

Abstract
Epithelial cells undergoing epithelial-to-mesenchymal transitions have often been shown to behave as cancer stem cells, but the precise molecular connection remains elusive. At the genetic level, stemness is governed by LIN28/let-7 double inhibition switch, whereas EMT/MET is controlled by miR-200/ZEB double inhibition circuit and LIN28 is inhibited by miR-200, coupling the two modules. Here, using a specially devised theoretical framework to investigate the dynamics of the LIN28/let-7 system, we show that it can operate as a three-way switch (between low, high and intermediate LIN28 levels termed the D, U and hybrid D/U states) similar to the three-way operation of the miR-200/ZEB circuit that allows for the existence of a hybrid epithelial/mesenchymal (E/M) phenotype. We find significant correspondence between the existence of the three states of the two circuits: E–D, M–U and E/M–D/U. Incorporating the activation of OCT4 by LIN28, we find that the hybrid E/M phenotype has high likelihood (when compared with either the E or M states) to gain stemness. Combining the LIN28/let-7 regulation by NF-κB and c-MYC, we find that NF-κB, but not c-MYC, elevates the likelihood of E/M phenotype to gain stemness. Our results are consistent with emerging concept that partial EMT can lead to stemness.
Keywords: epithelial–mesenchymal transition, stemness, cancer stem cells, LIN28/let-7, hybrid epithelial/mesenchymal phenotype
1. Introduction
Understanding cell fate decisions during tumour progression remains a major research challenge in modern biology. For instance, metastasis and relapse, the two deadliest aspects of cancer, continue to elude us and remain clinically insuperable [1]. Metastasis involves aberrant transitions between epithelial and mesenchymal phenotypes, whereas tumour relapse is caused by wake-up of a few dormant therapy-resistant cancer stem cells (CSCs) [2]. It is now established that the genetic regulatory network for epithelial–mesenchymal transitions (EMTs) is interconnected with the stemness regulatory network [2]. However, the underlying dynamics and operative principles of these interconnections between metastasis and stemness remain elusive. Understanding these principles is likely to provide important clues for assisting therapeutic advances.
Metastases of carcinomas typically begin when epithelial (E) cells of the primary tumour undergo EMT to lose their cell–cell adhesion and gain migratory and invasive mesenchymal (M) characteristics. These newly transformed motile cells navigate through the extracellular matrix and enter the bloodstream as circulating tumour cells (CTCs) [3]. These CTCs exit the bloodstream at multiple organs and undergo a mesenchymal-to-epithelial transition (MET), regaining their epithelial characteristics to establish micrometastases that later mature into fully developed secondary tumours. Recent theoretical work has indicated that the core decision network regulating carcinoma EMT/MET also allows for transition into a hybrid epithelial/mesenchymal (E/M) phenotype [4]. This hybrid E/M phenotype, often referred to as a partial EMT (pEMT) state, has also been observed experimentally in ovarian carcinoma cell lines [5]. Cells belonging to this phenotype have combined epithelial (cell–cell adhesion) and mesenchymal (motility) traits that enable them to migrate collectively, as seen during wound healing and branching morphogenesis [6]. In cancer, such cohort migration is manifested as the migration of clusters of CTCs in the bloodstream, as observed with lung, prostate and breast cancer patients [7–10]. Such collective migration obviates the need for all cells to detect external signals for migration and facilitates subsequent rounds of dissemination [11]. Therefore, collective migration of CTCs possibly plays an important functional role in metastatic progression [6].
To successfully complete the last step of metastasis, i.e. colonization, the CTCs that exit the bloodstream should self-renew as well as regenerate the heterogeneity of the parental tumour. These two traits are usually considered to be associated with stemness characterizing CSCs [2]. Recent studies have shown that cells undergoing EMT acquire these stem-cell- or tumour-initiating capabilities [12,13], through aberrant activation of several pathways such as NF-κB, Notch, Wnt and TGF-β [2]. However, how these signalling networks mediate this interconnection between EMT and stemness has not yet been elucidated. For example, it has been shown that mesenchymal cells can adopt stemness [12,13], yet the fundamental question, whether hybrid E/M cells can also adopt stem-cell characters, remains open. This fundamental question is also associated with a major dilemma in the clinic—whether to isolate single CSCs or a cluster of CSCs in the bloodstream to characterize the prognostic risk.
Addressing this question calls for understanding regulation of both EMT/MET and stemness and elucidating the operating principles of the connection between them. EMTs are regulated by the miR-200/ZEB mutually inhibitory circuit that operates as a three-way switch with the three possible states being the epithelial, mesenchymal and hybrid E/M phenotypes [4]. Similarly, stemness is regulated by a mutually inhibitory circuit between RNA binding factor LIN28 and microRNA let-7 [14]. The LIN28/let-7 system is connected to the miR-200/ZEB circuit in multiple ways—miR-200 inhibits LIN28 directly [15], and LIN28 activates the pluripotency marker OCT4 [16] that is linked to miR-200 [17]. Studies in both embryonic development and reprogramming indicate that cells with intermediate levels of OCT4 are closely associated with maximum stemness (or pluripotency). Consistently, both very high and very low levels of OCT4 lead to differentiation into specific phenotypes [18–20]. Here, we focus on the coupling of OCT4, LIN28/let-7 and miR-200/ZEB. In addition, because the LIN28/let-7 circuit receives external signals from the oncogene c-MYC and the inflammatory response regulator NF-κB [21–24] (figure 1a,b), we also investigate the effect of these signals on the coupling between LIN28/let-7 and miR-200/ZEB circuits.
Figure 1.
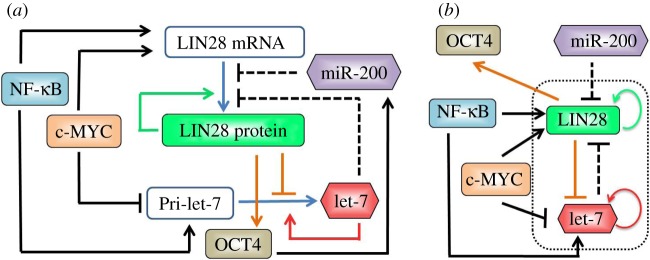
The regulatory network coupling EMT and stemness. (a) Novel modes of regulation in the LIN28/let-7 circuit. LIN28 promotes its own translation (green solid line), and inhibits let-7 processing (orange solid line); let-7 promotes its own processing (red solid line) and inhibits LIN28 translation (black dashed line). NF-κB activates both LIN28 and let-7, whereas c-MYC activates LIN28 and inhibits let-7. miR-200 inhibits LIN28, that translationally activates OCT4 (orange solid arrow) that activates miR-200. (b) Effective circuit showing the LIN28/let-7 double negative feedback loop that receives external input signals from miR-200, c-MYC and NF-κB; and has OCT4 as the output. The activation of miR-200 by OCT4 has not been considered in our current framework. A blue solid line shows conversion of one species to another, a solid black arrow shows transcriptional activation, a solid black bar denotes transcriptional repression, and a black dashed line shows microRNA-mediated translational repression.
Here, we have devised a specific theoretical framework to investigate the dynamics of the LIN28/let-7 double inhibitory circuit and show that it can operate as a three-way switch. Several previous studies show that mutually inhibitory circuits—either those comprising two transcription factors (canonical TF–TF toggle switch) or those comprising a transcription factor and a microRNA (chimera TF-miR toggle switch)—can act as three-way switches when one or two of the components are self-activating [4,25–29]. Motivated by these studies, we expected the LIN28/let-7 circuit will also allow tristability, because both LIN28 and let-7 are self-activating elements [30–32]. However, because the modes of regulation in the LIN28/let-7 circuit present unique dynamical properties that are different from those of the previously studied circuits, we had to devise a new theoretical framework. More specifically, modelling the dynamics of the LIN28/let-7 circuit poses a special theoretical challenge because it involves three unique regulatory mechanisms that have not been modelled before: (i) translational self-activation (of LIN28) [30,31], (ii) inhibition of microRNA processing (of let-7 by LIN28) [14], and (iii) self-activation of microRNA processing (of let-7) [32] (see table S1 in the electronic supplementary material, §S1, for circuit details).
To devise the theoretical framework presented here, we started from our previously devised framework for modelling miRNA-mediated translational repression [4,29]. We have generalized this framework to include the novel modes of regulation in the LIN28/let-7 circuit mentioned earlier. With the new framework at hand, first, we investigate the dynamics of the LIN28/let-7 system as a stand-alone circuit (i.e. when driven by miR-200 alone). Subsequently, we investigate the dynamics of the circuit when it is driven by miR-200 in combination with external inputs from c-MYC and NF-κB. It may be noted that we do not include the regulation of miR-200 by OCT4 in our current model.
Exploiting our new model, we find that LIN28/let-7 can operate as a three-way (ternary) switch between three possible states: (i) (high LIN28, low let-7) state—(1, 0) or up (U) state, (ii) (low LIN28, high let-7) state—(0, 1) or down (D) state, and (iii) (medium LIN28, medium let-7) state—(1/2, 1/2) or down/up (D/U) state. Importantly, we show that these three states can typically be expected to correspond to the mesenchymal, epithelial and hybrid E/M phenotypes, respectively. Analysing the effects of external signals on LIN28/let-7 circuit, we found that c-MYC and NF-κB drive the circuit to (1, 0) and (1/2, 1/2) states, respectively. We also show that intermediate levels of OCT4, the proposed readout for stemness, can be attained by intermediate levels of LIN28, i.e. (1/2, 1/2) or D/U state. Because the (1/2, 1/2) or D/U state can correspond to the hybrid E/M phenotype, especially under the influence of NF-κB, we indicate that E/M state is also likely to be associated with stemness.
2. Material and methods
2.1. Model formulation
We generalized and extended the recent theoretical framework devised by Lu et al. [29] to incorporate the special modes of regulation operative in the LIN28/let-7 circuit—inhibition and self-activation of microRNA processing, and translational self-activation. Effects of miR-200, c-MYC and NF-κB are incorporated as external signals. The indirect feedback of LIN28 on miR-200 through OCT4 is not included in the current framework. Equations (2.1) and (2.2) represent time evolution of the levels of mature microRNA let-7 (µ) and LIN28 protein (B), respectively.
We use shifted Hill functions to describe the effect of regulation of one species by the other. Shifted Hill functions [4] are defined as weighted sum of excitatory Hill function H+(X) and inhibitory Hill function H−(X), thus Hs(X,λ) = H−(X) + λH+(X), where the weight factor λ represents the fold-change in production rate owing to the regulation. Therefore, for activation, λ > 1; for repression, λ < 1; and for no effect, λ = 1. As per our notation, λX,Y denotes the weight factor (λ) for the regulation of X on Y.
More specifically, the concentration of mature miRNA let-7 (μ) depends on the innate degradation rate of mature let-7 (depicted by kμ), and innate processing rate of the precursors to mature miRNAs (depicted by gμ). This innate processing rate is affected in two ways—it is decreased due to the inhibition of processing by LIN28 (B), but it is increased owing to autocatalytic activity of mature let-7 (μ; figure 1a). Thus, two shifted Hill functions are multiplied to gµ: Hs(B,λB,μ) with λB,μ < 1 (denoting inhibition by LIN28 (B)), and Hs(μ,λμ,μ) with λμ,μ > 1 (denoting self-activation of let-7 (μ)).
Therefore, the dynamics of the level of mature let-7 (μ) is governed by equation (2.1) given as
| 2.1 |
Typically, miRNAs are more stable than the mRNAs [4], so we can assume that mRNA always reaches steady states. The steady-state level of the LIN28 mRNA (m) can be determined simply by m = gm/km, where gm and km are the production and degradation rates for LIN28 mRNA (m), respectively.
Next, the concentration of LIN28 protein (B) depends on the innate degradation rate of LIN28 (depicted by kB), and the steady-state mRNA (m) levels times the innate translation rate of m to B (depicted by gB). But, this innate translation rate is decreased owing to the interference of mature let-7 (μ) with the translation, and increased due to the binding of LIN28 (B) to its own mRNA promoting translation (figure 1a). Thus, two shifted Hill functions are multiplied to mgB: Hs(μ,λμ,B) with λμ,B < 1 (denoting inhibition by mature let-7 (μ)), and Hs(B,λB,B) with λB,B > 1 (denoting translational self-activation of LIN28 (B)).
Therefore, the dynamics of the level of LIN28 protein (B) is governed by equation (2.2) given as
| 2.2 |
2.2. Incorporating the effects of input from miR-200, NF-κB and c-MYC
When studying these effects, we incorporate in equations (2.1) and (2.2), using the proper Hill functions, inhibition of LIN28 by miR-200, activation of LIN28 by NF-κB and c-MYC, activation of let-7 by NF-κB and inhibition of let-7 by c-MYC. See the electronic supplementary material, §2, for details on model formulation.
2.3. Computational methods
Matlab continuation method MATCONT [33] was used to calculate the nullclines and bifurcations. All model parameters can be found in the electronic supplementary material, §2, and sensitivity analysis in the electronic supplementary material, §3.
3. Results
3.1. LIN28/let-7 can operate as a three-way switch
We start by analysing the LIN28/let-7 circuit when LIN28 is inhibited by a constant level of miR-200 as an input signal. A typical phase-space diagram (figure 2a) shows that the circuit can have three coexisting stable states (three green solid circles). These states are (i) high LIN28 with low let-7—(1, 0) or up (U) state, (ii) low LIN28 with high let-7—(0, 1) or down (D) state, and (iii) medium LIN28 with medium let-7—(1/2, 1/2) or down/up (D/U) state. This result is consistent with experiments that identify not only the cells with (high LIN28, low let-7) and (low LIN28, high let-7) expression, but also the cells with concomitant expression of both LIN28 and let-7—(medium LIN28, medium let-7) [30,31]. Therefore, the LIN28/let-7 circuit can operate as a three-way switch.
Figure 2.
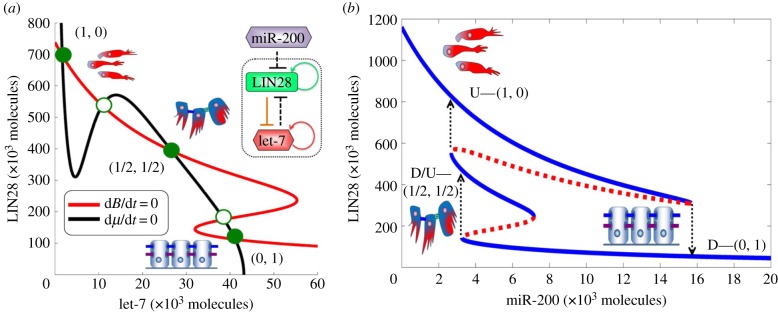
Dynamical behaviour of the LIN28/let-7 circuit. (a) Nullclines and three possible steady states as predicted by the model for fixed miR-200 = 5000 molecules. Red nullcline is for dB/dt = 0, and black nullcline is for dμ/dt = 0. Green solid dots denote possible states (stable fixed points), and green hollow circles denote unstable fixed points (details of stability analysis are given in electronic supplementary material, §6). The states are U—(1, 0) or (high LIN28, low let-7); D—(0, 1) or (low LIN28, high let-7); and D/U—(1/2, 1/2) or (medium LIN28, medium let-7). (b) Bifurcation of LIN28 protein levels in response to inhibition by miR-200. It illustrates the possible coexistence of three states between epithelial (high miR-200 levels) and mesenchymal (low miR-200 levels). Note that for the parameters used in this figure (c-MYC = 0 and NF-κB = 0), D/U state exists for miR-200 levels from 3000 to 7000 molecules, levels that are consistent with typical levels of miR-200 corresponding to the E/M phenotype (5000–15 000 molecules [4]). As is shown later, in the presence of NF-κB, the range of existence of the D/U state can increase from 4000 to 12 000 miR-200 molecules. Starting from the E phenotype, at bottom right part of the blue curve, and decreasing miR-200 levels, the circuit undergoes a transition from the D—(0, 1) state into the D/U—(1/2, 1/2) state. Further decrease in miR-200 to levels that correspond to M phenotype leads to a transition from the D/U state into the U state. Starting from U state and increasing miR-200 levels, the circuit directly undergoes a transition into the D state at miR-200 level that corresponds to the E phenotype.
The EMT regulatory circuit miR-200/ZEB can also operate as a three-way switch between epithelial (E), mesenchymal (M) and hybrid E/M phenotypes [4]. These phenotypes correspond to (high miR-200, low ZEB), (low miR-200, high ZEB) and (medium miR-200, medium ZEB) states, respectively [4]. Because miR-200 directly inhibits LIN28, we expect to discover a correspondence between (E, E/M, M), and the three states of the LIN28/let-7 circuit. To test the expectation, we have analysed the range of existence of the three possible states of LIN28/let-7 for miR-200 ranging from pure epithelial levels to pure mesenchymal levels (figure 2b). As seen in figure 2b, at very high levels of miR-200 corresponding to the E phenotype, only the D—(0, 1) state exists. Oppositely, at very low levels of miR-200 corresponding to the M phenotype, only the U—(1, 0) state exists. Conversely, at intermediate levels of miR-200 corresponding to the hybrid E/M phenotype (approx. 5000–15 000 molecules [4]), the D/U—(1/2, 1/2) state can coexist with the U and D states. In addition, for some range of intermediate miR-200 levels, the U and D states coexist. The bifurcation diagrams also explain the asymmetry between EMT (reducing miR-200 levels) and MET (increasing miR-200 levels) as observed in some experimental studies where the hybrid E/M phenotype was attained during EMT [34], but not during MET [35]. Concurrently, the bifurcation diagram shows that cells have a significantly lower likelihood to enter the D/U—(1/2, 1/2) state during METs.
The results are consistent with experimental observations that epithelial cells typically have low LIN28 levels with high let-7 levels; and mesenchymal cells typically have high LIN28 levels with low let-7 levels [15,36]. Therefore, we propose that cells belonging to the hybrid E/M phenotype are likely to be in the (medium LIN28, medium let-7) or D/U—(1/2, 1/2) state. To indicate this, we added the cartoon of the hybrid E/M cells near the D/U—(1/2, 1/2) state (for completeness, we added the cartoon of the mesenchymal cells near the U—(1, 0) state and that of the epithelial cells near the D—(0, 1) state). More specifically, we expect this to be the case when the miR-200 levels of the cells in the D/U state fall within the range of miR-200 levels that correspond to the hybrid E/M phenotype, i.e. 5000–15 000 molecules of miR-200 [4].
3.2. The effect of c-MYC
As is illustrated in figure 1b, c-MYC activates LIN28 and inhibits let-7. The effect of c-MYC at different levels of miR-200 is shown in figure 3. As can be seen, when miR-200 levels are set to be 7000 molecules, a typical level for the E/M phenotype, the D/U—(1/2, 1/2) state exists for finite range of c-MYC, and at higher c-MYC values, only the U state exists and the circuit becomes monostable (figure 3a). However, when the levels of miR-200 do not correspond to the E/M phenotype, the range of existence of the D/U state decreases significantly and the circuit becomes bistable or even monostable (electronic supplementary material, §4).
Figure 3.
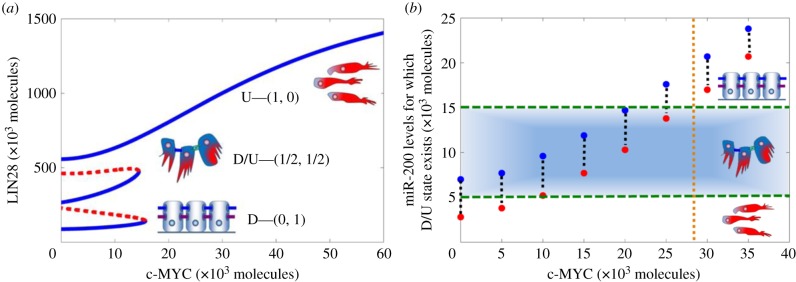
Circuit response to c-MYC and miR-200. (a) Bifurcation diagram showing the dependence of the level of LIN28 on c-MYC when LIN28 is inhibited by constant miR-200 level = 7000 molecules. Blue solid lines indicate stable states, and red dotted lines denote unstable states. Corresponding EMT phenotypes have been indicated for every stable state. (b) Required levels of miR-200 for the D/U—(1/2, 1/2) state to exist for different levels of c-MYC. Red dots show minimum miR-200 levels for which the D/U state can exist, and blue dots are for corresponding maximum values. The area within the green horizontal dashed lines represents the range of miR-200 levels that are associated with the hybrid E/M phenotype (5000–15 000 molecules) [4]. Corresponding phenotypes have been indicated for different states (E, M, E/M) depending on miR-200 levels. Brown vertical dotted line denotes the maximum estimated levels of c-MYC in a cell (approx. 30 000 molecules [37]).
Notably, even at intermediate levels of miR-200 (the blue region highlighted in figure 3b), the D/U state can be maintained only at low levels of c-MYC. As c-MYC increases, the miR-200 levels required to maintain the D/U state increase significantly (figure 3b). These required levels of miR-200 at high values of c-MYC, as suggested by the model, are much larger than the levels of miR-200 that define the E/M phenotype, i.e. 5000–15 000 molecules [4], and rather correspond to the epithelial phenotype. Therefore, at c-MYC levels higher than typical levels (approx. 30 000 molecules [37], vertical dotted brown line in figure 3b), D/U state corresponds to the epithelial phenotype and not to the hybrid E/M phenotype, i.e. high levels of c-MYC drive the cell into the U state. These results are consistent with experiments showing that overexpression of c-MYC impairs wound healing [38], a typical example of transition to E/M phenotype and then re-epithelialization.
3.3. The effect of NF-κB
As is illustrated in figure 1, NF-κB activates both LIN28 and let-7. The effect of NF-κB at different levels of miR-200 is shown in figure 4.
Figure 4.

Circuit response to external signals NF-κB and miR-200. (a) Bifurcation diagram shows the dependence of the level of LIN28 on NF-κB when LIN28 is inhibited by constant miR-200 level = 6000 molecules. (b) Complementary bifurcation diagram showing the dependence of the level of LIN28 on miR-200 when the circuit is driven by constant level of NF-κB = 25 000 molecules. Note that the required levels of miR-200 for the existence of D/U state (4000–12 000 molecules) overlap well with the levels of miR-200 for the existence of E/M phenotype (5000–15 000 molecules) [4]. Blue solid lines indicate stable states, and red dotted lines denote unstable states. Corresponding phenotypes (E, M, E/M) have been indicated for every stable state. (c) Range of miR-200 levels for which the D/U—(1/2, 1/2) state exists, for varying values of NF-κB. Red dots show minimum values of miR-200 for which this state exists, and blue dots for corresponding maximum values. Corresponding phenotypes have been indicated for different states (E, M, E/M) depending on miR-200 levels. (d) Range of NF-κB values for which D/U state exists for different values of miR-200. Red dots are for minimum values of NF-κB for which this state exists, and blue dots are for corresponding maximum values. Arrows denote that the corresponding maximum levels for NF-κB are greater than 60 000 molecules. Brown horizontal dotted line denotes the maximum estimated levels of NF-κB in a cell (50 000 molecules [39]). Areas within the green dashed lines—horizontal in (c) and vertical in (d)—represent miR-200 levels that are associated with the hybrid E/M phenotype.
For intermediate levels of miR-200, the D/U—(1/2, 1/2) state exists for a significant range of NF-κB levels as can be seen from the bifurcation diagram of the dependence of the LIN28 levels on NF-κB (figure 4a). Complementarily, for intermediate levels of NF-κB, a bifurcation diagram of the dependence of LIN28 levels on miR-200 points out that the D/U state exists for a significant range of miR-200 levels (4000–12 000 molecules; figure 4b). This bifurcation diagram also indicates an asymmetry between EMT (reducing the level of miR-200) and MET (increasing the level of miR-200), an asymmetry which has also been observed in previous experimental and theoretical analysis [4,34,35].
Figure 4c shows that with increasing NF-κB, the D/U state exists for an increasing range of miR-200 levels. Importantly, for larger NF-κB values, this range of miR-200 levels almost entirely overlaps with the range of miR-200 levels that defines the hybrid E/M phenotype [4], thereby implying that high levels of NF-κB tend to maintain cells in the D/U state, and promote the correspondence between the D/U state and the E/M phenotype. This result is consistent with experiments showing that overexpression of NF-κB, which maintains D/U state, promotes wound healing [40], an example of cells moving collectively in the E/M phenotype.
Figure 4d shows the range of NF-κB values for which the D/U state exists, at different fixed levels of miR-200. As can be seen, for higher levels of miR-200, the D/U—(1/2, 1/2) state exists for an increasing range of NF-κB levels. However, at very high miR-200 levels that are associated with the epithelial phenotype (more than 15 000 molecules [4]), the minimum NF-κB levels required to enable the existence of the D/U state are close to the maximum experimentally observed levels of NF-κB (35 000 molecules in normal cells and probably higher, up to 50 000 molecules, in cancer cells [39]).
3.4. The association between the hybrid E/M phenotype and stemness
To further reveal the dependence of stemness likelihood on both miR-200 and NF-κB, we construct the phase-space diagram (two-parameter bifurcation diagram) shown in figure 5a. Each phase corresponds to the coexistence of one or more phenotypes. More specifically, the possible states are (i) phases with only one phenotype—{D} and {U}, (ii) phases in which two phenotypes coexist—{D, U} and {D/U, U}, and (iii) a phase in which all three phenotypes can coexist—{D, D/U, U}. The diverse phases recapitulate the plasticity of cell phenotypes as driven differently by miR-200 and NF-κB. The phases containing the D/U state—{D, D/U, U} and {D/U, U}—exist for intermediate values of miR-200, and become more prominent with increasing levels of NF-κB, thus showing again that increasing values of NF-κB stabilize the D/U state for intermediate levels of miR-200 that correspond to the hybrid E/M phenotype.
Figure 5.

Cells in hybrid E/M state are highly likely to be stem-like. (a) Phase-space diagram of the response of LIN28/let-7 circuit to miR-200 and NF-κB. Each phase corresponds to a different combination of coexisting phenotypes and is denoted by a different colour. For example, in phase {D}, cells are in D state; and cells in phase {D, U, D/U} can adopt any of the three states—D, U or D/U. (b) Mapped regions for likelihood of stemness, as defined by a range of intermediate OCT4 levels (relative levels = 0.25–0.65). Cells in D/U state in the entire {D, U, D/U} phase and part of the {D/U, U} phase (as marked with black dotted lines, and bound by black solid lines in yellow region, and black solid lines at the boundary of pink and light green region) have high likelihood of stemness. In addition, cells in the U state are likely to be stem-like in a part of the {D, U} region (as marked with red dotted lines and bound by red solid lines in light green region, and red solid lines at the boundary of light green and blue region). Cells in both U and D/U states are both likely to be stem-like in the small area at the bottom (bounded with red and black lines) in the {D, U, D/U} phase.
It has been suggested that stemness is associated with intermediate levels of OCT4—both too high and too low levels of OCT4 lead the cell to different differentiation paths, thereby depriving the cell of pluripotency [18–20]. Motivated by these observations, we proceed to compute the dependence of the levels of the pluripotency (stemness) marker OCT4 on its activator LIN28. OCT4 levels were calculated as a Hill's function of LIN28 levels (see electronic supplementary material, §2). We chose a representative range of relative OCT4 levels that correspond to high likelihood for stemness (0.25–0.65 relative to the saturation level of OCT4 when it is activated by the maximum level of LIN28) and mapped regions of likelihood for stemness defined this way in the phase diagram. Note that the range of OCT4 levels is expected to be cancer and even patient specific. The results here for the specific ‘guessed range’ are presented to illustrate the concept. We find high stemness likelihood in the entire region of the {D, D/U, U} phase, in part of the region of the {D/U, U} phase and in a part of the region of the {D, U} phase (figure 5b). Notably, the area associated with stemness when cells are in the D/U state (area marked by black dotted lines in figure 5b) is significantly larger than that when cells are in the U state (area marked by red dotted lines in figure 5b), especially when let-7 levels are not too low (see the sensitivity analysis in the electronic supplementary material, §3). Therefore, we propose that cells in the hybrid D/U state are also likely to adopt stemness and not only cells in the U state. In addition, with increasing NF-κB, the area associated with stemness decreases for the U state, but not for the D/U state (figure 5b), thereby implying that NF-κB can facilitate the association of the D/U state and/or the E/M phenotype with stemness.
In the light of the above, we predict that for some cancer types, cells in the hybrid E/M phenotype might even be more likely to adopt stemness when compared with cells in the mesenchymal phenotype. This prediction is consistent with some non-cancer cases: recent studies found that adult stem cells such as epithelial stem cells (those that give rise to all three layers of the skin), hepatic stellate cells (epithelial progenitors in adult mouse livers) and trophoblast stem cells (precursors of differentiated cells of the placenta) co-express both epithelial and mesenchymal markers, and define the ‘metastable’ hybrid E/M phenotype with stemness characteristics [41–43].
3.5. Association between dynamics of epithelial-to-mesenchymal transition and stemness
We proceed to investigate the dynamics of LIN28/let-7 circuit in response to external signal miR-200 that decreases in time representing EMT (figure 6). The circuit starts in the D state, and exhibits a transition from this state into the D/U state as the levels of miR-200 decrease from levels corresponding to epithelial phenotype (more than 15 000 molecules) to levels corresponding to the E/M hybrid phenotype (5000–15 000 molecules). It stays at the D/U state for 4.25–6 days and then goes through another transition into the U state as the levels of miR-200 continue to decrease towards levels that correspond to the mesenchymal phenotype (figure 6a). More specifically, the miR-200 levels continue to decrease until t = 7 days (which is consistent with the typical time for EMT [12]); therefore, we see that LIN28 levels saturate only at t = 7 days, and that they continue to increase even after the cell enters the mesenchymal phenotype at t = 6 days. A similar treatment of the circuit with miR-200 at lower values of NF-κB significantly shortens the duration of stay in the hybrid state before transitioning into the mesenchymal state (figure 6c). This behaviour is explained by our previous observation that high levels of NF-κB maintain cells in the hybrid or the D/U—(1/2, 1/2) state.
Figure 6.

Temporal dynamics of partial and complete epithelial–hybrid–mesenchymal transition. Panels (a,c) show the temporal evolution of let-7 (blue) and LIN28 (red) for fixed NF-κB = 40 000 molecules and NF-κB = 10 000 molecules, respectively, as miR-200 (black) levels decrease over time. The area in the brown rectangle shows the transition from D state (low LIN28, high let-7) to D/U state (medium LIN28, medium let-7) and then to the U state (low LIN28, high let-7). Panels (b,d) denote temporal evolution of OCT4 (green) corresponding to LIN28 (red) levels for NF-κB = 40 000 molecules and NF-κB = 10 000 molecules, respectively. Horizontal dotted blue lines show the range of OCT4 for stemness, and the black rectangle highlights the corresponding values of LIN28 for which the cells are likely to adopt stemness.
We also plot the relative levels of OCT4 during EMT. Some cells in the E/M phenotype are likely to be in the D/U state and thus have intermediate levels of OCT4 that represent likelihood for stemness (pluripotency). On the other hand, cells in the mesenchymal phenotype are likely to be in the U state with quite high levels of OCT4 and hence might have lower likelihood for stemness (figure 6b,d). Because the exact levels of LIN28 (and hence OCT4) depend on the level of NF-κB, the latter determines the likelihood of cells in the hybrid E/M and mesenchymal phenotypes to adopt stemness. However, it is less probable that cells in the mesenchymal phenotype will retain pluripotency (stemness), at least for the ‘guessed range’ of OCT4 levels used here and along the specific path of circuit dynamics as taken during these dynamic simulations.
4. Discussion
The LIN28/let-7 mutually inhibitory circuit is a key player in connecting EMT and stemness. Although its recognized importance has led to intense experimental efforts [14–16], it has been given limited theoretical attention to date. We have constructed a theoretical framework to study the LIN28/let-7 circuit as a stand-alone module, including its response to other players connecting EMT and stemness: c-MYC and NF-κB.
We find that the LIN28/let-7 circuit can act as a three-way switch giving rise to three possible states—D, U and D/U that we typically associate with the epithelial, mesenchymal and hybrid E/M phenotypes, respectively. Experiments validate our association of epithelial and mesenchymal cells with D and U states, respectively [15,36]; however, direct measurements of both LIN28 and let-7 are needed to validate our proposed association of the D/U state with the E/M phenotype.
We also show that while c-MYC drives the cells towards a U state, NF-κB can help to maintain the cells in a D/U state, which corresponds to the hybrid E/M phenotype for high NF-κB levels. Further, NF-κB also activates both miR-200 [44] and ZEB [45], two sides of the EMT decision-making circuit, thereby possibly stabilizing the E/M phenotype. Therefore, we predict that NF-κB might facilitate the migration of CTCs as a cluster of hybrid E/M cells. Our predictions are consistent with the fact that triple negative breast cancer that has a constitutively higher expression of NF-κB among different breast cancer subtypes [46] has the highest percentage of hybrid E/M cells in the CTCs [5].
Based on intermediate OCT4 levels as the readout for pluripotency, we predict that cells belonging to the hybrid E/M phenotype or equivalently those in the D/U—(1/2, 1/2) state have relatively high likelihood to adopt stemness. Moreover, for some cancers, the cells in the hybrid E/M phenotype might even show higher stemness likelihood than cells of mesenchymal phenotype. Indeed, adult epithelial, trophoblast and hepatic stem/progenitor cells, as well as neoplastic stem cells, co-express epithelial and mesenchymal markers [41–43,47] in agreement with our model prediction. In addition, our predictions are consistent with experiments showing that clusters of CTCs (hybrid E/M phenotype) have higher metastatic potential than the individual CTCs (mesenchymal phenotype) [48] and the recent ‘EMT gradient model’ proposing that cells undergoing pEMT, but not necessarily those undergoing complete EMT, lie in the ‘stemness window’ [49].
Our proposed association of E/M phenotype with stemness likelihood is also consistent with the fact that: during metazoan development (type I EMT), glomerular epithelial cells in the kidney undergo pEMT to attain traits of bipotent kidney progenitors [50]; during tissue repair (type II EMT), hybrid E/M cells function as bipotent adult hepatic progenitors and repopulate the rat liver upon injury [51]; and during metastases in ovarian and breast cancer (type III EMT), some cells in hybrid E/M phenotype acquire stem-cell features and can drive in vivo tumour growth, but mesenchymal cells lose these properties significantly [52,53]. Furthermore, recent studies show that the bipotent CSCs in breast and squamous cell carcinoma often co-express both epithelial and mesenchymal markers [54,55] (see detailed discussion in the electronic supplementary material, §5). These results support our prediction that hybrid E/M cells can gain stemness.
Yet, to avoid possible confusion, it is important to clarify that our results, showing significant likelihood of the hybrid cells to adopt stemness, do not imply that epithelial phenotype cannot adopt stemness. More specifically, we show that for high c-MYC levels, the D/U state is associated with the epithelial phenotype, and not with the hybrid E/M one. Given the association of the D/U state with stemness, this result implies that epithelial cells are also likely to possess stemness, especially when c-MYC is overexpressed. Indeed, c-MYC is known to be overexpressed in cases where stemness is associated with the epithelial phenotype—embryonic stem cells [56] and induced pluripotent stem cells [35].
Our results indicate that tristability in LIN28/let-7 circuit affords higher plasticity to the cells in both their epithelial–hybrid–mesenchymal transitions as well as modulating their stemness. Because EMT is no longer considered to be a binary process [6,57,58], and stemness is being understood as a reversible trait of cells rather than their fixed state [59], such multistability, as also observed during transitions between amoeboid and mesenchymal phenotypes [60], might be used by cancer cells to adapt to their changing microenvironments.
Future theoretical studies of the circuits studying EMT and stemness should consider the effect of activation of OCT4 by miR-200 [17], inhibition of EMT by let-7 through HMGA2 [36] and mutual inhibition between ZEB1/2 and MET-inducing transcription factor OVOL1/2 [61], all of which can introduce feedback loops in the regulatory circuit we analysed and affect its dynamics.
To conclude, we present the first step towards computational modelling of the important interconnections between EMT and stemness. More external signals from the tumour microenvironment such as TGF-β and HIF-1α, along with cell–cell communication through Notch signalling pathway, can also be incorporated into our theoretical framework to elucidate their effect on this interconnection. A better understanding of these interconnections holds promise for improved therapeutic strategies to target therapy-resistant CSCs as well as CTCs [2].
Supplementary Material
Acknowledgements
We have benefitted from discussions with Profs Kenneth Pienta, Donald Coffey, José N. Onuchic and Cindy M. Farach-Carson. M.K.J. also thanks Drs Baotong Zhang and Sumaiyah K. Rehman for useful discussions.
Funding statement
H.L. and E.B.-J. were supported by NSF Center for Theoretical Biological Physics (NSF grant no. PHY-1308264) and by the Cancer Prevention and Research Institute of Texas (CPRIT) at Rice University. E.B.-J. was supported by a grant from the Tauber Family Funds and the Maguy-Glass Chair in Physics of Complex Systems at Tel Aviv University. S.A.M. was supported by a grant from the National Institutes of Health (5RO1CA155243) at MD Anderson.
Conflict of interests
The authors declare no conflict of interest.
References
- 1.Wan L, Pantel K, Kang Y. 2013. Tumor metastasis: moving new biological insights into the clinic. Nat. Med. 19, 1450–1464. ( 10.1038/nm.3391) [DOI] [PubMed] [Google Scholar]
- 2.May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. 2011. Epithelial–mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 13, 202 ( 10.1186/bcr2789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai JH, Yang J. 2013. Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 27, 2192–2206. ( 10.1101/gad.225334.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu M, Jolly MK, Levine H, Onuchic JN, Ben-Jacob E. 2013. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl Acad. Sci. USA 110, 18 144–18 149. ( 10.1073/pnas.1318192110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang RY-J, et al. 2013. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 4, e915 ( 10.1038/cddis.2013.442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Revenu C, Gilmour D. 2009. EMT 2.0: shaping epithelia through collective migration. Curr. Opin. Genet. Dev. 19, 338–342. ( 10.1016/j.gde.2009.04.007) [DOI] [PubMed] [Google Scholar]
- 7.Yu M, et al. 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 19, 349–361. ( 10.1126/science.1228522) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, Farace F. 2011. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 105, 1338–1341. ( 10.1038/bjc.2011.405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou J-M, et al. 2011. Circulating tumor cells as a window on metastasis biology in lung cancer. Am. J. Pathol. 178, 989–996. ( 10.1016/j.ajpath.2010.12.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong AJ, et al. 2011. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 9, 997–1007. ( 10.1158/1541-7786.MCR-10-0490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bednarz-Knoll N, Alix-Panabières C, Pantel K. 2012. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 31, 673–687. ( 10.1007/s10555-012-9370-z) [DOI] [PubMed] [Google Scholar]
- 12.Mani SA, et al. 2008. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. ( 10.1016/j.cell.2008.03.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morel A-P, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. 2008. Generation of breast cancer stem cells through epithelial–mesenchymal transition. PLoS ONE 3, e2888 ( 10.1371/journal.pone.0002888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, et al. 2010. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 70, 9463–9472. ( 10.1158/0008-5472.CAN-10-2388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, Sarkar FH. 2010. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 5, e12445 ( 10.1371/journal.pone.0012445) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu C, Ma Y, Wang J, Peng S, Huang Y. 2010. Lin28-mediated post-transcriptional regulation of Oct4 expression in human embryonic stem cells. Nucleic Acids Res. 38, 1240–1248. ( 10.1093/nar/gkp1071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang G, et al. 2013. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl Acad. Sci. USA 110, 2858–2863. ( 10.1073/pnas.1212769110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niwa H, Miyazaki J, Smith AG. 2000. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 24, 372–376. ( 10.1038/74199) [DOI] [PubMed] [Google Scholar]
- 19.Karwacki-Neisius V, et al. 2013. Reduced Oct4 expression directs a robust pluripotent state with distinct signaling activity and increased enhancer occupancy by Oct4 and Nanog. Cell Stem Cell 12, 531–545. ( 10.1016/j.stem.2013.04.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theunissen TW, van Oosten AL, Castelo-Branco G, Hall J, Smith A, Silva JCR. 2011. Nanog overcomes reprogramming barriers and induces pluripotency in minimal conditions. Curr. Biol. 21, 65–71. ( 10.1016/j.cub.2010.11.074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang T-C, et al. 2009. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc. Natl Acad. Sci. USA 106, 3384–3389. ( 10.1073/pnas.0808300106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, et al. 2011. C-myc inhibits the transcription of the microRNA cluster MC-let-7a-1∼let-7d via a non-canonical E-box. J. Biol. Chem. 286, 39 703–39 714. ( 10.1074/jbc.M111.293126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iliopoulos D, Hirsch HA, Struhl K. 2009. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706. ( 10.1016/j.cell.2009.10.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang DJ, Legesse-Miller A, Johnson EL, Coller HA. 2012. Regulation of the let-7a-3 promoter by NF-κB. PLoS ONE 7, e31240 ( 10.1371/journal.pone.0031240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macía J, Widder S, Solé R. 2009. Why are cellular switches Boolean? General conditions for multistable genetic circuits. J. Theor. Biol. 261, 126–135. ( 10.1016/j.jtbi.2009.07.019) [DOI] [PubMed] [Google Scholar]
- 26.Andrecut M, Halley JD, Winkler DA, Huang S. 2011. A general model for binary cell fate decision gene circuits with degeneracy: indeterminacy and switch behavior in the absence of cooperativity. PLoS ONE 6, e19358 ( 10.1371/journal.pone.0019358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang S, Guo Y-P, May G, Enver T. 2007. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev. Biol. 305, 695–713. ( 10.1016/j.ydbio.2007.02.036) [DOI] [PubMed] [Google Scholar]
- 28.Guantes R, Poyatos JF. 2008. Multistable decision switches for flexible control of epigenetic differentiation. PLoS Comput. Biol. 4, e1000235 ( 10.1371/journal.pcbi.1000235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu M, Jolly MK, Gomoto R, Huang B, Onuchic J, Ben-Jacob E. 2013. Tristability in cancer-associated microRNA-TF chimera toggle switch. J. Phys. Chem. B 117, 13 164–13 174. ( 10.1021/jp403156m) [DOI] [PubMed] [Google Scholar]
- 30.Hafner M, Max KEA, Bandaru P, Morozov P, Gerstberger S, Brown M, Molina H, Tuschl T. 2013. Identification of mRNAs bound and regulated by human LIN28 proteins and molecular requirements for RNA recognition. RNA 19, 613–626. ( 10.1261/rna.036491.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilbert ML, et al. 2012. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol. Cell 48, 195–206. ( 10.1016/j.molcel.2012.08.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zisoulis DG, Kai ZS, Chang RK, Pasquinelli AE. 2012. Autoregulation of microRNA biogenesis by let-7 and argonaute. Nature 486, 541–544. ( 10.1038/nature11134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhooge A, Govaerts W, Kuznetsov YA. 2003. MATCONT: a Matlab package for numerical bifurcation analysis of ODEs. ACM Trans. Math. Softw. 29, 141–164. ( 10.1145/779359.779362) [DOI] [Google Scholar]
- 34.Leroy P, Mostov KE. 2007. Slug is required for cell survival during partial epithelial–mesenchymal transition of HGF-induced tubulogenesis. Mol. Biol. Cell 18, 1943–1952. ( 10.1091/mbc.E06-09-0823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, et al. 2010. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63. ( 10.1016/j.stem.2010.04.014) [DOI] [PubMed] [Google Scholar]
- 36.Guo L, et al. 2013. Stat3-coordinated Lin-28–let-7–HMGA2 and miR-200–ZEB1 circuits initiate and maintain oncostatin M-driven epithelial–mesenchymal transition. Oncogene 32, 5272–5282. ( 10.1038/onc.2012.573) [DOI] [PubMed] [Google Scholar]
- 37.Penn LJ, Brooks MW, Laufer EM, Land H. 1990. Negative autoregulation of c-myc transcription. EMBO J. 9, 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, Merchant A, Galiano RD, Tomic-Canic M. 2005. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am. J. Pathol. 167, 59–69. ( 10.1016/S0002-9440(10)62953-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayot F, Jayaprakash C. 2006. NF-κB oscillations and cell-to-cell variability. J. Theor. Biol. 240, 583–591. ( 10.1016/j.jtbi.2005.10.018) [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Wu X, Shi T, Lu L. 2013. EGF-induced corneal epithelial wound healing though NF-κB subtype-regulated CTCF activation. J. Biol. Chem. 288, 24 363–24 371. ( 10.1074/jbc.M113.458141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conigliaro A, et al. 2013. Evidence for a common progenitor of epithelial and mesenchymal components of the liver. Cell Death Differ. 20, 1116–1123. ( 10.1038/cdd.2013.49) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi SS, Diehl AM. 2009. Epithelial-to-mesenchymal transitions in the liver. Hepatology 50, 2007–2013. ( 10.1002/hep.23196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abell AN, et al. 2011. MAP3K4/CBP-regulated H2B acetylation controls epithelial–mesenchymal transition in trophoblast stem cells. Cell Stem Cell 8, 525–537. ( 10.1016/j.stem.2011.03.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baud J, Varon C, Chabas S, Chambonnier L, Darfeuille F, Staedel C. 2013. Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PLoS ONE 8, e60315 ( 10.1371/journal.pone.0060315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. 2006. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 26, 711–724. ( 10.1038/sj.onc.1209808) [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi N, et al. 2009. Constitutive activation of nuclear factor-κB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 100, 1668–1674. ( 10.1111/j.1349-7006.2009.01228.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pattabiraman DR, Weinberg RA. 2014. Tackling the cancer stem cells: what challenges do they pose? Nat. Rev. Drug Discov. 13, 497–512. ( 10.1038/nrd4253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aceto N, et al. 2014. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. ( 10.1016/j.cell.2014.07.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ombrato L, Malanchi I. 2014. The EMT universe: space between cancer cell dissemination and metastasis initiation. Crit. Rev. Oncog. 19, 349–361. ( 10.1615/CritRevOncog.2014011802) [DOI] [PubMed] [Google Scholar]
- 50.Swetha G, Chandra V, Phadnis S, Bhonde R. 2011. Glomerular parietal epithelial cells of adult murine kidney undergo EMT to generate cells with traits of renal progenitors. J. Cell. Mol. Med. 15, 396–413. ( 10.1111/j.1582-4934.2009.00937.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yovchev MI, Grozdanov PN, Zhou H, Racherla H, Guha C, Dabeva MD. 2007. Identification of adult hepatic progenitor cells capable of repopulating injured rat liver. Hepatology 47, 636–647. ( 10.1002/hep.22047) [DOI] [PubMed] [Google Scholar]
- 52.Strauss R, et al. 2011. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 6, e16186 ( 10.1371/journal.pone.0016186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarrio D, Franklin CK, Mackay A, Reis-Filho JS, Isacke CM. 2012. Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells 30, 292–303. ( 10.1002/stem.791) [DOI] [PubMed] [Google Scholar]
- 54.Liu S, et al. 2014. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2, 78–91. ( 10.1016/j.stemcr.2013.11.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H, Costea DE, Mackenzie IC. 2011. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 71, 5317–5326. ( 10.1158/0008-5472.CAN-11-1059) [DOI] [PubMed] [Google Scholar]
- 56.Varlakhanova NV, Cotterman RF, deVries WN, Morgan J, Donahue LR, Murray S, Knowles BB, Knoepfler PS. 2010. myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation 80, 9–19. ( 10.1016/j.diff.2010.05.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu M, Jolly MK, Onuchic J, Ben-Jacob E. 2014. Toward decoding the principles of cancer metastasis circuits. Cancer Res. 74, 4574–4587. ( 10.1158/0008-5472.CAN-13-3367) [DOI] [PubMed] [Google Scholar]
- 58.Pinto CA, Widodo E, Waltham M, Thompson EW. 2013. Breast cancer stem cells and epithelial mesenchymal plasticity: implications for chemoresistance. Cancer Lett. 341, 56–62. ( 10.1016/j.canlet.2013.06.003) [DOI] [PubMed] [Google Scholar]
- 59.Antoniou A, Hébrant A, Dom G, Dumont JE, Maenhaut C. 2013. Cancer stem cells, a fuzzy evolving concept: a cell population or a cell property? Cell Cycle 12, 3743–3748. ( 10.4161/cc.27305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang B, Lu M, Jolly MK, Tsarfaty I, Onuchic J, Ben-Jacob E. 2014. The three-way switch operation of Rac1/RhoA GTPase-based circuit controlling amoeboid–hybrid–mesenchymal transition. Sci. Rep. 4, 6449 ( 10.1038/srep06449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roca H, et al. 2013. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 8, e76773 ( 10.1371/journal.pone.0076773) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.