

Abstract
Liposomes are vesicular structures made of lipids that are formed in aqueous solutions. Structurally, they resemble the lipid membrane of living cells. Therefore, they have been widely investigated, since the 1960s, as models to study the cell membrane, and as carriers for protection and/or delivery of bioactive agents. They have been used in different areas of research including vaccines, imaging, applications in cosmetics and tissue engineering. Tissue engineering is defined as a strategy for promoting the regeneration of tissues for the human body. This strategy may involve the coordinated application of defined cell types with structured biomaterial scaffolds to produce living structures. To create a new tissue, based on this strategy, a controlled stimulation of cultured cells is needed, through a systematic combination of bioactive agents and mechanical signals. In this review, we highlight the potential role of liposomes as a platform for the sustained and local delivery of bioactive agents for tissue engineering and regenerative medicine approaches.
Keywords: liposomes, scaffolds, delivery systems, bioactive agents, stem cells
1. Introduction
Lipids are hydrophobic or amphiphilic small molecules [1]. Therefore, they can be classified as fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids, saccharolipids and polyketides [2,3]. The amphiphilic nature of some lipids allows them to form organized structures such as vesicles or membranes, when immersed in an aqueous environment. They can be extracted from plant- or animal-derived tissues by low-polarity solvents such as chloroform.
Lipids play a vital role in physiological and pathophysiological events of living systems [4]. It is believed that life started when nucleic acids were enclosed within a membrane. The biological membrane separates nucleic acids from the external environment and controls the transfer of information and the transport of ions and molecules between the inside and outside of the cellular membrane. A cell membrane is a complex and dynamic system which consists of two lipid molecules held together by hydrophobic interactions, and self-assembled as a continuous bilayer with proteins embedded within the membrane or transiently associated with it (figure 1) [6].
Figure 1.

A cell membrane is a fluid with various proteins attached to the lipid bilayer. Adapted from [5]. (Online version in colour.)
This dynamic system is obviously necessary for life. The inner part of the cell membrane aggregates the hydrophobic moieties of the lipids, and the outside is hydrophilic. Integral proteins are usually in the hydrophobic core of the bilayer [5]. Peripheral proteins are bound to the surface of the membrane. The fluid mosaic model hypothesizes that the plasma membrane and organelle membranes consist of proteins embedded in a fluid phospholipid bilayer. The position of proteins is not static. Like the phospholipids in the bilayer, membrane proteins are in constant motion [5]. Therefore, the cell membrane allows the chemical reactions to occur much more efficiently in an enclosed area and protects the genetic information. The main lipid biosynthetic organelle is the endoplasmic reticulum which produces the bulk of the structural phospholipids and cholesterol [7]. The most important lipid function in the organism is their role in the plasma membrane.
Glycerophospholipids, or phospholipids, are key components of the lipid bilayer of cells [8–10]. Phospholipids, together with membrane proteins and cholesterol, are involved in many cell functions such as in the control of cell shape, compartmentalization, the storage of compounds, ion transport, metabolism, cell signalling processes and cell fusion processes [10]. Phospholipids consist of a glycerol which is linked to a phosphate group (PO42−) and to two fatty acids. In some cases, the phosphate group is bonded to another small organic molecule, such as a choline. Figure 2 shows an example of a phospholipid and the main parts of its composition. Further details of lipids and their properties will be discussed in §2.1.
Figure 2.
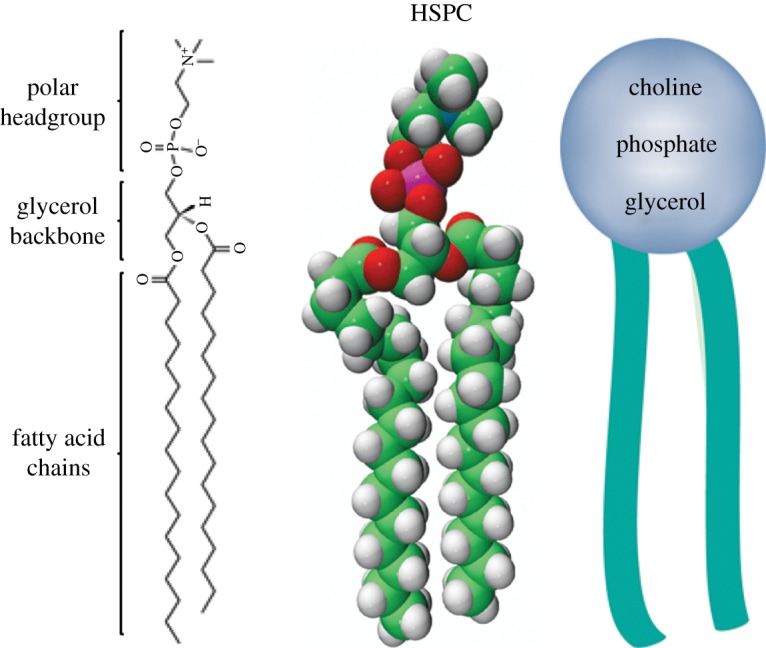
Chemical, three-dimensional and schematic structure of L-α-phosphatidylcholine, hydrogenated (soy) (HSPC) composed of fatty acid chains, glycerol backbone and the headgroup (choline). Adapted from [11]. (Online version in colour.)
Phospholipids may be classified as natural or synthetic. Natural phospholipids may be obtained from various sources such as soya bean or egg yolk. In terms of the polar headgroups, phospholipids are classified as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidylglycerol (PG) and phosphatidic acid (PA). PC and PE are the most abundant phosphatides in plants and animals and are also the most used to produce liposomes [12]. However, natural phospholipids are less stable than the synthetic phospholipids [13]. Synthetic phospholipids can be produced from natural lipids. The modification of the non-polar and polar regions of phospholipid molecules allows the creation of an unlimited variety of well-defined and characterized phospholipids [13]. Examples of synthetic lipids are dipalmitoyl phosphatidylcholine (DPPC), dimyristoyl phosphatidylcholine (DMPC), distearoyl phosphatidylcholine (DSPC) and hydrogenated soy phosphatidylcholine (HSPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), (1-palmitoyl-2-stearoyl(5-DOXYL)-sn-glycero-3phosphocholine (SLPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DSPG), 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG) and 1,2-distearoyl-sn-glycero-3-phosphate (DSPA).
Sterol lipids, such as cholesterol (Chol) and its derivatives, are hydrophobic lipids that have an important role in the animal cell membrane [7]. Steroids are a family of lipids distinguished by the four-ring structure shown in figure 3. The various steroids differ from one another by the functional groups or side groups attached to those rings. Chol is distinguished by a hydrocarbon ‘tail’ formed of isoprene subunits. It is an important component of plasma membranes in many organisms. In mammals, it is also used as the starting point for the synthesis of several of the signalling molecules called hormones. Oestrogen, progesterone and testosterone are examples of hormones derived from Chol [14].
Figure 3.
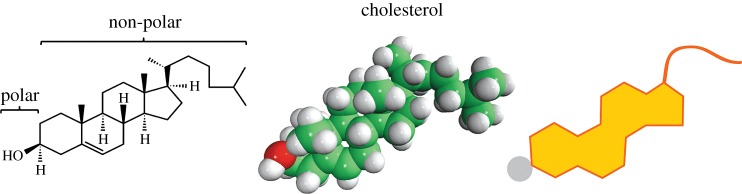
Chemical, three-dimensional and schematic structure of cholesterol (Chol). Adapted from [14]. (Online version in colour.)
Compounds that contain both hydrophilic and hydrophobic elements are referred as amphiphilic. The amphiphilic nature of phospholipids is of utmost importance for their biological function [14]. Specifically, this structure is responsible for their presence and functional role in cellular membranes. Phospholipids spontaneously self-assemble into ordered lyotropic liquid-crystalline phases in the presence of water [10]. The formation of these lipid-based structures depends on the phospholipid intrinsic factors, such as the nature and size of the lipid headgroup and the length and degree of unsaturation of the acyl chains, and on the extrinsic factors, such as temperature, pH, concentration and the presence of solutes and other lipids [7,10]. Examples of lipid-based structures are monolayers, lipid bilayers, micelles, liposomes and tubules [7,15]. Herein, we will review the use of lipids to form liposomes and discuss some of their applications, mainly focusing in tissue engineering (TE) and regenerative medicine.
2. Liposomes
Liposomes are self-assembled vesicles that have the ability to encapsulate aqueous solutions and hydrophobic compounds. They are considered the oldest nanocarrier systems, which were first discovered in the mid-1960s by A. D. Bangham [16]. Bangham performed experiments to determine how lipids behave when immersed in water. When transmission electron microscopes became available, Bangham was able to obtain high-resolution images of the lipid–water mixtures. The images showed that lipids form water-filled vesicles, resembling cells, which he called liposomes [14]. For further details about the development of liposomes, the reader is directed to a recent review that cites the significant contributions of the early pioneers in the liposome field [17]. Since their discovery, a large number of studies have been carried out to understand their biophysical and biochemical properties, and possible applications. They have been used as: model membrane systems to study the basic nature of cell membranes [6], in biochemistry and molecular biology [6,18], in analytical methods [18], in microfluidic technologies [19], as a template for the production of nanogels [20], in cosmetics and food technology [21,22], in imaging [23], as drug delivery systems in pharmacology [24–26], and in TE [27]. In this section, we will revisit the liposome properties, their formulation/functionalization, preparation methods and stability.
2.1. Liposome properties
Liposomes are spherical lipid bilayers with diameters ranging from the nanometre to the micrometre scale [28]. Liposomes have different advantages when compared with other alternative carrier systems [29,30]. One of the main advantages is the fact that they are made of natural materials, i.e. lipids, and they can be easily synthesized in the laboratory.
The properties of the liposomes are mainly dependent on the characteristics of the lipids. A phospholipid has a headgroup, a glycerol backbone and two fatty acid chains (the so-called tails), as described above and depicted in figure 2. One of the oxygen groups of phosphoric acid may be esterified to a variety of organic molecules including glycerol, choline, ethanolamine, serine and inositol. Examples of negative lipids are PG, PS, PI and PA. Other lipids, such as 1,2-dioleoyl-3-dimethylammonium-propane (DODAP) and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), are mixed with neutral phospholipids to produce positively charged liposomes [3]. The nature of the phospholipid is also related to the length of the fatty acid chains. Therefore, the fatty acids differ in the number of carbon atoms and the degree of unsaturation [3]. When a double bond exists between two carbon atoms (C = C) in a hydrocarbon chain, the chain is said to be unsaturated, whereas hydrocarbon chains without double bonds (i.e. C – C) are said to be saturated. The length and degree of saturation of the lipid chain influence the gel liquid-crystalline phase transition temperature, Tc (table 1). Considering the number of carbon atoms, the fatty acid can be named as lauric (C12), myristic (C14), palmitic (C16) and stearic (C18). In general, unsaturated fatty acids occur in natural phospholipids such as PC. DPPC, DMPC, DSPC and HSPC are the most common synthetic phospholipids.
Table 1.
Crystalline phase transition (Tc) of some phospholipids. Adapted from [31].
| phospholipid | acyl chain length, no. unsaturation | Tc (°C) |
|---|---|---|
| DSPC | 18 : 0, 18 : 0 | 55 |
| HSPC | 16–18 (mixture) | 52 |
| DPPC | 16 : 0, 16 : 0 | 42 |
| POPC | 16 : 0, 18 : 1 | −7 |
| SLPC | 18 : 0, 18 : 2 | −16.7 |
| DOPC | 18 : 1, 18 : 1 | −21 |
| DSPG | 18 : 0, 18 : 0 | 53 |
| DPPG | 16 : 0, 16 : 0 | 41.1 |
| DSPA | 18 : 0, 18 : 0 | 58 |
In aqueous environments, the lipids tend to self-assemble into vesicles. Interactions between themselves, hydrophilic interactions between polar headgroups, van der Waals interactions between hydrocarbon chains and also with water (hydrophilic interactions and hydrophobic effect) lead to the formation of lipid-based structures such as liposomes and micelles (figure 4) [32]. Micelles are tiny droplets created when the hydrophilic heads of phospholipids face the water, and the hydrophobic tails cluster together to hide from the water molecules. Phospholipids with compact and short tails tend to form micelles. Phospholipids with longer tails tend to form liposomes (lipid bilayer with two sheets of phospholipid molecules). The head of the phospholipid could contain highly polar covalent bonds, as well as positive and negative charges. Typically, these charges and polar bonds of the head interact with water molecules, when a phospholipid is placed in a solution, whereas the fatty acid tails of a phospholipid do not interact with water (non-polar), which means that they do not form hydrogen bonds with the hydrocarbon tail [14].
Figure 4.
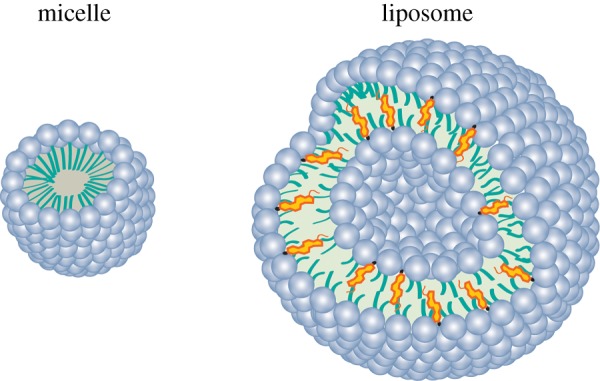
Alternative lipid-based particles: micelle and a liposome. (Online version in colour.)
The relative fluidity and the mobility of each lipid molecule within the bilayer constitute important properties of the liposomes. The lipid bilayer has the tendency to allow a given substance to pass across it—the so-called selective permeability [32]. This means that the internal environment of a liposome can become different from the outer space. Indeed, this capacity to keep different environments between external and internal space is also one of the main characteristics of cells [14,15].
As previously mentioned, the liposomes comprise highly selective membranes (figure 5). Small non-polar molecules move across the lipid bilayer quickly, whereas large molecules and charged substances cross the membrane slowly [14,33]. Additionally, water molecules and ions are also capable of moving across the lipid bilayer. Basically, water moves across lipid bilayers from regions of high concentration to regions of low concentration by osmosis. The solutes move by diffusion from a region of high concentration to a region of low concentration [14]. A comparison between glucose and sucrose permeability indicates that the smaller molecules (glucose) diffuses faster than larger molecules (sucrose) by two orders of magnitudes [33]. Comparing the diffusion of glucose and ions, glucose being a larger molecule, its permeability coefficient is approximately 6.40 to 250 times higher than Cl−, K+ and Na+. The permeability of the lipid bilayer is very sensitive to the charge of the ion, being larger in the case of Cl− by more than one order of magnitude compared with monovalent cations [33]. Moreover, liposomes have low permeability to hydrophilic molecules and high permeability to hydrophobic molecules [17]. More details about the release kinetics of bioactive agents from liposomes will be discussed in the section ‘Release of bioactive agents from liposomes’.
Figure 5.

Selective permeability of lipid bilayers. Adapted from [14]. (Online version in colour.)
The degree of fatty acid saturation also affects the permeability of the lipid bilayers to specific molecules (figure 6) [15]. Because C – H bonds have more free energy than C = C bonds, saturated fats have higher chemical energy than unsaturated fats [14]. The double bonds create spaces among the tightly packed tails. Consequently, lipid bilayers containing many unsaturated fatty acids have more gaps and may be more permeable than bilayers with fewer unsaturated fatty acids [14].
Figure 6.
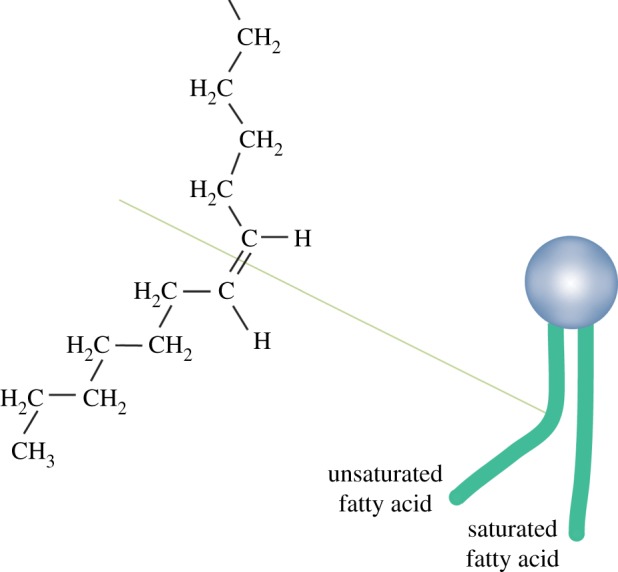
Unsaturated and saturated hydrocarbons. A double bond in a hydrocarbon chain creates spaces in the double layer. Adapted from [14]. (Online version in colour.)
Another parameter that can affect the mobility of the lipids within the bilayer is the temperature [34]. At a given temperature, a lipid bilayer can exist in a gel- or fluid-phase (figure 7). Depending on the lipid Tc, membranes composed of different lipids can exhibit different fluidity levels at the same temperature. The Tc of phospholipids depends on the following: (i) the length of the acyl chain in the lipid; (ii) the degree of saturation of the hydrocarbon chains in the lipid; (iii) the ionic strength of the suspension medium; and (iv) the type of the polar headgroup [36].
Figure 7.

Effect of temperature and Chol on phospholipid bilayer permeability. Chol eliminates the transition phase of the lipid bilayer. The Chol increases the lipid bilayer permeability in the gel phase and decreases it in the fluid phase. Adapted from [15,35]. (Online version in colour.)
Lipid bilayers dominated by phospholipids with long and saturated hydrocarbon tails should be stiffer and less permeable, because the interactions among the tails are stronger, leading to a high Tc. Indeed, hydrophobic interactions become stronger as saturated hydrocarbon tails increase in length [37]. Additionally, longer tail lipids have more area to interact [31,35,38]. Conversely, unsaturated lipids have a significantly lower Tc than saturated lipids [32].
After the formation of the liposomes, the movement of the molecules within and across the lipid bilayer is influenced by the temperature and the structure of the hydrocarbon tails [14]. As illustrated in figure 7, at a temperature below the Tc, phospholipids are in the gel phase, presenting low fluidity and permeability to encapsulated monovalent and divalent cations [15]. At this temperature, individual molecules within the bilayer move slowly. As a result, the hydrophobic tails in the interior of the lipid bilayer pack together more tightly [14]. At a temperature above the Tc, the phospholipids are in a fluid phase and have high fluidity, but also relatively low permeability [15]. Therefore, the individual molecules within the lipid bilayer move rapidly. At a temperature equal to Tc, the lipid bilayer increases the permeability by several orders of magnitude. This phenomenon is attributed to the presence of highly permeable interfacial regions between coexisting gel and fluid bilayer domains [15,39].
As previously described, cholesterol and its derivatives are typically included in the liposome preparation. Therefore, the presence of Chol has a great influence on the properties of the lipid bilayers [31,33]. Specifically, the addition of Chol to a lipid bilayer decreases its fluidity and permeability to water within the fluid phase. The Chol molecule orients itself among the phospholipid molecules, with its hydroxyl group facing the water phase, the tricyclic ring sandwiched between the first few carbons of the fatty acyl chains, into the hydrocarbon core of the lipid bilayer [3]. Because the steroid rings of Chol are dense, adding Chol to a lipid bilayer should increase the density of the hydrophobic section. This decreases the flexibility of the surrounding lipid chains, increases the mechanical rigidity of the fluid bilayers and decreases their lateral diffusion (figure 7). Additionally, Chol can inhibit the crystallization of the hydrocarbon chains of saturated lipids to form a gel-state system [33,36]. It was observed that the effect of Chol in decreasing the permeability of Na+, K+, Cl− and glucose at 36°C is independent of the surface charge and headgroups [33]. The permeability decreased by a factor of 4–18 for the cations, and by only 2 for glucose and Cl−. The authors concluded that Chol affects the process of dissolution more than the process of diffusion [33]. Therefore, the addition of Chol to the lipid bilayer modifies its molecular packing. The lipid bilayer becomes more condensed when compared with pure phospholipids above the Tc, and more fluid when compared with the pure phospholipids below the Tc [33].
2.2. Formulation and functionalization
One of the main concerns in designing a liposome formulation is the stability of the bioactive agent and of the lipid components, which can be affected by the lipid concentration, the environment, pH and temperature, and also their susceptibility to enzyme degradation. Additionally, the liposome formulation depends on the bioactive agent to be encapsulated, the preparation method, the phospholipid composition and the intended application. Liposome formulations composed of phospholipids and Chol are defined as ‘conventional liposomes’ (figure 8a). Conventional liposome pharmacology and tissue distribution depend on the size, surface charge and membrane packing density [31]. For encapsulation of hydrophilic bioactive agents, Chol and saturated phospholipids are the most important factors that allow the membrane permeability to be reduced [31]. Cholesterol is commonly used in combination with phospholipids, because Chol can make liposome membranes stronger, as previously explained. The mole percentage of Chol within the liposome composition is commonly not more than 50% [12]. The hydrophobic interactions of Chol with membrane lipids forces phospholipid headgroups to shield Chol from water [40]. Cholesterol is necessary for the stabilization and maintenance of the bioactive agent in the core of the liposome, and this effect decreases with the increase in the temperature [33].
Figure 8.

Cross section of a liposome: (a) conventional liposome; (b) SSL; (c) ligand-targeted liposome; (d) fluorescent liposome and charged liposomes. (Online version in colour.)
The discovery in 1990 of the steric stabilization was one of the main advances in the development of liposomes [17]. Coating the liposomes with hydrophilic polymers such as polyethylene glycol (PEG) changes their surface properties [41]. The liposome formulations composed of phospholipids, Chol and PEG are defined as ‘sterically stabilized liposomes’ (SSLs), also called ‘stealth’ liposomes, or PEGylated liposomes (figure 8b) [31,42]. It is known that PEG chains grafted onto lipid membranes act like polymeric chains grafted onto solid surfaces, stretching into brushes with increasing surface density. Based on this concept, it is believed that the PEGylated surface provides a steric barrier that prevents the adsorption of proteins onto the liposome surface [31,42]. Numerous formulations of SSLs have been described for the systemic delivery of bioactive agents [25]. However, this surface modification revealed some limitations due to its degradation under mechanical stress as a result of its ether structure and its non-biodegradability, as well as the resulting possible accumulation in the body [43]. Initially, it was believed that SSLs could be non-immunogenic, but an immune response can be elicited after subsequent administrations of PEGylated liposomes, leading to a rapid blood clearance or undesirable side effects [25,44]. Therefore, SSLs should be designed in a way that circumvents those PEG-associated limitations, mainly affecting the blood circulation half-life and the intracellular bioavailability [25,43,45]. A variety of synthetic and natural polymers such as poly(amino acid)s, heparin, dextran and chitosan were also proposed to replace PEG [43].
The effective delivery of bioactive agents towards target cells still represents an enormous challenge. One of the most promising strategies involves the covalent attachment of a ligand at the extremity of PEG chains grafted onto the liposome surface, which is intended to interact with antigens or receptors overexpressed at the surface of the target cells [46,47]. Antibodies and antibody fragments are the most widely used moieties/ligands due to the high specificity for their target antigens [28]. This has led to a new class of nanocarriers called ligand-targeted liposomes [48,49]. The conjugation of antibodies can be done directly either to the lipid bilayer of the liposomes in the presence or absence of PEG chains or to the distal end of the PEG chain (figure 8c). The former, antibody-targeted liposomes are rapidly cleared from the circulation, which limits their in vivo bio-distribution. PEG could eventually be used to overcome this limitation. However, when antibodies are attached at the liposome surface, their antigen binding may be masked by the presence of PEG in the same liposome, especially when longer chain PEG molecules are used. Thus, the latter strategy, coupling of ligands to the terminus of PEG molecules engrafted into the liposome surface, is the most used [17]. Trojan horse liposomes are brain transport vectors that include endogenous peptides, modified proteins and peptidomimetic monoclonal antibodies [49]. These liposomes target specific receptor/transport systems of the brain capillary endothelium and undergo receptor-mediated transcytosis through the blood–brain barrier.
Fluorescent lipids are also used in the liposome formulations (figure 8d). Liposomes carrying encapsulated fluorophores have been synthesized as novel fluorescent markers to image flow profiles in micro-fabricated structures [19]. Fluorescent-labelled lipids are incorporated within the lipid bilayer, and they have the following function: trigger rapid membrane fusion, enable fluorescence imaging of cell membranes and membrane traffic processes [50]. Moreover, negatively and positively charged lipids are also added to the liposome formulation to help in the liposome stabilization and avoid agglomeration [51].
Liposomes of DSPC/Chol (3 : 2) extruded through 400 nm filters were cleared 7.5 times faster than liposomes extruded through 200 nm filters, which in turn are cleared five times faster than small unilamellar vesicles (ULVs) [52]. The clearance of liposomes containing PEG-PE of different sizes (less than 70 nm, 150–200 nm and more than 300 nm) was investigated [53]. It was found that larger liposomes (more than 300 nm) and small liposomes (less than 70 nm) were more rapidly cleared from the circulation than the liposomes of size 150–200 nm [53]. In another study, the clearance was unaffected by the Chol content of the liposomes [54]. Liposomes of different composition with a particle size of about 90 nm were prepared using DSPC, Chol and cholesten-5-yloxy-N-(4-((1-imino-2-d-thiogalactosylethyl) amino) butyl) formamide (Gal-C4-Chol), and labelled with [3H] cholesterol hexadecyl ether [55]. DSPC/Chol/Gal-C4-Chol (60 : 35 : 5) liposomes exhibit extensive hepatic uptake when compared with DSPC/Chol (60 : 40) liposomes [55]. Kawakami et al. [56,57] synthesized a mannosylated cholesterol derivative (Man-C4-Chol) for mannose receptor-mediated gene transfection to macrophages, and they studied the effect of the lipid composition of mannosylated cationic liposomes on in vivo transfection of plasmid DNA (pDNA). pDNA complexed with Man-C4-Chol liposomes showed higher transfection activity than that complexed with conventional cationic liposomes using mouse peritoneal macrophages. Therefore, the transfection efficiency of pDNA complexed with Man-C4-Chol liposomes was inhibited in the presence of mannose, suggesting that the complexes of pDNA and mannosylated cationic liposomes are recognized and taken up by the mannose receptors on macrophages. The liposome formulations, Man-C4-Chol (1/0.5/0.5), Man-C4-Chol/DOPE (3/2) and DOTMA/Chol (1/1), complexed with pDNA-encoding luciferase gene (pCMV-Luc) were compared by intravenous and intra-portal injections in mice. The highest gene expression was observed in the lung using the control cationic DOPE/Chol liposomes with both routes. Man-C4-Chol/DOPE liposome/DNA complexes showed the highest gene expression in the liver after intravenous and intra-portal injection. DOTMA/Chol/Man-C4-Chol liposome showed the highest gene expression in the liver by intravenous injection, but intra-portal injection showed high expression in the lung [56,57].
2.3. Classification
Liposomes could be classified based on the method of their preparation, by the number of bilayers present in the vesicle, or by their size [3]. However, the classification of liposomes by the number of bilayers and size are the most commonly used, rather than by the method of their preparation. Based on the number of bilayers and vesicles, the liposomes are classified as ULVs (25 nm to 1 µm), or multi-lamellar vesicles (MLVs, 0.1–15 µm), or multi-vesicular vesicles (MVVs, 1.6–10.5 µm). Furthermore, based on their size, unilamellar liposomes are classified as large unilamellar vesicles (LUVs, 100 nm to 1 µm) and small unilamellar vesicles (SUVs, 25–50 nm) (figure 9) [18].
Figure 9.
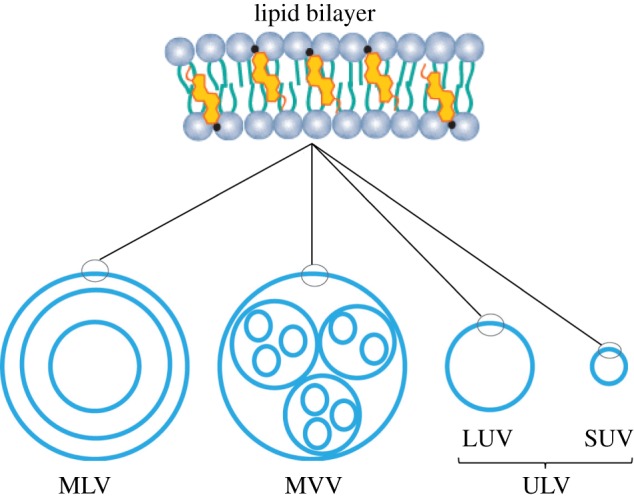
Lipid bilayer structure and types of liposomes: MLVs, MVVs, ULVs. Additionally, ULVs can be sub-classified as LUVs and SUVs. Adapted from [18]. (Online version in colour.)
2.4. Preparation methods
Many reports about the production of liposomes can be found in the literature [3,16,37,58–62]. Common liposome production methods include: thin-film hydration, reverse-phase evaporation, ethanol injection, polyol dilution, freeze–thaw, double emulsions, proliposome method, French press extrusion, detergent removal and high-pressure homogenization [12,37,51]. These methods typically produce LUVs or MLVs, depending on the selected method. Although all these methods can be used to manufacture liposomes, just three of them are usually used [63]: thin-film hydration, reverse-phase evaporation and the ethanol injection method, which are described below. One of the main concerns in liposome manufacturing is the toxicity related to the organic solvents used. Several techniques have been suggested for the removal of detergent and solvent traces from liposomes. These techniques include gel filtration, vacuum, centrifugation and dialysis [51]. A new method for the fast production of liposomes without the use of any hazardous chemicals or processes has been described. This method involves the hydration of the liposome components in an aqueous medium, followed by the heating (up to 120°C) of these components in the presence of glycerol (3% v/v) [64,65]. For more details on this method for liposome preparation, the reader is directed to the review in [51].
2.4.1. Thin-film hydration
The original method of Bangham is the simplest one when compared with the other methods mentioned above [16,61]. It involves the use of volatile organic solvents, mainly chloroform, ether or methanol, to dissolve or solubilize the lipids. The lipids are deposited as a thin film on the bottom wall of a round flask, while the solvent is evaporated by a rotary evaporation technique, under reduced pressure or a nitrogen stream. An aqueous buffer is added to the deposited lipids, allowing their hydration, at a temperature above the Tc of the lipid or of the highest melting component of the mixture [66]. Different MLVs can be produced depending on the hydration time, the re-suspension method, the lipid concentration and composition, and the volume of suspending the aqueous phase [12]. When comparing the thin-film hydration, the reverse-phase evaporation and the ethanol injection methods to prepare cationic liposomes, the results showed that liposomes prepared by the thin-film method were of the best quality and stability [63]. This liposome preparation method also presents some limitations such as low encapsulation ability and the difficulty in producing nano-size liposomes. However, sonication or extrusion through polycarbonate membranes can be employed to obtain ULVs [12,18,51].
2.4.2. Reverse-phase evaporation
In this method, the liposomes are formed from water-in-oil emulsions of phospholipids and buffer, in an excess of an organic phase. The phospholipids are firstly dissolved in organic solvents to form a film, then the solvents are removed by evaporation. The thin film is re-suspended in diethyl ether, followed by the addition of water. The preparation is then sonicated during a brief time period, forming a homogeneous emulsion. The organic solvents are removed under reduced pressure by continued rotary evaporation, resulting in the formation of a viscous gel-like intermediate phase characterized by a LUV dispersion. This method can encapsulate large macromolecules, with high encapsulation efficiency (EE) (20–68%). The main disadvantage of this method is the exposure of the material to be encapsulated to organic solvents and to sonication conditions, which may result in denaturation of sensitive molecules [12,66].
2.4.3. Ethanol injection
In this method, lipids dissolved in ethanol are rapidly injected into a buffer solution, where they spontaneously form SUVs, with a diameter of 30 nm [67]. However, the size of the liposomes can be increased by increasing the lipid concentration [66]. This liposome preparation method has the advantage of avoiding chemical or physical treatment of lipids. However, the concentration of vesicles produced is low and it involves an extra step to remove ethanol [12].
2.5. Liposome characterization techniques
Methods of characterization of liposomes after production and upon storage are required for an adequate quality control [3]. Additionally, the chemical and physical characteristics of the liposomes allow their in vitro and in vivo behaviour to be predicted [31,68,69]. The main properties used to characterize the liposomes include the average diameter and the polydispersity index, the EE, the ratio of phospholipids to drug concentration and the lamellarity determination [69]. The average size and size distribution of liposomes are important parameters especially when the liposomes are intended for therapeutic use by the inhalation or parenteral routes [69]. For instance, small liposomes can pass through the fenestrae of the liver sinusoids and can circulate in the body for long time periods. Conversely, large liposomes are quickly cleared by macrophages [31]. Therefore, the potential therapeutic application of a given liposome is highly influenced by its average size and, consequently, by the possible liposome–cell interactions at the target sites of action.
Table 2 lists some techniques used to physically characterize the liposomes, in terms of the size distribution and zeta potential. Dynamic light scattering (DLS), sometimes referred to as photon correlation spectroscopy or quasi-elastic light scattering, is a technique enabling the measurement of the size of particles in the sub-micrometre range [72]. Typically, particle size measurements are taken between 2 and 5 min, which allows DLS to be classified as a rapid technique. To proceed with DLS analysis, the particles (up to tens of thousands) are suspended in a solution and illuminated by light in order for the particles to scatter light in a given index of refraction, different from that of the suspending solvent [3]. Therefore, liposomes are measured in their natural state, without the need for dehydration or staining.
Table 2.
Liposome physical characterization techniques.
Microscopic techniques such as atomic force microscopy (AFM), environmental scanning electron microscopy (ESEM), transmission electron microscopy (TEM) and confocal laser scanning microscopy (CLSM) can give more information about the nanoscale structures of liposomes. They can provide information regarding shape and morphology (i.e. by AFM and TEM), dimensions (by AFM, ESEM, TEM and CLSM), surface properties (just by AFM) and internal structure (only by CLSM) [70]. When comparing all these microscopy-based techniques, electron microscopy (i.e. ESEM or TEM) requires the specialized freeze fracture technique, which is prone to introduce distortion and artefacts from sample preparation, and the need to have hundreds of photographs in order to provide comparable statistics, constituting time-consuming techniques [69].
The zeta (ζ)-potential is used to measure the intensity of the repulsive electrostatic interaction between naturally charged colloidal particles [73]. Indeed, measurements of zeta potential are commonly used to predict the stability of colloidal systems. If all the particles in a suspension have a large negative or positive zeta potential, they will tend to repel each other and, therefore, there will be no tendency to aggregate. Typically, particles with zeta potentials more positive than +30 mV or more negative than −30 mV are considered stable [69]. However, if the particles have low zeta potential values, then there will be no force to prevent the particles from flocculating [69].
Laser Doppler electrophoresis (LDE) is the most widely used technique to measure the zeta potential. In this technique, a laser is used to provide a light source, illuminating particles within the samples. The incident laser beam passes through the centre of the sample cell and the light is scattered at a determined angle, which is detected. When an electric field is applied to the cell, any particles moving through the measurement volume will lead to fluctuation of the detected light with a frequency proportional to the particle speed. This information is passed to a digital signal processor and, then, the zeta potential is calculated [69]. This technique has been used to investigate how particle size changes as a function of any parameter of the preparation. It is used to monitor the effect of medium changes, e.g. pH, temperature, surfactant, blood serum, adsorption of proteins, and to develop formulations that resist aggregation/flocculation. It also enables the thickness of coatings at the surface of the liposomes to be measured. It has been widely used to predict the effectiveness of the liposomes' coating against opsonization in vivo [74]. Moreover, it gives information about whether the active agent used is encapsulated or adsorbed at the surface of liposomes. For instance, one can obtain information about DNA–liposome interactions, DNA–peptide interactions and DNA condensation during liposomal encapsulation, tracking the changes in size of liposome–drug complexes [74–76]. However, there are several factors that can affect the zeta-potential, such as pH, conductivity and concentration of a formulation component.
2.6. Liposome limitations
Liposomes have been widely used as carriers to encapsulate and to protect bioactive agents from the surrounding environments. Indeed, liposome drug products were the first type of therapeutic nanoparticles being introduced in the market [77]. For instance, Doxil, a doxorubicin-loaded liposome, was approved by the US Food and Drug Administration in 1995 for treatment of Kaposi's sarcoma [77]. In spite of the successful clinical application and of the advantages of this drug carrier, liposomes also present limitations, namely their low stability, low solubility, short circulation half-life, susceptibility to lipid oxidation and hydrolysis, leakage and fusion, reproducibility, difficulties in scaling up, high production costs and sterilization issues [62,78].
Stability is the main concern in all steps of liposome production, storage and administration, and includes the examination of: (i) the chemical stability of the lipids; (ii) the maintenance of the vesicle size and structure; (iii) the retention of entrapped contents; and (iv) the influence of biological fluids on the integrity and permeability properties of the liposomes [66]. Chemical and physical stability are important parameters that affect the biological performance of liposomes [12]. The physical stability of liposomes is mainly related to possible aggregation/agglomeration, fusion and their content leakage to the surrounding environment [31]. Liposomes stored under sterile conditions, in phosphate-buffered saline under nitrogen, can retain their entrapped bioactive agent for extended periods of time [66].The relative retention of a bioactive agent depends on the liposome type (i.e. MLV > reverse-phase evaporation vesicle > SUV), the temperature (i.e. 4°C > 25°C > 35°C) and lipid composition (i.e. saturated phospholipids > saturated phospholipids with equimolar cholesterol > unsaturated phospholipids with equimolar cholesterol > unsaturated phospholipids) [66]. Lipids can suffer auto-oxidation, which is usually induced by light, metal ions or temperature. Additionally, the hydrolysis of the phospholipids to fatty acids and 1- and 2-acyl-lysophospholipids leads to the production of glycerol phosphor compounds and causes chemical degradation of the liposomes during storage [79]. Antioxidants, complexing agents (e.g. EDTA) and inert atmosphere (e.g. nitrogen) are commonly used to avoid hydrolysis of the phospholipids. Other ways to overcome problems concerning the chemical decomposition of liposomes is their storage in a dry state (i.e. freeze dried), using a cryoprotectant. For example, addition of trehalose, which replaces water during the freeze drying, is effective in preventing fusion and dehydration damage in phospholipid vesicles [80].
Initially, it was assumed that, as liposomes were primarily made of natural lipids such as PC, they would avoid the mononuclear phagocyte system clearance and would not be recognized as an antigen. However, it was realized that liposomes would be accumulated in organs such as liver and spleen, and cleared by macrophages (immune system), and the clearance was related to the composition and the size of the liposomes [31]. Therefore, this could be an advantage when those organs or cell populations are the intended target [36]. However, when other organs and cell populations are the target, this represents a main obstacle. Since this discovery, the goal has been to design new liposome formulations to bypass the immune system. Therefore, the physiological conditions of the body should be taken into consideration when designing a liposome formulation for a specific cell or organ target when the liposomes are injected intravenously [29].
Currently, there are numerous methods available for laboratory-scale production (§2.4), but only a few large-scale manufacturing techniques are available [62]. Liposome manufacturing involves numerous unit operations which are not easy to scale up to commercial production levels [78]. Therefore, all the large-scale manufacturing techniques have serious limitations in terms of entrapment of sensitive molecules, exposure to mechanical and/or chemical stress, temperature and sterilization conditions. A detailed review of liposome technology aimed at industrial-scale production can be found in [62]. Currently, there is no established consensus regarding the liposome manufacturing methods. Solvent extraction systems require significant investment and are very expensive to operate and maintain. The most used method to achieve sterility for pharmaceutical products is sterile filtration using a polycarbonate membrane [78]. However, this method has disadvantages; for example, it needs to be performed under aseptic conditions, it is relatively expensive as it operates under high pressure, it is time consuming and it is not effective in removing virus [78].
3. Bioactive agent delivery
3.1. Loading liposomes with bioactive agents
The main advantages of liposomes as a bioactive agent delivery system are: (i) they have a versatile structure which can be tailored for each application; (ii) they can accommodate any type of bioactive agents either in their inner compartment (i.e. hydrophilic molecules) or within the lipid bilayer (i.e. lipophilic molecules) or both (amphiphilic molecules); (iii) their nanoscale properties determine the solubility, diffusivity, bio-distribution and biological fate; (iv) they are minimally toxic, non-immunogenic and fully biodegradable; (v) they are flexible to link with site-specific ligands to achieve active targeting; and (vi) they increase the efficacy and therapeutic index of bioactive agents [12,37]. Therefore, the retention of bioactive agents within liposomes is especially important, not only during storage but also during in vivo administration [17].
The assembling of phospholipids into a hydrated bilayer structure is coupled with the entrapment of a portion of the aqueous medium within the continuous closed bilayer. Therefore, it may seem that the encapsulation of bioactive agents into liposomes should be a trivial process. However, there are some limitations to the encapsulation of molecules into liposomes, such as the type of bioactive agent (i.e. molecular weight and chemical properties), the liposome (i.e. size and lipid composition) and the manufacturing method. Selecting bioactive agents with physical characteristics that make them susceptible to retention into the liposomes is another approach to control the loading and release rate [17]. There are two ways to encapsulate bioactive agents into liposomes: passively, when the bioactive agent is encapsulated during the liposome formation; or actively, when the bioactive agent is encapsulated after liposome formation [37]. The advantage of active encapsulation is that the bioactive agent loading can be performed independently of the time and site of liposome production [3,17]. However, they can only be applied to a small number of bioactive agents with specific physico-chemical properties [12].
Pay load (PL) and EE are the terms used to determine the amount of bioactive agent incorporated into the liposomes. PL is the bioactive agent–lipid (mol mol−1) ratio and EE is the PL in the final liposome formulation compared with the initial PL used for liposome preparation. Sometimes, the EE is used to indicate the percentage of bioactive agent encapsulated in relation to the amount of bioactive agent offered for encapsulation during liposome preparation, but the amount of lipid is not quantified. This quantification method is misleading, as it highly depends on the initial amount of bioactive agent offered to a specific lipid quantity [12] (figure 10).
Figure 10.
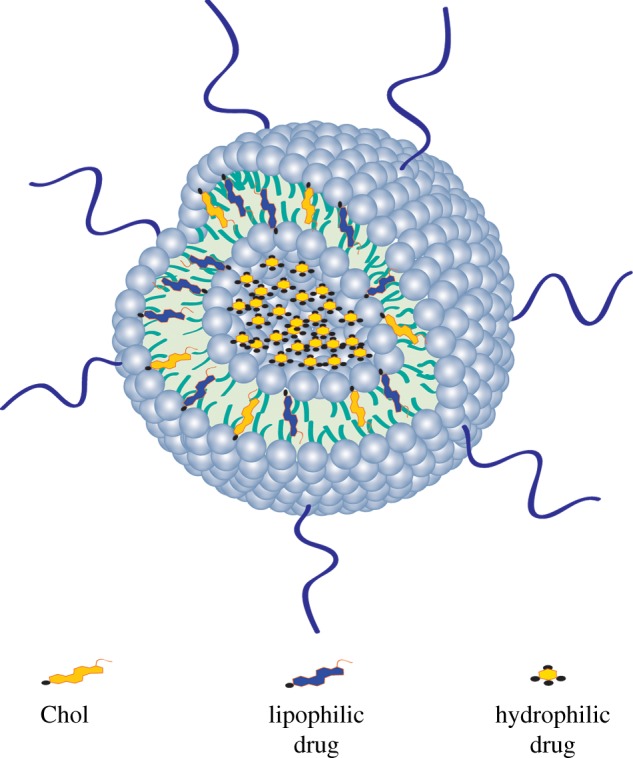
Representation of hydrophilic and lipophilic drug encapsulation into the liposome. Hydrophilic drug is encapsulated in the inner core of the liposome. Lipophilic drug is encapsulated into the lipid bilayer. Chol and the lipophilic drug compete for the same space in the lipid bilayer. (Online version in colour.)
3.1.1. Hydrophilic bioactive agents
These are dissolved in the external aqueous phase, during liposome preparation, and become entrapped within the inner compartment of the liposome after solvent evaporation [81]. Thin-film hydration is the simplest method for the encapsulation of hydrophilic bioactive agents. Encapsulation becomes more difficult and inefficient as the size of the bioactive agent increases, e.g. large plasmids [36]. Additionally, the EE and bioactive agent-to-lipid ratios (i.e. PL) achieved by thin-film hydration are low [36]. The percentage of encapsulation of hydrophilic bioactive agent depends on the size of liposomes: MLVs > LUVs > SUVs [82]. Also, the liposome charge affects the entrapment of hydrophilic bioactive agent: positively charged > negatively charged > neutral liposomes [83]. The preparation of liposomes using unsaturated phospholipids (e.g. DOPC) demonstrated a higher EE than those liposomes using saturated phospholipids (e.g. DPPC) [84]. The EE was more dependent on the number of unsaturated bonds than on the alkyl chain length of the phospholipid molecule [84].
To improve the EE of hydrophilic bioactive agents, it is necessary to use different methods of liposome preparation, such as reverse-phase evaporation, dehydration–rehydration of empty liposomes and freeze–thaw cycling [36]. There are other strategies to increase the EE of hydrophilic bioactive agents into liposomes: by changing the pH and the ions and creating ammonium sulfate gradients (also called active loading) [3,17,85]. The development of these strategies for improving encapsulation and retention of bioactive agents into liposomes relies on experimental studies aimed at understanding the molecular factors governing the barrier properties [85]. For example, bioactive agents that are weak base/acid can diffuse through the lipid membrane and accumulate into the inner compartment of the liposome [85,86]. The ammonium sulfate gradient approach differs from most other chemical approaches as it does not require liposomes with an acidic interior or an alkaline extra-liposome phase [86]. This approach has been used to encapsulate bioactive agents inside the liposomes at high efficiency (more than 90%) [86]. Moreover, to solve the problem of stabilization of bioactive agents, one may increase the rigidity of the liposome membrane, while concurrently decreasing their tendency for aggregation by properly selecting the liposome lipid components [12]. The use of a thin polymeric coating made of chitosan or alginate may be a strategy to increase the stability of bioactive agents into the liposomes [87–89]. However, the polymeric coatings may increase the size of the liposomes and may also interact with the phospholipid bilayer. Another strategy is to covalently cross-link inter-lipid bilayers. For instance, cross-linked MLVs can reduce systemic toxicity and improve therapeutic efficacy [90].
3.1.2. Lipophilic bioactive agents
In order to maximize the association of lipophilic bioactive agents with liposomes, the most common practice is to mix the lipids with the lipophilic bioactive agent and evaporate the solvent to form a thin-lipid film. This mixture is re-hydrated in the buffer and the liposome-associated bioactive agent is separated from the free bioactive agent [66]. The interaction of such bioactive agents within the lipid bilayer depends on the amount and structure of the lipophilic bioactive agent, which results in alterations in the vesicle properties such as permeability, size and stability of the bilayer [91]. The encapsulation of lipophilic bioactive agents decreases as the lipid chain length increases, and may destabilize the liposomes [92]. Conversely, the addition of Chol increases the rigidity and stability of liposome membranes, as was mentioned in the section on ‘Liposome properties’. However, if the bioactive agent is lipophilic, it might be displaced by adding Chol in the lipid bilayer. Therefore, the presence of Chol decreases the encapsulation of lipophilic bioactive agents [91,92]. A system has been proposed to enhance the EE of lipophilic bioactive agents [93]. This consists of combining liposomes and cyclodextrin–bioactive agent complexes by forming bioactive agent-in-cyclodextrins-in-liposomes [93]. Cyclodextrins are hydrophobic inside and hydrophilic outside, cavity-forming, water-soluble oligosaccharides that can accommodate water-insoluble bioactive agents into their cavities, increasing their water solubility. Therefore, the water-soluble bioactive agent–cyclodextrin complex is capable of being encapsulated within the aqueous compartments of the liposomes. An EE of 32.3 ± 11.9% for dehydroepiandrosterone and 31.9 ± 11.8% for hydroxypropyl-β-cyclodextrin was achieved by using this method [12,93].
3.2. Release of bioactive agents from liposomes
Another critical issue when designing liposomes as bioactive agent carriers is the control of the bioactive agents' release, which may dictate their therapeutic safety and efficacy. The release kinetics is affected by the nature of the bioactive agent, the liposome composition, the method of encapsulation (i.e. passive or active) and the intended application [12,94]. To understand the release of bioactive agents from liposomes, we have to understand the structure of the liposome and the possible interactions between the bioactive agent, the lipid bilayer and the surrounding environment. For example, encapsulation of dexamethasone (Dex) is higher in DSPC liposomes than in PC liposomes, and Dex is displaced from liposomes as the Chol content of liposome membranes increases [55,91]. Also, a kinetic release study performed in buffer or serum medium showed that the release of Dex was provoked by the dilution of liposome [91]. In another study, the release profile of Dex from the DSPE–PEG-coated liposomes was performed in phosphate-buffered saline using dialysis tubes for 21 days [55]. The release profile of Dex showed an initial burst release within 12 h. Following the initial release, a slower release was observed until day 6. Afterwards, Dex continued to be released at a slower but steady rate until day 21 [55].
Bioactive agents (i.e. molecules or ions) encapsulated into the liposomes tend to move spontaneously to the outer environment of the lipid bilayer. The movement of molecules and ions that results from their kinetic energy is known as diffusion [14]. Therefore, the bioactive agent tends to move from a region of high concentration to a region of low concentration. This process depends on the type of bioactive agent and the lipid composition [15,36,39]. For instance, when the initial concentration of Ca2+ is high inside the liposome, but low outside, heating to Tc results in immediate diffusion of the Ca2+ into the medium [15,39]. Therefore, phospholipids with high Tc may reduce the diffusion of ions and low molecular weight molecules. The influence of the liposomal composition on release of ibuprofen was investigated [95]. The long alkyl chain lipid enhanced ibuprofen EE and retention at 37°C: dilignoceroyl phosphatidylcholine (C24PC) > DSPC > DMPC > PC. After 30 min of incubation, PC liposomes released 15.5% of the ibuprofen load compared with 1.5% for C24PC liposomes, and major differences in release were evident after 24 h of incubation [95]. At 37°C, both PC (Tc < 0°C) and DMPC (Tc = 23°C) liposomes were in the fluid state resulting in an increasing amount of drug released compared with the higher transition temperature lipids DSPC (Tc = 55°C) and C24PC (Tc = 80°C). Therefore, C24PC liposomes released less ibuprofen than DSPC liposomes despite both systems being in the ordered gel phase. This behaviour may be explained by the increased van der Waals interactions between the longer lipid chains and the increased lipid phase area within the liposomes enhancing the drug binding and bilayer stability [95]. Moreover, the inclusion of charged lipids in the liposome formulations may influence bioactive agents' release. The addition of 2 mol of the anionic lipid dicetylphosphate (DCP) reduced the EE and increased the release of ibuprofen (69.0 ± 3.7%), and this was explained by the electrostatic repulsive forces between the carboxyl group of ibuprofen and the anionic headgroup of DCP [95]. The addition of 2 mol of the cationic lipid stearylamine (SA) to the liposome formulation (PC : Chol—16 mol : 4 mol) increased the EE by approximately 8–47% and also increased the release of ibuprofen (71.2 ± 2.8%) when compared with PC : Chol [95]. The authors suggested that the electrostatic attraction between the positively charged head group in SA and the carboxyl group present in dissociated ibuprofen appears to have little effect on ibuprofen association with MLVs. Moreover, addition of SA to the liposome formulation also resulted in a reversal of surface charge (due to the cationic headgroup of SA) and an increase in vesicle size of approximately 0.8 µm compared with the unmodified PC : Chol formulation. This could be the reason for the increase in the release of the drug. These results indicate that the presence of charged lipids (anionic or cationic) within the liposome bilayer increases the permeability of the lipid bilayer and lipid–drug binding.
A successful treatment using bioactive agent-loaded liposomes depends also on the route of administration (e.g. subcutaneously, orally and intravenously). Conventional liposomes, subcutaneously administrated, aim to target the lymphatic system for imaging, distribution of therapeutic agents or vaccination [12]. Moreover, conventional liposomes may be developed to circumvent the endoplasmic reticular system or to mask the toxic side effects of bioactive agents [12]. Liposomes administered intravenously face barriers such as the endothelial lining of the vasculature and the blood–brain barrier. In those particular situations, it is important to keep the bioactive agent in liposomes until they reach the target site [36]. If the bioactive agent leaks out of the liposome at a rapid rate, it will be lost before reaching the site of action, and no therapeutic benefit will be obtained. On the other hand, if the bioactive agent leaks out of the liposomes slowly, it will be able to reach the site of action, but the levels of released bioactive agent will never reach the desired therapeutic concentrations [36]. The low integrity of liposomes, after in vivo administration and in contact with the blood components, may result in removal of some lipid molecules and consequent opening of pores into the liposome bilayer, through which the loaded bioactive agent may leak out. Also, the physical instability of liposomes that results in aggregation and fusion may release the bioactive agent from liposomes [36]. For example, neutral liposomes tend to aggregate and increase the size of the liposomes [96].
The interaction of liposomes with cells is a critical aspect of bioactive agents' efficacy. In some clinical applications, the bioactive agent has to be released inside the cells (e.g. tumour cells) to have a beneficial therapeutic effect. In vitro and in vivo studies have shown that the main interactions of liposomes with cells are simple adsorption (by specific interactions with cell-surface components, electrostatic forces, or by non-specific weak hydrophobic forces) or following endocytosis [37]. Many attempts to activate the release of bioactive agents from liposomes in the vicinity of or inside the cells have been described [17,97]. Active release relies on developing mechanisms to increase the permeability or to destabilize the liposome bilayer once it reaches the target site. A variety of stimuli such as pH [46,98], temperature [39,99], ultrasonic waves [100], magnetic fields [101,102] and light [103,104] are currently being investigated to improve the target release of bioactive agent. Although the concept of triggered release is very promising, more studies have to be performed to prove its application in humans [17].
4. Applications of liposomes in tissue engineering
In living organisms, the lipids are considered basic building blocks, because they are formed of a polar headgroup, one or more hydrophobic tail regions and a backbone structure that connects the two [15]. Therefore, researchers have been taking advantage of the characteristics and versatility of lipids to find new applications [12,15]. Liposomes and micelles are the most widely used lipid-based nanoparticles [105]. Other lipid structures can be obtained such as cubic-, hexagonal- or sponge-phase structures [106]. They offer the advantage of stability and could be used as novel biomaterials mimicking biological membranes. It is believed that phospholipid-based materials may be increasingly used as tools for the manipulation of cell and tissue responses to biomaterials, namely for the controlled release of bioactive agents and for reconstructive surgery [15].
Nowadays, the most clinically used applications of liposome-based therapy are in the treatment of cancer and of systemic fungal infections [36]. However, the future of liposomes is not limited to those therapeutic applications. Liposomes, being a highly flexible platform, have been used in different areas of research including the production of vaccines, imaging, cosmetics and TE [27,36]. Herein, we will focus on lipids as a biomaterial and review some applications of the liposomes, mainly in TE and regenerative medicine. It is expected that therapeutic nanoparticles, such as liposomes, offer the potential to dramatically improve the effectiveness and side-effect profile of new and existing bioactive agents [77]. Moreover, many tissue-engineered scaffolds have already been approved for human use [27]. Therefore, combining liposomes with scaffolds has the potential for clinical translation in the near future.
4.1. Combining liposomes with scaffolds
The field of biomaterials has been advancing towards the nanoscale design of bioactive systems of drug release or scaffolding, aimed at TE and regenerative medicine strategies [107]. The combination of drug-loaded nanoparticles with scaffolds can be used to spatially and temporally control the release of bioactive agents, leading to a sustained and local delivery [108]. Over recent years, there have been an increasing number of studies aiming to direct stem cell fate through the delivery of growth/differentiation factors, DNA or interference RNA (RNAi). The encapsulation of the bioactive agents into nanoparticles such as liposomes has many advantages, as described in the section ‘Loading liposomes with bioactive agents’. However, the single use of liposomes is limited due to the absence of a three-dimensional mechanical support frequently required to promote tissue regeneration [107].
Many types of scaffolds can be made of natural and/or synthetic polymers, using different processing techniques. For an extensive analysis of the topic, the reader is directed to reviews of TE scaffolding in [109–112]. TE scaffolds can be designed to physically and chemically control the release pattern of any incorporated bioactive agents [27]. Particularly, hydrogels are a set of scaffolds that present an enormous potential for application as smart and stimuli-responsive biomaterials [113]. The delivery of bioactive agents from scaffolds to promote regeneration is often challenging due to the complex fabrication processes. For instance, bioactive agents can be incorporated in the matrix or encapsulated into hollow nanofibres [114]. When the bioactive agent is incorporated in the matrix, there is a strong burst release within the first few hours [115]. Therefore, when the bioactive agent is encapsulated into hollow nanofibres, there is a better control of the bioactive agent release profile, but some of the complications during nanofibre preparation do not allow for easy large-scale production [116].
The combination of liposomes with scaffolds has been already reviewed elsewhere [15,27,108,117]. All these strategies aspire to combine the advantageous properties of liposomes and polymer matrices, aiming at developing materials that can sequester and maintain liposomes at a local tissue site. Stimuli-responsive liposomes have also been used as devices to control chemical reactions, which result in the rapid formation of a biomaterial such as the in situ formation of minerals, polymers or mineral/polymer composite biomaterials [15,118–121]. This exploits the release of entrapped substances from liposomes at temperatures near the lipid Tc (i.e. approx. 37°C) (see figure 7; see also the ‘Liposome properties’ section) [39]. The liposome structure is very sensitive to organic solvents, temperature and pH. Therefore, many ways to immobilize liposomes at the scaffold surface have been proposed [27,122–125]. There are two ways to immobilize liposomes at the surface of the scaffolds: (i) non-specific immobilization, which means that the liposomes are adsorbed at the surface of the scaffold and are easily removed during the cell culture at each medium exchange; (ii) specific immobilization, which means that the liposomes are covalently bound at the surface of the scaffold, increasing their stability. A scaffold system that uses naturally occurring interactions between liposomes and the fibrinogen was used to obviate the need for chemical conjugation [126]. To facilitate liposome adsorption, scaffold surfaces were coated with various extracellular matrix proteins, which were able to transfect a higher number of cells, while at the same time reducing the amount of DNA required [127]. Recently, a chemical modification of electrospun polycaprolactone (PCL) nanofibre meshes (NFMs) was reported enabling the immobilization of bioactive-loaded liposomes onto their surfaces [124,125]. To achieve this, initial UV-ozone irradiation was used to generate reactive free radicals that were immediately subjected to aminolysis. These modified surfaces were reacted with 2IT to generate sulfide (SH) pendant groups. Dexamethasone and pDNA-encoding RUNX2-loaded liposomes were covalently bonded to the SH groups present at the surface of electrospun NFMs [124,125]. The availability of the drug-release vehicle at the surface of the NFMs (where initial cellular contact occurs) enables a sustainable release of the Dex in the vicinity of the cells in culture and, consequently, increases its efficacy and bioavailability [125]. It was concluded that the amount of liposomes immobilized is specifically controlled by the amount of SH groups available at the nanofibres' surface [124,125]. Another strategy to combine liposomes and nanofibres is to use coaxial electrospinning. This technique enables the incorporation of liposomes into nanofibres [128]. Table 3 shows the applications of liposomes combined with scaffolds for TE approaches.
Table 3.
Examples of lipid/liposome-based strategies for bioactive agent release in TE and regenerative medicine approaches. Signal transducers and activators of transcription (STAT), human bone marrow-derived mesenchymal stem cells (hBMSCs), luciferase (Luc), enhanced green fluorescent protein (EGFP), encoding β-galactosidase (βGal), vascular endothelial growth factor (VEGF), human bone morphogenetic protein 2 (hBMP-2), internal ribosome entry site (IRES), Dickkopf-related protein 1 (DKK1), transforming growth factor β1 (TGF), insulin-like growth factor 1 (IGF1), doxorubicin (DOX), model protein lysozyme (LYZ), hydroxyapatite (HA), nerve growth factor (NGF), fibroblast growth factor (FGF), glial cell line-derived neurotrophic factor (GDNF), gene-activated matrix (GAM).
| material | bioactive agent | applications | comments | year/references |
|---|---|---|---|---|
| cell culture substrate | pDNA-Luc-EGFP | gene delivery | absorbed lipoplex transfected primary cells with improved cellular viability relative to bolus delivery | 2005 [127] |
| microRNA and pre-mRNA | neural regeneration | cells transfected with miR-124 using lipofectamine played a key role in the differentiation of progenitor cells to mature neurons | 2007 [129] | |
| siRNA knockdown chordin | bone regeneration | endogenously produced chordin constrains the osteogenic differentiation of human MSCs. MSCs were transfected with siRNA using lipofectamine | 2008 [130] | |
| pDNA-Luc-EGFP | gene delivery | enhancing lipoplex delivery should target the material design to increase internalization | 2009 [131] | |
| pDNA-βGal, pDNA-Luc-EGFP | gene delivery | the presence of the peptide in the lipoplex increased internalization efficiency | 2009 [132] | |
| pDNA-BMP2-VEGF-BMP-2-VEGF | bone regeneration | BMSCs transfected with reconstructed plasmid p-hBMP-2-VEGF-loaded liposomes could secrete a high level of BMP-2 and hVEGF | 2010 [133] | |
| siRNA-STAT3 | bone regeneration | elimination of the STAT3 signalling pathway by the inhibitors, STAT3 siRNA, resulted in the acceleration and augmentation of BMP-induced osteogenic differentiation in MSCs | 2010 [134] | |
| miR-335-5p | bone regeneration | miRNA activates Wnt signalling and promotes osteogenic differentiation by downregulating DKK1 | 2011 [135] | |
| Dex | bone regeneration | Dex-loaded liposomes do not have any cytotoxic effect, and they are able to promote an earlier induction of hBMSC differentiation into the osteogenic lineage | 2012 [55] | |
| chitosan–fibrin matrix | quinacrine | drug delivery system | liposome immobilized in fibrin chitosan matrix released quinacrine for 9 days for neutral liposomes, 5 days for PEGylated liposomes and 17 days after cross linking of the matrix | 2006 [136] |
| chitosan scaffolds | pDNA-TGF-β1 | cartilage regeneration | autologous MSCs transfected with the TGF-β1 gene using lipofectamine, and seeded into chitosan scaffolds filled a defect with regenerated hyaline-like cartilage tissue | 2007 [137] |
| chitosan scaffold–fibrin gel | tirofiban | drug delivery system | the release periods of tirofiban from chitosan scaffold-fibrin gel loaded with liposomes can sustain 20% longer and significantly less burst release than with liposomes | 2008 [138] |
| collagen scaffold | pDNA-IGF-1 | cartilage regeneration | chondrocyte-seeded scaffolds prolonged and elevated IGF-1 expression by using the cross-linking method of plasmid incorporation along with the addition of the lipid transfection reagent | 2007 [139] |
| collagen scaffold | pDNA-GDNF | neural regeneration | combination of scaffold and lipid transfection of MSCs is a promising approach for the long-term production of glial cell line-derived neurotrophic factor | 2008 [140] |
| collagen/HA scaffold | DOX, LYZ, BMP2 | bone regeneration | thiol bisphosphonate-decorated liposomes increased retention in the collagen/HA scaffolds after subcutaneous implantation in rats | 2012 [122] |
| fibrin scaffold | tobramycin | treatment of Pseudomonas keratitis | tobramycin-loaded liposomes immobilized in fibrin scaffold inhibit colonies of Pseudomonas in albino rabbit corneas | 1992 [141] |
| fibrin scaffold | protein horseradish peroxidase | drug delivery | protein-filled liposomes were absolutely stable within the fibrin scaffold | 2000 [142] |
| fibrin clots | TGF-β1 | cartilage regeneration | fibrin containing TGF-β1-loaded liposome released TGF-β1 at a slower rate than the one containing the free TGF-β1 | 2003 [143] |
| fibronectin-coated PLG | pDNA-NGF | neural regeneration | sustained lipoplex release was obtained for up to 50 days, with rates controlled by the fabrication conditions | 2006 [144] |
| fibrin sealant | pDNA-VEGF | wound healing | topical fibrin-mediated administration of a VEGF-A plasmid using lipofectamine may serve as an alternative in treating ischaemic skin flaps | 2007 [145] |
| fibrin hydrogel | pDNA-βGal | gene delivery | cell transfection depended strongly on the local cell-pDNA microenvironment (two dimensions versus three dimensions) | 2009 [146] |
| fibrin scaffold | pDNA-βGal-EGFP | multiple gene delivery | the pDNA-loaded lipoplexes immobilized in fibrin showed significantly higher transfection efficiency for two reporter genes at 7 days than pDNA-loaded lipoplexes alone in a rabbit ear ulcer model | 2009 [126] |
| fibrin scaffold–fibrin microsphere | pDNA | gene delivery | fibrin microspheres were shown to degrade and release DNA differentially compared with fibrin scaffold | 2011 [147] |
| GAM | pDNA-encoding BMP4 | bone regeneration | transfection efficiencies improved in vitro when liposomes where released from resorbable GAM | 2005 [148] |
| PCL/PVA NFM | TGF-β, bFGF, IGF-I | cartilage regeneration | coaxial electrospinning enables the incorporation of liposomes into nanofibres and enhanced MSC proliferation | 2012 [128] |
| PCL NFM | Dex | bone regeneration | Dex-loaded liposomes immobilized at the surface of electrospun PCL NFMs did not exhibit any cytotoxic effect, being able to successfully promote the osteogenic differentiation of hBMSCs | 2013 [125] |
| PHEMA fibrous scaffolds | fetal bovine serum | cartilage regeneration | attached fetal bovine serum-loaded liposomes significantly improved both chondrocyte adhesion and proliferation | 2012 [123] |
| PLG scaffold | pDNA-βGal | spinal cord injury | in vivo, lipoplexes immobilized at PLG scaffolds increased transgene expression levels for at least three weeks in a rat spinal cord hemisection model | 2009 [149] |
| scaffold with tendon cell sheets | collagen V siRNA | tendon regeneration | downregulation of collagen V α1 or α2 chains by siRNAs using lipofectamine had different effects on collagen I and decorin gene expressions of tenocytes | 2011 [150] |
| PCL NFM | DNA (RUNX2) | bone regeneration | RUNX2-loaded liposomes immobilized at the surface of electrospun PCL NFMs induce a long-term gene expression of eGFP and RUNX2 by cultured hBMSCs | 2014 [124] |
Tissue regeneration depends not only on the bioactive agent itself such as growth factors (GFs), but also on the various parameters associated with its presentation, including concentration, spatio-temporal gradients, combination with other GFs and the target cell type [151–154]. Bioactive agent-loaded liposomes combined with scaffolds offer various intrinsic benefits such as: (i) effective concentration; (ii) stable concentration gradients; (iii) multiple bioactive agent delivery; and (iv) spatial patterning [27]. The bioactive agent delivered by the liposome–scaffold device can be of two types: (i) growth/differentiation factor, by the incorporation of the proper bioactive agent-loaded liposomes into the scaffold; and (ii) nucleic acid delivery, by the incorporation of the DNA (or RNAi) into liposomes that encodes (or silences) a specific protein or by cell delivery, in which cells act as ‘GF factories’. These types of local bioactive agent release system will be discussed in §4.2.
4.2. Growth/differentiation factor delivery
The tissue regeneration process involves complex cascades of bioactive agents such as GFs, cytokines and other molecules. GFs are endogenous polypeptides that act through the cell-surface receptors to regulate cellular activities such as proliferation, migration and differentiation [155]. The outcome of GF therapeutics mainly depends on their delivery mode due to their rapid clearance in vivo [156]. Furthermore, one of the main challenges in TE is to find the best way to induce the correct differentiation of stem cells. So far, the most common approach relies on the use of cocktails of growth/differentiation factors supplemented in the culture medium. For instance, bone morphogenetic protein 2 (BMP-2) is added to induce the osteogenic differentiation, whereas transforming growth factor β (TGF-β) is used to promote the chondrogenic differentiation. An adequate combination of signalling molecules should be provided by controlled release systems in order to promote the desired regenerative outcome [112,157–159]. Therefore, liposomes can be used as carriers for the spatio-temporal controlled delivery of GFs, improving stem cell proliferation and differentiation in vitro [160,161].
Liposomes were used with some success in an animal model for cartilage repair [143]. They were used as release systems of TGF-β1 over a period of some weeks, avoiding the typical side effects of systemic administration, due to its direct injection into the joint cavity. Moreover, the conjugation of TGF-β1-loaded liposomes with a scaffold improved its release kinetics and local efficacy [143]. Bisphosphonate-coated liposomes displayed a strong binding to a collagen/hydroxyapatite (HA) composite scaffold, increasing their retention in the collagen/HA scaffolds after their subcutaneous implantation in rats [122]. The bisphosphonate-coated liposomes were able to entrap BMP-2 and deliver it locally [122]. The immobilization of fetal bovine serum-loaded liposomes in a poly(2-hydroxyethyl methacrylate) fibrous scaffold significantly improved chondrocyte adhesion and proliferation [123]. The aim of this study was to investigate the interaction between liposomes and fibre scaffolds and to develop a novel drug delivery system. The release profile of Dex showed an initial burst release, although Dex continued to be released at a slower but steady rate until day 21 [55]. This time frame was selected in accordance with the culture time usually required to obtain a complete osteogenic differentiation of mesenchymal stem cells (MSCs) in vitro. Dex-loaded liposomes did not have any cytotoxic effect on human bone marrow-derived mesenchymal stem cells (hBMSCs). They were able to promote an earlier induction of differentiation of hBMSCs into the osteogenic lineage [55]. Biological assays showed that Dex-loaded liposomes immobilized at the surface of electrospun PCL NFMs did not exhibit any cytotoxic effect, being able to successfully promote the osteogenic differentiation of hBMSCs [125].
4.3. Therapeutic gene delivery
The use of growth/differentiation factors to induce stem cell differentiation in vivo has some limitations such as short half-lives, denaturation during the encapsulation processes, time-consuming, long time periods to obtain the differentiated cells, use of cocktails of GFs and difficulty to differentiate the cells into one specific lineage [108,162]. Therefore, gene therapy, encoding transcription factors or encoding for a specific or to a set of proteins may be a good approach to overcome these limitations and to control stem cell differentiation [124]. Transcription factors would ensure that expression of all natural splice variants occurs in a coordinated time and sequence, and may regulate a cascade of multiple different genes. Gene therapy was initially envisioned as the insertion of a functioning gene into the host cell genome to replace a hereditary genetic defect or, more recently, to provide a new function in a cell such as overexpressing GFs or even killing cancer cells [36,148]. When the pDNA enters into the nucleus, it is intercalated in the DNA of the host cell and, then, transcribed into messenger RNA (mRNA). Therefore, therapeutic proteins or GFs are produced using the cell machinery outside of the nucleus.
Another way to control stem cell differentiation relies on the delivery of RNAi, as previously mentioned [162]. RNAi acts by binding to nucleic acids, inhibiting gene transcription and translation; in other words, RNAi acts by silencing genes of interest through the eradication of target mRNAs through the introduction of small interfering RNAs (siRNAs), small hairpin RNAs (shRNAs) or micro-RNAs (miRNAs) [25,108,162]. The main advantage of this strategy is that the RNAi induces the silencing of targeted genes without integration into the host genome. Therefore, this field has grown considerably in the last decades, due to its huge potential to treat diseases by replacing defective or missing genes or silencing unwanted gene expression [163]. Notable progress has been achieved by the delivery of miRNAs to reprogramme somatic cells into pluripotent stem cells (iPSCs), thus obviating the need to introduce pDNA into donor cells [164].
The delivery of genes can be made by an ex vivo approach or directly into a target cell or tissue. The ex vivo method generally uses autologous cells that are recovered from the patient's body. The cells are generally transduced with viral vectors, containing recombinant genes, and re-inserted/transplanted into the target tissue [148]. Likewise, pDNA or RNAi is poorly taken up by the host cells, being subjected to degradation when exposed to blood proteins [165]. Viral and non-viral vectors are examples of carriers used to transfect cells [166–168]. The widely used method to transfer genes to stem cells is performed through viral vectors (i.e. lentivirus and retrovirus), because of their higher transgene expression and transduction efficiency. Thus, when stem cells are used to correct a genetic pathology and to express the therapeutic gene for the duration of a patient's life, viral vectors are preferred [169]. Conversely, when stem cells are used to treat non-inherited diseases and are only required to express the therapeutic gene for a short period of time, non-viral vectors are preferred [169]. Although viral vectors are more efficient, they possess some limitations such as high production cost, safety issues including mutagenesis, the immunogenicity of the virus proteins, lack of desired tissue selectivity and generation of infectious viruses due to recombination [170–172]. Therefore, there is a need for a delivery system that not only protects the pDNA/RNAi and facilitates its cellular uptake, but additionally enhances the potential for a targeted and effective delivery [165,169]. Non-viral delivery systems (i.e. liposomes, cationic lipids, polymers and proteins) have comparatively lower transfection efficiency, but they have been proposed due to their safety, easy production and higher pDNA size encapsulation [27,108,162,172–174]. Moreover, non-viral vectors provide flexibility in formulation design which can be tailored to interact with the DNA cargo. Also, by incorporating targeting ligands, it is possible to specify the route of vector administration and enhance the delivery to specific tissues or cells.
Liposomes are considered the first non-viral delivery system used in cell biology [27]. Cationic lipids have emerged as the primary option for gene delivery [77,175]. Various cationic lipids were synthesized and evaluated for the transfection of cells [175]. These cationic liposomes show high transfection efficiency, which can be attributed partly to their interactions with negatively charged cell membranes [27]. Lipofectamine, a leading commercial reagent, is a cationic liposome formulation commonly used to transfect cells [176]. However, their success in vivo as a gene therapy strategy has been limited due to the lack of colloidal stability, short duration of gene expression and cytotoxicity [175,177,178]. Cationic polymers have also been proposed as non-viral gene delivery systems, due to their flexible properties, facile synthesis, robustness and proven gene delivery efficiency. For further details about the most recent scientific advances in cationic polymers and their derivatives not only for gene delivery purposes but also for various alternative therapeutic applications, the reader is directed to the review in [179]. The use of liposomes combined with scaffolds may contribute to overcoming the issues of toxicity, long-term expression and silencing efficiency. Specifically, the topical delivery of gene-loaded liposomes via a biomaterial scaffold may reduce their exposure to immune cells, enhance cellular uptake and possibly allow for a sustained delivery [27,162].
Liposome–scaffold systems may be used as a biomaterial to deliver genes in an efficient, cell-controlled and spatially localized manner for TE applications. Bone and cartilage regeneration with a gene therapy strategy is one of the clinically relevant applications [108]. Those strategies have focused on the delivery of genes encoding BMPs and TGF-β that initiate bone and cartilage progenitor cell differentiation, and coordinate the pathways of newly formed bone ossification and cartilage maturation [133,137,148,180]. Tissue vascularization was exploited by the delivery of genes encoding VEGF using bone marrow stromal cells [181]. Liposomes loaded with DNA encoding the 165 amino acid form of VEGF were injected into rat skin stimulating wound healing [182]. The flap survival was enhanced by 14%, and the histological analysis showed new vessel formation [182]. A plasmid expression vector containing VEGF was constructed to be administered to the wound bed of rat abdominal skin flaps in a fibrin sealant [145]. The topical fibrin-mediated administration of a VEGF-A plasmid increased flap survival by 7 days. pDNA-encoding RUNX2-loaded liposomes were covalently immobilized at the surface of PCL nanofibres [124]. The biological result using hBMSCs showed that hBMSCs cultured on RUNX2-loaded liposomes immobilized at the surface of electrospun PCL NFM showed enhanced levels of metabolic activity and total protein synthesis. RUNX2-loaded liposomes immobilized at the surface of electrospun PCL NFMs induce a long-term gene expression of eGFP and RUNX2 by cultured hBMSCs. Furthermore, osteogenic differentiation of hBMSCs was also achieved by the overexpression of other osteogenic markers in medium free of osteogenic supplementation.
4.4. Magnetite cationic liposomes
The main properties of magnetic particles are that they are non-toxic and biocompatible, they can be injected and they can eventually accumulate in the target tissue or organ by the application of an external magnetic field [183]. They are attracted to high magnetic flux density, which is used for bioactive agent targeting and bio-separation including cell sorting. Because of their small size (i.e. approx. 10 nm), they can be encapsulated into the inner compartment of liposomes (figure 11). Magnetite cationic liposomes (MCLs) are magnetic nanoparticles, positively charged, that can interact with the negatively charged cell membranes. They find many applications such as in hyperthermic treatments and in TE and regenerative medicine strategies [183]. Table 4 compiles some of the studies in which MCLs were used in TE approaches. The application of magnetic nanoparticles and magnetic force for TE is termed ‘magnetic force-based tissue engineering (Mag-TE)’ [183]. For instance, MSC sheets created by magnetic nanoparticle-containing liposomes may represent a new modality for therapeutic angiogenesis and bone tissue regeneration [193]. MCLs successfully facilitated cell seeding into the interior space of the scaffolds [187].
Figure 11.
Magnetite cationic liposome. (Online version in colour.)
Table 4.
Applications of MCLs.
| applications | achievements | year/references |
|---|---|---|
| cell sheet with RGD-MCLs in TE | coating the culture surface with RGD-MCLs facilitated cell growth, cell sheet construction and cell sheet harvesting, using magnetic force without enzymatic treatment | 2005 [184] |
| three-dimensional tissue-engineered tubular structures | tubular structures were constructed using magnetic force. Two types of tissue were used to create tubular structures: urinary tissue and vascular tissue | 2005 [185] |
| cell sheet approach for choroidal neovascularization | MCLs were used to construct and deliver retinal pigment epithelium cell sheets in vitro. 15-layered cell sheets were formed after 24 h of culture | 2005 [186] |
| enhancement of cell seeding into the deep internal space of the scaffolds (collagen sponges and polylactic acid sponge) | fibroblasts labelled with MCLs were seeded onto scaffolds. Cell-seeding efficiency increased significantly in all scaffolds when compared with those without magnetic force | 2006 [187] |
| bone TE using bone marrow stromal cells and three-dimensional HA scaffolds | magnetically labelled BMSCs were successfully seeded into the internal space of scaffolds with a high cell density. The levels of alkaline phosphatase and osteocalcin were significantly higher than those by static seeding | 2007 [188] |
| in vitro reconstruction of three-dimensional bone tissues without the use of scaffolds | MSCs magnetically labelled with MCLs formed multi-layered sheet-like structures after a 24 h culture period and maintained the ability to differentiate into osteoblasts, adipocytes or chondrocytes after a 21 day culture period. Transplantation of the MSC sheets into the bone defect in the crania of nude rats showed new bone formation | 2007 [189] |
| incorporation of capillary-like structures into dermal cell sheets | human umbilical vein endothelial cells co-cultured with magnetically labelled normal human dermal fibroblast (NHDF) sheets showed tube-like formation of human aortic endothelial cells, resembling early capillaries, within or on the surface NHDF sheets | 2007 [190] |
| effective cell seeding onto decellularized blood vessels for vascular TE | cells labelled with MCLs increased the cell seeding into porcine decellularized carotid artery scaffold. The scaffold was successfully constructed with two human cells, smooth muscle cells and dermal fibroblasts | 2007 [191] |
| viral vector (VEGF) labelled with MCLs for fabrication of angiogenic cell sheets | a retroviral vector encoding VEGF was labelled with MCLs, to magnetically attract the particles onto a monolayer of mouse myoblast C2C12 cells. Subcutaneous transplantation of C2C12/VEGF cell sheets into nude mice produced thick tissues, with a high cell density, and promoted vascularization | 2010 [192] |
| MSC sheet for therapeutic angiogenesis and tissue regeneration | human MSCs incubated with MCLs formed multi-layered cell sheets according to magnetic force. MSC sheets layered onto the ischaemic tissues of nude mice before skin closure showed greater angiogenesis than the control and MSC injected | 2011 [193] |
| construction of three-dimensional artificial skeletal muscle tissues | MCLs were used to magnetically label C2C12 myoblast cells for the construction of artificial skeletal muscle tissues by an applied magnetic force. Elongated and multi-nucleated myotubes were observed within the tissue | 2011 [194] |
| iPS cell sheets for reparative angiogenesis | mouse iPS cell-derived Flk-1(+) cells were incubated with MCLs. Implantation of the Flk-1(+) cell sheet accelerated revascularization of ischaemic hindlimbs relative to the contralateral limbs in nude mice and increased the expressions of VEGF and bFGF in ischaemic tissue | 2013 [195] |
4.5. Liposomes as templates
We have focused herein on the use of liposomes as a device for the release of bioactive agents. However, liposomes can be used as templates to produce polymeric nanoparticles and nanogels (figure 12) [20,196,197]. In this approach, the inner compartment of the liposome can be used as the reaction vessel [15,196]. For example, the encapsulation of sodium alginate was performed by the thin-film method. Liposomes were extruded through a polycarbonate membrane of uniform pore size followed by incubation with high concentrations of calcium chloride. The diffusion of calcium into the liposome interior resulted in alginate gelation within the liposome of size 748 ± 280 nm [197]. Another study reported the production of alginate nanogels with a size distribution between 120 and 200 nm [196]. Liposomes were also used as a template to prepare monodisperse PEG hydrogel nanoparticles [20]. The procedure for the preparation of PEG nanoparticles using liposomes consisted of the encapsulation of a photo-polymerizable PEG hydrogel solution into the cavity of the liposomes, extrusion through a polycarbonate membrane and photopolymerization of the contents inside the liposomes by UV irradiation (figure 12b). The size distribution of the prepared particles was 94 ± 12 nm after extrusion through the membrane with pore size of 100 nm. This approach also enables the surface of the hydrogel nanoparticles with functional groups to be modified in a one-step procedure [20].
Figure 12.

Strategies to produce nanogels using liposomes as a template. (a) Ca2+ calcium alginate gelation and (b) UV-photopolymerization. (Online version in colour.)
The ability to encapsulate a gel in the inner compartment of liposomes using specific polymers has particular implications beyond drug delivery [198]. Specifically, it can mimic a eukaryotic cell, which is considered to be a gel enclosed within a lipid bilayer membrane. The gel may be formed by the polymerization of cytoskeletal proteins such as actin, filament and tubulin. Therefore, a liposome with a gelled core might be a better study model of a biological cell [199]. Moreover, it could also be used as a container for single molecule fluorescence studies, e.g. for localizing a single DNA or protein molecule within the interior.
The lipid–polymer hybrid (LPH) nanoparticles have also been investigated to deliver therapeutic compounds in a controlled manner [200]. These hybrid nanoparticles combine the unique strengths of liposome and polymeric nanoparticles. They may be used to overcome their limitations in terms of bioactive agent EE, storage stability and release [200]. LPH nanoparticle synthesis requires the use of microfluidics technology to improve the mixing process, but is restricted by a low throughput. However, a pattern-tunable micro-vortex platform can allow mass production and size control of LPH nanoparticles, with superior reproducibility and homogeneity.
5. Concluding remarks
Liposomes are vesicular structures made of lipids that are formed in aqueous solutions, and they resemble the lipid membrane of cells. Since their discovery, a lot of research has been carried out to achieve effective delivery of therapeutic bioactive agents, mainly in cancer research, with some products now coming onto the market. However, there are some limitations to overcome. For example, new liposome-based formulations have to be developed to overcome the rapid blood clearance of PEGylated liposomes. The concept of triggered release is very promising and more studies are needed to validate its applicability in vivo, in humans. Also, large-scale manufacturing methods and technologies are required for marketed products providing sterile, well-characterized and stable products.
In this review, we have highlighted some applications of liposomes in TE and regenerative medicine. Liposomes can be viewed as a platform to induce the differentiation of stem cells through the release of bioactive agents (i.e. GFs or nucleic acids). One of the main challenges in stem cell research is the successful differentiation of stem cells into a specific lineage. However, conventional methods to induce the overexpression of lineage-specific proteins relies on the use of growth/differentiation factor cocktails with sub-optimal outcomes. Liposomes can control growth/differentiation factor release and avoid their side effects. Another possible alternative relies on the delivery of nucleotides (i.e. pDNA and RNAi). The true potential of gene delivery is the possibility to guide the stem cell fate in the absence of inductive factors. The incorporation of pDNA into the host cells' genome raises biocompatibility issues in the context of TE, where differentiated cells are used for subsequent in vivo applications. RNAi may be a possible alternative to guide stem cells' fate without incorporation of pDNA into the host cells' genome. Cationic liposomes are a possible alternative to transfect cells, but they have a lower transfection efficiency [169]. The combination of liposomes with scaffolds represents a good approach to overcome this limitation.
The efficacy of using liposome–scaffolds in TE and regenerative medicine is clear from the literature. However, the potential of this strategy has to be investigated in order to optimize liposome formulations and the best material for the specific application. We believe that liposome–scaffolds may enhance stem cell differentiation and bring novelties in TE and regenerative medicine.
Funding statement
The authors thank the Portuguese Foundation for Science and Technology for the PhD grant to N.S.M. (SFRH/BD/62465/2009), the post-doctoral grants of A.M. (SFRH/BPD/73663/2010). This study was also partly supported by POLARIS (FP7-REGPOT-2012-2013-1), RL3—TECT—NORTE-01-0124-FEDER-000020, co-financed by the North Portugal Regional Operational Programme (ON.2—O Novo Norte), under the National Strategic Reference Framework (NSRF), through the European Regional Development Fund (ERDF), the OsteoGraphy (PTDC/EME-MFE/2008) and MaxBone (PTDC/SAU-ENB/115179/2009) projects.
References
- 1.Fahy E, et al. 2009. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50, S9–S14. ( 10.1194/jlr.R800095-JLR200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LIPID-MAPS. 2014. Lipidomics gateway See http://www.lipidmaps.org.
- 3.Vemuri S, Rhodes CT. 1995. Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm. Acta Helv. 70, 95–111. ( 10.1016/0031-6865(95)00010-7) [DOI] [PubMed] [Google Scholar]
- 4.Subramaniam S, Fahy E, Gupta S, Sud M, Byrnes RW, Cotter D, Dinasarapu AR, Maurya MR. 2011. Bioinformatics and systems biology of the lipidome. Chem. Rev. 111, 6452–6490. ( 10.1021/cr200295k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mader S. 2010. Inquiry into life, 13, ilustrada edn New York, NY: McGraw-Hill Education. [Google Scholar]
- 6.Richter RP, Berat R, Brisson AR. 2006. Formation of solid-supported lipid bilayers: an integrated view. Langmuir 22, 3497–3505. ( 10.1021/la052687c) [DOI] [PubMed] [Google Scholar]
- 7.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. ( 10.1038/nrm2330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singer SJ, Nicolson GL. 1972. The fluid mosaic model of the structure of cell membranes. Science 175, 720–731. ( 10.1126/science.175.4023.720) [DOI] [PubMed] [Google Scholar]
- 9.Berridge MJ, Irvine RF. 1989. Inositol phosphates and cell signaling. Nature 341, 197–205. ( 10.1038/341197a0) [DOI] [PubMed] [Google Scholar]
- 10.Messersmith PB, Vallabhaneni S, Nguyen V. 1998. Preparation of calcium-loaded liposomes and their use in calcium phosphate formation. Chem. Mat. 10, 109–116. ( 10.1021/cm970251f) [DOI] [Google Scholar]
- 11.Avanti-Polar-Lipids. See http://avantilipids.com/.
- 12.Antimisiaris SG, Kallinteri P, Fatouros DG. 2007. Liposomes and drug delivery. In Pharmaceutical manufacturing handbook (ed. S Cox Gad), pp. 443–533. Hoboken, NJ: John Wiley and Sons, Inc. [Google Scholar]
- 13.Eibl H, Kaufmann-Kolle P. 1995. Medical application of synthetic phospholipids as liposomes and drugs. J. Liposome Res. 5, 131–148. ( 10.3109/08982109509039914) [DOI] [Google Scholar]
- 14.Freeman S. 2011. Biological science—lipids, membranes, and the first cells. San Fransicso, CA: Benjamin Cummings. [Google Scholar]
- 15.Collier JH, Messersmith PB. 2001. Phospholipid strategies in biomineralization and biomaterials research. Annu. Rev. Mater. Res. 31, 237–263. ( 10.1146/annurev.matsci.31.1.237) [DOI] [Google Scholar]
- 16.Bangham A, Standish M, Watkins J. 1965. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 13, 238–252. ( 10.1016/S0022-2836(65)80093-6) [DOI] [PubMed] [Google Scholar]
- 17.Allen TM, Cullis PR. 2013. Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48. ( 10.1016/j.addr.2012.09.037) [DOI] [PubMed] [Google Scholar]
- 18.Gómez-Hens A, Manuel Fernández-Romero J. 2005. The role of liposomes in analytical processes. Trends Anal. Chem. 24, 9–19. ( 10.1016/j.trac.2004.07.017) [DOI] [Google Scholar]
- 19.Singh AK, Cummings EB, Throckmorton DJ. 2001. Fluorescent liposome flow markers for microscale particle image velocimetry. Anal. Chem. 73, 1057–1061. ( 10.1021/ac001159x) [DOI] [PubMed] [Google Scholar]
- 20.An SY, et al. 2009. Preparation of monodisperse and size-controlled poly(ethylene glycol) hydrogel nanoparticles using liposome templates. J. Colloid Interface Sci. 331, 98–103. ( 10.1016/j.jcis.2008.11.022) [DOI] [PubMed] [Google Scholar]
- 21.Reineccius Gary A. 1995. Liposomes for controlled release in the food industry. In Encapsulation and controlled release of food ingredients (eds SJ Risch, GA Reineccius), pp. 113–131. Washington, DC: American Chemical Society. [Google Scholar]
- 22.Xia S, Xu S. 2005. Ferrous sulfate liposomes: preparation, stability and application in fluid milk. Food Res. Int. 38, 289–296. ( 10.1016/j.foodres.2004.04.010) [DOI] [Google Scholar]
- 23.Boerman OC, Laverman P, Oyen WJG, Corstens FHM, Storm G. 2000. Radiolabeled liposomes for scintigraphic imaging. Prog. Lipid Res. 39, 461–475. ( 10.1016/S0163-7827(00)00013-8) [DOI] [PubMed] [Google Scholar]
- 24.Voinea M, Simionescu M. 2002. Designing of ‘intelligent’ liposomes for efficient delivery of drugs. J. Cell. Mol. Med. 6, 465–474. ( 10.1111/j.1582-4934.2002.tb00450.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomes-da-Silva LC, Fonseca NA, Moura V, de Lima MCP, Simoes S, Moreira JN. 2012. Lipid-based nanoparticles for siRNA delivery in cancer therapy: paradigms and challenges. Acc. Chem. Res. 45, 1163–1171. ( 10.1021/ar300048p) [DOI] [PubMed] [Google Scholar]
- 26.Ulbrich W, Lamprecht A. 2010. Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases. J. R. Soc. Interface 7, S55–S66. ( 10.1098/rsif.2009.0285.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulkarni M, Greiser U, O'Brien T, Pandit A. 2010. Liposomal gene delivery mediated by tissue-engineered scaffolds. Trends Biotechnol. 28, 28–36. ( 10.1016/j.tibtech.2009.10.003) [DOI] [PubMed] [Google Scholar]
- 28.Banerjee R. 2001. Liposomes: applications in medicine. J. Biomater. Appl. 16, 3–21. ( 10.1106/RA7U-1V9C-RV7C-8QXL) [DOI] [PubMed] [Google Scholar]
- 29.Fenske DB, Chonn A, Cullis PR. 2008. Liposomal nanomedicines: an emerging field. Toxicol. Pathol. 36, 21–29. ( 10.1177/0192623307310960) [DOI] [PubMed] [Google Scholar]
- 30.Immordino ML, Dosio F, Cattel L. 2006. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 1, 297–315. ( 10.2217/17435889.1.3.297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. 1999. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 51, 691–744. [PubMed] [Google Scholar]
- 32.Frézard F. 1999. Liposomes: from biophysics to the design of peptide vaccines. Braz. J. Med. Biol. Res. 32, 181–189. ( 10.1590/S0100-879X1999000200006) [DOI] [PubMed] [Google Scholar]
- 33.Papahadjopoulos D, Nir S, Ohki S. 1972. Permeability properties of phospholipid membranes: effect of cholesterol and temperature. BBA Biomembr. 266, 561–583. ( 10.1016/0005-2736(72)90354-9) [DOI] [PubMed] [Google Scholar]
- 34.Koynova R, Caffrey M. 1998. Phases and phase transitions of the phosphatidylcholines. BBA Rev. Biomembr. 1376, 91–145. [DOI] [PubMed] [Google Scholar]
- 35.Frézard F, Schettini DA, Rocha OGF, Demicheli C. 2005. Liposomes: physicochemical and pharmacological properties, applications in antimony-based chemotherapy. Quim. Nova 28, 511–518. ( 10.1590/S0100-40422005000300025) [DOI] [Google Scholar]
- 36.Maurer N, Fenske DB, Cullis PR. 2001. Developments in liposomal drug delivery systems. Expert Opin. Biol. Ther. 1, 923–947. ( 10.1517/14712598.1.6.923) [DOI] [PubMed] [Google Scholar]
- 37.Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Nejati-Koshki K. 2013. Liposome: classification, preparation, and applications. Nanoscale Res. Lett. 8, 102 ( 10.1186/1556-276X-8-102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Batista CM, Carvalho CMBd, Magalhães NSS. 2007. Liposomes and their therapeutic: state of art applications. Rev. Farm. Bioquim. Univ. Sao Paulo 43, 167–179. [Google Scholar]
- 39.Murphy WL, Messersmith PB. 2000. Compartmental control of mineral formation: adaptation of a biomineralization strategy for biomedical use. Polyhedron 19, 357–363. ( 10.1016/S0277-5387(99)00366-6) [DOI] [Google Scholar]
- 40.Huang JY, Feigenson GW. 1999. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys. J. 76, 2142–2157. ( 10.1016/S0006-3495(99)77369-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodle MC, Lasic DD. 1992. Sterically stabilized liposomes. Biochim. Biophys. Acta 1113, 171–199. ( 10.1016/0304-4157(92)90038-C) [DOI] [PubMed] [Google Scholar]
- 42.Tsukanova V, Salesse C. 2004. On the nature of conformational transition in poly(ethylene glycol) chains grafted onto phospholipid monolayers. J. Phys. Chem. 108, 10 754–10 764. ( 10.1021/jp036992n) [DOI] [Google Scholar]
- 43.Knop K, Hoogenboom R, Fischer D, Schubert US. 2010. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 49, 6288–6308. ( 10.1002/anie.200902672) [DOI] [PubMed] [Google Scholar]
- 44.Ishida T, Harada M, Wang XY, Ichihara M, Irimura K, Kiwada H. 2005. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control Release 105, 305–317. ( 10.1016/j.jconrel.2005.04.003) [DOI] [PubMed] [Google Scholar]
- 45.Kale AA, Torchilin VP. 2007. ‘Smart’ drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J. Liposome Res. 17, 197–203. ( 10.1080/08982100701525035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moura V, Lacerda M, Figueiredo P, Corvo ML, Cruz MEM, Soares R, de Lima MCP, Simoes S, Moreira JN. 2012. Targeted and intracellular triggered delivery of therapeutics to cancer cells and the tumor microenvironment: impact on the treatment of breast cancer. Breast Cancer Res. Treat. 133, 61–73. ( 10.1007/s10549-011-1688-7) [DOI] [PubMed] [Google Scholar]
- 47.Shi N, Pardridge WM. 2000. Noninvasive gene targeting to the brain. Proc. Natl Acad. Sci. USA 97, 7567–7572. ( 10.1073/pnas.130187497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paszko E, Senge MO. 2012. Immunoliposomes. Curr. Med. Chem. 19, 5239–5277. ( 10.2174/092986712803833362) [DOI] [PubMed] [Google Scholar]
- 49.Pardridge WM. 2002. Drug and gene targeting to the brain with molecular Trojan horses. Nat. Rev. Drug Discov. 1, 131–139. ( 10.1038/nrd725) [DOI] [PubMed] [Google Scholar]
- 50.Kleusch C, Hersch N, Hoffmann B, Merkel R, Csiszar A. 2012. Fluorescent lipids: functional parts of fusogenic liposomes and tools for cell membrane labeling and visualization. Molecules 17, 1055–1073. ( 10.3390/molecules17011055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mozafari MR. 2005. Liposomes: an overview of manufacturing techniques. Cell. Mol. Biol. Lett. 10, 711–719. [PubMed] [Google Scholar]
- 52.Senior J, Crawley JCW, Gregoriadis G. 1985. Tissue distribution of liposomes exhibiting long half-lives in the circulation after intravenous injection. Biochim. Biophys. Acta 839, 1–8. ( 10.1016/0304-4165(85)90174-6) [DOI] [PubMed] [Google Scholar]
- 53.Litzinger DC, Buiting AMJ, van Rooijen N, Huang L. 1994. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. BBA Biomembr. 1190, 99–107. ( 10.1016/0005-2736(94)90038-8) [DOI] [PubMed] [Google Scholar]
- 54.Barza M, Stuart M, Szoka F. 1987. Effect of size and lipid-composition on the pharmacokinetics of intravitreal liposomes. Invest. Ophthalmol. Vis. Sci. 28, 893–900. [PubMed] [Google Scholar]
- 55.Murao A, Nishikawa M, Managit C, Wong J, Kawakami S, Yamashita F, Hashida M. 2002. Targeting efficiency of galactosylated liposomes to hepatocytes in vivo: effect of lipid composition. Pharm. Res. 19, 1808–1814. ( 10.1023/A:1021433206081) [DOI] [PubMed] [Google Scholar]
- 56.Kawakami S, Sato A, Yamada M, Yamashita F, Hashida M. 2001. The effect of lipid composition on receptor-mediated in vivo gene transfection using mannosylated cationic liposomes in mice. Stp Pharma. Sci. 11, 117–120. [Google Scholar]
- 57.Kawakami S, Sato A, Nishikawa M, Yamashita F, Hashida M. 2000. Mannose receptor-mediated gene transfer into macrophages using novel mannosylated cationic liposomes. Gene Ther. 7, 292–299. ( 10.1038/sj.gt.3301089) [DOI] [PubMed] [Google Scholar]
- 58.Lichtenberg D, Barenholz Y. 1988. Liposomes—preparation, characterization, and preservation. Methods Biochem. Anal. 33, 337–462. ( 10.1002/9780470110546.ch7) [DOI] [PubMed] [Google Scholar]
- 59.Olson F, Hunt CA, Szoka FC, Vail WJ, Papahadjopoulos D. 1979. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta 557, 9–23. ( 10.1016/0005-2736(79)90085-3) [DOI] [PubMed] [Google Scholar]
- 60.Vanrooijen N, Sanders A. 1994. Liposome-mediated depletion of macrophages—mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 174, 83–93. ( 10.1016/0022-1759(94)90012-4) [DOI] [PubMed] [Google Scholar]
- 61.Bangham AD. 1993. Liposomes: the Babraham connection. Chem. Phys. Lipids 64, 275–285. ( 10.1016/0009-3084(93)90071-A) [DOI] [PubMed] [Google Scholar]
- 62.Wagner A, Vorauer-Uhl K. 2011. Liposome technology for industrial purposes. J. Drug Deliv. 2011, 1–9. ( 10.1155/2011/591325) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang SY, Chen JY, Zhao D, Han DE, Chen XJ. 2012. Comparative study on preparative methods of DC-Chol/DOPE liposomes and formulation optimization by determining encapsulation efficiency. Int. J. Pharm. 434, 155–160. ( 10.1016/j.ijpharm.2012.05.041) [DOI] [PubMed] [Google Scholar]
- 64.Mozafari MR, Reed CJ, Rostron C. 2002. Development of non-toxic liposomal formulations for gene and drug delivery to the lung. Technol. Health Care 10, 342–344. [Google Scholar]
- 65.Mozafari MR, Reed CJ, Rostron C, Martin DS. 2004. Transfection of human airway epithelial cells using a lipid-based vector prepared by the heating method. J. Aerosol Med. 17, 100. [Google Scholar]
- 66.Szoka F, Papahadjopoulos D. 1980. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 9, 467–508. ( 10.1146/annurev.bb.09.060180.002343) [DOI] [PubMed] [Google Scholar]
- 67.Brunner J, Skrabal P, Hauser H. 1976. Single bilayer vesicles prepared without sonication physicochemical properties. Biochim. Biophys. Acta 455, 322–331. ( 10.1016/0005-2736(76)90308-4) [DOI] [PubMed] [Google Scholar]
- 68.Torchilin VP, Weissig V. 2003. Liposomes: a practical approach, 2nd edn Oxford, UK: Oxford University Press. [Google Scholar]
- 69.Laouini A, Jaafar-Maalej C, Limayem-Blouza I, Sfar S, Charcosset C, Fessi H. 2012. Preparation, characterization and applications of liposomes: state of the art. J. Colloid Sci. 1, 147–168. [Google Scholar]
- 70.Ruozi B, Belletti D, Tombesi A, Tosi G, Bondioli L, Forni F, Vandelli MA. 2011. AFM, ESEM, TEM, and CLSM in liposomal characterization: a comparative study. Int. J. Nanomed. 6, 557–563. ( 10.2147/IJN.S14615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Franzen U, Vermehren C, Jensen H, Ostergaard J. 2011. Physicochemical characterization of a PEGylated liposomal drug formulation using capillary electrophoresis. Electrophoresis 32, 738–748. ( 10.1002/elps.201000552) [DOI] [PubMed] [Google Scholar]
- 72.Chu B, Lin FL. 1974. Laser light-scattering study of a ternary liquid-mixture—ethanol water chloroform. J. Chem. Phys. 61, 5132–5146. ( 10.1063/1.1681858) [DOI] [Google Scholar]
- 73.Uskokovic V, Odsinada R, Djordjevic S, Habelitz S. 2011. Dynamic light scattering and zeta potential of colloidal mixtures of amelogenin and hydroxyapatite in calcium and phosphate rich ionic milieus. Arch. Oral Biol. 56, 521–532. ( 10.1016/j.archoralbio.2010.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malvern-Instrument-Company. 2013. Dynamic light scattering and zeta potential See http://www.malvern.com/.
- 75.Phillips NC, Heydari C. 1996. Modulation of cationic liposomal DNA zeta potential and liposome–protein interaction by amphiphilic poly (ethylene glycol)*. Pharm. Pharmacol. 2, 73–76. [Google Scholar]
- 76.Perrie Y, Gregoriadis G. 2000. Liposome-entrapped plasmid DNA: characterisation studies. Biochim. Biophys. Acta 1475, 125–132. ( 10.1016/S0304-4165(00)00055-6) [DOI] [PubMed] [Google Scholar]
- 77.Burgess P, Hutt PB, Farokhzad OC, Langer R, Minick S, Zale S. 2010. On firm ground: IP protection of therapeutic nanoparticles. Nat. Biotech. 28, 1267–1270. ( 10.1038/nbt.1725) [DOI] [PubMed] [Google Scholar]
- 78.Toh M-R, Chiu GNC. 2013. Liposomes as sterile preparations and limitations of sterilisation techniques in liposomal manufacturing. Asian J. Pharm. Sci. 8, 88–95. ( 10.1016/j.ajps.2013.07.011) [DOI] [Google Scholar]
- 79.Grit M, Crommelin JA. 1993. Chemical-stability of liposomes—implications for their physical stability. Chem. Phys. Lipids 64, 3–18. ( 10.1016/0009-3084(93)90053-6) [DOI] [PubMed] [Google Scholar]
- 80.Cabral ECM, Zollner RL, Santana MHA. 2004. Preparation and characterization of liposomes entrapping allergenic proteins. Braz. J. Chem. Eng. 21, 137–146. ( 10.1590/S0104-66322004000200002) [DOI] [Google Scholar]
- 81.Xu X, Khan MA, Burgess DJ. 2012. Predicting hydrophilic drug encapsulation inside unilamellar liposomes. Int. J. Pharm. 423, 410–418. ( 10.1016/j.ijpharm.2011.12.019) [DOI] [PubMed] [Google Scholar]
- 82.Maestrelli F, González-Rodríguez ML, Rabasco AM, Mura P. 2006. Effect of preparation technique on the properties of liposomes encapsulating ketoprofen–cyclodextrin complexes aimed for transdermal delivery. Int. J. Pharm. 312, 53–60. ( 10.1016/j.ijpharm.2005.12.047) [DOI] [PubMed] [Google Scholar]
- 83.Ma L, Ramachandran C, Weiner ND. 1991. Partitioning of a homologous series of alkyl paraaminobenzoates in dipalmitoylphosphatidylcholine liposomes—effect of liposome type. Int. J. Pharm. 77, 127–140. ( 10.1016/0378-5173(91)90310-K) [DOI] [Google Scholar]
- 84.Sakai H, Gotoh T, Imura T, Sakai K, Otake K, Abe M. 2008. Preparation and properties of liposomes composed of various phospholipids with different hydrophobic chains using a supercritical reverse phase evaporation method. J. Oleo Sci. 57, 613–621. ( 10.5650/jos.57.613) [DOI] [PubMed] [Google Scholar]
- 85.Joguparthi V, Anderson BD. 2008. Liposomal delivery of hydrophobic weak acids: enhancement of drug retention using a high intraliposomal pH. J. Pharm. Sci. 97, 433–454. ( 10.1002/jps.21135) [DOI] [PubMed] [Google Scholar]
- 86.Haran G, Cohen R, Bar LK, Barenholz Y. 1993. Transmembrane ammonium-sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta 1151, 201–215. ( 10.1016/0005-2736(93)90105-9) [DOI] [PubMed] [Google Scholar]
- 87.Gonçalves MCF, Mertins O, Pohlmann AR, Silveira NP, Guterres SS. 2012. Chitosan coated liposomes as an innovative nanocarrier for drugs. J. Biomed. Nanotechnol. 8, 240–250. ( 10.1166/jbn.2012.1375) [DOI] [PubMed] [Google Scholar]
- 88.Li N, Zhuang C-Y, Wang M, Sui C-G, Pan W-S. 2012. Low molecular weight chitosan-coated liposomes for ocular drug delivery: in vitro and in vivo studies. Drug Deliv. 19, 28–35. ( 10.3109/10717544.2011.621994) [DOI] [PubMed] [Google Scholar]
- 89.Haidar ZS. 2010. Bio-inspired/-functional colloidal core-shell polymeric-based nanosystems: technology promise in tissue engineering, bioimaging and nanomedicine. Polymers 2, 323–352. ( 10.3390/polym2030323) [DOI] [Google Scholar]
- 90.Joo K-I, Xiao L, Liu S, Liu Y, Lee C-L, Conti PS, Wong MK, Li Z, Wang P. 2013. Crosslinked multilamellar liposomes for controlled delivery of anticancer drugs. Biomaterials 34, 3098–3109. ( 10.1016/j.biomaterials.2013.01.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsotas VA, Mourtas S, Antimisiaris SG. 2007. Dexamethasone incorporating liposomes: effect of lipid composition on drug trapping efficiency and vesicle stability. Drug Deliv. 14, 441–445. ( 10.1080/10717540701603530) [DOI] [PubMed] [Google Scholar]
- 92.Bhardwaj U, Burgess DJ. 2010. Physicochemical properties of extruded and non-extruded liposomes containing the hydrophobic drug dexamethasone. Int. J. Pharm. 388, 181–189. ( 10.1016/j.ijpharm.2010.01.003) [DOI] [PubMed] [Google Scholar]
- 93.McCormack B, Gregoriadis G. 1994. Drugs-in-cyclodextrins-in liposomes: a novel concept in drug delivery. Int. J. Pharm. 112, 249–258. ( 10.1016/0378-5173(94)90361-1) [DOI] [Google Scholar]
- 94.Lindner LH, Hossann M. 2010. Factors affecting drug release from liposomes. Curr. Opin. Drug Discov. Devel. 13, 111–123. [PubMed] [Google Scholar]
- 95.Mohammed AR, Weston N, Coombes AGA, Fitzgerald M, Perrie Y. 2004. Liposome formulation of poorly water soluble drugs: optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 285, 23–34. ( 10.1016/j.ijpharm.2004.07.010) [DOI] [PubMed] [Google Scholar]
- 96.Ahl PL, Bhatia SK, Meers P, Roberts P, Stevens R, Dause R, Perkins WR, Janoff AS. 1997. Enhancement of the in vivo circulation lifetime of l-α-distearoylphosphatidylcholine liposomes: importance of liposomal aggregation versus complement opsonization. BBA Biomembr. 1329, 370–382. ( 10.1016/S0005-2736(97)00129-6) [DOI] [PubMed] [Google Scholar]
- 97.Samoshin AV, Veselov IS, Chertkov VA, Yaroslavov AA, Grishina GV, Samoshina NM, Samoshin VV. 2013. Fliposomes: new amphiphiles based on trans-3,4-bis(acyloxy)-piperidine able to perform a pH-triggered conformational flip and cause an instant cargo release from liposomes. Tetrahedron Lett. 54, 5600–5604. ( 10.1016/j.tetlet.2013.07.156) [DOI] [Google Scholar]
- 98.Fonseca C, Moreira JN, Ciudad CJ, de Lima MCP, Simoes S. 2005. Targeting of sterically stabilised pH-sensitive liposomes to human T-leukaemia cells. Eur. J. Pharm. Biopharm. 59, 359–366. ( 10.1016/j.ejpb.2004.08.012) [DOI] [PubMed] [Google Scholar]
- 99.Sasaki Y, Iwamoto S, Mukai M, Kikuchi J-I. 2006. Photo- and thermo-responsive assembly of liposomal membranes triggered by a gemini peptide lipid as a molecular switch. J. Photochem. Photobiol. A Chem. 183, 309–314. ( 10.1016/j.jphotochem.2006.05.022) [DOI] [Google Scholar]
- 100.Herbst SM, et al. 2009. Delivery of stem cells to porcine arterial wall with echogenic liposomes conjugated to antibodies against CD34 and intercellular adhesion molecule-1. Mol. Pharm. 7, 3–11. ( 10.1021/mp900116r) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanaka H, Sugita T, Yasunaga Y, Shimose S, Deie M, Kubo T, Murakami T, Ochi M. 2005. Efficiency of magnetic liposomal transforming growth factor-beta 1 in the repair of articular cartilage defects in a rabbit model. J. Biomed. Mater. Res. A 73, 255–263. ( 10.1002/jbm.a.30187) [DOI] [PubMed] [Google Scholar]
- 102.Matsuo T, Sugita T, Kubo T, Yasunaga Y, Ochi M, Murakami T. 2003. Injectable magnetic liposomes as a novel carrier of recombinant human BMP-2 for bone formation in a rat bone-defect model. J. Biomed. Mater. Res. A 66, 747–754. ( 10.1002/jbm.a.10002) [DOI] [PubMed] [Google Scholar]
- 103.Dai Y-Q, Qin G, Geng S-Y, Yang B, Xu Q, Wang J-Y. 2012. Photo-responsive release of ascorbic acid and catalase in CDBA-liposome for commercial application as a sunscreen cosmetic. RSC Adv. 2, 3340–3346. ( 10.1039/c2ra01171a) [DOI] [Google Scholar]
- 104.Randles EG, Bergethon PR. 2013. A photodependent switch of liposome stability and permeability. Langmuir 29, 1490–1497. ( 10.1021/la303526k) [DOI] [PubMed] [Google Scholar]
- 105.Buse J, El-Aneed A. 2010. Properties, engineering and applications of lipid-based nanoparticle drug-delivery systems: current research and advances. Nanomedicine 5, 1237–1260. ( 10.2217/nnm.10.107) [DOI] [PubMed] [Google Scholar]
- 106.Shanmugam T, Banerjee R. 2011. Nanostructured self assembled lipid materials for drug delivery and tissue engineering. Ther. Deliv. 2, 1485–1516. ( 10.4155/tde.11.105) [DOI] [PubMed] [Google Scholar]
- 107.Santo VE, Gomes ME, Mano JF, Reis RL. 2012. From nano- to macro-scale: nanotechnology approaches for spatially controlled delivery of bioactive factors for bone and cartilage engineering. Nanomedicine 7, 1045–1066. ( 10.2217/nnm.12.78) [DOI] [PubMed] [Google Scholar]
- 108.Storrie H, Mooney DJ. 2006. Sustained delivery of plasmid DNA from polymeric scaffolds for tissue engineering. Adv. Drug Deliv. Rev. 58, 500–514. ( 10.1016/j.addr.2006.03.004) [DOI] [PubMed] [Google Scholar]
- 109.Dawson E, Mapili G, Erickson K, Taqvi S, Roy K. 2008. Biomaterials for stem cell differentiation. Adv. Drug Deliv. Rev. 60, 215–228. ( 10.1016/j.addr.2007.08.037) [DOI] [PubMed] [Google Scholar]
- 110.Burg KJL, Porter S, Kellam JF. 2000. Biomaterial developments for bone tissue engineering. Biomaterials 21, 2347–2359. ( 10.1016/S0142-9612(00)00102-2) [DOI] [PubMed] [Google Scholar]
- 111.Martins A, Araújo JV, Reis RL, Neves NM. 2007. Electrospun nanostructured scaffolds for tissue engineering applications. Nanomedicine 2, 929–942. ( 10.2217/17435889.2.6.929) [DOI] [PubMed] [Google Scholar]
- 112.Santo VE, Gomes ME, Mano JF, Reis RL. 2012. Controlled release strategies for bone, cartilage and osteochondral engineering. Part I: recapitulation of tissue healing and variables for the design of delivery systems. Tissue Eng. Part B Rev. 19, 308–326. ( 10.1089/ten.teb.2012.0138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alexander C. 2008. Stimuli-responsive hydrogels—Drugs take control. Nat. Mater. 7, 767–768. ( 10.1038/nmat2281) [DOI] [PubMed] [Google Scholar]
- 114.Yoo HS, Kim TG, Park TG. 2009. Surface-functionalized electrospun nanofibers for tissue engineering and drug delivery. Adv. Drug Deliv. Rev. 61, 1033–1042. ( 10.1016/j.addr.2009.07.007) [DOI] [PubMed] [Google Scholar]
- 115.Thakur RA, Florek CA, Kohn J, Michniak BB. 2008. Electrospun nanofibrous polymeric scaffold with targeted drug release profiles for potential application as wound dressing. Int. J. Pharm. 364, 87–93. ( 10.1016/j.ijpharm.2008.07.033) [DOI] [PubMed] [Google Scholar]
- 116.Sirc J, et al. 2012. Controlled gentamicin release from multi-layered electrospun nanofibrous structures of various thicknesses. Int. J. Nanomed. 7, 5315–5325. ( 10.2147/IJN.S35781) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Adler AF, Leong KW. 2010. Emerging links between surface nanotechnology and endocytosis: impact on nonviral gene delivery. Nano Today 5, 553–569. ( 10.1016/j.nantod.2010.10.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.DiTizio V, Ferguson GW, Mittelman MW, Khoury AE, Bruce AW, DiCosmo F. 1998. A liposomal hydrogel for the prevention of bacterial adhesion to catheters. Biomaterials 19, 1877–1884. ( 10.1016/S0142-9612(98)00096-9) [DOI] [PubMed] [Google Scholar]
- 119.Sanborn TJ, Messersmith PB, Barron AE. 2002. In situ crosslinking of a biomimetic peptide-PEG hydrogel via thermally triggered activation of factor XIII. Biomaterials 23, 2703–2710. ( 10.1016/S0142-9612(02)00002-9) [DOI] [PubMed] [Google Scholar]
- 120.Pederson AW, Ruberti JW, Messersmith PB. 2003. Thermal assembly of a biomimetic mineral/collagen composite. Biomaterials 24, 4881–4890. ( 10.1016/S0142-9612(03)00369-7) [DOI] [PubMed] [Google Scholar]
- 121.Zhang ZY, Shum P, Yates M, Messersmith PB, Thompson DH. 2002. Formation of fibrinogen-based hydrogels using phototriggerable diplasmalogen liposomes. Bioconjug. Chem. 13, 640–646. ( 10.1021/bc015580j) [DOI] [PubMed] [Google Scholar]
- 122.Wang G, Mostafa NZ, Incani V, Kucharski C, Uludag H. 2012. Bisphosphonate-decorated lipid nanoparticles designed as drug carriers for bone diseases. J. Biomed. Mater. Res. Part A 100A, 684–693. ( 10.1002/jbm.a.34002) [DOI] [PubMed] [Google Scholar]
- 123.Rampichova M, et al. 2012. A simple drug anchoring microfiber scaffold for chondrocyte seeding and proliferation. J. Mater. Sci. Mater. Med. 23, 555–563. ( 10.1007/s10856-011-4518-x) [DOI] [PubMed] [Google Scholar]
- 124.Monteiro N, Ribeiro D, Martins A, Faria S, Fonseca NA, Moreira JN, Reis RL, Neves NM. 2014. Instructive nanofibrous scaffold comprising runt-related transcription factor 2 gene delivery for bone tissue engineering. ACS Nano 8, 8082–8094. ( 10.1021/nn5021049) [DOI] [PubMed] [Google Scholar]
- 125.Monteiro N, Martins A, Pires R, Faria S, Fonseca NA, Moreira JN, Reis RL, Neves NM. 2014. Immobilization of bioactive factor-loaded liposomes at the surface of electrospun nanofibers targeting tissue engineering. Biomater. Sci. 2, 1195–1209. ( 10.1039/C4BM00069B) [DOI] [PubMed] [Google Scholar]
- 126.Kulkarni M, Breen A, Greiser U, O'Brien T, Pandit A. 2009. Fibrin−lipoplex system for controlled topical delivery of multiple genes. Biomacromolecules 10, 1650–1654. ( 10.1021/bm900248n) [DOI] [PubMed] [Google Scholar]
- 127.Bengali Z, Pannier AK, Segura T, Anderson BC, Jang J-H, Mustoe TA, Shea LD. 2005. Gene delivery through cell culture substrate adsorbed DNA complexes. Biotechnol. Bioeng. 90, 290–302. ( 10.1002/bit.20393) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mickova A, Buzgo M, Benada O, Rampichova M, Fisar Z, Filova E, Tesarova M, Lukas D, Amler E. 2012. Core/shell nanofibers with embedded liposomes as a drug delivery system. Biomacromolecules 13, 952–962. ( 10.1021/bm2018118) [DOI] [PubMed] [Google Scholar]
- 129.Makeyev EV, Zhang J, Carrasco MA, Maniatis T. 2007. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative Pre-mRNA splicing. Mol. Cell 27, 435–448. ( 10.1016/j.molcel.2007.07.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kwong FNK, Richardson SM, Evans CH. 2008. Chordin knockdown enhances the osteogenic differentiation of human mesenchymal stem cells. Arthritis Res. Ther. 10, R65 ( 10.1186/ar2436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bengali Z, Rea JC, Gibly RF, Shea LD. 2009. Efficacy of immobilized polyplexes and lipoplexes for substrate-mediated gene delivery. Biotechnol. Bioeng. 102, 1679–1691. ( 10.1002/bit.22212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rea JC, Gibly RF, Barron AE, Shea LD. 2009. Self-assembling peptide–lipoplexes for substrate-mediated gene delivery. Acta Biomater. 5, 903–912. ( 10.1016/j.actbio.2008.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo-ping W, Xiao-chuan H, Zhi-hui Y, Li G. 2010. Influence on the osteogenic activity of the human bone marrow mesenchymal stem cells transfected by liposome-mediated recombinant plasmid pIRES-hBMP2-hVEGF165 in vitro. Ann. Plast. Surg. 65, 80–84. ( 10.1097/SAP.0b013e3181b4bc5d) [DOI] [PubMed] [Google Scholar]
- 134.Levy O, Ruvinov E, Reem T, Granot Y, Cohen S. 2010. Highly efficient osteogenic differentiation of human mesenchymal stem cells by eradication of STAT3 signaling. Int. J. Biochem. Cell Biol. 42, 1823–1830. ( 10.1016/j.biocel.2010.07.017) [DOI] [PubMed] [Google Scholar]
- 135.Zhang J, Tu Q, Bonewald LF, He X, Stein G, Lian J, Chen J. 2011. Effects of miR-335–5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J. Bone Miner. Res. 26, 1953–1963. ( 10.1002/jbmr.377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chung TW, Yang MC, Tsai WJ. 2006. A fibrin encapsulated liposomes-in-chitosan matrix (FLCM) for delivering water-soluble drugs—influences of the surface properties of liposomes and the crosslinked fibrin network. Int. J. Pharm. 311, 122–129. ( 10.1016/j.ijpharm.2005.12.038) [DOI] [PubMed] [Google Scholar]
- 137.Guo C-A, Liu X-G, Huo J-Z, Jiang C, Wen X-J, Chen Z-R. 2007. Novel gene-modified-tissue engineering of cartilage using stable transforming growth factor-beta 1-transfected mesenchymal stem cells grown on chitosan scaffolds. J. Biosci. Bioeng. 103, 547–556. ( 10.1263/jbb.103.547) [DOI] [PubMed] [Google Scholar]
- 138.Wang SS, Yang MC, Chung TW. 2008. Liposomes/chitosan scaffold/human fibrin gel composite systems for delivering hydrophilic drugs—release behaviors of tirofiban in vitro. Drug Deliv. 15, 149–157. ( 10.1080/10717540801952456) [DOI] [PubMed] [Google Scholar]
- 139.Capito RM, Spector M. 2007. Collagen scaffolds for nonviral IGF-1 gene delivery in articular cartilage tissue engineering. Gene Ther. 14, 721–732. ( 10.1038/sj.gt.3302918) [DOI] [PubMed] [Google Scholar]
- 140.Bolliet C, Bohn MC, Spector M. 2008. Non-viral delivery of the gene for glial cell line-derived neurotrophic factor to mesenchymal stem cells in vitro via a collagen scaffold. Tissue Eng. Part C Methods 14, 207–219. ( 10.1089/ten.tec.2008.0168) [DOI] [PubMed] [Google Scholar]
- 141.Fruchtperry J, Assil KK, Ziegler E, Douglas H, Brown SI, Schanzlin DJ, Weinreb RN. 1992. Fibrin-enmeshed tobramycin liposomes—single application topical therapy of pseudomonas keratitis. Cornea 11, 393–397. ( 10.1097/00003226-199209000-00006) [DOI] [PubMed] [Google Scholar]
- 142.Meyenburg S, Lilie H, Panzner S, Rudolph R. 2000. Fibrin encapsulated liposomes as protein delivery system—studies on the in vitro release behavior. J. Control. Release 69, 159–168. ( 10.1016/S0168-3659(00)00295-9) [DOI] [PubMed] [Google Scholar]
- 143.Giannoni P, Hunziker EB. 2003. Release kinetics of transforming growth factor-beta 1 from fibrin clots. Biotechnol. Bioeng. 83, 121–123. ( 10.1002/bit.10639) [DOI] [PubMed] [Google Scholar]
- 144.Whittlesey KJ, Shea LD. 2006. Nerve growth factor expression by PLG-mediated lipofection. Biomaterials 27, 2477–2486. ( 10.1016/j.biomaterials.2005.11.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Michlits W, Mittermayr R, Schaefer R, Redl H, Aharinejad S. 2007. Fibrin-embedded administration of VEGF plasmid enhances skin flap survival. Wound Repair Regen. 15, 360–367. ( 10.1111/j.1524-475X.2007.00238.x) [DOI] [PubMed] [Google Scholar]
- 146.Lei P, Padmashali RM, Andreadis ST. 2009. Cell-controlled and spatially arrayed gene delivery from fibrin hydrogels. Biomaterials 30, 3790–3799. ( 10.1016/j.biomaterials.2009.03.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kulkarni MM, Greiser U, O'Brien T, Pandit A. 2011. A temporal gene delivery system based on fibrin microspheres. Mol. Pharm. 8, 439–446. ( 10.1021/mp100295z) [DOI] [PubMed] [Google Scholar]
- 148.Winn SR, Chen JC, Gong X, Bartholomew SV, Shreenivas S, Ozaki W. 2005. Non-viral-mediated gene therapy approaches for bone repair. Orthod. Craniofac. Res. 8, 183–190. ( 10.1111/j.1601-6343.2005.00332.x) [DOI] [PubMed] [Google Scholar]
- 149.De Laporte L, Yan AL, Shea LD. 2009. Local gene delivery from ECM-coated poly(lactide-co-glycolide) multiple channel bridges after spinal cord injury. Biomaterials 30, 2361–2368. ( 10.1016/j.biomaterials.2008.12.051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu P, Zhang GR, Song XH, Zou XH, Wang LL, Ouyang HW. 2011. Col V siRNA engineered tenocytes for tendon tissue engineering. PLoS ONE 6, e21154 ( 10.1371/journal.pone.0021154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tayalia P, Mooney DJ. 2009. Controlled growth factor delivery for tissue engineering. Adv. Mater. 21, 3269–3285. ( 10.1002/adma.200900241) [DOI] [PubMed] [Google Scholar]
- 152.Schroeder JE, Mosheiff R. 2011. Tissue engineering approaches for bone repair: concepts and evidence. Injury 42, 609–613. ( 10.1016/j.injury.2011.03.029) [DOI] [PubMed] [Google Scholar]
- 153.Kock L, van Donkelaar CC, Ito K. 2012. Tissue engineering of functional articular cartilage: the current status. Cell Tissue Res. 347, 613–627. ( 10.1007/s00441-011-1243-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Balmayor ER, Pashkuleva I, Frias AM, Azevedo HS, Reis RL. 2011. Synthesis and functionalization of superparamagnetic poly-epsilon-caprolactone microparticles for the selective isolation of subpopulations of human adipose-derived stem cells. J. R. Soc. Interface 8, 896–908. ( 10.1098/rsif.2010.0531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Quaglia F. 2008. Bioinspired tissue engineering: the great promise of protein delivery technologies. Int. J. Pharm. 364, 281–297. ( 10.1016/j.ijpharm.2008.04.030) [DOI] [PubMed] [Google Scholar]
- 156.Zhang SF, Uludag H. 2009. Nanoparticulate systems for growth factor delivery. Pharm. Res. 26, 1561–1580. ( 10.1007/s11095-009-9897-z) [DOI] [PubMed] [Google Scholar]
- 157.Santo VE, Gomes ME, Mano JF, Reis RL. 2012. Controlled release strategies for bone, cartilage and osteochondral engineering. Part II: challenges on the evolution from single towards multiple bioactive factor delivery. Tissue Eng. Part B Rev. 19, 327–352. ( 10.1089/ten.teb.2012.0727) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kelleher CM, Vacanti JP. 2010. Engineering extracellular matrix through nanotechnology. J. R. Soc. Interface 7, S717–S729. ( 10.1098/rsif.2010.0345.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee K, Silva EA, Mooney DJ. 2011. Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J. R. Soc. Interface 8, 153–170. ( 10.1098/rsif.2010.0223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tehrany EA, Kahn CJF, Baravian C, Maherani B, Belhaj N, Wang X, Linder M. 2012. Elaboration and characterization of nanoliposome made of soya; rapeseed and salmon lecithins: application to cell culture. Colloid Surf. B Biointerfaces 95, 75–81. ( 10.1016/j.colsurfb.2012.02.024) [DOI] [PubMed] [Google Scholar]
- 161.Monteiro N, Martins A, Ribeiro D, Faria S, Fonseca NA, Moreira JN, Reis RL, Neves NM. 2013. On the use of dexamethasone-loaded liposomes to induce the osteogenic differentiation of human mesenchymal stem cells. J. Tissue Eng. Regen. Med. 7 ( 10.1002/term.1817) [DOI] [PubMed] [Google Scholar]
- 162.Yau WWY, Rujitanaroj P-O, Lam L, Chew SY. 2012. Directing stem cell fate by controlled RNA interference. Biomaterials 33, 2608–2628. ( 10.1016/j.biomaterials.2011.12.021) [DOI] [PubMed] [Google Scholar]
- 163.Mao S, Sun W, Kissel T. 2010. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 62, 12–27. ( 10.1016/j.addr.2009.08.004) [DOI] [PubMed] [Google Scholar]
- 164.Sun X, Fu X, Han W, Zhao Y, Liu H. 2010. Can controlled cellular reprogramming be achieved using microRNAs? Ageing Res. Rev. 9, 475–483. ( 10.1016/j.arr.2010.06.002) [DOI] [PubMed] [Google Scholar]
- 165.Podesta JE, Kostarelos K. 2009. Engineering cationic liposome: sirna complexes for in vitro and in vivo delivery. Methods Enzymol. 464, 343–354. ( 10.1016/S0076-6879(09)64017-9) [DOI] [PubMed] [Google Scholar]
- 166.von Groll A, Levin Y, Barbosa MC, Ravazzolo AP. 2006. Linear DNA low efficiency transfection by liposome can be improved by the use of cationic lipid as charge neutralizer. Biotechnol. Prog. 22, 1220–1224. ( 10.1021/bp060029s) [DOI] [PubMed] [Google Scholar]
- 167.Jang JH, Houchin TL, Shea LD. 2004. Gene delivery from polymer scaffolds for tissue engineering. Expert Rev. Med. Devices 1, 127–138. ( 10.1586/17434440.1.1.127) [DOI] [PubMed] [Google Scholar]
- 168.Pornpattananangkul D, Olson S, Aryal S, Sartor M, Huang C-M, Vecchio K, Zhang L. 2010. Stimuli-responsive liposome fusion mediated by gold nanoparticles. ACS Nano 4, 1935–1942. ( 10.1021/nn9018587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Santos JL, Pandita D, Rodrigues J, Pego AP, Granja PL, Tomas H. 2011. Non-viral gene delivery to mesenchymal stem cells: methods, strategies and application in bone tissue engineering and regeneration. Curr. Gene Ther. 11, 46–57. ( 10.2174/156652311794520102) [DOI] [PubMed] [Google Scholar]
- 170.Vo TN, Kasper FK, Mikos AG. 2012. Strategies for controlled delivery of growth factors and cells for bone regeneration. Adv. Drug Deliv. Rev. 64, 1292–1309. ( 10.1016/j.addr.2012.01.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Barua S, Ramos J, Potta T, Taylor D, Huang HC, Montanez G, Rege K. 2011. Discovery of cationic polymers for non-viral gene delivery using combinatorial approaches. Comb. Chem. High Throughput Screen. 14, 908–924. ( 10.2174/138620711797537076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kawakami S, Higuchi Y, Hashida M. 2008. Nonviral approaches for targeted delivery of plasmid DNA and oligonucleotide. J. Pharm. Sci. 97, 726–745. ( 10.1002/jps.21024) [DOI] [PubMed] [Google Scholar]
- 173.Pack DW, Hoffman AS, Pun S, Stayton PS. 2005. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 4, 581–593. ( 10.1038/nrd1775) [DOI] [PubMed] [Google Scholar]
- 174.Newland B, Dowd E, Pandit A. 2013. Biomaterial approaches to gene therapies for neurodegenerative disorders of the CNS. Biomater Sci 1, 556–576. ( 10.1039/c3bm60030k) [DOI] [PubMed] [Google Scholar]
- 175.Woodle MC, Scaria P. 2001. Cationic liposomes and nucleic acids. Curr. Opin. Colloid Interface Sci. 6, 78–84. ( 10.1016/S1359-0294(00)00091-1) [DOI] [Google Scholar]
- 176.Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ, Ciccarone VC. 2004. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 33, 95–103. ( 10.1016/j.ymeth.2003.11.023) [DOI] [PubMed] [Google Scholar]
- 177.Newland B, Moloney TC, Fontana G, Browne S, Abu-Rub MT, Dowd E, Pandit AS. 2013. The neurotoxicity of gene vectors and its amelioration by packaging with collagen hollow spheres. Biomaterials 34, 2130–2141. ( 10.1016/j.biomaterials.2012.11.049) [DOI] [PubMed] [Google Scholar]
- 178.Yang F, et al. 2009. Genetic engineering of human stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles. Proc. Natl Acad. Sci. USA 107, 3317–3322. ( 10.1073/pnas.0905432106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Samal SK, Dash M, Van Vlierberghe S, Kaplan DL, Chiellini E, van Blitterswijk C, Moroni L, Dubruel P. 2012. Cationic polymers and their therapeutic potential. Chem. Soc. Rev. 41, 7147–7194. ( 10.1039/c2cs35094g) [DOI] [PubMed] [Google Scholar]
- 180.Lei L, Liao W, Sheng P, Huang G, Jiang L. 2006. Self-induction of rabbit marrow stromal stem cells into chondrocytes by transfection with reconstructed PGL3-transforming growth factor beta1 gene in vitro. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 20, 134–138. [PubMed] [Google Scholar]
- 181.Bai X-F, Tian W-D, Chen X-Z, Li Z-Y. 2005. Expression of human VEGF165 gene in rat bone marrow stromal cells in vitro. J. Sichuan Univ. 36, 468–470. [PubMed] [Google Scholar]
- 182.Liu PY, Tong WJ, Liu K, Han SH, Wang XT, Badiavas E, Rieger-Christ K, Summerhayes I. 2004. Liposome-mediated transfer of vascular endothelial growth factor cDNA augments survival of random-pattern skin flaps in the rat. Wound Repair Regen. 12, 80–85. ( 10.1111/j.1067-1927.2004.012114.x-1) [DOI] [PubMed] [Google Scholar]
- 183.Ito A, Shinkai M, Honda H, Kobayashi T. 2005. Medical application of functionalized magnetic nanoparticles. J. Biosci. Bioeng. 100, 1–11. ( 10.1263/jbb.100.1) [DOI] [PubMed] [Google Scholar]
- 184.Ito A, Ino K, Kobayashi T, Honda H. 2005. The effect of RGD peptide-conjugated magnetite cationic liposomes on cell growth and cell sheet harvesting. Biomaterials 26, 6185–6193. ( 10.1016/j.biomaterials.2005.03.039) [DOI] [PubMed] [Google Scholar]
- 185.Ito A, Ino K, Hayashida M, Kobayashi T, Matsunuma H, Kagami H, Ueda M, Honda H. 2005. Novel methodology for fabrication of tissue-engineered tubular constructs using magnetite nanoparticles and magnetic force. Tissue Eng. 11, 1553–1561. ( 10.1089/ten.2005.11.1553) [DOI] [PubMed] [Google Scholar]
- 186.Ito A, Hibino E, Kobayashi C, Terasaki H, Kagami H, Ueda M, Kobayashi T, Honda H. 2005. Construction and delivery of tissue-engineered human retinal pigment epithelial cell sheets, using magnetite nanoparticles and magnetic force. Tissue Eng. 11, 489–496. ( 10.1089/ten.2005.11.489) [DOI] [PubMed] [Google Scholar]
- 187.Shimizu K, Ito A, Honda H. 2006. Enhanced cell-seeding into 3D porous scaffolds by use of magnetite nanoparticles. J. Biomed. Mater. Res. Part B 77B, 265–272. ( 10.1002/jbm.b.30443) [DOI] [PubMed] [Google Scholar]
- 188.Shimizu K, Ito A, Honda H. 2007. Mag-seeding of rat bone marrow stromal cells into porous hydroxyapatite scaffolds for bone tissue engineering. J. Biosci. Bioeng. 104, 171–177. ( 10.1263/jbb.104.171) [DOI] [PubMed] [Google Scholar]
- 189.Shimizu K, Ito A, Yoshida T, Yamada Y, Ueda M, Honda H. 2007. Bone tissue engineering with human mesenchymal stem cell sheets constructed using magnetite nanoparticles and magnetic force. J. Biomed. Mater. Res. Part B 82B, 471–480. ( 10.1002/jbm.b.30752) [DOI] [PubMed] [Google Scholar]
- 190.Ino K, Ito A, Kumazawa H, Kagami H, Ueda M, Honda H. 2007. Incorporation of capillary-like structures into dermal cell sheets constructed by magnetic force-based tissue engineering. J. Chem. Chem. Eng. Jpn 40, 51–58. ( 10.1252/jcej.40.51) [DOI] [Google Scholar]
- 191.Shimizu K, Ito A, Arinobe M, Murase Y, Iwata Y, Narita Y, Kagami H, Ueda M, Honda H. 2007. Effective cell-seeding technique using magnetite nanoparticles and magnetic force onto decellularized blood vessels for vascular tissue engineering. J. Biosci. Bioeng. 103, 472–478. ( 10.1263/jbb.103.472) [DOI] [PubMed] [Google Scholar]
- 192.Akiyama H, Ito A, Kawabe Y, Kamihira M. 2010. Genetically engineered angiogenic cell sheets using magnetic force-based gene delivery and tissue fabrication techniques. Biomaterials 31, 1251–1259. ( 10.1016/j.biomaterials.2009.11.017) [DOI] [PubMed] [Google Scholar]
- 193.Ishii M, Shibata R, Numaguchi Y, Kito T, Suzuki H, Shimizu K, Ito A, Honda H, Murohara T. 2011. Enhanced angiogenesis by transplantation of mesenchymal stem cell sheet created by a novel magnetic tissue engineering method. Arterioscler. Thromb. Vasc. Biol. 31, 2210–2215. ( 10.1161/ATVBAHA.111.231100) [DOI] [PubMed] [Google Scholar]
- 194.Yamamoto Y, Ito A, Fujita H, Nagamori E, Kawabe Y, Kamihira M. 2011. Functional evaluation of artificial skeletal muscle tissue constructs fabricated by a magnetic force-based tissue engineering technique. Tissue Eng. Part A 17, 107–114. ( 10.1089/ten.tea.2010.0312) [DOI] [PubMed] [Google Scholar]
- 195.Kito T, et al. 2013. iPS cell sheets created by a novel magnetite tissue engineering method for reparative angiogenesis. Sci. Rep. 3, 1418 ( 10.1038/srep01418) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hong JS, Vreeland WN, DePaoli Lacerda SH, Locascio LE, Gaitan M, Raghavan SR. 2008. Liposome-templated supramolecular assembly of responsive alginate nanogels. Langmuir 24, 4092–4096. ( 10.1021/la7031219) [DOI] [PubMed] [Google Scholar]
- 197.Monshipouri M, Rudolph AS. 1995. Liposome-encapsulated alginate—controlled hydrogel particle formation and release. J. Microencapsul. 12, 117–127. ( 10.3109/02652049509015282) [DOI] [PubMed] [Google Scholar]
- 198.Joo V, Ramasamy T, Haidar ZS. 2011. A novel self-assembled liposome-based polymeric hydrogel for cranio-maxillofacial applications: preliminary findings. Polymers 3, 967–974. ( 10.3390/polym3020967) [DOI] [Google Scholar]
- 199.Viallat A, Dalous J, Abkarian M. 2004. Giant lipid vesicles filled with a gel: shape instability induced by osmotic shrinkage. Biophys. J. 86, 2179–2187. ( 10.1016/S0006-3495(04)74277-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kim Y, Lee Chung B, Ma M, Mulder WJM, Fayad ZA, Farokhzad OC, Langer R. 2012. Mass production and size control of lipid–polymer hybrid nanoparticles through controlled microvortices. Nano Lett. 12, 3587–3591. ( 10.1021/nl301253v) [DOI] [PMC free article] [PubMed] [Google Scholar]