

Abstract
Background
Rhinovirus infection or dsRNA stimulation increased thymic stromal lymphopoietin (TSLP), an upstream pro-allergic cytokine, in asthmatic bronchial epithelial cells. We hypothesized that dsRNA challenges superimposed on established experimental allergic asthma constitute a useful exacerbation model. We further hypothesized that TSLP is induced at dsRNA- and rhinoviral infection-induced exacerbations.
Methods
Allergic mice were challenged with OVA followed by three daily intranasal challenges with dsRNA or saline. Bronchoalveolar lavage fluid (BALF) was analysed for total protein, lactate dehydrogenase (LDH), CXCL1/KC, CCL2/MCP-1 and differential cell counts. Lung tissue histology, neutrophils and TSLP, TNF-α, IFN-β and IFN-λ mRNA were examined. Alternatively, allergen-challenged mice received intranasal rhinovirus-(RV)-1B followed by lung TSLP immunostaining.
Results
In mice with allergic airway inflammation, dsRNA challenges caused a significant exacerbation increasing lung tissue inflammation score and tissue neutrophilia. Bronchoalveolar lavage fluid neutrophils, total protein, LDH, CXCL1/KC and CCL2/MCP-1 were also increased (P < 0.01), and so were lung tissue expressions of TNF-α, IFN-λ and TSLP (P < 0.01), but IFN-β was not increased. TSLP, IFN-λ and LDH were not increased by allergen or dsRNA challenges alone, but increased exclusively at exacerbations. RV1B infection-induced exacerbation also increased lung tissue TSLP (P < 0.05).
Conclusions
dsRNA-induced exacerbation in mice with experimental asthma involved general inflammation, cytokines and interferons, in agreement with previous observations in exacerbating human asthma. Additionally, both dsRNA and RV1B infection increased lung TSLP exclusively at exacerbations. Our data suggest that dsRNA challenges superimposed on allergic inflammation are suited for pharmacological studies of asthma exacerbations including the regulation of lung tissue TSLP, TNF-α, IFN-β and IFN-λ.
Keywords: allergy, asthma, exacerbation, neutrophils, TLR3
The clinical importance of common cold-induced exacerbations of asthma has been increasingly established during the last 150 years, with rhinoviral (RV) infections occurring in the majority of exacerbations 1,2. Inflammatory cytokines and chemokines, such as TNF-α, CXCL8/IL8 and CCL2/MCP-1, are induced in the lung during asthma exacerbations 3. The exacerbations also exhibit cell lysis/necrosis. Hence, lactate dehydrogenase (LDH), an indicator of cell lysis, is a sputum marker of RV-induced exacerbations 4. Mechanisms involved in RV-induced asthma exacerbations and in the development of severe asthma remain to be defined in several respects, and novel treatments are needed for these severe conditions. Hence, experimental exacerbation models are warranted.
In vivo experiments with live RV infections require laboratory facilities dedicated to viral handling and validated methodology for viral propagation and assessment of infection titres to control infection take rate. The opportunity of using a mimic of RV-induced effects in cell systems and in vivo is therefore an attractive alternative. To this end, administration of dsRNA is increasingly employed. dsRNA is a biologically active intermediate molecule formed in many types of viral infections including RV-induced common cold. dsRNA and its synthetic mimic, poly(I:C), are archetypical TLR3 ligands increasingly utilized in experimental studies of mechanisms of inflammation and host defence 5–7. As a challenge agent, dsRNA has advantages of well-controlled dose–response evaluations and repeatability. Effects of dsRNA have recently been studied in murine models of allergic asthma with focus on dsRNA treatment during sensitization and challenge of the animals with allergen 5,8,9. However, less is known about the effects of dsRNA on lungs with established experimental asthma. Additionally, dsRNA may mimic not only viral infection but also the effects of necrotic cells 10 that also characterize viral-induced exacerbations of asthma 4.
Using primary bronchial epithelial cells from asthma and COPD donors, we observed that RV infection as well as dsRNA stimulation increased the expression of thymic stromal lymphopoietin (TSLP), TNF-α and CXCL8/IL-8 11–13. Thymic stromal lymphopoietin was of particular interest because this cytokine was overproduced in diseased compared to healthy cells 11,13. Previous studies had found that bronchial TSLP mRNA is increased in severe asthma 14,15. Thymic stromal lymphopoietin has also been proposed as a ‘master switch’ of asthma in mouse models 16. The actions of TSLP, involving direct effects on target immune cells and indirect effects through priming of dendritic cells, have made this cytokine a compelling asthma drug target 17. The experimental actions of TSLP together with an increased production of TSLP in asthmatic bronchial epithelial cells exposed to RV or dsRNA underpin our hypothesis that TSLP is potentially important in exacerbation-induced worsening of asthma at and beyond the acute inflammatory effects 11,12. However, there is no information on the induction of TSLP expression in the animal models of asthma exacerbations.
The purpose of this study was to develop a model of asthma exacerbation that involves increased lung expression of such cytokines that are potential targets for pharmacological inhibition. Selectivity is important to spare host defence. Hence, novel drugs aimed at treatment for exacerbations of asthma should probably not inhibit antiviral defence mechanisms such as the production of interferon beta (IFN-β) and IFN lambda (IFN-λ) by lung cells 18. However, these IFNs are also discussed as potential inducers; thus, Holt and Sly have advanced the possibility that IFN-β promotes IgE production in asthma 19, and IFN-λ stood out amongst mediators by being the only one increased at asthma exacerbations in children 20. A new model of exacerbation of asthma should therefore include inducement of these IFNs to allow the discovery of different action profiles of novel drugs.
Here, we have examined the effects of repeated airway administrations of dsRNA to mouse allergic lungs with and without already established allergic inflammation. We demonstrate that dsRNA induces an exacerbation characterized by eosinophilic–neutrophilic–lymphocytic lung inflammation with an increased expression of inflammatory cytokines, such as CXCL1/KC (corresponding to CXCL8), TNF-α and IFN-λ. Lung tissue expression of TSLP and IFN-λ and bronchoalveolar lavage fluid (BALF) LDH levels were increased only at exacerbation. In a separate study, we confirmed that lung TSLP is increased also at exacerbations induced by RV1B infection [RV1B infection superimposed on allergic inflammation is otherwise reported elsewhere 21,22].
Materials and methods
Animals and experimental study design
BALB/c mice (Taconic, Lille Skensved, Denmark) of 8 weeks old were used in the study. The animal ethics committee at Lund University approved the animal experiments. The first part of this study was addressed by using repeated intranasal dsRNA challenges (polyinosine–polycytidylic acid) (poly(I:C); Invitrogen Ltd, Paisley, UK) to naïve mice with the aim to find a suitable dose regimen for the administration of intranasal dsRNA to evoke exacerbation. Next dsRNA treatment or saline control was administered to mice with allergic asthma, the latter according to a validated protocol in our laboratory 23. For experimental design, see Fig. 1. The second part of the study included mice immunized and challenged with ovalbumin followed by infection with RV1B or challenge with UV-inactivated virus as described previously 21,22. Briefly, mice received RV1B, UV-RV1B or PBS intranasally, and the experiment was terminated day 2 or 7 postinfection when lung tissue was collected for immunohistochemistry.
Figure 1.
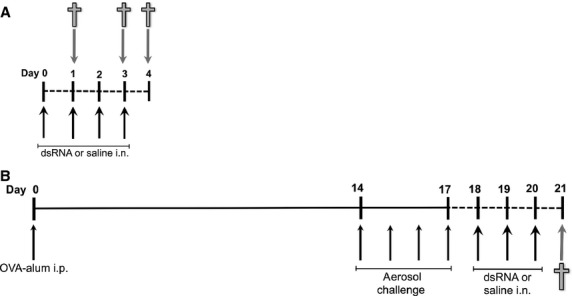
Study design involving dsRNA challenges to mice with and without established asthma. Mice received up to four challenges with dsRNA, 100 μg intranasally every 24 h, and were killed 24, 72 and 96 h after first dsRNA administration (A). The study design shown in (B) was chosen for the exacerbation model. Mice were sensitized with OVA–alum at day 0, followed by four consecutive days of aerosol challenge starting day 14. Following the sensitization and allergen challenges, the mice received three dsRNA challenges, days 18–20 (B). Animals were killed 24 h after the final dsRNA administration. i.p. = intraperitoneal; i.n. = intranasal.
Bronchoalveolar lavage, differential cell count and lung dissection
Bronchoalveolar lavage fluid was collected and total cell count was performed, using an automatic cell counter (NucleoCounter, Chemometec Lillerød, Denmark), and the cell suspension was cytospin-centrifuged onto microscope slides, stained and counted as previously described 23. Following BAL, the lungs were dissected and the left lung lobe was fixed in 4% paraformaldehyde, while the right lung was weighed and frozen in −80°C, until used for RT-qPCR analysis.
Analysis of BALF
Total protein concentration was analysed using bicinchoninic acid (BCA) assay (Pierce® BCA Protein Assay Kit detection; Fisher Scientific AB, Gothenburg, Sweden), according to the manufacturer's instructions. Release of cytokines CXCL1/KC and CCL2/MCP-1 in BALF supernatant was detected using enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Abingdon, UK), according to the manufacturer's instructions.
Lung histology and immunostaining for neutrophils and TSLP
Paraffin-embedded tissues were sectioned into 4 μm thick and stained with haematoxylin and eosin (H&E) staining. The H&E slides were analysed using a light microscope (Nikon ECLIPSE 80i, Nikon Corporation, Tokyo, Japan), and a score (1–6) was given reflecting the degree of lung inflammation. Main features taken into account were the amount of perivascular and peribronchial inflammatory infiltrates, alveolar wall thickness and degree of epithelial hypertrophy. Sections with no obvious infiltrates were scored 0. All slides were analysed blindly by two independent observers. Additional sections for each animal were incubated with antibodies for detecting either neutrophils (NIMP-R14; Abcam, Cambridge, UK) or TSLP (GeneTex Inc., Irvine, CA, USA). Sections were incubated with appropriate biotinylated secondary antibodies (ABC Vectastain; Vector Laboratories, Burlingame, CA, USA) and visualized using 3,3′-diaminobenzidine (DAB, Vectastain; Vector Laboratories) followed by an H&E counterstain. Sections were scanned for image analysis (Oxford with software Aperio ScanScope™; Aperio Technologies, Vista, CA, USA), and the total parenchymal area was measured using ImageJ 1.44 (National Institute of Health, Bethesda, MD, USA). Neutrophil counts were quantified as total number of cells per mm2 lung tissue. Thymic stromal lymphopoietin immunostaining was quantified as peribronchial TSLP-positive cells per μm2, and the area was analysed by Aperio ImageScope software. One section per animal was analysed.
RNA extraction and quantification of TSLP, TNF-α, IFN-β and IFN-λ gene expression with RT-qPCR
Frozen lung tissue was homogenized mechanically (OmniPrep Rotor Stator Generator; Omni International, Kennesaw, GA, USA), and total RNA was extracted using RNA extraction kit Nucleospin® RNA II (Machery-Nagel, Düren, Germany), according to the manufacturer's instructions. 1 μg of RNA was reverse-transcribed to cDNA using a RT-kit (PrimerDesign, Southampton, UK), and quantitative PCR was performed using the reagents from PrimerDesign. Subsequently, thermocycling and real-time detection of PCR products were performed on an IcyclerIQ sequence detection system (Mx3000P; Stratagene, La Jolla, CA, USA) with standard cycling parameters. Genes of interest were normalized to house keeping gene 18S using the delta–delta Ct method. The following primer sequences were used for TSLP, TNF-α, IFN-β and IFN-λ.
TSLP: AAACTGAGAGAAATGACGGTACT (forward) and TCTGGAGATTGCATGAAGGAATA (reverse).
TNF-α: AGCCAGGAGGGAGAACAGA (forward) and CAGTGAGTGAAAGGGACAGAAC (reverse).
IFN-β: ATGGAAAGATCAACCTCACCTAC (forward) and GGATGGCAAAGGCAGTGTAA (reverse).
IFN-λ: ACCAGGCTCCCAGTGGAA (forward) and TTTTTGAAGGCCTGCAGCTCT (reverse).
Statistical analysis
Data are expressed as mean and standard error of the mean (SEM). All data were analysed using unpaired t-test after assumed Gaussian distribution, which was tested for normal distribution with Kolmogorov–Smirnov test. P-values of <0.05 were considered statistically significant. Comparisons between the groups were analysed using one-way anova. Statistics were performed with the software GraphPadPrism version 5.0 (GraphPad Software, San Diego, CA, USA, http://www.graphpad.com).
Results
Effects of intranasal dsRNA administration to naïve mice
To optimize the dsRNA-induced exacerbation model, we first studied the effects of several once-daily dsRNA (100 μg) challenges in naïve mice and evaluated the effects at different time points. We found that the total protein concentration in BALF, reflecting general tissue response to inflammation, was increased after 72 h (involving three dsRNA challenges) and increased even further at 96 h (involving four dsRNA challenges) compared to saline control (Fig. 2A). We measured the cell necrosis marker LDH, known to increase during viral-induced asthma exacerbations 4. There was a trend that LDH was increased by dsRNA challenges at 72 and 96 h (P = 0.09 compared with saline control at 96 h, Fig. 2B). The cellular inflammation was characterized by neutrophilia already 24 h after one dsRNA challenge and was sustained by repeated challenges at all time points analysed (Fig. 2C). Bronchoalveolar lavage fluid lymphocytes increased gradually after dsRNA exposures for 24–96 h (Fig. 2C). Bronchoalveolar lavage fluid concentrations of the chemoattractants CXCL1/KC and CCL2/MCP-1 increased significantly 24 h after dsRNA exposure and remained increased at later time points (Fig. 2D,E). Our data show that dsRNA challenges to naïve mice, without prior lung inflammation, induce chemoattractants and cellular inflammation already after one challenge (24 h), whereas the increase in total protein and possibly LDH requires at least three dsRNA challenges (72 h). Collectively, these data are in accordance with previous studies 6 and suggest that three days' challenges are appropriate. Three days is also the approximate time period during which human airway inflammation and symptoms develop after rhinoviral infection. We further carried out preliminary experiments using half the dose of dsRNA (50 μg) and three days of daily challenges. As indicated by total cells, total protein and LDH in BALF, the 100 μg dose of dsRNA produced a more robust response with relatively little variation in data (Fig. S1). Both dose levels were well tolerated by the animals. Hence, three daily administrations of 100 μg dsRNA and the 72-h time point were selected for the evaluation of effects in exacerbation experiments using our established mouse model of experimental asthma.
Figure 2.
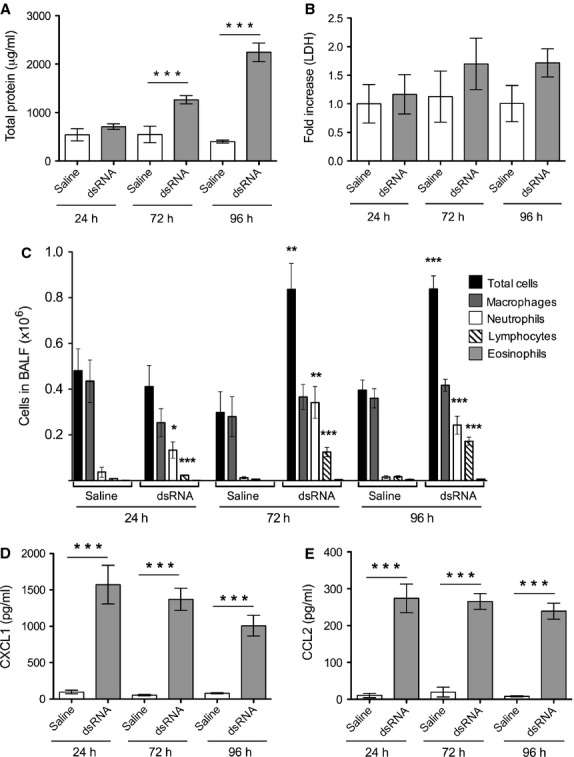
Effects of intranasal dsRNA administration to naïve mice. Bronchoalveolar lavage fluid data indicated that dsRNA increased the total protein concentration (A), tended to increase lactate dehydrogenase (B), increased neutrophils and lymphocytes (C) and increased concentrations of CXCL1/KC (D) and CCL2/MCP-1 (E) compared to saline control. Data are presented as mean ± SEM, n = 6–18 in each group, ***P < 0.001 compared to saline control, **P < 0.01 compared to saline control, *P < 0.05 compared to saline control.
DsRNA administration to mice with established experimental asthma exacerbates several features of lung inflammation
Intranasal dsRNA or saline (control) was administered to mice with established allergic inflammation beginning 24 h after the final OVA challenge (Fig. 1). Similar to naïve mouse lungs, dsRNA increased total protein content in the BALF of previously sensitized and allergen-challenged animals (Fig. 3A). However, the dsRNA-induced increase in total protein concentration was markedly augmented in mice with already established allergic eosinophilic airway inflammation (Fig. 3A). Lactate dehydrogenase levels in BALF were also increased after dsRNA administration to the lungs with established allergic inflammation (compared to baseline allergic inflammation, Fig. 3B). Furthermore, three days of local airway administration of dsRNA changed the OVA-induced eosinophilic inflammation into a largely neutrophilic inflammation (Fig. 3C). Yet, release into the BALF of the neutrophilic chemoattractant and IL-8-like cytokine, CXCL1/KC, was similarly induced by dsRNA, irrespective of established allergic inflammation (Fig. 3D). CCL2/MCP-1 levels in BALF were also increased by dsRNA administration, independent of the allergic inflammation (Fig. 3E).
Figure 3.
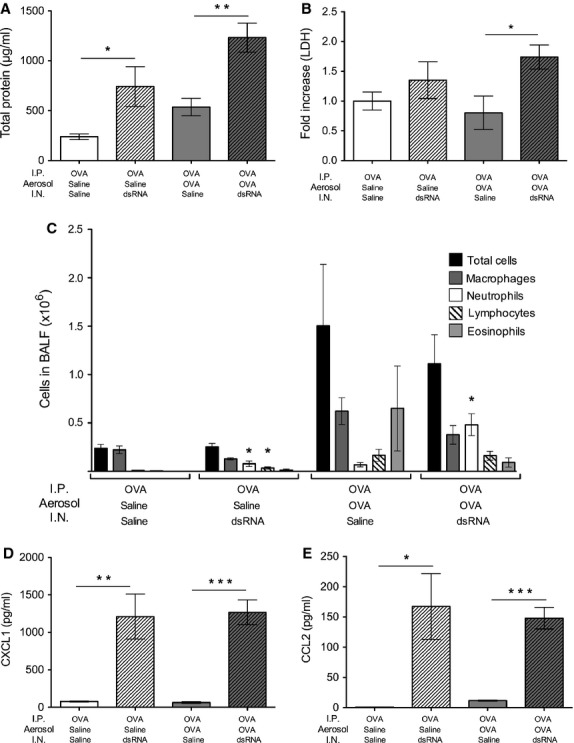
Intranasal dsRNA administration caused exacerbation of lung inflammation in mice with established allergic inflammation. dsRNA challenges to mice with established allergic inflammation increased total protein concentration (A), lactate dehydrogenase release (B), neutrophils (C), CXCL1/KC (D) and CCL2/MCP-1 (E) compared to allergic inflammation alone (OVA/OVA/saline). Data are presented as mean ± SEM, n = 4–9 in each group. ***P < 0.001 compared to saline control, **P < 0.01 compared to saline control *P < 0.05 compared to saline control.
dsRNA exacerbated lung tissue inflammation and neutrophil numbers in mice with established allergic asthma
Because BALF data may not always reflect inflammatory processes occurring in the lung tissue 24, we analysed paraffin-embedded lung tissue stained with H&E and immunostaining for neutrophils. The highest lung tissue inflammation score was recorded in dsRNA-treated mice with an already established allergic eosinophilic airway inflammation (Fig. 4A–E). Similarly, the lung tissue infiltration of neutrophils was greatest in mice with established allergic inflammation receiving the additional dsRNA treatment (Fig. 4F–H).
Figure 4.
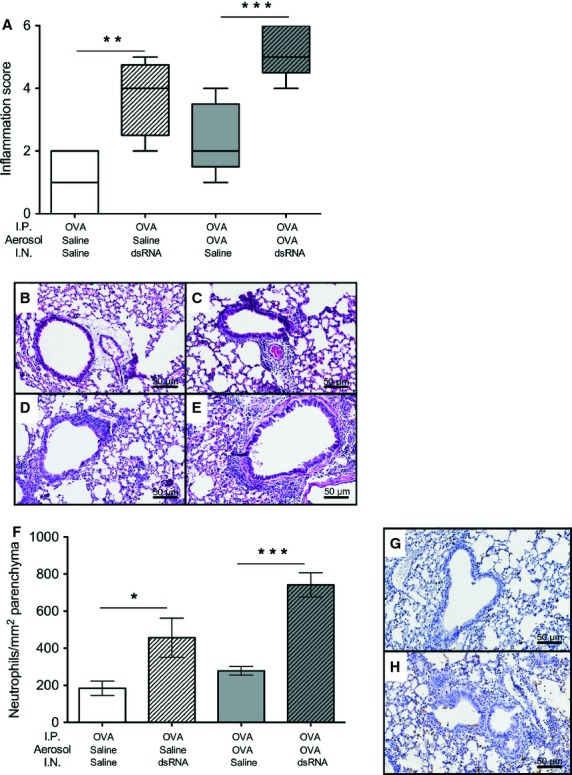
dsRNA challenges of mice with established allergic inflammation produced exacerbations involving neutrophilic lung tissue inflammation. Inflammation scored in lung tissue (A) indicated that cell infiltrates and epithelial hypertrophy were most pronounced in allergic inflamed lungs subjected to dsRNA-induced exacerbations. Representative illustrations of H&E-stained lung sections after different treatments: OVA/saline/saline (B), OVA/saline/dsRNA (C), OVA/OVA/saline (D) and OVA/OVA/dsRNA (E). Infiltration of neutrophils was greatest at dsRNA-induced exacerbations (F), as also illustrated by representative micrographs: OVA/OVA/saline (G) and OVA/OVA/dsRNA (H). n = 4–9 in each group. ***P < 0.001 compared to saline control, **P < 0.01 compared to saline control *P < 0.05 compared to saline control.
dsRNA increased lung TSLP expression only in mice with established allergic airway inflammation
Tissue mRNA levels of TSLP (Fig. 5A) were increased only in mice with established allergic eosinophilic inflammation subjected to dsRNA-induced exacerbation (P < 0.01 compared to allergic baseline inflammation). We measured TSLP in BALF but found no detectable levels of TSLP, suggesting that TSLP was not secreted into the lumen. We hypothesized that basolateral epithelial secretion of TSLP could occur and thus performed immunostaining for peribronchial TSLP and observed increased TSLP staining after dsRNA-induced exacerbation (Fig. 5C,D) although the computer-assisted evaluation of the staining difference did not reach a statistical significance (Fig. 5B). We also expanded our study to determine whether TSLP is induced by actual rhinoviral infection. We thus analysed TSLP immunostaining days 2 and 7 postinfection with RV1B. We found an increased TSLP positivity in the bronchial tissue of animals with established allergic inflammation with superimposed rhinoviral infection compared to nonallergic lungs day 2 postinfection (P < 0.05; Fig. 5E–I). There was an insignificant trend at increased TSLP staining in animals with allergic inflammation receiving UV-treated virus that affected the difference between this control and the exacerbating animals (P = 0.17; Fig. 5E).
Figure 5.
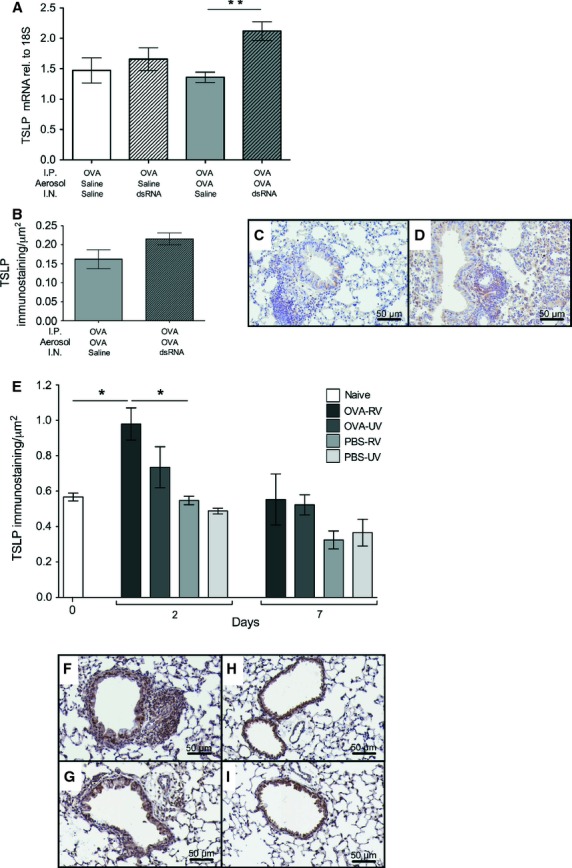
Increased thymic stromal lymphopoietin (TSLP) in response to dsRNA challenges or RV1B infection in lungs with established allergic inflammation. dsRNA increased lung tissue TSLP mRNA only in mice with experimental asthma (A). This was confirmed using immunostaining for TSLP where lungs subjected to dsRNA-induced exacerbation had more peribronchial TSLP-positive cells (D) compared to saline (C). However, the computer-assisted evaluation of TSLP immunostaining did not reach a statistical significance (B). n = 4–9 in each group. Two days postinfection with RV1B, lungs with already established allergic inflammation exhibited increased peribronchial TSLP positivity compared to nonallergic lungs in response to the viral infection (E). Photomicrographs of TSLP staining in allergic (F) and nonallergic (G) lungs two days after viral RV1B infection. TSLP staining in UV-irradiated RV1B-treated allergic (H) and nonallergic (I) lungs after two days is also illustrated. n = 3 in each group, data are presented as mean ± SEM, **P < 0.01 compared to saline control, *P < 0.05 compared to saline control. Scale bar = 50 μm.
dsRNA increased lung mRNA levels of TNF-α and IFN-λ, but not IFN-β, in mice with established allergic airway inflammation
We also analysed lung expression of three other important mediators known to be dysregulated in asthma exacerbations. TNF-α known to correlate with asthma severity and interferons demonstrated to be deficient in asthma patients undergoing exacerbations. We found that the pro-inflammatory cytokine TNF-α was significantly induced by dsRNA alone and further significantly increased after dsRNA-induced exacerbation (Fig. 6A). mRNA expression of type III interferon, IFN-λ, was increased exclusively during dsRNA-induced exacerbation (Fig. 6B). By contrast, IFN-β was induced by either allergen (P < 0.001) or dsRNA challenges (P < 0.05) alone. However, in mice with established allergic inflammation, dsRNA challenges did not increase IFN-β (Fig. 6C).
Figure 6.
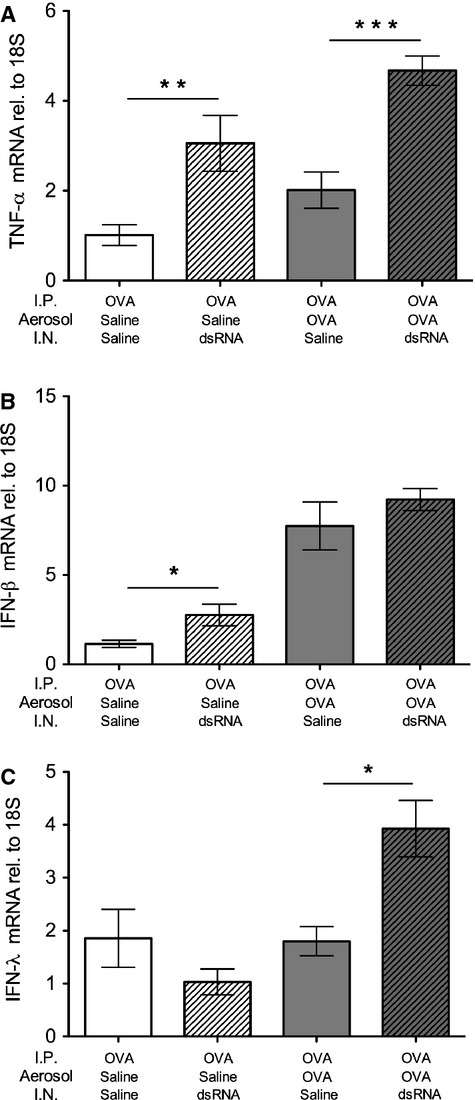
dsRNA challenges increased lung mRNA levels of TNF-α and IFN-λ, but not IFN-β, in mice with established allergic airway inflammation. Tissue mRNA expression of TNF-α (A) was induced by dsRNA and was further increased at dsRNA-induced exacerbation, whereas IFN-λ was exclusively expressed at exacerbation (B). IFN-β was induced by either dsRNA or allergen alone, but not further increased at exacerbation (C). Data are presented as mean ± SEM, n = 4–9 in each group. ***P < 0.001 compared to saline control, **P < 0.01 compared to saline control, *P < 0.05 compared to saline control.
Discussion
We demonstrated that airway dsRNA challenges of mice with allergic experimental asthma produced an exacerbation-like condition with increased lung tissue inflammation and increased BALF neutrophilia, protein concentration and LDH levels. In contrast to previous work involving mice receiving dsRNA during the development of allergic inflammation 5,8,9, the present dsRNA exposure after allergen-induced inflammation did not increase the lung eosinophilia but instead added a neutrophilic inflammation in lung tissues and air spaces. Furthermore, it was only in mice subjected to combined allergic and dsRNA-induced inflammation that we observed increased lung expression of the cytokine TSLP, a potential drug target in the development of severe asthma 11. Similarly, in the present complementary RV1B infection study, it was only when the infection was superimposed on an established allergic inflammation that lung tissue staining of TSLP was increased [other aspects on RV1B-induced exacerbations have been reported elsewhere 22]. The present pulmonary effects of allergen challenge followed by dsRNA exposures exhibit similarities to the observations made in exacerbations of human asthma. These include, beyond common lung tissue and BALF inflammation features, increased LDH and IFN-λ. Several aspects of the present model thus advance the field of experimental exacerbations of asthma.
dsRNA is not only a viral infection surrogate; its actions can also represent the effects of necrotic cells as they alert the immune system to danger. The functional importance of dsRNA-like actions of necrotic cells is supported by the findings in in vivo and in vitro systems 10,25. Necrotic cells, including eosinophils undergoing primary lysis 24, likely occur at exacerbations of human asthma. Cell lysis, evidenced by increased levels of LDH in induced sputum samples 4, is also a distinct biomarker of viral-induced exacerbations of asthma. The present observation that dsRNA challenge of mice with established allergic inflammation increased BALF levels of LDH thus strengthens the similarities between our dsRNA challenge model and exacerbations of asthma. Microvascular–epithelial plasma exudation is a measure of general severity of lung inflammation 26. The plasma exudation response is a nonsieved process letting out and activating potent protein systems of circulating blood. It is evoked by viral infection of human airways 27 and characterizes severe and acute asthma 26. In accord, we observed an agreement between augmented BALF total protein levels and lung tissue inflammatory score in this study. Similar to RV infection of mouse airways 21,28,29, dsRNA-induced lung inflammation is neutrophilic 6,7. Pronounced neutrophilia in response to dsRNA (this study) and RV infection 29 is evident in both BALF and lung tissues. Expectedly, the neutrophilic response was associated with the increased BALF levels of the neutrophil attractant CXCL1/KC. TNF-α, a potent promoter of pulmonary neutrophilia and increased in asthma exacerbations, was strongly induced by dsRNA and associated with tissue neutrophilia (this study). We also found that dsRNA-induced exacerbation increased the production of CXCL1/KC and CCL2/MCP-1, known to be increased in the lungs of patients with exacerbation of asthma 3. Hence, pharmacology of several potentially important target molecules in neutrophilic exacerbations can be examined in this model.
Neither dsRNA alone nor RV1B infection alone was sufficient to increase lung expression of TSLP in this study. Oxidative stress may enhance airway responses to dsRNA 30 potentially contributing to the present increased lung tissue TSLP expression exclusively at exacerbations. However, the exacerbation in this study was not sufficient for increasing TSLP in BALF to detectable levels. Yet, the present increase in lung tissue expression of TSLP provides in vivo opportunities to determine the effects of experimental drugs that in vitro inhibit dsRNA-induced generation of TSLP 18,31,32. Repeated viral-induced exacerbations may be involved in the progression of bronchial obstructive diseases, but there is little information as yet from animal models in this regard 33. The increase in TSLP makes it also of interest to explore expression and potential roles of this cytokine in repeated dsRNA-induced exacerbations of mouse asthma.
It is currently argued that defect generation of IFN-β and IFN-λ contributes to exacerbations of asthma 34,35. However, there is no known exacerbation animal model that has demonstrated reduced IFN-β or IFN-λ generation. Both allergen and dsRNA challenges alone were sufficient to increase the lung tissue expression of IFN-β in this study. Interestingly, IFN-β was not further increased at dsRNA-induced exacerbation, supporting the possibility that this exacerbation model mimics defect generation of this IFN in asthma. By contrast, IFN-λ was exclusively increased at dsRNA-induced exacerbations in this study. Interestingly, IFN-λ was also exclusively associated with viral infection-induced exacerbations of asthma in children 20. The present finding that allergen challenge alone increased IFN-β is also novel and needs to be further explored especially in view of the potential of this interferon to stimulate the pathways for increased production of IgE 19. As with several other cytokines, IFNs may have dual roles and it cannot be excluded that IFNs could be drug targets at exacerbations. The present dsRNA-exacerbation model provides an opportunity for future pharmacological studies on type I and type III IFN responses.
In conclusion, this study identifies a mouse model of asthma exacerbations harbouring several inflammatory characteristics of severe acute stages of the human disease. Thus, by giving repeated doses of dsRNA, an acknowledged mimic of pathogenic actions of viral infection and a potential mimic of effects of necrotic cells, an already established allergic inflammation was considerably worsened. The present exacerbation was indicated by the histological score and increased release of proteins, including several cytokines, as well as LDH in the airway lumen. Intriguingly, in the present dsRNA-exacerbation model, the IFN-β response was not increased compared to the baseline allergic inflammation. The present exacerbation evoked by dsRNA induced lung expression of TSLP, an upstream, pro-inflammatory cytokine. We also found that TSLP was increased in a mouse model where allergic inflammation was superimposed by RV1B infection. These data supported our hypothesis that TSLP may be involved in the development of severe asthma caused by frequent exacerbations. In accord with the findings in human asthma, we further demonstrated that LDH and IFN-λ were increased exclusively at exacerbation in the present dsRNA challenge model. The present observations on TSLP, LDH and IFNs are novel and constitute potentially important advance of this experimental research niche. We suggest that the present approach, involving dsRNA challenge-induced worsening and expansion of allergic lung inflammation, complements RV infection-induced exacerbation studies and is useful for the study of mechanisms and pharmacology of single and repeated exacerbations of experimental mouse asthma.
Acknowledgments
This work was supported in part by the Swedish Medical Research Council, Heart and Lung Foundation, VINNOVA – Sweden's Innovation Agency, and Chair from Asthma UK (CH11SJ) and by MRC Centre Grant G1000758, ERC FP7 Advanced Grant 233015, the Predicta FP7 Collaborative Project Grant 260895 and the Wellcome Trust sponsored Centre for Respiratory Infection (CRI).
Author contributions
LU conceived the design of this study and supervised the scientific work except for the part involving RV1B infection. IMP, HA, AB, LU, NB and NG performed experiments. LU, IMP and HA analysed the data. SJ supervised RV1B experiments. LU wrote first drafts of the manuscript. All authors contributed and approved the full manuscript.
Conflicts of interest
The authors have nothing to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Effects of intranasal (i.n.) dsRNA administration to mouse lungs.
References
- 1.Johnston SL. Natural and experimental rhinovirus infections of the lower respiratory tract. Am J Respir Crit Care Med. 1995;152:S46–S52. doi: 10.1164/ajrccm/152.4_Pt_2.S46. [DOI] [PubMed] [Google Scholar]
- 2.Persson C, Uller L. HH Salter (1860s): taking cold as original cause and provocative of attacks of asthma. Thorax. 2013;68:489. doi: 10.1136/thoraxjnl-2012-203110. [DOI] [PubMed] [Google Scholar]
- 3.Daldegan MB, Teixeira MM, Talvani A. Concentration of CCL11, CXCL8 and TNF-alpha in sputum and plasma of patients undergoing asthma or chronic obstructive pulmonary disease exacerbation. Braz J Med Biol Res. 2005;38:1359–1365. doi: 10.1590/s0100-879x2005000900010. [DOI] [PubMed] [Google Scholar]
- 4.Wark PA, Johnston SL, Moric I, Simpson JL, Hensley MJ, Gibson PG. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur Respir J. 2002;19:68–75. doi: 10.1183/09031936.02.00226302. [DOI] [PubMed] [Google Scholar]
- 5.Torres D, Dieudonne A, Ryffel B, Vilain E, Si-Tahar M, Pichavant M, et al. Double-stranded RNA exacerbates pulmonary allergic reaction through TLR3: implication of airway epithelium and dendritic cells. J Immunol. 2010;185:451–459. doi: 10.4049/jimmunol.0902833. [DOI] [PubMed] [Google Scholar]
- 6.Stowell NC, Seideman J, Raymond HA, Smalley KA, Lamb RJ, Egenolf DD, et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir Res. 2009;10:43. doi: 10.1186/1465-9921-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aeffner F, Traylor ZP, Yu EN, Davis IC. Double-stranded RNA induces similar pulmonary dysfunction to respiratory syncytial virus in BALB/c mice. Am J Physiol Lung Cell Mol Physiol. 2011;301:L99–L109. doi: 10.1152/ajplung.00398.2010. [DOI] [PubMed] [Google Scholar]
- 8.Jeon SG, Oh SY, Park HK, Kim YS, Shim EJ, Lee HS, et al. TH2 and TH1 lung inflammation induced by airway allergen sensitization with low and high doses of double-stranded RNA. J Allergy Clin Immunol. 2007;120:803–812. doi: 10.1016/j.jaci.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 9.Reuter S, Dehzad N, Martin H, Bohm L, Becker M, Buhl R, et al. TLR3 but not TLR7/8 ligand induces allergic sensitization to inhaled allergen. J Immunol. 2012;188:5123–5131. doi: 10.4049/jimmunol.1101618. [DOI] [PubMed] [Google Scholar]
- 10.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uller L, Leino M, Bedke N, Sammut D, Green B, Lau L, et al. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax. 2010;65:626–632. doi: 10.1136/thx.2009.125930. [DOI] [PubMed] [Google Scholar]
- 12.Brandelius A, Yudina Y, Calven J, Bjermer L, Andersson M, Persson C, et al. dsRNA-induced expression of thymic stromal lymphopoietin (TSLP) in asthmatic epithelial cells is inhibited by a small airway relaxant. Pulm Pharmacol Ther. 2011;24:59–66. doi: 10.1016/j.pupt.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Calven J, Yudina Y, Hallgren O, Westergren-Thorsson G, Davies DE, Brandelius A, et al. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG-I-like helicases. J Innate Immun. 2012;4:86–99. doi: 10.1159/000329131. [DOI] [PubMed] [Google Scholar]
- 14.Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 15.Shikotra A, Choy DF, Ohri CM, Doran E, Butler C, Hargadon B, et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J Allergy Clin Immunol. 2012;129:104–111. doi: 10.1016/j.jaci.2011.08.031. [DOI] [PubMed] [Google Scholar]
- 16.Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203:269–273. doi: 10.1084/jem.20051745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int. 2012;61:3–17. doi: 10.2332/allergolint.11-RAI-0395. [DOI] [PubMed] [Google Scholar]
- 18.Brandelius A, Mahmutovic Persson I, Calven J, Bjermer L, Persson CG, Andersson M, et al. Selective inhibition by simvastatin of IRF3 phosphorylation and TSLP production in dsRNA-challenged bronchial epithelial cells from COPD donors. Br J Pharmacol. 2013;168:363–374. doi: 10.1111/j.1476-5381.2012.02131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat Med. 2012;18:726–735. doi: 10.1038/nm.2768. [DOI] [PubMed] [Google Scholar]
- 20.Miller EK, Hernandez JZ, Wimmenauer V, Shepherd BE, Hijano D, Libster R, et al. A mechanistic role for type III IFN-lambda1 in asthma exacerbations mediated by human rhinoviruses. Am J Respir Crit Care Med. 2012;185:508–516. doi: 10.1164/rccm.201108-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14:199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glanville N, Message SD, Walton RP, Pearson RM, Parker HL, Laza-Stanca V, et al. gammadeltaT cells suppress inflammation and disease during rhinovirus-induced asthma exacerbations. Mucosal Immunol. 2013;6:1091–1100. doi: 10.1038/mi.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uller L, Lloyd CM, Rydell-Tormanen K, Persson CG, Erjefalt JS. Effects of steroid treatment on lung CC chemokines, apoptosis and transepithelial cell clearance during development and resolution of allergic airway inflammation. Clin Exp Allergy. 2006;36:111–121. doi: 10.1111/j.1365-2222.2006.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uller L, Persson CG, Erjefalt JS. Resolution of airway disease: removal of inflammatory cells through apoptosis, egression or both? Trends Pharmacol Sci. 2006;27:461–466. doi: 10.1016/j.tips.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Lim DM, Wang ML. Toll-like receptor 3 signaling enables human esophageal epithelial cells to sense endogenous danger signals released by necrotic cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G91–G99. doi: 10.1152/ajpgi.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Persson C, Uller L. Roles of plasma exudation in asthma and COPD. Clin Exp Allergy. 2009;39:1626–1629. doi: 10.1111/j.1365-2222.2009.03373.x. [DOI] [PubMed] [Google Scholar]
- 27.Greiff L, Andersson M, Svensson C, Linden M, Myint S, Persson CG. Allergen challenge-induced acute exudation of IL-8, ECP and alpha2-macroglobulin in human rhinovirus-induced common colds. Eur Respir J. 1999;13:41–47. doi: 10.1034/j.1399-3003.1999.13a09.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newcomb DC, Sajjan US, Nagarkar DR, Wang Q, Nanua S, Zhou Y, et al. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am J Respir Crit Care Med. 2008;177:1111–1121. doi: 10.1164/rccm.200708-1243OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagarkar DR, Wang Q, Shim J, Zhao Y, Tsai WC, Lukacs NW, et al. CXCR2 is required for neutrophilic airway inflammation and hyperresponsiveness in a mouse model of human rhinovirus infection. J Immunol. 2009;183:6698–6707. doi: 10.4049/jimmunol.0900298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koarai A, Sugiura H, Yanagisawa S, Ichikawa T, Minakata Y, Matsunaga K, et al. Oxidative stress enhances toll-like receptor 3 response to double-stranded RNA in airway epithelial cells. Am J Respir Cell Mol Biol. 2010;42:651–660. doi: 10.1165/rcmb.2008-0345OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahmutovic-Persson I, Johansson M, Brandelius A, Calven J, Bjermer L, Yudina Y, et al. Capacity of capsazepinoids to relax human small airways and inhibit TLR3-induced TSLP and IFNbeta production in diseased bronchial epithelial cells. Int Immunopharmacol. 2012;13:292–300. doi: 10.1016/j.intimp.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Uller L. Interferon regulatory factor 3 activation mediates viral stimulus-induced bronchial production of thymic stromal lymphopoietin. J Allergy Clin Immunol. 2013;131:926. doi: 10.1016/j.jaci.2012.11.034. [DOI] [PubMed] [Google Scholar]
- 33.Wedzicha JA, Donaldson GC. Natural history of successive COPD exacerbations. Thorax. 2012;67:935–936. doi: 10.1136/thoraxjnl-2012-202087. [DOI] [PubMed] [Google Scholar]
- 34.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effects of intranasal (i.n.) dsRNA administration to mouse lungs.