

Abstract
Objective
Iron may contribute to the pathogenesis and progression of multiple sclerosis (MS) due to its accumulation in the human brain with age. Our study focused on nonheme iron distribution and the expression of the iron-related proteins ferritin, hephaestin, and ceruloplasmin in relation to oxidative damage in the brain tissue of 33 MS and 30 control cases.
Methods
We performed (1) whole-genome microarrays including 4 MS and 3 control cases to analyze the expression of iron-related genes, (2) nonheme iron histochemistry, (3) immunohistochemistry for proteins of iron metabolism, and (4) quantitative analysis by digital densitometry and cell counting in regions representing different stages of lesion maturation.
Results
We found an age-related increase of iron in the white matter of controls as well as in patients with short disease duration. In chronic MS, however, there was a significant decrease of iron in the normal-appearing white matter (NAWM) corresponding with disease duration, when corrected for age. This decrease of iron in oligodendrocytes and myelin was associated with an upregulation of iron-exporting ferroxidases. In active MS lesions, iron was apparently released from dying oligodendrocytes, resulting in extracellular accumulation of iron and uptake into microglia and macrophages. Iron-containing microglia showed signs of cell degeneration. At lesion edges and within centers of lesions, iron accumulated in astrocytes and axons.
Interpretation
Iron decreases in the NAWM of MS patients with increasing disease duration. Cellular degeneration in MS lesions leads to waves of iron liberation, which may propagate neurodegeneration together with inflammatory oxidative burst.
Multiple sclerosis (MS) is a chronic disease of the central nervous system (CNS) leading to oligodendrocyte destruction, demyelination, remyelination, astrocytic scar formation, and neurodegeneration, all being associated with inflammation.1 Effective immunomodulatory therapies target inflammation and subsequent clinical relapses in patients with relapsing–remitting MS (RRMS).2 In contrast, current therapeutic options in primary progressive MS (PPMS) or secondary progressive MS (SPMS) largely remain limited to symptomatic relief. Several factors might prevent therapeutic efficacy,3 among which abnormal iron deposition has recently gained particular interest.4,5 Iron accumulates with increasing age in the healthy human brain, being most prominent after the age of 40 to 50 years,6 which is the time window for patients starting with either PPMS or SPMS.7 Most iron found in the human brain parenchyma is stored as nonheme iron in oligodendrocytes and myelin.8 Iron within the catalytic center of various enzymes is essential for normal brain metabolism, for example, oxidative phosphorylation and myelination.9 In liberated form, however, ferrous iron ions may generate toxic reactive oxygen species (ROS).10 ROS lead to harmful oxidation of lipids and DNA within their immediate vicinity, which is termed oxidative damage. Moreover, mitochondria are both vulnerable to and, if injured, a source of elevated ROS.11,12 Mitochondrial injury is related to oxidative damage in MS.13–15 Oligodendrocytes, which are besides myelin the primary target of inflammatory attacks in MS, are especially vulnerable to such injury.16 Several pathological studies have focused on iron in MS.17–19 We present a study of 63 well-characterized MS and control autopsy cases, examining nonheme iron load as well as the expression of proteins involved in iron metabolism. Our current data on the altered distribution of iron in the brains of MS patients suggest that its liberation within active lesions may amplify demyelination and neurodegeneration.
Materials and Methods
Sample Characterization
The ethics committee of the Medical University of Vienna approved the study (Ethik Kommission No. 535/2004). It was performed on formalin-fixed, paraffin-embedded (FFPE) autopsy brain material from 30 controls without neurological disease or brain lesions and 33 MS cases (Table 1). The MS samples included 7 acute MS (AMS) cases, defined by a clinical course leading to death within 12 months after disease onset. Three patients died during a disease exacerbation in the RRMS stage. Twelve patients presented with SPMS and 9 with PPMS. One case was classified as benign MS. In 1 case, the disease course remained uncertain. Clinical and pathological information (see Table 1) was derived from a database that served as the basis for an earlier study.1
Table 1.
Clinical Data of the Study Cohort
| Case | Age, yr, Median (range) | Sex, F:M Ratio | Clinical Course | Disease Duration, mo, Median (range) |
|---|---|---|---|---|
| Controls 1–30 | 38 (7–97) | 16:14 | — | — |
| Controls 1–18 | 67.5 (30–97) | 12:6 | — | — |
| Controls 19–30 | 19 (7–29) | 4:8 | — | — |
| MS 1–33 | 57 (34–83) | 21:12 | — | 156 (0.2–444) |
| MS 1a | 45 | M | AMS | 0.2 |
| MS 2a | 45 | M | AMS | 0.6 |
| MS 3 | 35 | M | AMS | 1.5 |
| MS 4 | 52 | M | AMS | 1.5 |
| MS 5 | 78 | M | AMS | 2.0 |
| MS 6a | 69 | F | AMS | 2.0 |
| MS 7a | 34 | F | AMS | 4.0 |
| MS 8 | 40 | F | RRMS | 120 |
| MS 9 | 57 | F | RRMS | 156 |
| MS 10 | 44 | F | RRMS | 262 |
| MS 11 | 66 | F | SPMS | 96 |
| MS 12 | 34 | M | SPMS | 120 |
| MS 13 | 41 | M | SPMS | 137 |
| MS 14 | 62 | F | SPMS | 144 |
| MS 15 | 53 | F | SPMS | 241 |
| MS 16 | 61 | F | SPMS | 288 |
| MS 17 | 64 | F | SPMS | 336 |
| MS 18 | 56 | M | SPMS | 372 |
| MS 19 | 78 | F | SPMS | 372 |
| MS 20 | 76 | M | SPMS | 372 |
| MS 21 | 81 | F | SPMS | 432 |
| MS 22 | 46 | F | SPMS | 444 |
| MS 23 | 55 | F | PPMS | 60 |
| MS 24 | 54 | F | PPMS | 72 |
| MS 25 | 67 | M | PPMS | 87 |
| MS 26 | 53 | M | PPMS | 168 |
| MS 27 | 77 | F | PPMS | 168 |
| MS 28 | 34 | F | PPMS | 204 |
| MS 29 | 71 | F | PPMS | 264 |
| MS 30 | 83 | F | PPMS | 360 |
| MS 31 | 75 | F | PPMS | 372 |
| MS 32 | 68 | M | Benign | 372 |
| MS 33 | 68 | F | Uncertain | 120 |
Indicates inclusion of case in microarray study.
AMS = acute MS; F = female; M = male; MS = multiple sclerosis; PPMS = primary progressive MS; RRMS = relapsing–remitting MS; SPMS = secondary progressive MS.
Whole-Genome Microarrays
Technical issues of the presented microarray data have been published previously.13 Shortly, RNA quality of FFPE material was assessed by in situ hybridization for proteolipid protein (PLP) mRNA as described.20 Tissue blocks showing a strong PLP hybridization signal within oligodendrocytes were selected for RNA extraction. Under RNase-free conditions, 6- to 10μm thick sections were mounted on glass slides. Initial (prephagocytic) lesions,21,22 active lesions with early myelin degradation products, and periplaque white matter (PPWM) of MS cases were microdissected. Normal white matter (NWM) from controls was likewise collected. Labeling of RNA, hybridization to microarrays as well as microarray scanning, and quantile normalization of the raw data were done externally (Source BioScience imaGenes, Berlin, Germany). For data analysis, a literature-based search for genes involved in iron metabolism was performed. All microarray data have been deposited in the NIH National Center for Biotechnology Information’s Gene Expression Omnibus (accession number GSE32915). Technical obstacles resulting from formalin fixation–induced strand breaks in the mRNA of FFPE tissue were discussed in previous publications.13,23 Due to the position of some microarray probes, insufficient hybridization with fragmented RNA could have led to false-negative results.
Neuropathology, Iron Histochemistry, and Immunohistochemistry
Tissue blocks (n = 130) were stained with hematoxylin & eosin and Luxol fast blue–periodic acid Schiff myelin stain and inspected for exclusion of confounding pathology. Lesion staging based on earlier characterization1 and was performed with immunohistochemistry for myelin of MS tissue blocks (n = 89). Demyelinating activity was evaluated by assessing myelin degradation products within lysosomes of macrophages.24 For detection of total (ferrous and ferric) as well as ferrous nonheme iron, we applied diaminobenzidine (DAB)-enhanced Turnbull blue staining (Supplementary Materials and Methods).25,26 Immunohistochemistry using DAB as chromogen was performed as described.27 All primary antibodies were incubated overnight at 4°C. Primary antibodies and antigen retrieval methods are listed in Table 2. Iron, ferritin, its subunit ferritin light polypeptide (FTL), hephaestin, and ceruloplasmin were detected on consecutive sections. Oligodendrocytes, microglia, and macrophage numbers were evaluated by analyzing sections stained for TPPP/p25, Iba-1, and CD68, respectively. Immunohistochemistry for oxidized phospholipids (E06 epitope) was performed as described.28 Double and triple immunolabeling for light and confocal fluorescence microscopy are described in the Supplementary Materials and Methods.
Table 2.
Antibodies, Dilutions, and Antigen Retrieval Methods for Immunohistochemistry
| Target | Antibody Type | Dilution | Antigen Retrieval | Source (product number) | Protocol |
|---|---|---|---|---|---|
| Ferritin | Pc rabbit | 1:1,000 | 60 minutes steaming with EDTA pH 8.5 | Sigma, St Louis, MO (F5012) | SL-LM |
| FTL | Pc rabbit | 1:100 | None | Proteintech, Chicago, IL (10727-1-AP) | SL-LM |
| Hephaestin | Pc goat | 1:50 | 60 minutes steaming in citrate buffer, pH 6.0 | Santa Cruz Biotechnology, Santa Cruz, CA (sc-49969) | DL-LM, TL-FCM |
| Ceruloplasmin | Pc rabbit | 1:25 | 60 minutes steaming in citrate buffer, pH 6.0 | Sigma (HPA001834) | SL-LM, DL-FCM |
| E06 | Mc mouse | 10μg/ml | None | Palinski et al 199654 | SL-LM |
| Iba-1 | Pc rabbit | 1:3,000 | 60 minutes steaming with EDTA, pH 9.0 | Wako Chemicals, Richmond, VA (019-19741) | SL-LM, DL-LM |
| CD68 | Mc mouse | 1:100 | 60 minutes steaming with EDTA, pH 9.0 | Dako, Carpinteria, CA (M0814) | SL-LM |
| PLP | Mc mouse | 1:1,000 | 60 minutes steaming with EDTA, pH 9.0 | AbD Serotec, Oxford, UK (MCA839G) | SL-LM |
| MBP | Pc rabbit | 1:2,500 | None | Dako (A0623) | SL-LM |
| CAII | Pc sheep | 1:500 | 30 minutes steaming with EDTA, pH 8.5 | Binding Site, Birmingham, UK (PC076) | TL-FCM |
| TPPP/p25 | Pc rabbit | 1:500 | 30 minutes steaming with EDTA, pH 9.0 | Provided by G. G. Kovacs, Vienna | DL-LM |
| GFAP | Mc mouse | 1:100 | 60 minutes steaming in citrate buffer, pH 6.0 | Thermo Scientific, Waltham, MA (MS-1376-P1) | DL-LM, DL-FCM, |
| GFAP | Pc rabbit | 1:1,500 | 30 minutes steaming with EDTA, pH 8.5 | Dako (Z0334) | TL-FCM |
| Fibrinogen | Mc mouse | 1:500 | 60 minutes steaming in citrate buffer, pH 6.0 | Abcam, Cambridge, MA (ab58207) | DL-FCM |
CAII = carbonic anhydrase II; DL = double-labeling; EDTA = ethylenediaminetetraacetic acid; FCM = fluorescence confocal microscopy; FTL = ferritin light polypeptide; GFAP = glial fibrillary acidic protein; LM = light microscopy; MBP = myelin basic protein; Mc = monoclonal; Pc = polyclonal; PLP = proteolipid protein; SL = single-labeling; TL = triple-labeling; TPPP/p25 = tubulin-polymerization promoting protein.
Quantification
Two different methods of digital optical densitometry were applied on TIFF images of sections. One led to an overall gray value integration of the images stained for total nonheme iron, labeled “iron optical density” (Fig 1); the other set a threshold to obtain cellular staining (all other densitometry results are shown in Table 3 and Supplementary Figs 2 and 3). In addition, immunostained cells were also counted manually. Further details concerning the quantification methods and the selected regions of interest (ROIs) are described in the Supplementary Materials and Methods.
Figure 1.
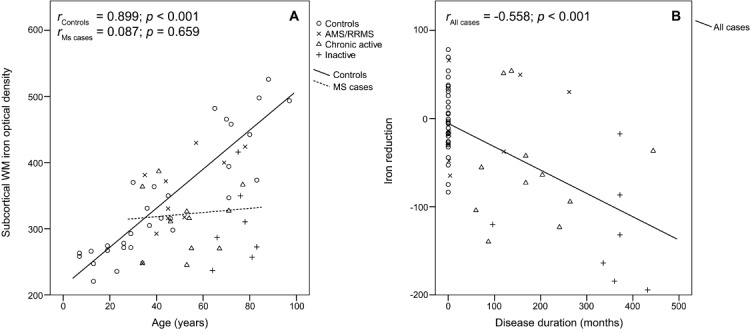
Correlation of whole tissue iron with age (A) and disease duration (B). (A) Whole tissue iron, as quantified by digital optical densitometry, correlated with age (in years) at the nonlesioned subcortical white matter (WM). Lines represent separate linear regressions of controls and multiple sclerosis (MS) cases. (B) Whole tissue iron is subtracted from the iron levels predicted by the age-dependent linear regression equation derived from controls (continuous line in A). There is a significant correlation between the reduction of iron load and disease duration. Furthermore, a significant negative partial correlation between whole tissue iron and disease duration (in months) was found when pooling all data points and setting disease duration of controls to zero (rpartial = −0.497, p < 0.001). Analysis of whole tissue iron in the deep WM led to comparable results (not shown). AMS = acute MS; RRMS = relapsing–remitting MS.
Table 3.
Quantitative Data on Iron, Iron-Related Proteins, Cellular Markers, and Oxidative Damage
| Staining | Quantification Method | NWM | NAWM | PPWM | Early Active | Late Active | Inactive | Remyelinated |
|---|---|---|---|---|---|---|---|---|
| Total nonheme iron | Ds | 0.13 (0.62) | 0.03 (0.7)a | 0.22 (2.37)b | 0.09 (3.45) | 0.61 (3.04) | 0.07 (0.53) | 0.03 (1.09) |
| Ferritin | Ds | 0.89 (2.23) | 1.47 (3.89) | 3.41 (13.54)a,b | 11.26 (9.11)a,b | 4.69 (13.07)a,b | 1.06 (4.7) | 2.32 (3.07)a,b |
| FTL | Ds | 0.2 (1.13) | 0.25 (1.95) | 1.5 (14.53)a,b | 1.78 (13.62)a,b | 1.6 (15.23) | 0.22 (1.5) | 0.23 (2.54) |
| Oligodendrocytes | ||||||||
| TPPP | Ct of TPPP+ cells | 283.5 (399) | 163 (452) | 140 (407) | 66 (215)a,b,c | 0 (0)a,b | ND | |
| Ferritin+ OG | Cta,b | 128.5 (188) | 98.83 (180) | 24.5 (207)a,b | 1.67 (48)a,b | 1 (6)a,b | 0 (53)a,b | 41 (166) |
| FTL+ OG | Cta,b | 58 (225) | 50.5 (157) | 9.5 (217)a,b | 2 (18)a,b | 1.17 (9)a,b | 0 (56)a,b | 18 (188) |
| Heph+ OG | Ct of TPPPa,cHeph+ cells | 27.5 (25) | 14.5 (22)a | 24.5 (55)b | 5 (23)a,c | 0 (0)a,b | ND | |
| % Heph+ OG | TPPP+Heph+ cells / TPPP+ cells × 100 | 11 (16) | 7 (12) | 18 (26)b | 10 (10)c | NA | ND | |
| Microglia/macrophages | ||||||||
| Iba-1 | Ds | 1.69 (2.28) | 1.54 (4.13) | 3.72 (13.42)a,b | 8.5 (7.81)a,b | 4.32 (7.77)a,b | 0.83 (3.1) | 1.3 (4.16) |
| CD68 | Ds | 1.39 (1.68) | 1.89 (5.08) | 3.74 (19.32)a,b | 9.7 (12.55)a,b | 5 (7.84)a,b | 1.14 (6.4) | 1.56 (3.17) |
| Iba-1 | Ct | 95.5 (155) | 88 (285) | 217 (824)a,b | 573 (620.33)a,b | 175 (502)a,b | 38 (283)a,b | 94 (187) |
| CD68 | Ct | 88.5 (45) | 113 (153) | 235.33 (602)a,b | 424.5 (522)a,b | 306 (369)a,b | 61 (259)a,b | 108 (120) |
| Ferritin+ MG | Ct | 26 (34) | 30 (97.5) | 128.89 (568)a,b | 425 (477)a,b | 147 (504)a,b | 17.5 (108) | 36 (76) |
| FTL+ MG | Ct | 18 (35) | 18 (78) | 87.5 (359)a,b | 334 (321.5)a,b | 148.42 (436)a,b | 6 (69) | 40 (89) |
| Ferritin+ Dys MG | Ct | 1.5 (14) | 4 (36)a | 19 (117)a,b | 14 (58)a,b | 10 (27)a | 9.5 (52)a | 3 (8) |
| FTL+ Dys MG | Ct | 0 (9) | 1.69 (19.5) | 12.94 (65)a,b | 7.25 (10)a,b | 9 (38.33)a,b | 3.5 (45.5)a | 1 (7) |
| Astrocytes | ||||||||
| Ferritin+ AG | Ct | 0 (0) | 0 (6) | 2.88 (40)a,b | 2 (17)a,b | 17.5 (44)a,b | 15.5 (81)a,b | 4 (12)a,b |
| FTL+ AG | Ct | 0 (0) | 0 (4)a | 2 (20.33)a,b | 0.83 (38)a,b | 4.5 (57)a,b | 4 (64)a,b | 1 (14)a,b |
| Heph+ AG | Ct of GFAP+Heph+ cells | 18 (30) | 2 (15)a | 7.50 (40) | 12 (67)b,c | 4 (9) | ND | |
| Oxidative damage | ||||||||
| E06+ axonal spheroids | Ct | 0 (0) | 0 (2)a | 2 (15)a,b | 5.25 (10)a,b | 1 (6)a | 0 (5)a | 0 (1) |
Quantitative data are given as median (range in parentheses).
Significant versus NWM.
Significant versus NAWM.
Indicates no discrimination between early and late active regions of interest in the quantification.
AG = astroglia; Ct = manual counting; Ds = densitometry; Dys MG = dystrophic microglia; FTL = ferritin light polypeptide; GFAP = glial fibrillary acidic protein; Heph = hephaestin; MG = microglia/macrophages; NA = not applicable; NAWM = normal-appearing WM; ND = not determined; NWM = normal WM; OG = oligodendroglia; PPWM = periplaque WM; TPPP = tubulin-polymerization promoting protein; WM = white matter.
Statistics
IBM (Armonk, NY) SPSS Statistics Version 20 was used for data analysis. Nonparametric tests were applied for group comparisons between different ROIs. Data of multiple ROIs of the same type per case were averaged to finally represent each case with 1 value per ROI. Kruskal–Wallis group testing was followed by Mann–Whitney U post hoc tests and Bonferroni–Holm correction. Each ROI was compared to both NWM of controls and normal-appearing white matter (NAWM) of MS patients. Pearson coefficient was done for all correlation analyses except for correlation of cellular iron with E06 optical density, which was calculated with the Spearman procedure due to the presence of an outlier. For partial Pearson correlation analysis (see Fig 1A) between iron and disease duration, we controlled for age and set disease duration of controls to zero. To visualize the effect of disease duration on iron load, the difference between iron optical density and the linear regression equation derived from the controls was plotted against disease duration (see Fig 1B).
Results
Iron in the NWM Increases with Age in Controls but Not in MS Cases
Applying the DAB-enhanced Turnbull blue staining,25,26 the highest total nonheme iron contents were observed in deep gray matter nuclei and the leukocortical boundary (Fig 2). Within the deep white matter (WM), iron content was comparatively low.6 In line with previous findings,8,26 most iron in control NWM and MS NAWM was stored in oligodendrocytes and myelin within ferritin (Supplementary Fig 1). In NWM, nonheme iron was detected only exceptionally in other cells such as microglia. Iron has been shown to increase with age in human brains.6 Densitometric analysis of the iron content revealed a significant correlation of iron load with age in the 30 controls in the subcortical (see Fig 1A) and deep WM (not shown). No correlation with age was observed in all MS cases in both WM regions. In acute MS patients with short disease duration, the brain iron content was similar to that of age-matched controls. However, we found a significant decrease of iron load in the NAWM (see Table 3) in patients with chronic progressive disease, correlating with disease duration (see Fig 1B). Compared to the NWM, the NAWM showed a low degree of inflammation, microglia activation, and acute axonal injury, which increased toward the edges of lesions.1,3 This was associated with a lower iron load and decreased numbers of ferritin+ and FTL+ oligodendrocytes, but an increase in iron- and ferritin-containing microglia in the PPWM29 close to lesions. Hephaestin and ceruloplasmin are membrane-bound ferroxidases cooperating with ferroportin to export intracellular iron.30 In the NWM and NAWM, hephaestin was expressed in oligodendrocytes and astrocytes, whereas ceruloplasmin was detected in astrocytes only. Counting of hephaestin+ oligodendrocytes and astrocytes revealed a significant reduction in NAWM compared to NWM. In the PPWM, however, hephaestin+ oligodendrocytes were more numerous. Hephaestin expression was pronounced in individual oligodendrocytes, especially in PPWM close to active lesions, where in addition many astrocytes expressed ceruloplasmin. In summary, our data indicate that the global expression of hephaestin is decreased in the NAWM of MS patients in association with oligodendrocyte loss and with a decrease of iron load in the remaining cells. In PPWM close to active lesion edges, however, the expression of hephaestin and ceruloplasmin is increased within individual cells, suggesting active iron export.
Figure 2.

Iron and the expression of iron-related proteins in the white matter (WM) of controls and multiple sclerosis (MS) patients. (A) Luxol fast blue–periodic acid Schiff myelin staining (blue) and (B) total nonheme iron (brown) of consecutive double-hemispheric slides of MS Subject 25. High iron load is visible in the basal ganglia and the leukocortical boundary. Multiple demyelinated WM lesions, which harbor less iron than the normal-appearing WM (NAWM), are indicated by arrows. (C–Q) Presence of total nonheme iron and iron-related proteins in the normal WM (NWM) of controls (left panel of images; C, F, I, L, O), in the NAWM of MS patients (middle panel; D, G, J, M, P), and in the periplaque WM (PPWM) of MS patients close to the edge of an active lesion (right panel; E, H, K, N, Q). (C–E) In the NWM (C), iron is mainly present in myelin and cells with oligodendrocytic morphology (inset in C) and is reduced in the NAWM of MS patients (D). Inset in D shows small round cells indicative of oligodendrocytes and a larger cell indicative of activated microglia morphology. Close to active plaques, iron is largely lost from oligodendrocytes and myelin, but is present in activated microglia cells (E). (F–H) Immunohistochemistry for ferritin (brown) shows a similar cellular distribution as iron; however, more cells are detected with ferritin immunohistochemistry than with total nonheme iron staining. (I–N) Hephaestin (blue) is expressed in the NWM in oligodendrocytes (I; oligodendrocyte marker TPPP/p25 = red in I–K) and in astrocytes (L; astrocyte marker glial fibrillary acidic protein = red in L–N). In the MS NAWM, both hephaestin+ oligodendrocytes (J) and astrocytes (M) are reduced. In the PPWM close to active lesions, hephaestin is upregulated in relation to the NAWM in oligodendrocytes (K), but not in astrocytes (N). (O–Q) Ceruloplasmin (brown) is mainly observed in astrocytes. Its expression is consistently low in the NWM (O), elevated in the NAWM of some MS patients (P), and consistently strong at the edge of active lesions (Q). AG = astroglia; OG = oligodendroglia; scale bars = 100μm; inset scale bars = 10μm (C, I–N) or 25μm (D–H).
Changes of Iron Content and of Iron-Related Molecules in MS Lesions
The expression of iron-related genes was analyzed using microdissected brain tissue of 4 patients with AMS (indicated as included in microarray study in Table 1) and 3 controls. PPWM, initial lesions,21,22 and actively demyelinating lesions were compared to NWM of controls. We found an upregulated transcription of ferritin genes (FTL, FTH1, and FTMT) involved in intracellular iron storage and detoxification (Table 4). Upregulation of FTMT (mitochondrial ferritin) was accompanied by downregulation of the mitochondrial iron exporter frataxin in initial lesions, suggestive of iron-related mitochondrial injury31 in early MS lesions. Molecules involved in cellular iron import (eg, transferrin receptor, DMT1, ZIP14) as well as cellular iron export and oxidation of iron (hephaestin, APP) showed elevated transcription, being most pronounced in PPWM. Our data suggest an upregulation of cellular defense against iron toxicity in active MS lesions, and upregulation of glial iron shuttling in the PPWM around active lesions.
Table 4.
mRNA Expression Levels of Iron-Related Genes in Multiple Sclerosis Compared to Control White Matter
| Gene Symbol | Fold Changes Compared to Controls | Gene Name | Synonyms | Gene Function | Accession Number | ||
|---|---|---|---|---|---|---|---|
| PPWM | Initial Lesion | Active Lesion | |||||
| FTHa | 1.45 | 6.04 | 1.41 | Ferritin, heavy polypeptide 1 | Intracellular iron storage | NM_002032 | |
| FTL | 5.02 | 3.34 | 3.47 | Ferritin, light polypeptide | Intracellular iron storage | NM_000146 | |
| FTMT | 1.93 | 2.76 | 2.44 | Ferritin, mitochondrial | Mitochondrial iron storage | NM_177478 | |
| TF | 0.40 | 1.17 | 0.15 | Transferrin | Iron transport within cells | NM_001063 | |
| TFRC | 2.01 | 0.90 | 1.41 | Transferrin receptor | Import of transferrin-bound iron | NM_003234 | |
| SCARA5 | 0.73 | 1.74 | 2.39 | Scavenger receptor class A, member 5 | Import of ferritin-bound iron | NM_173833 | |
| SLC11A2 | 3.20 | 0.54 | 1.02 | Solute carrier family 11, member 2 | DMT1, NRAMP2 | Iron importer | NM_000617 |
| SLC11A1 | 0.48 | 0.56 | 0.73 | Solute carrier family 11, member 1 | NRAMP1 | Lysosomal iron transporter | NM_000578 |
| SLC39A14a | 2.33 | 1.75 | 0.99 | Solute carrier family 39, member 14 | ZIP14 | Iron importer | NM_015359 |
| TRPC6a | 1.93 | 5.96 | 1.21 | Transient receptor potential cation channel, subfamily C, member 6 | Iron importer | NM_004621 | |
| MCOLN1 | 1.17 | 0.56 | 0.38 | Mucolipin 1 | TRPML1 | Endosomal/lysosomal iron transporter | NM_020533 |
| CYBRD1 | 0.38 | 0.52 | 0.61 | Cytochrome b reductase 1 | DCYTB | Ferrireductase | NM_024843 |
| STEAP3 | 1.56 | 1.96 | 0.80 | STEAP family member 3 | Ferrireductase | NM_182915 | |
| SLC40A1a | 0.97 | 0.99 | 1.05 | Solute carrier family 40, member 1 | Ferroportin | Iron exporter | NM_014585 |
| CPa | 1.05 | 1.16 | 0.99 | Ceruloplasmin | Ferroxidase | NM_000096 | |
| HEPH | 3.65 | 1.88 | 3.39 | Hephaestin | Ferroxidase | NM_014799 | |
| APP | 2.16 | 1.74 | 1.92 | Amyloid beta precursor protein | Ferroxidase | NM_000484 | |
| HAMP | 1.52 | 1.04 | 0.78 | Hepcidin | Regulation of ferroportin localization | NM_021175 | |
| MON1A | 0.85 | 0.52 | 0.84 | MON1 homolog A | Regulation of ferroportin localization | NM_032355 | |
| SLC25A28 | 0.53 | 0.66 | 0.72 | Solute carrier family 25, member 28 | Mitoferrin-2 | Mitochondrial iron transporter | NM_031212 |
| FXNa | 0.94 | 0.08 | 0.56 | Frataxin | Mitochondrial iron metabolism | NM_181425 | |
False-negative results cannot be ruled out due to position of probes.
PPWM = periplaque white matter.
Iron Accumulates at the Edges and Within a Subset of Active Lesions
In MS lesions, the highest iron load was seen at the edges of classic active,29 slowly expanding, and some inactive lesions, but not of remyelinated plaques (Fig 3; see Table 3). Within lesion centers, the iron content varied between lesions from the same case and even more between lesions of different cases. Studying 100 WM lesions of our sample, we observed on a case-based average 8% to contain more iron, whereas 27% contained equal and 65% contained less iron than the surrounding NAWM. Lesions with increased iron load were in the late active stage, whereas inactive or remyelinated lesions invariably showed equal or less iron load. In active lesions, demyelination and oligodendrocyte destruction occur in a zone of variable size in the PPWM at the lesion border.29 Thus, iron-containing myelin and oligodendrocytes gradually decreased from the PPWM toward the lesion centers. Correlation of cellular iron with cellular ferritin, as quantified by densitometry, showed the presence of iron being invariably accompanied by the presence of ferritin (see Supplementary Fig 2). Ferritin, however, was also detected in macrophages and microglia in the absence of iron, especially in classic active lesions, explaining the discrepancies of iron and ferritin densitometry in early and late active lesion centers. In comparison to total ferritin, FTL staining was less abundant, but showed a similar cellular distribution.26 In addition, early active lesions contained abundant small iron-reactive granules in the extracellular space. In contrast to total nonheme iron, which was most prominently seen in cytoplasmic compartments, ferrous iron reactivity was mainly present in extracellular granules and within endosomes or lysosomes of macrophages. Extracellular ferrous iron was mainly observed in aged patients with acute MS, where active lesions formed in brains with high iron load. Toward the plaque center, the density of iron-reactive macrophages and microglia decreased, but these cells were frequently encountered in perivascular spaces. Within the center of inactive lesions, iron-containing oligodendrocytes were rare or absent, reflecting demyelination and oligodendrocyte loss.32 Iron, when present at all, was seen within astrocytes, axons, and occasional macrophages. In contrast to the PPWM, only sparse remaining oligodendrocytes but more astrocytes expressed hephaestin in active lesions, where ceruloplasmin was predominantly seen in astrocytes and axons. Ceruloplasmin reactivity in astrocytes and axons partly colocalized with fibrin in active lesions, indicating uptake of soluble serum ceruloplasmin from the extracellular space under conditions of blood–brain barrier damage.
Figure 3.

Iron and iron-related molecules in different types of MS lesions. (A–F) There are 3 different types of MS lesions regarding iron content. (A, D) In slowly expanding lesions with demyelinating activity at the lesion edge, and less frequently and less pronounced in inactive lesions, a rim of iron within microglia and macrophages is seen at the lesion edge. The iron content within the lesion is reduced, but perivascular accumulation of iron is occasionally seen within lesions. (A) Proteolipid protein (PLP) immunohistochemistry (IHC). (D) Total nonheme iron staining. (B, E) Some classic late active MS lesions contain more iron compared to the normal-appearing white matter (NAWM). The iron is found within macrophages, astrocytes, and axons in these lesions. (B) Luxol fast blue–periodic acid Schiff myelin staining (blue). (E) Total nonheme iron staining. (C, F) In the majority of MS lesion, iron is reduced compared to the NAWM, and total nonheme iron staining closely matches the staining for myelin. (C) PLP IHC. (F) Total nonheme iron staining. (G–U) Iron and iron-related proteins are shown in early active lesions (left panel; G, J, M, P, S), late active lesions (middle panel; H, K, N, Q, T), and inactive lesions (right panel; I, L, O, R, U). The figure documents an extreme example (MS Subject 5, who developed acute MS at the age of 78 years; the lesions formed in a brain with a high age-related iron load). Lesional activity was defined according to Brück et al.24 In early active lesions, total nonheme iron (G, brown) is mainly seen in the cytoplasm of macrophages and microglia and to a lesser extent as fine extracellular granules. In contrast, ferrous nonheme iron (J, brown) is largely detected as extracellular granules or in lysosomes or endosomes of macrophages and microglia. Ferritin expression (M, brown) additionally reflects the massive macrophage and microglia activation. Few oligodendrocytes (P; TPPP/p25 = red in P–R) and many astrocytes (S; glial fibrillary acidic protein [GFAP] = red in S to U) express hephaestin (blue in P–U) in this early active lesion. In late active lesions, there is a shift of iron-containing macrophages toward the perivascular space (H). Extracellular total (H, brown) and ferrous nonheme iron (K, brown) are sparse in comparison to early active lesions. Ferritin (N; brown) is predominantly expressed in macrophages. Oligodendrocytes are largely lost (Q), and there is sparse hephaestin expression in astrocytes (T). In inactive lesions, total nonheme iron is seen in few scattered astrocytes (I), whereas ferrous nonheme iron (L) is not detectable. Ferritin expression (O; brown) is only seen in some cells, predominantly astrocytes. Oligodendrocytes are lost from the lesions (R), and there is profound fibrillary gliosis (U). Hephaestin expression is minor or absent (R, U). (V) Confocal laser microscopy shows the expression of hephaestin (green) in oligodendrocytes (carbonic anhydrase II = red) and astrocytes (GFAP = blue). In this area of MS periplaque white matter, hephaestin is detected predominantly in oligodendrocytes. In active lesions (W–Y), ceruloplasmin (green in W–Y) is detected mainly in astrocytes (W; GFAP = red in W) and axons (X). However, in many cells and axons, ceruloplasmin is colocalized with fibrin (red in X and Y), suggesting nonspecific uptake of soluble serum ceruloplasmin from the extracellular space under conditions of severe blood–brain barrier damage. Nonetheless, some cells with astrocyte morphology are stained for ceruloplasmin in the absence of fibrin reactivity, suggesting autochthonous expression of ceruloplasmin in at least some astrocytes. AG = astroglia; OG = oligodendroglia; scale bars = 4mm (A–F), 100μm (G–U); inset scale bars = 10μm.
Iron-Related Microglial Dystrophy in MS Lesions and Microglial Loss in Inactive Lesion Centers
At lesion edges and less prominently in lesion centers, a substantial proportion of ferritin- and iron-containing microglia displayed dystrophic features such as focal swellings, fragmentation of cell processes, or cytorrhexis (Fig 4A, B). These features of dystrophy, indicating microglia degeneration, have been described in the ageing brain, Alzheimer disease, and Huntington disease.33–35 The highest numbers of dystrophic microglia were found at lesion edges, which showed high iron load within microglia. Subsequent counting of the 2 markers Iba-1 and CD68 confirmed a significant reduction of microglia cells in inactive lesion centers compared to the NAWM (see Table 3). In contrast, remyelinated lesions showed only sparse microglial dystrophy or loss and no iron deposition at their edges. When microglia and macrophages were lost, iron accumulation was seen in astrocytes and axons (see Fig 4C, D) within a subset of active plaques or plaque edges.
Figure 4.
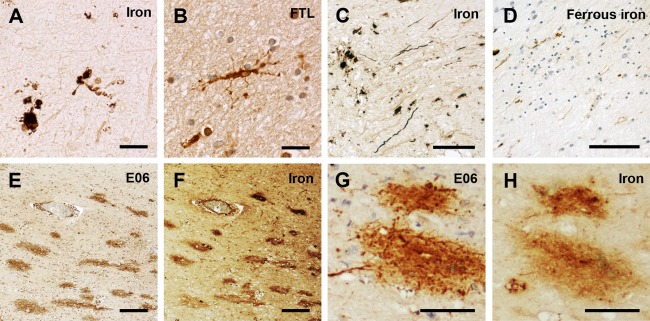
Dystrophic microglia, axonal iron, and oxidized phospholipids in multiple sclerosis (MS) lesions. Iron-loaded microglia and macrophages in active MS lesions show signs of degeneration (dystrophy) with process beading, retraction, and fragmentation (A), which is also visible in microglia stained for ferritin light polypeptide (FTL; B, brown). At active lesion edges, total (C) and rarely also ferrous nonheme iron (D, brown) accumulates in axons. (E–H) Oxidized phospholipids (E06 reactivity; E and G; brown) are detected in lesions with high iron content (total nonheme iron staining in F and H). Scale bars = 20μm (A, B); 100μm (C, D); 200μm (E, F); 75μm (G, H).
Correlation between Iron Deposition and Presence of Oxidized Phospholipids
For evaluation of oxidative damage, oxidized phospholipids were stained (E06 epitope).28 E06+ axonal spheroids, indicating acute axonal damage linked to oxidative damage, were most abundant in actively demyelinating lesions (see Table 3), where iron-containing oligodendrocytes or microglia were progressively lost and ferrous iron was observed within the extracellular space. Optical E06 density was weakly but significantly correlated (r = 0.259, p < 0.001) with cellular iron load (see Supplementary Fig 3). Hence, we reason that iron contributes to but is not the only factor leading to oxidative damage in MS. Nevertheless, some lesions with high iron load displayed a 1-to-1 colocalization of iron and E06 (see Fig 4E–H).
Discussion
Our study shows alterations in iron metabolism that differ between MS lesions and the NAWM. In the NAWM, age-related physiological iron increase in oligodendrocytes and myelin is apparently opposed by iron release from these structures, correlating with disease duration in chronic MS. Accordingly, a recent in vivo magnetic resonance study applying susceptibility-weighted imaging (R2′ sequence) found reduced signal intensity in the NAWM of MS patients in comparison to controls.36 According to the authors, such a reduction could be due to either a decrease of myelin, nonheme tissue iron, or hemoglobin in the brain vasculature. Our data show that in addition to a loss of oligodendrocytes, there is a reduction of nonheme iron within oligodendrocytes and myelin. This loss of iron may be related to the chronic inflammatory process, which is present not only in lesions, but also in the NAWM.1 A recent study addressed the effects of proinflammatory (tumor necrosis factor [TNF]-α) and anti-inflammatory (transforming growth factor [TGF]-β1) cytokines on microglia and astrocytes.37 Both cytokines led to iron uptake and retention in microglia. In astrocytes, however, TNF-α triggered iron retention, whereas TGF-β1 induced its release. Iron-laden oligodendrocytes are more vulnerable to the proinflammatory cytokines TNF-α and interferon-γ in vitro.38,39 Hence, inflammatory cytokines could trigger iron release from oligodendrocytes, similar to the effect of TGF-β1 on astrocytes. We found the iron export ferroxidases hephaestin and ceruloplasmin30 to be upregulated in oligodendrocytes and astrocytes in particular close to inflamed lesion edges. This increase may in part explain the reduction of the iron load in MS NAWM.
In MS lesions, oxidative stress appears to play a major pathogenic role. This hypothesis is supported by accumulation of oxidized DNA and phospholipids in degenerating oligodendrocytes, axons and neurons in active MS lesions,28 upregulation of antioxidative defense mechanisms,40 and effective antioxidative treatment for RRMS patients.41 Potential sources of oxygen and nitric oxide radicals include oxidative burst13,42,43 and mitochondrial injury,12 which has been demonstrated in neurons, demyelinated axons,14,15 oligodendrocytes, and astrocytes31 in MS cortex and WM lesions. However, radical injury can be further amplified by transition metals such as iron or copper,44 and iron toxicity has been suggested to participate in neurodegenerative diseases,45 including MS.46 Pathological and magnetic resonance imaging studies have revealed iron accumulation at the edges of chronic MS lesions.17,26,47 Iron was present in oligodendrocytes in the NAWM and accumulated in microglia and macrophages at lesion edges.26,47 We found that iron-containing oligodendrocytes and myelin were destroyed in active MS lesions,21,22 which presumably led to a wave of iron liberation from intracellular stores into the extracellular space. We detected extracellular iron, including ferrous iron, especially in active lesions of aged patients with acute MS and short disease duration, where new active lesions form against the background of high tissue iron load. These events may be followed by a shift of cellular iron storage from oligodendrocytes to microglia and macrophages. MS lesions with increased iron load in comparison to the surrounding NAWM were late active, characterized by the abundance of macrophages with late myelin degradation products throughout the demyelinated area. Iron was located within these macrophages, but to a lower degree also in astrocytes and axons. Iron accumulation in these particular lesions may additionally be related to active inflammation. Blood–brain barrier damage with subsequent passive transferrin-bound iron leakage into the lesions may increase the iron content. In addition, hypoxia-induced upregulation of iron regulatory proteins (IRP1 and 2) and subsequently elevated translation of transferrin receptors48 could be involved in increased cellular iron storage.46 MS lesions display hypoxialike tissue injury,49 which has been linked to inflammation-induced mitochondrial injury,31 a state called virtual hypoxia.50 Nonetheless, this iron is apparently cleared from the lesions, because inactive demyelinated lesions showed on average a lower iron load than the surrounding NAWM. The perivascular accumulation of iron-containing macrophages suggests that they remove iron from the lesions through perivascular drainage into the cervical lymph nodes, as shown for macrophages containing myelin and neuronal antigens51 or ultrasmall superparamagnetic particles of iron oxide.52
Additionally, iron-containing microglia and macrophages showed signs of process fragmentation and cytorrhexis, termed microglial dystrophy or senescence,33 which has been linked to iron accumulation, ferritin upregulation, and oxidative stress.34 Degradation of dystrophic microglia at MS lesion edges and centers may lead to a second wave of iron release and subsequent accumulation within other microglia, astrocytes, or axons. Extracellular and axonal iron might promote neurodegeneration in MS, as we found E06+ axonal spheroids predominantly at sites of demyelination and cellular iron liberation in active lesions. Thus, 2 sources of radicals, namely oxidative burst from activated microglia13 and iron within axons or the extracellular space, may act synergistically to promote neurodegeneration.
However, iron is also essential for cellular function and homeostasis in the CNS. Recently, iron-containing ferritin has been shown to promote oligodendrogenesis and oligodendrocyte differentiation from precursor cells when injected into the rat spinal cord.53 Thus, iron liberated in the lesions may augment demyelination and neurodegeneration, and the observed overall iron loss in the NAWM may be both protective against inflammatory attacks and detrimental by reducing the capacity of the MS brain for remyelination and tissue repair. Iron-chelating therapies for MS patients can therefore, based on our current knowledge, not be recommended. Nevertheless, blocking harmful downstream effects of iron liberation, such as oxidation of lipids and DNA, might be beneficial for MS patients.
Acknowledgments
This study was funded by the Austrian Science Fund (Project P24245-B19).
We thank U. Köck, A. Kury, and M. Leisser for excellent technical assistance.
Potential Conflicts of Interest
S.P.: travel expenses, Biogen Idec, Teva. W.B.: board membership, Teva Pharma, Biogen Idec, Genzyme, Novartis; consultancy, Teva Pharma, Biogen Idec, Novartis, Genzyme; grants/grants pending, Teva Pharma, Biogen Idec, Novartis; speaking fees, Teva Pharma, Biogen Idec, Merck Serono, Novartis, Bayer Vital, Sanofi, Genzyme. H.L.: consultancy, Amgen, Biogen, Baxter; speaking fees, Novartis, Biogen Idec, Serono, Teva.
Supporting Information
References
- Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(pt 5):1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8:647–656. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- Khalil M, Langkammer C, Ropele S, et al. Determinants of brain iron in multiple sclerosis: a quantitative 3T MRI study. Neurology. 2011;77:1691–1697. doi: 10.1212/WNL.0b013e318236ef0e. [DOI] [PubMed] [Google Scholar]
- Singh AV, Zamboni P. Anomalous venous blood flow and iron deposition in multiple sclerosis. J Cereb Blood Flow Metab. 2009;29:1867–1878. doi: 10.1038/jcbfm.2009.180. [DOI] [PubMed] [Google Scholar]
- Hallgren B, Sourander P. The effect of age on the non-haemin iron in the human brain. J Neurochem. 1958;3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x. [DOI] [PubMed] [Google Scholar]
- Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343:1430–1438. doi: 10.1056/NEJM200011163432001. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL. Cellular management of iron in the brain. J Neurol Sci. 1995;134(suppl):33–44. doi: 10.1016/0022-510x(95)00206-h. [DOI] [PubMed] [Google Scholar]
- Todorich B, Pasquini JM, Garcia CI, et al. Oligodendrocytes and myelination: the role of iron. Glia. 2009;57:467–478. doi: 10.1002/glia.20784. [DOI] [PubMed] [Google Scholar]
- Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics. 2009;2:2. doi: 10.1186/1755-8794-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roede JR, Jones DP. Reactive species and mitochondrial dysfunction: mechanistic significance of 4-hydroxynonenal. Environ Mol Mutagen. 2010;51:380–390. doi: 10.1002/em.20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MT, Sharma R, Lim JL, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012;135(pt 3):886–899. doi: 10.1093/brain/aws012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Ziabreva I, Reeve AK, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011;69:481–492. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- Ziabreva I, Campbell G, Rist J, et al. Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia. 2010;58:1827–1837. doi: 10.1002/glia.21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craelius W, Migdal MW, Luessenhop CP, et al. Iron deposits surrounding multiple sclerosis plaques. Arch Pathol Lab Med. 1982;106:397–399. [PubMed] [Google Scholar]
- Adams CW. Perivascular iron deposition and other vascular damage in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1988;51:260–265. doi: 10.1136/jnnp.51.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine SM. Iron deposits in multiple sclerosis and Alzheimer’s disease brains. Brain Res. 1997;760:298–303. doi: 10.1016/s0006-8993(97)00470-8. [DOI] [PubMed] [Google Scholar]
- Breitschopf H, Suchanek G, Gould RM, et al. In situ hybridization with digoxigenin-labeled probes: sensitive and reliable detection method applied to myelinating rat brain. Acta Neuropathol. 1992;84:581–587. doi: 10.1007/BF00227734. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, et al. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130(pt 11):2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MT, Wimmer I, Hoftberger R, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136(pt 6):1799–1815. doi: 10.1093/brain/awt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brück W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol. 1995;38:788–796. doi: 10.1002/ana.410380514. [DOI] [PubMed] [Google Scholar]
- Meguro R, Asano Y, Odagiri S, et al. Nonheme-iron histochemistry for light and electron microscopy: a historical, theoretical and technical review. Arch Histol Cytol. 2007;70:1–19. doi: 10.1679/aohc.70.1. [DOI] [PubMed] [Google Scholar]
- Bagnato F, Hametner S, Yao B, et al. Tracking iron in multiple sclerosis: a combined imaging and histopathological study at 7 tesla. Brain. 2011;134(pt 12):3602–3615. doi: 10.1093/brain/awr278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Elger CE, Hans VH, et al. Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol. 2007;62:67–80. doi: 10.1002/ana.21148. [DOI] [PubMed] [Google Scholar]
- Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134(pt 7):1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H. The architecture of inflammatory demyelinating lesions: implications for studies on pathogenesis [review] Neuropathol Applied Neurobiol. 2011;37:698–710. doi: 10.1111/j.1365-2990.2011.01189.x. [DOI] [PubMed] [Google Scholar]
- Schulz K, Vulpe CD, Harris LZ, David S. Iron efflux from oligodendrocytes is differentially regulated in gray and white matter. J Neurosci. 2011;31:13301–13311. doi: 10.1523/JNEUROSCI.2838-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131(pt 7):1722–1735. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews I, Bergmann M, Bunkowski S, et al. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult Scler. 1998;4:55–62. doi: 10.1177/135245859800400203. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes KO, Sparks DL, Streit WJ. Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia. 2008;56:1048–1060. doi: 10.1002/glia.20678. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- Paling D, Tozer D, Wheeler-Kingshott C, et al. Reduced R2′ in multiple sclerosis normal appearing white matter and lesions may reflect decreased myelin and iron content. J Neurol Neurosurg Psychiatry. 2012;83:785–792. doi: 10.1136/jnnp-2012-302541. [DOI] [PubMed] [Google Scholar]
- Rathore KI, Redensek A, David S. Iron homeostasis in astrocytes and microglia is differentially regulated by TNF-alpha and TGF-beta1. Glia. 2012;60:738–750. doi: 10.1002/glia.22303. [DOI] [PubMed] [Google Scholar]
- Zhang X, Haaf M, Todorich B, et al. Cytokine toxicity to oligodendrocyte precursors is mediated by iron. Glia. 2005;52:199–208. doi: 10.1002/glia.20235. [DOI] [PubMed] [Google Scholar]
- Zhang X, Surguladze N, Slagle-Webb B, et al. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia. 2006;54:795–804. doi: 10.1002/glia.20416. [DOI] [PubMed] [Google Scholar]
- van Horssen J, Drexhage JA, Flor T, et al. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic Biol Med. 2010;49:1283–1289. doi: 10.1016/j.freeradbiomed.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- Gray E, Thomas TL, Betmouni S, et al. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci Lett. 2008;444:195–198. doi: 10.1016/j.neulet.2008.08.035. [DOI] [PubMed] [Google Scholar]
- Liu Y, Hao W, Letiembre M, et al. Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J Neurosci. 2006;26:12904–12913. doi: 10.1523/JNEUROSCI.2531-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Nunez MT, Urrutia P, Mena N, et al. Iron toxicity in neurodegeneration. Biometals. 2012;25:761–776. doi: 10.1007/s10534-012-9523-0. [DOI] [PubMed] [Google Scholar]
- Williams R, Buchheit CL, Berman NE, LeVine SM. Pathogenic implications of iron accumulation in multiple sclerosis. J Neurochem. 2012;120:7–25. doi: 10.1111/j.1471-4159.2011.07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt D, Boster A, Pei W, et al. Imaging cortical lesions in multiple sclerosis with ultra-high-field magnetic resonance imaging. Arch Neurol. 2010;67:812–818. doi: 10.1001/archneurol.2010.148. [DOI] [PubMed] [Google Scholar]
- Rathnasamy G, Ling EA, Kaur C. Iron and iron regulatory proteins in amoeboid microglial cells are linked to oligodendrocyte death in hypoxic neonatal rat periventricular white matter through production of proinflammatory cytokines and reactive oxygen/nitrogen species. J Neurosci. 2011;31:17982–17995. doi: 10.1523/JNEUROSCI.2250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboul-Enein F, Rauschka H, Kornek B, et al. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62:25–33. doi: 10.1093/jnen/62.1.25. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- van Zwam M, Huizinga R, Melief MJ, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med (Berl) 2009;87:273–286. doi: 10.1007/s00109-008-0421-4. [DOI] [PubMed] [Google Scholar]
- Oude Engberink RD, Blezer EL, Dijkstra CD, et al. Dynamics and fate of USPIO in the central nervous system in experimental autoimmune encephalomyelitis. NMR Biomed. 2010;23:1087–1096. doi: 10.1002/nbm.1536. [DOI] [PubMed] [Google Scholar]
- Schonberg DL, Goldstein EZ, Sahinkaya FR, et al. Ferritin stimulates oligodendrocyte genesis in the adult spinal cord and can be transferred from macrophages to NG2 cells in vivo. J Neurosci. 2012;32:5374–5384. doi: 10.1523/JNEUROSCI.3517-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palinski W, Ylä-Herttuala S, Rosenfeld ME, Butler SW, Socher SA, Parthasarathy S, Curtiss LK, Witztum JL. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis. 1990;10:325–335. doi: 10.1161/01.atv.10.3.325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.