

Abstract
Purpose
Copy-number variations as a mutational mechanism contribute significantly to human disease. Approximately one-half of the patients with Charcot–Marie–Tooth (CMT) disease have a 1.4 Mb duplication copy-number variation as the cause of their neuropathy. However, non-CMT1A neuropathy patients rarely have causative copy-number variations, and to date, autosomal-recessive CMT disease has not been associated with copy-number variation as a mutational mechanism.
Methods
We performed Agilent 8 × 60K array comparative genomic hybridization on DNA from 12 recessive Turkish families with CMT disease. Additional molecular studies were conducted to detect breakpoint junctions and to evaluate gene expression levels in a family in which we detected an intragenic duplication copy-number variation.
Results
We detected an ~6.25 kb homozygous intragenic duplication in NDRG1, a gene known to be causative for recessive HMSNL/CMT4D, in three individuals from a Turkish family with CMT neuropathy. Further studies showed that this intragenic copy-number variation resulted in a homozygous duplication of exons 6–8 that caused decreased mRNA expression of NDRG1.
Conclusion
Exon-focused high-resolution array comparative genomic hybridization enables the detection of copy-number variation carrier states in recessive genes, particularly small copy-number variations encompassing or disrupting single genes. In families for whom a molecular diagnosis has not been elucidated by conventional clinical assays, an assessment for copy-number variations in known CMT genes might be considered.
Keywords: autosomal recessive, Charcot–Marie–Tooth disease, CMT4D, CNV, NDRG1
INTRODUCTION
Charcot–Marie–Tooth (CMT) and related peripheral neuropathies represent a heterogeneous group of genetic disorders of the peripheral nervous system with an estimated frequency of 1 in 2,500 individuals, making it one of the most common inherited neurological diseases. CMT usually presents clinically as a distal symmetric polyneuropathy.1 During the past two decades, mutations in >40 genes were found to be causative for CMT; however, the molecular etiology is still not elucidated in ~10–20% of cases.
Copy-number variation (CNV) of the PMP22 gene region, due to the CMT1A duplication that frequently occurs de novo by the mechanism of nonallelic homologous recombination, is the most common underlying etiology; duplication of PMP22 gene region represents 70% of demyelinating, CMT1, neuropathy cases.2,3 Furthermore, rare CNVs that occur by mechanisms other than nonallelic homologous recombination and either change PMP22 copy number or disrupt PMP22 function can also be observed in some patients.4,5 Deletion CNVs of the X-linked Cx32 (GJB1) gene have also been reported.6 However, CNVs of other neuropathy genes are rarely reported.7 In this article, we describe a family with an intragenic duplication CNV in one of the autosomal-recessive (AR)–CMT genes, NDRG1, with three affected individuals born to consanguineous parents from a first-degree cousin marriage. To the best of our knowledge, this is the first report of AR duplication CNV causing a CMT disease phenotype and only the third mutation reported in the NDRG1 gene.
MATERIALS AND METHODS
Patients
This study was approved by the Institutional Review Board of both collaborating institutions, and informed consent was obtained before participation in this study. Twelve families with distal symmetric polyneuropathy referred to the Department of Neurology, Istanbul Medical Faculty at Istanbul University were included in this study. Deletion/duplication of the PMP22 gene region, point mutations in PMP22, myelin protein zero (MPZ), GJB1, and MPZ were excluded before the application of CMT gene targeted whole-genome array comparative genomic hybridization (aCGH). In a family in which we detected duplication CNV (HOU 1463), extended clinical studies including hearing tests, ophthalmologic examinations, and brain imaging studies were conducted on both apparently healthy and affected subjects.
CNV analysis
We designed a high-density oligonucleotide-based Agilent 8 × 60K custom microarray for investigating CNVs in CMT gene regions and linkage regions which potentially host CMT genes by using the Agilent eArray website (http://earray.chem.agilent.com/earray). We included 67 genes and their flanking 50 kb of upstream and downstream regions with an average genomic resolution of ~1 probe/200 bp. We also included 10 CMT linkage regions for which the responsible gene has not yet been identified with an average genomic resolution of ~1 probe/5 kb. Experiments were performed according to the manufacturer’s protocol and previously described methods.8
Briefly, parameters for digestion, labeling, purification of the labeled product, hybridization with gender-matched male (NA10851) or female (NA15510) control DNAs (obtained from Coriell Cell Repositories; http://ccr.coriell.org), washing, and scanning were conducted per the manufacturer’s protocol (version 6.0). Slides were scanned on Agilent Technologies’ DNA Microarray Scanner with a Surescan High-Resolution Technology scanner. Computational analyses including data extraction, background subtraction, and normalization were performed using Agilent Feature Extraction Software 10_7_3_1 (Agilent Technologies, Santa Clara, CA). These data were subsequently imported into aCGH analytics software (Genomic Workbench Standard Edition 5.0.14; Agilent Technologies). The genomic copy number was defined by analysis of the normalized log2 (Cy5/Cy3) ratio average of the CGH signal.
Breakpoint junction analyses
To detect the breakpoint junctions of the duplication, primers were designed at the apparent boundaries of duplication based on aCGH analysis and the genomic coordinates of interrogating probes demarcating transitions from normal copy to apparent copy-number gain consistent with duplication. Primers used for breakpoint junction included: 3724-2F1: 5′-AGGACTGGACAGAGACCTCGACCTT -3′ and 3724-2R1 5′-TGGGGCCGGTATTAGAACCTATGAA -3′. From comparisons with the reference haploid human genome sequence (http://genome.ucsc.edu/cgi-bin/hgTracks), the duplicated segment was expected to yield a polymerase chain reaction (PCR) product that is small enough (~12.5 kb) to be amplified by long-range PCR. We therefore performed long-range PCR to amplify the entire duplicated segment using primers flanking this region—3724-F1: 5′-CTGGAGCAGTAAGAAGCAAGGACGA-3′ and 3724-R1: 5′-GGATGCGAAGGGACTAGACTTGGG-3′—that amplify an ~14.8 kb segment encompassing the duplication and an ~7.4 kb segment in a wt allele. Experiments were performed as previously described.8
Quantitative real-time reverse transcriptase–PCR
To study the mRNA expression of the NDRG1 gene, we performed quantitative real-time reverse transcriptase–PCR in homozygous/heterozygous duplicated and wild-type individuals from the family. Total RNA was prepared from blood by using the PAXgene Blood RNA Kit (Qiagen, Valencia, CA), and cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad Hercules, CA). The real-time PCR assay was conducted with the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) using TaqMan Fast universal PCR master mix (Applied Biosystems). The cDNA samples were subsequently analyzed as triplicates by quantitative RT-PCR using TaqMan Gene Expression Assay (NDRG1: Hs 00608387_ m1, Applied Biosystems) and Pre-Developed TaqMan Assay Reagents for TBP (Applied Biosystems) as a control gene. Experiments were carried out according to the manufacturer’s protocol. Relative fold of mRNA expression changes were calculated using the comparative threshold cycle method (ΔΔCT).
Genome-wide SNP array genotyping
To investigate whether there are any other large CNVs in the genome and/or regions of homozygosity that may contribute to the phenotype, we performed genome-wide single-nucleotide polymorphism (SNP) array genotyping in all the members of this family using the Illumina OmniExpress Beadchip (Illumina, San Diego, CA) that interrogates 730,525 markers across the genome. Analysis of the SNP array data was performed using the Illumina Genome Studio software (Illumina).
RESULTS
Clinical evaluation
The proband (BAB3724 = II-2) was a 30-year-old male who had delayed motor milestones. Gait disturbance and frequent falls were first noted at 3 years of age. Involvement of the upper limbs became apparent at the age of 10 years. Orthopedic correction (achilloplasty) of both feet was carried out at 12 years of age. He also had hearing loss, which was noted at ~4 years of age. All neurological symptoms and signs were progressive.
The physical examination revealed severe distal and mild proximal muscle weakness and distal wasting with areflexia. Light touch, vibration, and position senses were severely diminished distally. Severe deformities including kyphoscoliosis, pes cavus, hammer toes, and claw hands were observed. Nerve conduction studies showed the absence of sensory nerve action potentials in all limbs (Table 1). Compound motor nerve potentials (CMAPs) could not be obtained. Musculocutaneous nerve (recorded from biceps muscle) motor conduction study revealed low CMAP amplitudes whereas axillary CMAPs (recorded from deltoid muscle) were normal. Brain magnetic resonance imaging was normal. Audiometry showed moderate sensorineural hearing loss especially at low frequencies. Detailed ocular examination raised suspicion of glaucoma. Intraocular pressure was normal with normal visual fields and so was the gonioscopic examination. Optical coherence tomography showed that the right retinal nerve fiber layer had a borderline thickness.
Table 1.
Sensory and motor nerve conduction studies of three patients and one healthy member of the family
| Patient (age in years) |
BAB3724 (30) |
BAB5658 (28)a |
BAB4149 (24) |
BAB4152 (17) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II-2 |
II-2 |
II-4 |
II-7 |
Normal values |
|||||||||||
| Motor NCSs | dL (ms) | V (m/s) | Amp (mV) |
dL (ms) | V (m/s) | Amp (mV) | dL (ms) | V (m/s) | Amp (mV) |
dL (ms) | V (m/s) | Amp (mV) | dL (ms) | CV (m/s) |
Amp (mV) |
| R axillary | 14.5 | N/A | 5.8 | N/A | N/A | N/A | 15.2 | N/A | 0.2 | N/A | N/A | N/A | <6.0 | >50 | >4.0 |
| R musculocutaneus | 7.1 | N/A | 0.9 | N/A | N/A | N/A | 7.2 | N/A | 1.5 | N/A | N/A | N/A | <6.0 | >50 | >4.0 |
| R median | NR | NR | NR | 4.7 | 56 | 12.9/11.1 | NR | NR | NR | 2.8 | 58 | 6.5/5.6 | <4.0 | >50 | 5.0 |
| R ulnar | NR | NR | NR | 3.2 | 65/64 | 10.8/9.2/9.2 | NR | NR | NR | 2.2 | 58/73 | 5.1/4.8/4.8 | <3.5 | >50 | 5.0 |
| R common peroneal | NR | NR | NR | 3.2 | 47 | 6.1/5.4 | NR | NR | NR | NRb | NRb | NRb | <4.5 | >40 | 2.0 |
| R tibialis | NR | NR | NR | 4.7 | 49 | 11.2/10.3 | NR | NR | NR | 5.5 | 40 | 0.8/0.4 | <5.0 | >40 | 4.0 |
| Sensory NCSs | dL (ms) | V (m/s) |
Amp
(μV) |
dL (ms) | V (m/s) | Amp (μV) | dL (ms) | V (m/s) |
Amp
(μV) |
dL (ms) | V (m/s) | Amp (μV) | dL (ms) |
CV
(m/s) |
Amp
(μV) |
|
| |||||||||||||||
| R median | NR | NR | NR | 2.7 | 54 | 17.0 | NR | NR | NR | 2.8 | 67 | 18 | <3.2 | >45 | >12.0 |
| R ulnar | NR | NR | NR | 2.2 | 52 | 9.4 | NR | NR | NR | 2.8 | 55 | 16 | <3.0 | >45 | >8.0 |
| R radial | NR | NR | NR | N/A | N/A | N/A | NR | NR | NR | N/A | N/A | N/A | <3.0 | >50 | >12 |
| R superficial peroneal | NR | NR | NR | 2.3 | 53 | 8.6 | NR | NR | NR | 3.0 | 42 | 5.7 | <3.5 | >40 | >5.0 |
| L superficial peroneal | N/A | N/A | N/A | 2.3 | 53 | 4.6 | N/A | N/A | N/A | N/A | N/A | N/A | <3.5 | >40 | >5.0 |
| R sural | NR | NR | NR | 2.7 | 53 | 20.8 | NR | NR | NR | 3.7 | 45 | 12 | <3.8 | >40 | >10.0 |
| L sural | N/A | N/A | N/A | 2.7 | 52 | 23 | N/A | N/A | N/A | N/A | N/A | N/A | <3.5 | >40 | >5.0 |
Amp, amplitude; CV, conduction velocity; dL, distal latency; L, left; N/A, not available; NCSs, nerve conduction studies; NR, not recordable; R, right.
Healthy member.
Recorded from extensor digitorum brevis and tibialis anterior muscles.
The proband’s affected 26-year-old sister (BAB4148 = II-3) was not examined however, she was said to have delayed motor milestones and deafness noted at the age of 2 years. She also had pes cavus, hammer toes, and claw hands. Audiometric tests revealed sensorineural hearing loss at both low and high frequencies.
The 24-year-old sister (BAB4149 = II-4) was more seriously affected than her siblings. She walked at the age of 1 year but had an unsteady gait. At the age of 5 years, weakness in her hands became apparent. She underwent orthopedic surgery (achilloplasty) at the age of 6 years. At the age of 18 years, she was diagnosed with glaucoma and myopia. She also had bilateral deafness. Symptoms progressed rapidly, and she was unable to walk without support in her teens. Her clinical examination showed severe distal and moderate proximal muscle weakness. All tendon reflexes were absent. Light touch, vibration, and position senses were severely diminished in the distal portions of her limbs. She had pes cavus, hammer toes, and claw hands.
Sensory nerve action potentials were absent in both upper and lower limbs (Table 1). CMAPs recorded from distal muscles were also absent. The amplitudes of musculocutaneous (biceps) and axillary CMAPs (deltoid) were reduced. Needle electromyography showed no activity in distal and proximal muscles of the lower limbs and distal muscles of the upper limbs; chronic denervation findings were present in proximal muscles of the upper limbs. Brain magnetic resonance imaging was normal. Audiometry tests could not be performed in this subject.
Healthy individuals BAB4150 (II-5) and BAB4151 (II-6) could not be examined, but they were reported to be normal. The clinical examination and motor and sensory nerve conduction studies were unremarkable (Table 1) in the other male healthy sibling BAB5658: II-1. His ocular examination and audiometry test were normal.
Subject BAB4152 (II-7) has a different clinical picture from other affected individuals in this family; in particular, this subject has a distinct neurological phenotype. She was born with pes equino varus deformity. She underwent orthopedic correction at the age of 1 and 3 years. Motor milestones were delayed, and she could never walk normally. Her hands were weak since early childhood. But unlike the others, the disease was slowly progressive. She did not complain of hearing or vision problems. She was treated for generalized tonic-clonic seizures since the age of 6 years. Her clinical examination revealed severe distal and moderate proximal muscle weakness in the lower limbs and moderate distal muscle weakness in the upper limbs. All tendon reflexes were present except at the achilles tendon. Vibration sensation was mildly diminished distally. She had mild-to-moderate intellectual disability, difficulty in performing daily activities, slurred speech, and difficulty in understanding commands. Furthermore, she was unable to complete elementary school due to consecutive failures in the first grade.
Sensory nerve action potentials and sensory conduction velocities were normal in all limbs (Table 1). Peroneal CMAPs (recorded from extensor digitorum brevis and tibialis anterior muscles) were absent, tibial CMAP amplitudes were reduced and tibial motor conduction velocities were normal. In the upper limbs, CMAP amplitudes and motor conduction velocities were within the normal limits. Needle electromyography showed that the chronic neurogenic changes were prominent in distal muscles of the lower limbs. Brain magnetic resonance imaging was normal. Detailed ocular examination showed a high suspicion of glaucoma. Intraocular pressure was normal with normal visual fields and normal gonioscopic examination for both eyes. Optical coherence tomography exposed generalized thinning in both retinal nerve fiber layer. Audiometry test was within the normal range.
NDRG1 intragenic CNV
We designed a targeted aCGH platform to investigate all known CMT gene regions of the human genome for CNV as small as <1 kb in length. We detected an apparent ~6.25 kb region of increased copy number (chr8: 134265065–134271319) encompassing exons 6–8 of the NDRG1 gene in the index case who presented with distal symmetric polyneuropathy and was given a diagnosis of CMT (BAB3724 = II-2); however, the observed aCGH gain (0.63–0.76) in affected individuals was more consistent with four copies of the locus, potentially reflecting either homozygous duplication or triplication. An extended family study showed that both unaffected parents and presumed heterozygous siblings have an intermediate copy number of NDRG1 with an average probe intensity of 0.37 (0.33–0.43) consistent with three copies at the locus. Affected siblings carry the same CNV gain as the index case level, with an average probe intensity of 0.71 (0.63–0.76), and unaffected siblings have an apparent normal copy number with an average probe intensity of 0.025 (0.00–0.05) (Figure 1a). These family segregation studies support the notion that a homozygous duplication is associated with the known NDRG1 mutation–associated neuropathy, whereas heterozygous duplication represents a carrier state for this recessive trait (Figure 1a), a contention further supported by follow-up PCR studies (see below).
Figure 1. Array comparative genomic hybridization (aCGH) results of the NDRG1 gene region in family HOU1463 and breakpoint junction analysis.
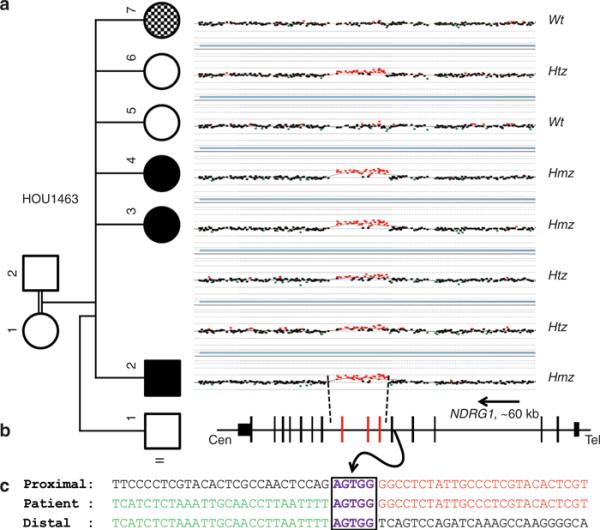
(a) aCGH segregation study within the family. Left column shows the pedigree with four affected (three black boxes indicate CMT4D, and the checkered box indicates the different, more severe neuropathy, and the intellectual disability of the phenotypically distinct sister) and three unaffected individuals born from apparently healthy individuals. Right column shows the figure of custom microarray (Agilent 8 × 60K) for all individuals available for study (II-1 was unavailable for molecular studies). Wt indicates wild type, Hmz indicates homozygous, and Htz indicates heterozygous alleles. (b) Graphic view of 16 exons (vertical black and red bars) of NDRG1. Red bars indicate the duplicated exons (exon 6 through 8). Size and orientation of NDRG1 is shown above the exons. (c) Breakpoint sequence analysis of the duplication. The proximal and distal sequences refer to reference sequence and to their position from the centromere. Proximal reference sequence and patient breakpoint sequences that match with proximal reference sequence are shown in red and distal reference sequence and patient breakpoint sequences that match with distal reference sequence are shown in green. Boxed sequences (purple) correspond to regions of microhomology and emphasize the sequence structure at breakpoint junction.
Breakpoint junction analysis revealed that five base pairs (AGTGG) of microhomology was shared between proximal and distal reference sequences: the proximal breakpoint mapped within a 229 bp short local repeat. Breakpoint junction studies showed that individuals, who carry the duplication in either the homozygous or heterozygous state, have a 1 kb junction amplification product. As anticipated, individuals who have normal alleles do not show any PCR junction band, consistent with the array results and the Mendelian expectation for a recessive model of inheritance (Figure 2a-c).
Figure 2. Polymerase chain reaction (PCR) results for copy-number variation (CNV) gain and breakpoint junction.
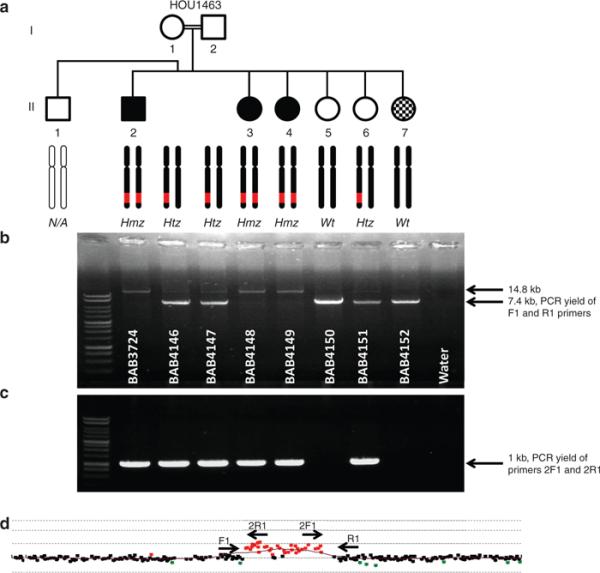
(a) Pedigree of family HOU1463. Each individual is coded with a BAB number (white in respective gel lanes). The array results for each individual are represented by two black chromosome schematics, in which a red region on the q arm designates the duplication predicted by array, and a black chromosome represents no rearrangement. The presumed array genotype is under the chromosome schematics, as well. Wt indicates wild type, Hmz indicates Homozygous, and Htz indicates heterozygous alleles. (b) Long-range PCR for duplicated region by primer pair 3724-F1 and 3724-R1. Individuals with a homozygous duplication allele (BAB3724, BAB4148, and BAB4149) show a single band migrating at ~14.8 kb in size, individuals with heterozygous duplication (BAB4146, BAB4147, and BAB4151) have two bands at 14.8 and 7.4 kb, and individuals who only carry the wild-type allele (BAB4150 and BAB4152) have one 7.4 kb band. (c) Long-range PCR assay results for the breakpoint junction by primer pair 3724-2F1 and 3724-2R1. Individuals who carry at least one duplicated segment have a 1 kb amplicon and individuals who are wild type do not, as expected, show any breakpoint junction fragment. (d) Array comparative genomic hybridization image showing location and orientation of primers to detect breakpoint junction and to amplify the entire duplicated segment. F1 and R1 primers amplify the entire product. 2F1 and 2R1 primers are placed inside the duplication in outward-facing orientation to obtain the breakpoint junction.
To be able to amplify the entire duplicated segment, oppositely oriented primers flanking the apparent duplication CNV were designed for a long-range PCR assay. The flanking primers amplify an ~14.8 kb band encompassing the ~12.5 kb duplicated region delineated from aCGH, therefore both array and breakpoint junction PCR results support the duplication event. We observed a single 14.8 kb PCR band in suspected homozygous duplicated individuals, two PCR bands of 7.4 and 14.8 kb in heterozygous individuals, and a single 7.4 kb PCR band in normal or wild-type individuals (Figure 2a,b). These studies clearly demonstrate a duplication structure and homozygous duplication, not triplication, in affected CMT individuals in this family. In addition, breakpoint PCR only generated 1 kb amplicons in individuals with suspected heterozygous or homozygous duplication (Figure 2c).
To assess how this intragenic genomic CNV affected gene function, after reverse transcriptase-PCR we performed cDNA sequencing of lymphoblastoid cell line RNA from both affected and unaffected individuals. As predicted by conceptual translation and shown by RT-PCR, we observed that the duplication of exon 6–8 in affected individuals leads to a nonsense mutation at codon 223 in the mRNA (Figure 3a,b).
Figure 3. Sequence analysis for cDNA and quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) study for NDRG1.
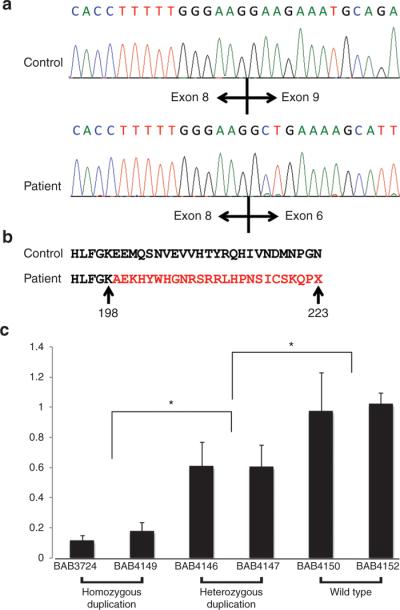
(a) Sanger sequence results of cDNA from the control and affected individuals. In healthy individuals, exon 8 is followed by exon 9 and in affected individuals exon 8 is followed by exon 6. (b) Analysis of the resultant sequence at the amino acid level. The duplication causes a frame shift mutation, altering the amino acid sequence for several residues before a premature termination codon. (c) Expression levels in blood were measured by quantitative RT-PCR using the TaqMan gene expression assay in triplicate and normalized to TBP. We observed a decrease in expression, 0.12- to 0.18-fold in patient samples (BAB3724 and BAB4149) and 0.6- to 0.61-fold in carriers (BAB4146 and BAB4147), compared with normal expression controls. We found an obviously decreased expression in both patients and carriers compared with control healthy individuals. *P < 0.01.
We compared the expression levels of NDRG1 in two individuals with homozygous duplication (BAB3724 and BAB4149), heterozygous duplication (BAB4146 and BAB4147), and wild type (BAB4150 and BAB4152) by using TaqMan Q-PCR. We observed a decrease of 0.12-fold (BAB3724) and 0.18-fold (BAB4149) in homozygous duplication individuals and 0.61-fold (BAB4146) and 0.6-fold (BAB4147) in unaffected heterozygous duplication individuals compared with a healthy family member (P < 0.001) (Figure 3c).
Regions of homozygosity and other potential CNVs
We performed whole-genome SNP arrays on DNA samples from all available family members to assess the genome-wide extent of absence of heterozygosity (AOH) including the precise region and size of AOH encompassing NDRG1. We then determined whether there are other CNVs in the genome that could potentially contribute to the phenotype in the family or perhaps explain the clinical variability of affected family members. We did not observe any additional large CNV detected by SNP array elsewhere in the genome or in other CMT gene regions or elsewhere that segregates with the CMT phenotype investigated in this report, nor did we identify any large de novo CNV that might explain the intellectual disability in family member II-7.
As expected due to the consanguinity in the family, the proband and other siblings have several regions of homozygosity, or AOH, throughout their genome. In total, 33 AOH regions spanning from 1.0 to 44.3 Mb in size were identified in the proband. The NDRG1 gene is encompassed within the largest 44.3 Mb region of AOH in this individual; and comparing multiple affected individuals, the minimum region of overlap between AOH regions spanning NDRG1 is 21.9 Mb (Figure 4).
Figure 4. NDRG1 resides in an ~22 Mb region of absence of heterozygosity (AOH) shared by all affected individuals.
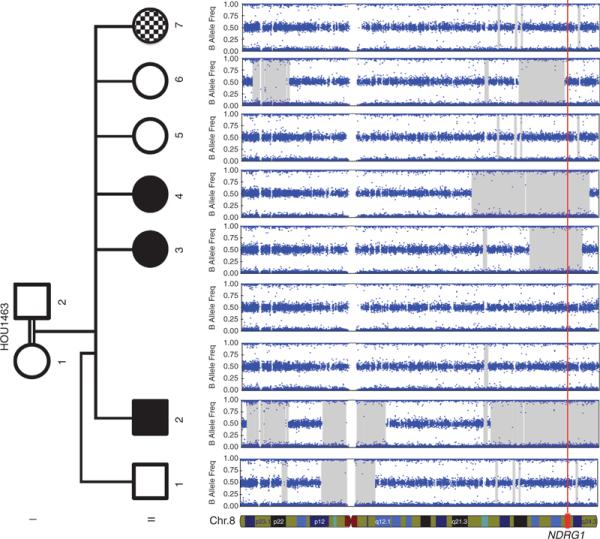
The figure depicts single-nucleotide polymorphism array data for the entirety of chromosome 8. Illumina Genome Studio software predicted AOH regions >0.25 Mb (shaded in gray). All affected individuals in the pedigree share a minimum overlapping region of ~21.9 Mb of AOH that encompasses the NDRG1 gene locus. All unaffected family members lacked a predicted AOH region that overlapped this gene. The location of NDRG1 is depicted at the bottom of the figure, and its location for each individual in the pedigree is denoted with the red vertical line.
DISCUSSION
In this article, we report the first AR duplication CNV of NDRG1 causing hereditary motor and sensory neuropathy-Lom (HMSN-Lom)/CMT4D (MIM: 601455) in a large Turkish pedigree. AR-CMT accounts for only 4% of the families in the European CMT population.9,10 By contrast, in communities with a higher rate of consanguineous marriage, for example, Middle Eastern countries, AR-CMT accounts for ~30–50% of all CMT cases.10,11
Approximately one-half of all patients with CMT12 and 70% of all patients with the demyelinating type (CMT1) have a 1.4 Mb duplication as the cause of their disease.13-15 In addition, rare smaller sized CNVs involving PMP22 have also been reported.5 However, CNVs in non-CMT1A patients are rarely reported. Hoyer et al.16 and Maeda et al.17 reported dominant CNVs in the gene encoding MPZ. In the X-linked recessive form of CMT, whole-gene deletions of GJB1 have been reported.6,17 Huang et al.7 screened 97 CMT patients for CNVs, and detected a novel shorter form of PMP22 duplication and two other CNVs in MTMR2 (CMT4B1, MIM: 603557) and ARHGEF10 (autosomal-dominant slowed nerve conduction velocity, MIM: 608136), which were determined to be non-disease-causing variants.
In the NDRG1 intragenic CNV reported in this article, breakpoint junction analysis reveals a 5 bp microhomology at the breakpoint, supporting a replicative mechanism such as fork stalling and template switching/microhomology-mediated breakage-induced replication as the underlying mechanism for generation.4 This is different from nonallelic homologous recombination, the underlying mechanism for 1.4 Mb recurrent duplication of PMP22 in CMT1A patients. Breakpoint junctions were studied in detail in GJB1 deletion and MPZ duplication cases also.6,17 In other CMT families that are caused by CNV, Alu-mediated deletion in GJB1 and duplication in MPZ have been observed, as well as microhomology-mediated deletion in GJB1. These data show that mechanisms other than nonallelic homologous recombination can be responsible for the formation of CNVs in non-PMP22 duplication patients.
HMSN-Lom/CMT4D was initially described by Kalaydjieva et al.18 as an AR peripheral neuropathy with normal intellect, deafness, and unusual neuropathologic features. It was identified in 14 affected individuals from the Gypsy community of Lom, a small town on the Danube River in the northwest of Bulgaria and referred to as “hereditary motor and sensory neuropathy-Lom” (HMSN-Lom). The disorder was subsequently reported in families from Italy, Slovenia, Germany, Spain, France, and Romania.19-22 To date, two disease-causing mutations in NDRG1 have been reported in humans: R148X and IVS8-1 G>A (g.2290787G>A) (http://www.molgen.ua.ac.be/CMTMutations/). In addition, a 10 bp deletion leading to a frameshift (p.R361SerfsX60) and a single-nucleotide variant mutation (causing aa change p.G98V) have been described in both inbred Greyhound and Alaskan Malamute pedigrees with polyneuropathy.23,24 AR peripheral neuropathies are relatively rare, but can be clinically more severe than autosomal-dominant forms of CMT. Clinically, HMSN-Lom/CMT4D presents with muscle weakness and wasting, tendon areflexia, skeletal and foot deformities, and sensory loss affecting all modalities. Deafness is an invariant feature of the phenotype and usually develops in the third decade.19,20,22,25,26 The subjects reported herein showed typical features, including muscle weakness and wasting, tendon areflexia, skeletal and foot deformities, and sensorineural loss with deafness.
The index case of this family (BAB3724) had additional clinical findings suggestive of possible glaucoma. Glaucoma with CMT is a rare feature and was reported in CMT4B2 caused by mutations of SBF2/MTMR13 (MIM: 607697).27-29 We initially considered the possibility that glaucoma may be another clinical finding in addition to the peripheral neuropathy and hearing loss in NDRG1 mutation cases. We performed eye examinations in BAB4152 and found that this individual, who has normal copy number in NDRG1, also has glaucoma findings. We then checked an AR glaucoma locus (MIM: 601771) for a possible AOH region spanning the CYP1B1 gene and found no large region of AOH. Improper segregation of NDRG1 and lack of AOH within the AR glaucoma region suggest that glaucoma may be associated with another currently undefined locus in this family. Individual BAB4152 has a history of epilepsy and intellectual disability in addition to a more severe neuropathy than her siblings. The patient is homozygous wild type at the NDRG1 locus, lacks a region of AOH spanning this gene, has a normal level of NDRG1 expression, and has additional clinical features (Figures 2 and 4). Therefore, we suggest this individual has a different clinical entity than that which we observe in BAB3724, BAB4148, or BAB4149.
Homozygous rare variants (either single-nucleotide variant or small CNV) within regions of AOH may account for variability in the clinical presentation and other features in the differentially affected individuals, such as the glaucoma present in the proband but not in other affected individuals or the intellectual disability and more severe neurological disorder of II-7/BAB4152. However, this hypothesis requires additional analyses and testing, as well as potentially exome sequencing of the differentially affected sibling. Of note, no further large CNVs in CMT gene loci were detected by SNP genotyping, indicating the effectiveness of our targeted array platform.
In this article, we reported the first recessive duplication CNV causing CMT and the third mutation in the NDRG1 gene. Although affected individuals primarily present with features explained by the AR duplication of NDRG1, further findings and clinical variability within the family may potentially be explained by de novo mutations or additional rare variant alleles that arose in recent ancestors and became reduced to homozygosity–a concept embodied in the term “clan genomics.”30
ACKNOWLEDGMENTS
We thank the patients and family for their contribution to this study. This work was supported in part by the US National Institute of Neurological Disorders and Stroke grant R01NS058529 and the US National Human Genome Research Institute (NHGRI) grant U54HG006542 to J.R.L. and the NHGRI grant U54-HG003273 to R.A.G. C.R.B. is a Howard Hughes Medical Institute fellow of the Damon Runyon Cancer Research Foundation (DRG 2155-13). This study was partially funded by Bogazici University Research Fund project number 09B101 and Neuroimmunology Association (NIMDER).
Footnotes
DISCLOSURE
J.R.L. is a paid consultant for Athena Diagnostics, has stock ownership in 23andMe and Ion Torrent Systems, and is a co-inventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. R.A.G. is a consultant to GE-Clarient. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis and clinical exome sequencing offered in the Medical Genetics Laboratory (http://www.bcm.edu/geneticlabs/). Other authors have no disclosures relevant to the manuscript.
REFERENCES
- 1.England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology. American Association of Neuromuscular and Electrodiagnostic Medicine. American Academy of Physical Medicine and Rehabilitation Practice parameter: the evaluation of distal symmetric polyneuropathy: the role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R. 2009;1:5–13. doi: 10.1016/j.pmrj.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Szigeti K, Lupski JR. Charcot-Marie-Tooth disease. Eur J Hum Genet. 2009;17:703–710. doi: 10.1038/ejhg.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiszniewski W, Szigeti K, Lupski JR. Hereditary Motor and Sensory Neuropathies. 6th Vol. 3. Elsevier; San Francisco, CA: 2013. [Google Scholar]
- 4.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Seeman P, Liu P, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzaga-Jauregui C, Zhang F, Towne CF, Batish SD, Lupski JR. GJB1/Connexin 32 whole gene deletions in patients with X-linked Charcot-Marie-Tooth disease. Neurogenetics. 2010;11:465–470. doi: 10.1007/s10048-010-0247-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Wu X, Montenegro G, et al. Copy number variations are a rare cause of non-CMT1A Charcot-Marie-Tooth disease. J Neurol. 2010;257:735–741. doi: 10.1007/s00415-009-5401-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pehlivan D, Hullings M, Carvalho CM, et al. NIPBL rearrangements in Cornelia de Lange syndrome: evidence for replicative mechanism and genotype-phenotype correlation. Genet Med. 2012;14:313–322. doi: 10.1038/gim.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubourg O, Tardieu S, Birouk N, et al. The frequency of 17p11.2 duplication and Connexin 32 mutations in 282 Charcot-Marie-Tooth families in relation to the mode of inheritance and motor nerve conduction velocity. Neuromuscul Disord. 2001;11:458–463. doi: 10.1016/s0960-8966(00)00222-4. [DOI] [PubMed] [Google Scholar]
- 10.Dubourg O, Azzedine H, Verny C, et al. Autosomal-recessive forms of demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:75–86. doi: 10.1385/nmm:8:1-2:75. [DOI] [PubMed] [Google Scholar]
- 11.4th Workshop of the European CMT-Consortium Neuromuscul Disord; 62nd ENMC International Workshop: rare forms of Charcot-Marie-Tooth Disease and Related Disorders; Soestduinen, The Netherlands. 16–18 October 1998; 1999. pp. 279–287. [DOI] [PubMed] [Google Scholar]
- 12.Szigeti K, Garcia CA, Lupski JR. Charcot-Marie-Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management. Genet Med. 2006;8:86–92. doi: 10.1097/01.gim.0000200160.29385.73. [DOI] [PubMed] [Google Scholar]
- 13.Wise CA, Garcia CA, Davis SN, et al. Molecular analyses of unrelated Charcot-Marie-Tooth (CMT) disease patients suggest a high frequency of the CMTIA duplication. Am J Hum Genet. 1993;53:853–863. [PMC free article] [PubMed] [Google Scholar]
- 14.Nelis E, Van Broeckhoven C, De Jonghe P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4:25–33. doi: 10.1159/000472166. [DOI] [PubMed] [Google Scholar]
- 15.Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39(7):S43–S47. doi: 10.1038/ng2084. Suppl. [DOI] [PubMed] [Google Scholar]
- 16.Høyer H, Braathen GJ, Eek AK, Skjelbred CF, Russell MB. Charcot-Marie-Tooth caused by a copy number variation in myelin protein zero. Eur J Med Genet. 2011;54:e580–e583. doi: 10.1016/j.ejmg.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Maeda MH, Mitsui J, Soong BW, et al. Increased gene dosage of myelin protein zero causes Charcot-Marie-Tooth disease. Ann Neurol. 2012;71:84–92. doi: 10.1002/ana.22658. [DOI] [PubMed] [Google Scholar]
- 18.Kalaydjieva L, Hallmayer J, Chandler D, et al. Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nat Genet. 1996;14:214–217. doi: 10.1038/ng1096-214. [DOI] [PubMed] [Google Scholar]
- 19.Merlini L, Villanova M, Sabatelli P, et al. Hereditary motor and sensory neuropathy Lom type in an Italian Gypsy family. Neuromuscul Disord. 1998;8:182–185. doi: 10.1016/s0960-8966(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 20.Baethmann M, Göhlich-Ratmann G, Schröder JM, Kalaydjieva L, Voit T. HMSNL in a 13-year-old Bulgarian girl. Neuromuscul Disord. 1998;8:90–94. doi: 10.1016/s0960-8966(98)00011-x. [DOI] [PubMed] [Google Scholar]
- 21.Colomer J, Iturriaga C, Kalaydjieva L, Angelicheva D, King RH, Thomas PK. Hereditary motor and sensory neuropathy-Lom (HMSNL) in a Spanish family: clinical, electrophysiological, pathological and genetic studies. Neuromuscul Disord. 2000;10:578–583. doi: 10.1016/s0960-8966(00)00149-8. [DOI] [PubMed] [Google Scholar]
- 22.Kalaydjieva L, Gresham D, Gooding R, et al. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet. 2000;67:47–58. doi: 10.1086/302978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drögemüller C, Becker D, Kessler B, et al. A deletion in the N-myc downstream regulated gene 1 (NDRG1) gene in Greyhounds with polyneuropathy. PLoS ONE. 2010;5:e11258. doi: 10.1371/journal.pone.0011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruun CS, Jäderlund KH, Berendt M, et al. A Gly98Val mutation in the N-Myc downstream regulated gene 1 (NDRG1) in Alaskan Malamutes with polyneuropathy. PLoS ONE. 2013;8:e54547. doi: 10.1371/journal.pone.0054547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalaydjieva L, Nikolova A, Turnev I, et al. Hereditary motor and sensory neuropathy–Lom, a novel demyelinating neuropathy associated with deafness in gypsies. Clinical, electrophysiological and nerve biopsy findings. Brain. 1998;121:399–408. doi: 10.1093/brain/121.3.399. Pt 3. [DOI] [PubMed] [Google Scholar]
- 26.Butinar D, Zidar J, Leonardis L, et al. Hereditary auditory, vestibular, motor, and sensory neuropathy in a Slovenian Roma (Gypsy) kindred. Ann Neurol. 1999;46:36–44. doi: 10.1002/1531-8249(199907)46:1<36::aid-ana7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 27.Senderek J, Bergmann C, Weber S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003;12:349–356. doi: 10.1093/hmg/ddg030. [DOI] [PubMed] [Google Scholar]
- 28.Azzedine H, Bolino A, Taïeb T, et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72:1141–1153. doi: 10.1086/375034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirano R, Takashima H, Umehara F, et al. SET binding factor 2 (SBF2) mutation causes CMT4B with juvenile onset glaucoma. Neurology. 2004;63:577–580. doi: 10.1212/01.wnl.0000133211.40288.9a. [DOI] [PubMed] [Google Scholar]
- 30.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]