LRRTM proteins [6], as well as regulating some calcium channels [7]; β-NRXNs have a single LNS domain, lack EGF-like sequences and contain fewer laminin G domains [8]. Nrxn1 also undergoes extensive alternative splicing, which is temporally and spatially controlled by neuronal activity via calcium/calmodulin-dependent kinase IV signalling [9].
Deletions within NRXN1 have been identified in individuals diagnosed with a range of neurodevelopmental disorders (NDD): including intellectual disability, developmental delay, speech and language delay [10, 11] autism spectrum disorders (ASD) [12–17] schizophrenia [18–21], and when homozygously deleted, early-onset epilepsy [22] or Pitt-Hopkins-like Syndrome [23]. A family with NRXN1 deletions, schizophrenia and type 1 diabetes has been described [24], which is plausible because neurexin 1 is expressed in β -cells of pancreas [25]. Although deletions in NRXN2 have not been reported, rare deletions in NRXN3 have been identified in ASD [26].
The above studies have mainly focused on the detection of CNVs in groups of patients with specific phenotypes. However, it is increasingly recognised that NRXN1 deletions may be risk factors for a variety of clinical disorders. In the present study, we report intragenic NRXN1 deletions detected during diagnostic cytogenetic testing of sequential referrals using a custom aCGH platform including dense coverage of the NRXN1 locus, and describe the phenotype of these patients and the size and position of their deletions.
Materials
Diagnostic referral cases
The tested cohort consisted of patients referred to Guy’s and St Thomas NHS Foundation Trust from regional paediatricians and other health specialists, as well as from genetics centres both in and outside the region (SE Thames). Array CGH analysis was initiated to determine the causes of developmental delay, neurocognitive disability, learning difficulties, behavioural abnormalities or birth defects or to confirm a clinical diagnosis of a suspected syndrome. All patient tests were carried out as part of standard clinical care, either as clinical referrals for array CGH testing following a normal karyotype, or those having array CGH as a first-line test in place of karyotyping. All data were anonymised.
Array CGH analysis
Testing was carried out at a regional cytogenetics CPA accredited laboratory, according to published protocols [27], using an Agilent oligonucleotide 60 K array platform (designs 028469 and 017457) with a total imbalance detection rate of 24%. There are 215 NRXN1 probes on the array, with a median spacing of 4.9 kb.
Genomic data and referral phenotype information was recorded in a clinical database, which at the time of analysis contained 10,397 clinical referrals, including 1368 patients referred for ASD, 360 of whom were female. Copy number variants in this population are available in the Brain and Body Genetics Resource Exchange (BB-GRE; bbgre.org).
Results and discussion
We found 34/10,397 patients with deletions within NRXN1 (0.33% of referrals). The majority of these were within the region of the gene encoding the alpha isoform but two included exons which are part of one of the beta isoforms of NRXN1 (exons numbered 19–24 in the present study). Details of patients and deletions are shown in Table 1, and the positions of the deletions with respect to the exons are shown in Figure 1. The frequency of deletion of each exon is shown in Figure 2. Eighteen of the patients in the present study had at least one exon deleted (0.17%); the first three exons were deleted in seven patients, all with NDD. Patients 10–16 had deletions including exon 4, the most commonly deleted exon; five of these patients had NDD. Patient 16 had a deletion of exon 4 alone and had no noted neurodevelopmental problems, although the age at presentation (see Table 1) was such that neurobehavioural problems would not yet be evident. There is evidence that exon 4 is not expressed in murine brain tissue and therefore isoforms of NRXN1 including exon 4 may not impact on neurodevelopment [28]. We found deletions in more distal exons, from 6 to 19, in patients with developmental delay, autism or severe speech delay. The three patients with deletions of exons which also form part of the beta isoform of NRXN1 (patients 1 [Developmental delay], and 34 [Developmental delay], autism) did not have unusual referral phenotypes. As noted below, patient 34 also has a second potentially pathogenic CNV.
Table 1.
Deletions in the NRXN1 gene in clinical referrals for array CGH analysis in the present study
| Patient | Start (hg19) | Stop (hg19) | Size (kb) | Exons | Inheritance | Other imbalances | Referral indication | Age (yrs) |
|---|---|---|---|---|---|---|---|---|
| 1 | 50,318,520 | 51,260,612 | 942 | 1-20 | Paternal | - | Developmental delay | 3 |
| 2 | 51,116,137 | 51,260,612 | 144 | 1-5 | De novo | - | Developmental delay, failure to thrive, pulmonary stenosis, hearing disorder, microcephaly, short stature, ?Noonan syndrome | 1 |
| 3 | 51,205,906 | 51,260,612 | 55 | 1-3 | N.A. | - | Learning difficulties | 9 |
| 4 | 51,245,074 | 51,260,612 | 16 | 1-3 | N.A. | 13q31.1(85,376,208-86,370,409)x1 | ADHD, learning disability | 41 |
| 5 | 51,245,074 | 51,260,612 | 16 | 1-3 | N.A. | - | Developmental delay | 4 |
| 6 | 51,251,498 | 51,260,612 | 9 | 1-3 | N.A. | - | Delayed (atypical) cognitive development, speech & language development disorder, motor skills development disorder | 1 |
| 7 | 51,251,498 | 51,260,612 | 9 | 1-3 | N.A. | - | Marked developmental delay, marked hypotonia (generalised), hypoplastic nails | 2 |
| 8 | 51,221,421 | 51,230,518 | 9 | (intron 3) | N.A. | - | ?Peutz-Jegher syndrome | 11 |
| 9 | 51,180,561 | 51,199,026 | 18 | (intron 3) | N.A. | - | Developmental delay, autism | 2 |
| 10 | 50,850,691 | 51,153,106 | 302 | 4-7 | N.A. | - | Developmental delay, speech delay | 3 |
| 11 | 50,957,455 | 51,199,026 | 242 | 4-5 | De novo | 7q35(145,650,395-146,558,801)x1 pat (CNTNAP2) | Early-onset epilepsy, myoclonic seizures, speech delay | 5 |
| 12 | 50,937,444 | 51,166,725 | 229 | 4-5 | Maternal | 14q21.1(41,234,592-41,532,307)x1 mat | Developmental delay | 6 |
| 13 | 51,037,104 | 51,153,106 | 116 | 4-5 | N.A. | - | Developmental delay, epilepsy, dystonia, microcephaly, squint | 2 |
| 14 | 51,037,104 | 51,153,106 | 116 | 4-5 | N.A. | - | Autism | 7 |
| 15 | 51,072,302 | 51,172,182 | 100 | 4-5 | N.A. | - | Amenorrhoea, premature ovarian failure | 32 |
| 16 | 51,153,052 | 51,189,385 | 36 | 4 | N.A. | - | Anterior anus | 0 |
| 17 | 50,902,782 | 51,148,567 | 246 | (intron 5) | Maternal | 7q11.23(72,700,414-73,777,326)x1 (Williams Beuren syndrome) | Developmental delay, microcephaly, behaviour problems | 4 |
| 18 | 51,008,023 | 51,122,150 | 114 | (intron 5) | N.A. | - | Developmental delay, speech delay, small mouth, microcephaly | 10 |
| 19 | 51,075,491 | 51,148,567 | 73 | (intron 5) | N.A. | - | Congenital heart defect | 0 |
| 20 | 51,043,498 | 51,109,749 | 66 | (intron 5) | N.A. | - | Developmental delay, speech delay, social communication difficulties | 3 |
| 21 | 51,043,498 | 51,088,201 | 45 | (intron 5) | N.A. | - | Behavioural problems | 6 |
| 22 | 51,021,452 | 51,049,704 | 28 | (intron 5) | N.A. | - | Autism, moderate learning difficulties | 11 |
| 23 | 51,049,645 | 51,066,637 | 17 | (intron 5) | Maternal | - | Dysmorphic, ?bronchiolitis | 0 |
| 24 | 51,049,645 | 51,066,637 | 17 | (intron 5) | Maternal | - | Interuterine growth retardation, pitting oedema, duplex right kidney, undescended right testis, single palmar creases | 0 |
| 25 | 51,100,412 | 51,113,311 | 13 | (intron 5) | N.A. | - | Myoclonic epilepsy episodes since six weeks of age | 0 |
| 27 | 50,902,782 | 50,943,419 | 41 | (intron 5) | Paternal | - | Speech delay, social communication difficulties | 5 |
| 28 | 50,982,113 | 51,003,663 | 22 | (intron 5) | N.A. | 22q11.21(18,896,971-21,377,825)x3 (22q11.2 duplication syndrome) | Autism | 10 |
| 29 | 50,982,113 | 51,003,663 | 22 | (intron 5) | N.A. | Dysplastic kidneys, ventral-septal defect,cryptorchidism | 0 | |
| 30 | 50,918,448 | 50,933,351 | 15 | (intron 5) | Maternal | 22q11.21(18,896,972-21,440,514)x3 dn (22q11.2 duplication syndrome) | Epilepsy with focal seizures | 11 |
| 31 | 50,943,360 | 50,957,514 | 14 | (intron 5) | N.A. | - | Learning difficulties, ADHD, autism | 7 |
| 32 | 50,505,606 | 50,909,824 | 404 | 6-18 | De novo | - | Developmental delay, severe speech delay | 5 |
| 26 | 50,775,890 | 51,037,163 | 261 | 6-10 | De novo | - | Developmental delay, hypotonia, speech delay | 6 |
| 33 | 50,744,594 | 50,831,617 | 87 | 10-13 | N.A. | - | Developmental delay | 1 |
| 34 | 50,450,675 | 50,600,302 | 150 | 19 | N.A. | 8q23.3(113,960,008-114,131,155)x1 (CSMD3) | Developmental delay, autism | 4 |
“Patient” refers to numbering in the text. “Inheritance” indicates if the imbalance was inherited (and if so from which parent, or de novo. N.A. is not assessed). Age (yrs) is age at testing.
Figure 1.
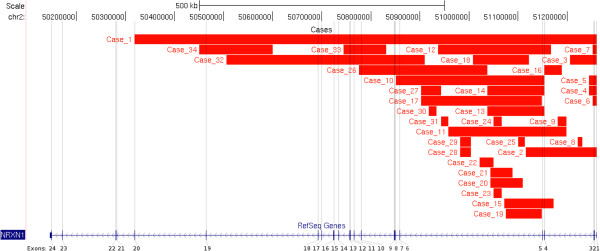
Deletions in the NRXN1 gene (HG19) in the present series. Exons are numbered according to refseq (Homo sapiens neurexin 1 (NRXN1), transcript variant alpha2, mRNA NCBI Reference Sequence: NM_001135659.1). Two of our patients have deletions that overlap with the beta isoform, patients 1, 34.
Figure 2.
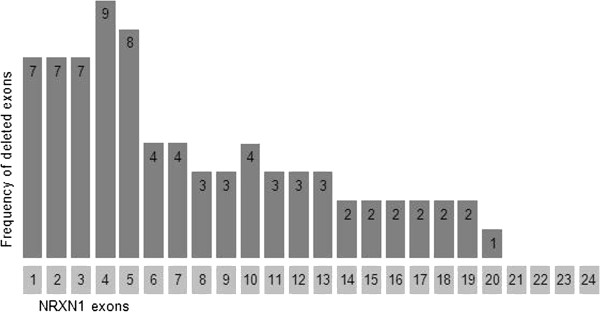
Frequency of exonic deletions in the present series.
NDDs were present in 27 out of the 34 patients, and included developmental delay, learning difficulties, ADHD, autism, speech delay, social communication difficulties, epilepsy, behaviour problems and microcephaly; of the remaining seven patients, five were below the age of 1 year, and would not necessarily be expected to present with such clinical features; follow-up studies on these children may provide further information, Sixteen of the patients had intronic deletions, two with deletions within intron 3 (one with NDD) and ten with intron 5 deletions (seven with NDD). Two of our subjects (19 and 29), both with deletions in intron 5, had cardiac defects. Three patients with intronic deletions had other, syndromic imbalances (Williams-Beuren syndrome (patient 17) and 22q11.2 duplication syndrome (patients 28 and 30)). Patient 11 (exons 4–5 deletion) also had a paternally inherited CNTNAP2 deletion and patient 34 (exon 19 deletion) had an intragenic deletion of CSMD3. Two other patients (4 and 12) carried additional copy number variants of uncertain significance.
Our present study describes a large series of NRXN1 deletions identified through a clinical genetics service. This data adds to the understanding of the genotype/phenotype correlation of NRXN1 deletions. Previously, Rujescu et al. [21] described the detection of twelve NRXN1 deletions in 2,977 patients with schizophrenia, of which seven (0.24% of the patient population) were exonic. These predominantly affected the 5’ exons of the gene, consistent with ablation of the alpha-isoform being the most common type of pathogenic mutation in NRXN1. They also found that ~ 0.14% of normal controls carried intronic NRXN1 deletions, and since this frequency did not differ significantly from that of intronic deletions in cases, they concluded that these were not likely to be pathogenic. Subsequently, Ching et al. [10] reported 12 exonic NRXN1 deletions in a group of 3,540 patients referred for array CGH testing for a variety of different phenotypes, giving a prevalence of 0.34% in this group. A further recent study [29] described 24 patients with intragenic deletions of NRXN1. Seventeen of these deletions involved exons of NRXN1, including four cases with involvement of exons encoding β-neurexin.
In the present study, 18 deletions in the case series were exonic (including two deletions which encompass both the alpha and beta isoforms of NRXN1), and the remainder intronic. The lower prevalence of exonic deletions in our study group (0.17%) than that of other studies [10, 20, 21] may reflect the wide range of referral indications in our cohort, and is very similar to the overall prevalence of 14/8798 (0.16%) calculated from combining totals from a number of published studies of schizophrenia [19].
The rate of intronic deletions we found in our case sample is similar to that seen by Rujescu et al. [21], and although approximately two thirds of our 13 patients with “intronic only” NRXN1 deletions had overt neurodisability phenotypes, this is most likely a result of the nature of the clinical referral population, as about two-thirds of referrals have this class of phenotype. For three of the patients with intronic NRXN1 deletions in our cohort, a second CNV was present (one Williams-Beuren deletion, and two 22q11.2 duplications), likely to have been contributory factors in their neurodisability.
However it is still possible that some intronic deletions in NRXN1 are pathogenic through the deletion of essential regulatory elements in the gene, such as alternative promoters, enhancers, or sequences involved in the complex splicing which generates various isoforms of NRXN1 mRNA. For example, Iijima et al., [9] found that alternative NRXN1 splicing (inclusion of the cassette exon 20; this is exon 21 using the refseq numbering in the present study) is dependent on the presence of AU-rich recognition sequences for SAM68 and related RNA-binding proteins in the flanking introns, and that the deletion of these has functional consequences. However we did not identify mutations encompassing the exon-21 introns in the present study.
There are many examples of pathogenic intronic deletions in the literature. For example intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria [30], in the PKD1 gene cause Rothmund-Thomson syndrome [31], and in the dihydropyrimidine dehydrogenase gene cause 5-fluorouracil toxicity [32]. Several mechanisms that could account for this [33], including deletion of an unknown exon, deletion of an alternative promoter and effects on regulatory sequences such as those controlling splicing, causing intron retention, exon skipping and cryptic splice site activation through the deletion of cis-acting elements, or constraining the intron size to below that required for proper splicing [34].
In the present study, while we did not find deletions extending as far as the exon-21 flanking introns sequences, our finding of intron 5 deletions in 14 affected individuals is interesting. The potential importance of intron 5 deletions has previously been suggested by Ching and colleagues, who reported a de novo intron 5 deletion in an individual with PDD-NOS [10]. In order to show that intron 5 deletions are pathogenic, a case–control analysis and demonstration of the presence of functional sequence would be required.
We also attempted to determine how many of the NRXN1 mutations we found were de novo in origin. In only eleven of our cohort could inheritance be established; of these, four had apparently arisen de novo; however, the neurocognitive status of the carrier parents of the other seven patients was not known.
It has also become evident that a subset of patients with neurodevelopmental disorders may have more than one pathogenic mutation [35]. In the present study, patient 11 had a paternally inherited contactin-associated protein 2 (CNTNAP2) deletion in addition to a de novo deletion of exons 4 to 5 of NRXN1. CNTNAP2 is a member of the neurexin superfamily, and deletions of CNTNAP2 have also been associated with NDDs [11, 36]; it is therefore entirely consistent that compromise of both NRXN1 and CNTNAP2 would lead to a severe NDD, as seen in our case (early-onset epilepsy, myoclonic seizures and speech delay). Heterozygous deletions of either NRXN1 or CNTNAP2 have been found to be associated with severe intellectual disability [11]. Patient 34 had developmental delay and autism, and in addition to an exon 19 NRXN1 deletion, also carried a chromosome 8 deletion resulting in intragenic deletion of CSMD3, a gene whose function is currently unknown, but which has been shown to be expressed in adult and fetal brain [37]. Floris et al. [38] described two patients with autistic disorder and balanced translocations with breakpoints close to CSMD3 suggesting that this is a candidate gene for autism. Apart from infants less than 1 year old, only one patient (patient 15) had an exonic deletion with no reported neurodevelopmental problems: this patient was tested for amenorrhea and premature ovarian failure, and had a deletion of exons 4 to 5, suggesting that this specific deletion may not result in fully penetrant functional compromise of NRXN1.
Pleiotropy is also an important feature of most pathogenic copy number mutations [39]. NRXN1 has been associated with a wide range of disorders, including schizophrenia, autism, epilepsy, and intellectual disability. The reasons behind this phenotypic variability are unknown; however, the presence of a second hit - mutations in other genes in brain development pathways - is very likely to modulate the effects of NRXN1 deletions, and environmental factors may also play a part in the differential expression and penetrance of NDDs. Further insight into this process will be obtained from genome sequencing, which will be able to identify ‘second hits’ including point mutations and other rare variants.
Conclusions
In the present study, we found a series of exonic deletions in the NRXN1 gene in patients referred for clinical diagnostic cytogenetic analysis. These deletions were found in patients with a range of neurodevelopmental disorders, including attention deficit hyperactivity disorder (ADHD), which until recently was not considered to have a genomic basis. NRXN1, like all neurexins, has two main isoforms, alpha and beta [4]. In common with other structural studies of NRXN1, our series of deletions predominantly affect the alpha isoform. Ullrich et al. [40] and Rowen et al. [41] have suggested that the presence of alternative promoters and the process of alternative splicing leads to the production of many different NRXN1 proteins. The structure and functional importance of these different proteins has not yet been elucidated, and further information on the phenotypic consequences, or lack thereof, of different NRXN1 deletions is therefore essential to the understanding of NDDs, and thence to the processes leading to normal brain development and function. Our data adds a further 18 exonic and 16 intronic deletions to the catalogue of previously published NRXN1 deletions. Interestingly, we found a high frequency of intron 5 deletions in our cohort; this finding may be worthy of further investigation.
Acknowledgements
The Brain and Body Genetic Resource Exchange (BB-GRE) is a service developed thanks to the strategic support from the National Institutes of Health Research, England (NIHR) comprehensive BRC at Guy’s and St Thomas, and the specialist Mental Health BRC at SLaM (http://www.slam.nhs.uk/research/biomedical-research-centre/brc-home). DAC is supported by PsychCNVs (Grant agreement number HEALTH-2007-2.2.1-10-223423).
Abbreviations
- NRXN1
Neurexin 1
- NRXN2
Neurexin 2
- NRXN3
Neurexin 3
- aCGH
Array comparative genome hybridisation
- HG19
The February 2009 human reference sequence (GRCh37/HG19).
Footnotes
Competing interests
The author declare that they have no competing interest.
Authors' contributions
SC, JWA, DAC, CMO conceived and designed the study, performed the data analysis and interpreted the results. SC, JWA, HG, DAC, CMO contributed to the drafting and/or editing of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Sarah Curran, Email: sarah.curran@kcl.ac.uk.
Joo Wook Ahn, Email: joowook.ahn@nhs.net.
Hannah Grayton, Email: hannah.grayton@kcl.ac.uk.
David A Collier, Email: david.collier@kcl.ac.uk.
Caroline Mackie Ogilvie, Email: Caroline.Ogilvie@genetics.kcl.ac.uk.
References
- 1.Lelyanova VG, Thomson D, Ribchester RR, Tonevitsky EA, Ushkaryov YA. Activation of alpha-latrotoxin receptors in neuromuscular synapses leads to a prolonged splash acetylcholine release. Bull Exp Biol Med. 2009;147:701–703. doi: 10.1007/s10517-009-0600-5. [DOI] [PubMed] [Google Scholar]
- 2.Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC. Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- 3.Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 4.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Missler M, Hammer RE, Sudhof TC. Neurexophilin binding to alpha-neurexins. A single LNS domain functions as an independently folding ligand-binding unit. J Biol Chem. 1998;273:34716–34723. doi: 10.1074/jbc.273.52.34716. [DOI] [PubMed] [Google Scholar]
- 6.Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudanova I, Sedej S, Ahmad M, Masius H, Sargsyan V, Zhang W, Riedel D, Angenstein F, Schild D, Rupnik M, Missler M. Important contribution of alpha-neurexins to Ca2 + −triggered exocytosis of secretory granules. J Neurosci. 2006;26:10599–10613. doi: 10.1523/JNEUROSCI.1913-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reissner C, Klose M, Fairless R, Missler M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci U S A. 2008;105:15124–15129. doi: 10.1073/pnas.0801639105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, Scheiffele P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell. 2011;147:1601–1614. doi: 10.1016/j.cell.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ching MS, Shen Y, Tan WH, Jeste SS, Morrow EM, Chen X, Mukaddes NM, Yoo SY, Hanson E, Hundley R. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:937–947. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregor A, Albrecht B, Bader I, Bijlsma EK, Ekici AB, Engels H, Hackmann K, Horn D, Hoyer J, Klapecki J. Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med Genet. 2011;12:106. doi: 10.1186/1471-2350-12-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wisniowiecka-Kowalnik B, Nesteruk M, Peters SU, Xia Z, Cooper ML, Savage S, Amato RS, Bader P, Browning MF, Haun CL. Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:983–993. doi: 10.1002/ajmg.b.31064. [DOI] [PubMed] [Google Scholar]
- 17.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zahir FR, Baross A, Delaney AD, Eydoux P, Fernandes ND, Pugh T, Marra MA, Friedman JM. A patient with vertebral, cognitive and behavioural abnormalities and a de novo deletion of NRXN1alpha. J Med Genet. 2008;45:239–243. doi: 10.1136/jmg.2007.054437. [DOI] [PubMed] [Google Scholar]
- 19.Kirov G, Gumus D, Chen W, Norton N, Georgieva L, Sari M, O'Donovan MC, Erdogan F, Owen MJ, Ropers HH, Ullmann R. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet. 2008;17:458–465. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 20.Vrijenhoek T, Buizer-Voskamp JE, Van der Stelt I, Strengman E, Sabatti C, Geurts Van Kessel A, Brunner HG, Ophoff RA, Veltman JA. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am J Hum Genet. 2008;83:504–510. doi: 10.1016/j.ajhg.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rujescu D, Ingason A, Cichon S, Pietilainen OP, Barnes MR, Toulopoulou T, Picchioni M, Vassos E, Ettinger U, Bramon E. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison V, Connell L, Hayesmoore J, McParland J, Pike MG, Blair E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A. 2011;155A:2826–2831. doi: 10.1002/ajmg.a.34255. [DOI] [PubMed] [Google Scholar]
- 23.Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MA, Ekici AB, Reis A. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85:655–666. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suckow AT, Comoletti D, Waldrop MA, Mosedale M, Egodage S, Taylor P, Chessler SD. Expression of neurexin, neuroligin, and their cytoplasmic binding partners in the pancreatic beta-cells and the involvement of neuroligin in insulin secretion. Endocrinology. 2008;149:6006–6017. doi: 10.1210/en.2008-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duong L, Klitten LL, Moller RS, Ingason A, Jakobsen KD, Skjodt C, Didriksen M, Hjalgrim H, Werge T, Tommerup N. Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:354–358. doi: 10.1002/ajmg.b.32036. [DOI] [PubMed] [Google Scholar]
- 26.Vaags AK, Lionel AC, Sato D, Goodenberger M, Stein QP, Curran S, Ogilvie C, Ahn JW, Drmic I, Senman L. Rare Deletions at the Neurexin 3 Locus in Autism Spectrum Disorder. Am J Hum Genet. 2012;90:133–141. doi: 10.1016/j.ajhg.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahn JW, Mann K, Walsh S, Shehab M, Hoang S, Docherty Z, Mohammed S, Mackie Ogilvie C. Validation and implementation of array comparative genomic hybridisation as a first line test in place of postnatal karyotyping for genome imbalance. Mol Cytogenet. 2010;3:9. doi: 10.1186/1755-8166-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mozhui K, Wang X, Chen J, Mulligan MK, Li Z, Ingles J, Chen X, Lu L, Williams RW. Genetic regulation of Nrnx1 expression: an integrative cross-species analysis of schizophrenia candidate genes. Transl Psychiatry. 2011;1:e38. doi: 10.1038/tp.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaaf CP, Boone PM, Sampath S, Williams C, Bader PI, Mueller JM, Shchelochkov OA, Brown CW, Crawford HP, Phalen JA. Eur J Hum Genet. 2012. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91:4022–4027. doi: 10.1210/jc.2005-2840. [DOI] [PubMed] [Google Scholar]
- 31.Wang LL, Worley K, Gannavarapu A, Chintagumpala MM, Levy ML, Plon SE. Intron-size constraint as a mutational mechanism in Rothmund-Thomson syndrome. Am J Hum Genet. 2002;71:165–167. doi: 10.1086/341234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Kuilenburg AB, Meijer J, Mul AN, Meinsma R, Schmid V, Dobritzsch D, Hennekam RC, Mannens MM, Kiechle M, Etienne-Grimaldi MC. Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Hum Genet. 2010;128:529–538. doi: 10.1007/s00439-010-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. 2005;42:737–748. doi: 10.1136/jmg.2004.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Yang Z, Kibukawa M, Paddock M, Passey DA, Wong GK. Minimal introns are not "junk". Genome Res. 2002;12:1185–1189. doi: 10.1101/gr.224602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA, Faas BH, Knoers NV, Cahn W, Kahn RS, Edelmann L. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry. 2008;13:261–266. doi: 10.1038/sj.mp.4002049. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu A, Asakawa S, Sasaki T, Yamazaki S, Yamagata H, Kudoh J, Minoshima S, Kondo I, Shimizu N. A novel giant gene CSMD3 encoding a protein with CUB and sushi multiple domains: a candidate gene for benign adult familial myoclonic epilepsy on human chromosome 8q23.3-q24.1. Biochem Biophys Res Commun. 2003;309:143–154. doi: 10.1016/S0006-291X(03)01555-9. [DOI] [PubMed] [Google Scholar]
- 38.Floris C, Rassu S, Boccone L, Gasperini D, Cao A, Crisponi L. Two patients with balanced translocations and autistic disorder: CSMD3 as a candidate gene for autism found in their common 8q23 breakpoint area. Eur J Hum Genet. 2008;16:696–704. doi: 10.1038/ejhg.2008.7. [DOI] [PubMed] [Google Scholar]
- 39.Grayton HM, Fernandes C, Rujescu D, Collier DA. Copy number variations in neurodevelopmental disorders. Prog Neurobiol. 2012;99:81–91. doi: 10.1016/j.pneurobio.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Ullrich B, Ushkaryov YA, Sudhof TC. Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron. 1995;14:497–507. doi: 10.1016/0896-6273(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 41.Rowen L, Young J, Birditt B, Kaur A, Madan A, Philipps DL, Qin S, Minx P, Wilson RK, Hood L, Graveley BR. Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]