

Abstract
Previously, we have shown that p-cyanophenylalanine (PheCN) and tryptophan (Trp) constitute an efficient fluorescence resonance energy transfer (FRET) pair that has several advantages over commonly used dye pairs. Here, we aim to examine the general applicability of this FRET pair in protein folding–unfolding studies by applying it to the urea-induced unfolding transitions of two small proteins, the villin headpiece subdomain (HP35) and the lysin motif (LysM) domain. Depending on whether PheCN is exposed to solvent, we are able to extract either qualitative information about the folding pathway, as demonstrated by HP35, which has been suggested to unfold in a stepwise manner, or quantitative thermodynamic and structural information, as demonstrated by LysM, which has been shown to be an ideal two-state folder. Our results show that the unfolding transition of HP35 reported by FRET occurs at a denaturant concentration lower than that measured by circular dichroism (CD) and that the loop linking helix 2 and helix 3 remains compact in the denatured state, which are consistent with the notion that HP35 unfolds in discrete steps and that its unfolded state contains residual structures. On the other hand, our FRET results on the LysM domain allow us to develop a model for extracting structural and thermodynamic parameters about its unfolding, and we find that our results are in agreement with those obtained by other methods. Given the fact that PheCN is a non-natural amino acid and, thus, amenable to incorporation into peptides and proteins via existing peptide synthesis and protein expression methods, we believe that the FRET method demonstrated here is widely applicable to protein conformational studies, especially to the study of relatively small proteins.
Fluorescence resonance energy transfer (FRET)1 is one of the most commonly used methods for probing the distance between two sites in biological systems (1). In particular, FRET has proven to be very useful and informative in probing the conformation, conformational change, and conformational dynamics of proteins (2, 3). However, the application of FRET to protein conformational studies is not without its limitations. For example, the commonly available dye FRET pairs are bulky in comparison to amino acid side chains and, therefore, may introduce undesirable structural perturbations to the protein. For this reason, dye attachment is often restricted to solvent-exposed or relatively unstructured sites, which in turn may limit the amount of information that can be extracted from FRET. In addition, dye labeling reactions are often incomplete and/or nonspecific, leading to skewed FRET results (4). Recently, it has been shown that a nitrile-derivatized phenylalanine residue, p-cyanophenylalanine (PheCN), and tryptophan (Trp) constitute an efficient donor–acceptor pair for FRET measurements and were used to investigate the conformational distribution of a 14-residue peptide in aqueous solution and also the membrane-mediated helix folding kinetics of an antimicrobial peptide (5–7). Here we further demonstrate that this amino acid FRET pair can also be used to characterize, sometimes quantitatively, protein folding and unfolding transitions.
Compared to commonly used dye fluorophores, the PheCN–Trp FRET pair offers several advantages in protein conformational studies, especially for proteins that are less tolerant to structural perturbations. First, PheCN is a non-natural amino acid that can be easily incorporated into peptides through standard solid-phase peptide synthesis or into proteins using modified expression protocols (8). Second, PheCN is considered a derivative of both phenylalanine (Phe) and tyrosine (Tyr), so it only minimally perturbs the native structure, especially when used to substitute for a Phe or Tyr residue. For example, PheCN has recently been incorporated into the hydrophobic core of protein NTL9, and the resultant mutant (Phe19PheCN) exhibited folding thermodynamic properties almost identical to those of the parent protein (9). Third, the Förster distance of the PheCN–Trp pair is ~16 Å, making it well-suited for probing relatively short separation distances along a certain polypeptide sequence. Finally, the C≡N stretching vibration of PheCN is sensitive to environment (10); thus, it can also be used independently as an infrared (IR) probe (8, 10, 11). While it is expected that the PheCN–Trp FRET pair is well-suited for protein folding and unfolding studies, its general applicability has yet to be demonstrated. Herein, we apply it to study the urea-induced unfolding of two small proteins, the chicken villin headpiece subdomain, HP35 (12, 13), and the LysM domain from Escherichia coli membrane-bound lytic murein transglycosylase D (MltD) (14). We aim to show that this FRET pair is useful for probing distance information from within the hydrophobic core of proteins and also to extract thermodynamic information.
Because of its small size and very fast folding rate, the folding mechanism of HP35 has been extensively studied using both experimental (15–20) and computational (21–33) methods. As shown (Figure 1a), HP35 (sequence, L42SDEDF-KAVF-GMTRSAFANL-PLWKQQNLKK-EKGLF76) folds into a three-helix bundle, which is stabilized by a hydrophobic core composed of primarily three Phe residues (i.e., Phe47, Phe51, and Phe58) and Val50 (13). In addition, the single Trp residue in HP35 is conveniently located at a solvent-exposed position and is approximately 12 Å from Phe58 based on the NMR structure. The latter is well within the Förster distance of the PheCN–Trp pair, making the Phe58/PheCN mutant (hereafter called HP35-P) a good model system for monitoring the unfolding of the loop linking helix 2 and helix 3.
Figure 1.

(a) NMR structure of HP35 (PDB entry 1VII). (b) NMR structure of the LysM domain (PDB entry 1E0G). In both cases, the Trp side chain and the side chain of the residue which was replaced with PheCN in this study are shown.
The LysM domain is a widespread protein module originally identified in enzymes that degrade bacterial cell walls and is primarily associated with peptidoglycan binding (34). As shown (Figure 1b), the LysM domain of MltD folds into a βααβ motif in which the two helices pack onto the same side of a two-stranded antiparallel β-sheet. In addition, it contains a single Trp residue (sequence, SITYRVRKG-DSLSSIAKRH-GVNIKDVMRW-NSDTANLQPG-DKLTLFVK), thus making it convenient to employ the proposed FRET study. More importantly, Nickson et al. (35) have recently used extensive Φ-value analyses to show that LysM is an ideal two-state folder and the ΔG for unfolding at 10 °C is 3.0 kcal/mol. To conduct the proposed FRET study, we replaced the C-terminal Lys residue with PheCN and the resultant mutant is called LysM-P hereafter.
In both cases, our results show that the global unfolding transitions can be followed by PheCN–Trp FRET. However, depending on the protein in question and also on the locations of the donor and acceptor, this FRET pair can reveal subtle details that may be obscured in measurements employing other conformational probes. Thus, this study demonstrates the utility of the PheCN–Trp FRET pair as a versatile conformational marker in protein folding studies.
MATERIALS AND METHODS
Materials
HP35-P and LysM-P were synthesized on a peptide synthesizer (Protein Technologies) using Rink resin. Peptides were then purified to homogeneity by reverse-phase HPLC. The identity of the peptide was further verified by matrix-assisted laser desorption ionization mass spectrometry.
CD Measurements
The far-UV circular dichroism (CD) spectra of HP35-P were measured on a 62A DS spectropolarometer (Aviv Associates) with a 1 mm sample cell. The HP35-P concentration was 23.9 μM as determined by its absorbance at 280 nm.
Absorption Measurement
All UV–vis spectra were measured on a Lambda 25 UV–vis spectrometer (Perkin-Elmer). The Fourier transform infrared (FTIR) spectrum of HP35-P in the C≡N stretching region was recorded at 26 °C on a Magna-IR 860 spectrometer (Nicolet) at 2 cm−1 resolution using a two-compartment CaF2 sample cell.
Fluorescence Measurements
The fluorescence spectra at 2 nm resolution were obtained on a Fluorolog 3.10 spectrofluorometer (Jobin Yvon Horiba) using a 1 cm quartz sample cell. All LysM-P urea titrations were conducted at 20 °C and in 20 mM phosphate buffer (pH 5.8), and the peptide concentration was 10 μM. Similarly, all HP35-P urea titrations were conducted at 20 °C and in 50 mM phosphate buffer (pH 7), and the peptide concentration was 7 μM. Temperature was regulated using a TLC 50 Peltier temperature controller (Quantum Northwest). To minimize self-quenching, the optical density (OD) of each sample at the excitation wavelength was adjusted to be in the range of 0.1–0.2. To achieve a high signal-to-noise (S/N) ratio, an integration time of 2 s/nm was used in HP35-P experiments and 1.5 s/nm for the LysM-P experiments. Although the relatively long integration time could result in sample photobleaching, control experiments showed that the emission intensity changed less than 5% during an hour of measurement, demonstrating that the effect of photobleaching was minimal in these measurements.
RESULTS AND DISCUSSION
As an amino acid FRET pair, the PheCN–Trp pair is expected to offer distinct advantages over commonly used dye fluorophores, especially in studies involving conformational changes of peptides and/or small proteins or conformational events occurring over a relatively short distance. For example, this FRET pair has recently been used to reveal the conformational distribution of short unstructured peptides in solution (5). Here we use two small proteins, HP35 and the LysM domain, to show that the PheCN–Trp FRET pair not only is broadly applicable to protein folding–unfolding studies but also can reveal either local or global folding–unfolding transitions, depending on the locations of the donor and acceptor and also the protein in question.
Application to HP35
HP35 provides an attractive model system for testing the utility of the PheCN–Trp FRET pair in protein folding–unfolding studies because it conveniently consists of a single Trp residue and three Phe residues that are structurally important and also tolerable to mutation (36). Here, we have chosen to mutate Phe58 to PheCN based on earlier studies that suggested it to be one of the most critical residues for defining the native fold of this protein (31, 33). Moreover, Phe58 and Trp64 are located on two helices that are separated by a short loop. Thus, this mutation might also prove to be useful in providing information about the conformational flexibility of the aforementioned loop, the formation of which has been suggested to be an important step in villin headpiece folding (33, 37, 38).
As shown (Figure 2), the CD spectrum of HP35-P shows characteristics of helical proteins, indicating that the mutation does not lead to significant disruption of the fold. In addition, the fluorescence spectrum of HP35-P in buffer, obtained by using an excitation wavelength of 240 nm where the absorbance of Trp is much smaller than that of PheCN (5), exhibits the characteristics of FRET (Figure 3). In particular, the donor fluorescence is quenched almost completely, indicating that the distance between the donor and acceptor is well within the Förster distance of the FRET pair. This is in agreement with the fact that in the folded state of HP35, the distance from Phe58 to Trp64 (Cα to Cα) is only 12 Å, significantly shorter than the R0 of the PheCN–Trp FRET pair (~16 Å). As expected (Figure 3), addition of urea causes the PheCN fluorescence to increase, indicating that the separation distance between the donor and acceptor increases as a result of urea-induced protein unfolding. Interestingly, however, the Trp fluorescence also shows a concomitant increase, indicative of deviation from a simple scenario in which the FRET signal is a function of only the donor–acceptor distance (see below).
Figure 2.

CD spectrum of HP35-P collected at 25 °C. The peptide concentration was 24 μM [in 50 mM phosphate buffe (pH 7)]. The inset shows the C≡N stretching vibration of HP35-P at 26 °C.
Figure 3.

Fluorescence spectra of HP35-P at different urea concentrations, as indicated in the figure. λex = 240 nm.
To provide a more quantitative assessment of the acquired FRET data, we decomposed the measured fluorescence spectra into their constituent spectral components, i.e., the fluorescence spectra of the donor and acceptor. This was accomplished by fitting an individual spectrum (i.e., the spectrum obtained at a certain urea concentration), Fobs(λ), to the following equation:
| (1) |
where FDA(λ) represents the fluorescence spectrum of the donor in the presence of the acceptor and is calculated as αI(λ), where I(λ) is the emission spectrum of free PheCN measured under the same experimental conditions and α is a constant which was allowed to vary during the fitting process. In addition, FAD(λ), the fluorescence spectrum of the acceptor in the presence of the donor, was modeled by two Gaussian functions whose amplitude, position, and width were allowed to vary in the fit. As shown (Figure 4a), the integrated areas of the emission spectra of PheCN and Trp increase with an increase in urea concentration, suggesting that either the quantum yield of the donor, that of the acceptor, or both are changing upon denaturation. Indeed, the Trp fluorescence of HP35-P, measured using an excitation wavelength of 290 nm where the absorbance of Trp overwhelms that of PheCN, shows a monotonic increase as a function of urea concentration (data not shown). In addition, it has been shown that the quantum yield of PheCN is environment-dependent, with a lower quantum yield in a more hydrophobic environment (9, 39). Thus, in the case presented here, the quantum yield of PheCN is expected to increase upon protein unfolding as it becomes more hydrated in the denatured state. Indeed, the C≡N stretching vibration of HP35-P in its folded state is centered around 2235 cm−1 (Figure 2, inset), indicating that the PheCN residue is only partially exposed to solvent (10).
Figure 4.

(a) Fluorescence intensities (integrated area) of PheCN (blue) and Trp (red) of HP35-P as a function of urea concentration. (b) Ratio of the PheCN fluorescence intensity (FDA) to the Trp fluorescence intensity (FAD) vs urea concentration. Also shown is the mean residue ellipticity of HP35-P at 222 nm as a function of urea concentration.
These factors make it difficult to extract any quantitative information (e.g., distance) from the corresponding FRET measurements. However, the ratio of donor to acceptor fluorescence intensity (hereafter termed RDA) at each urea concentration reports an apparent FRET efficiency which is determined by several factors, including the donor-to-acceptor distance, the Förster distance, which changes as a function of urea concentration, and the quantum yield of the acceptor (Trp). Despite potential complications arising from these factors, such a plot is advantageous in that it eliminates the need for a reference protein in which the acceptor is absent and intrinsically corrects for the uncertainty arising from variation in protein concentration and excitation energy at each urea concentration. As shown (Figure 4b), the RDA of HP35-P exhibits a monotonic increase with an increase in urea concentration and reaches a plateau at ~5 M urea, indicating that once unfolded the distance between PheCN and Trp in HP35-P is not sensitive to a further increase in the concentration of the denaturant. The FRET efficiency (E) can be determined from the ratio of FDA/FD, where FD is the donor fluorescence intensity when the acceptor is absent. At 7.5 M urea, the C≡N stretching frequencies of HP35-P and free PheCN are within 1 cm−1 of each other (data not shown), suggesting that the PheCN side chain in the unfolded protein is largely exposed to solvent. Thus, we have estimated E and consequently the separation distance between the donor and acceptor in the unfolded state of HP35-P using the fluorescence intensity of free PheCN. Assuming that the quantum yield of free PheCN is similar to that of PheCN in the unfolded HP35-P without the presence of Trp, the ensemble-averaged separation distance between PheCN and Trp at 8 M urea is estimated to be ~13.1 ± 2.0 Å, which indicates that the loop sandwiched between PheCN and Trp is rigid and also suggests that the unfolded state of HP35 is rather compact. The latter is consistent with the notion that a significant amount of residual structure exists in the denatured state of the villin headpiece (18, 40) and also the simulation studies of Duan and co-workers (33, 37), which concluded that this loop plays an important role in initiating the folding of HP35 by locking down the movements of helices harboring the FRET pair. Thus, the rigidity of this loop may contribute to the ultrafast folding rate of HP35 (16, 17) as it has been shown that loop formation plays an important role in controlling the folding kinetics of helix–turn–helix motifs (41). In support of this, NMR and CD experiments have shown that one highly conserved residue in the turn (42) is critical for folding (38).
Interestingly, the urea-induced unfolding transition reported by FRET (i.e., RDA vs [urea]) shows measurable difference from that reported by CD which monitors the total helical content of the protein (Figure 4b). Specifically, the change in FRET signal occurs at a urea concentration lower than that reported by CD. These results suggest that the two helical segments harboring the FRET pair undergo an expansion prior to the unfolding of individual helical structures. This observation is corroborated by the study of Brewer et al. (19), which showed that Ala57 becomes more solvent-exposed prior to global unfolding, and also the NMR study of Tycko and co-workers (43), which indicates a higher secondary structure content in protein molecules with disrupted tertiary structure. Finally, our result is also in accord with the molecular dynamics simulation of Bandyopadhyay et al. (31), where the Phe58 residue was shown to be the nucleation site for the unfolding process of HP35.
Application to the LysM Domain
The quantum yield of PheCN in an unstructured peptide environment is not very sensitive to urea concentration (44). Thus, it is feasible to extract more quantitative folding–unfolding information using the PheCN–Trp FRET pair, provided the PheCN is placed in a solvent-exposed position. To verify the feasibility of this, we have conducted similar FRET studies on a mutant of the LysM domain, Lys48PheCN (LysM-P). We chose the LysM domain because it is an ideal two-state folder, thus making it easier to compare thermodynamic results obtained from different spectroscopic methods. In addition, the LysM domain contains a single Trp residue located at a solvent-exposed position, making the proposed FRET experiment convenient. Furthermore, the C-terminal residue Lys48 is not only solvent-exposed but also located in an unstructured region of the protein. Thus, the Lys to PheCN mutation is expected not to perturb in any significant manner the structure and stability of the LysM domain.
As shown (Figure 5), the fluorescence spectra of LysM-P at different urea concentrations, which were obtained by exciting the PheCN residue at 240 nm, show characteristics of FRET between PheCN and Trp29. Following the protocols discussed above (i.e., eq 1), we further decompose these fluorescence spectra into their constituent PheCN and Trp fluorescence spectra. As shown (Figure 6a), the fluorescence intensity (or the integrated area of the fluorescence spectrum, FDA or FAD) of the donor and acceptor increases and decreases, respectively, monotonically with an increasing urea concentration, indicating that unfolding enlarges the separation distance between the donor and acceptor. As expected, the ratio of FDA to FAD (Figure 6b), which is related to the FRET efficiency, shows a sigmoidal dependence on denaturant concentration, typical for a cooperative or two-state folding–unfolding transition, which is in agreement with the study of Nickson et al. (35).
Figure 5.

Fluorescence spectra of LysM-P at different urea concentrations, as indicated in the figure. λex = 240 nm.
Figure 6.
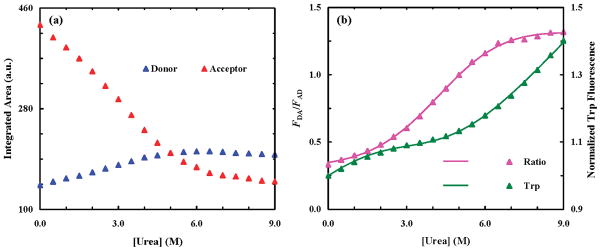
(a) Fluorescence intensity (integrated area) of PheCN (blue) and Trp (red) of LysM-P as a function of urea concentration. (b) Ratio of the PheCN fluorescence intensity (FDA) to the Trp fluorescence intensity (FAD) vs urea concentration. Also shown is the normalized fluorescence intensity of Trp, measured upon direct excitation at 290 nm, as a function of urea concentration. Smooth lines are global fits to these data according to the methods discussed in the text.
It is easy to show that for a two-state folding–unfolding transition, the FRET signal or the ratio of FDA to FAD is determined by the following equation:
| (2) |
where QD and QA are the quantum yields of the donor and acceptor, respectively, while subscripts f and u refer to the folded and unfolded states, respectively. In addition, r is the distance between the donor and acceptor and R0 is the Förster distance for the FRET pair, which is defined as (45)
| (3) |
where η is the refractive index of the medium, NA is Avogadro’s number, κ is the orientation factor, and J(λ) represents the overlap integral of the donor emission and the acceptor absorption profiles. Furthermore, Keq is the equilibrium constant for unfolding, which is defined as
| (4) |
with
| (5) |
where ΔG°(0) is the free energy change for unfolding in water and m is a constant (46, 47).
Equation 2 indicates that to determine Keq, one needs to know both QD and QA. However, if we assume that QD does not change (or R0 is a constant) upon unfolding of the LysM domain, which is valid in the current case because the PheCN is located in a solvent-exposed position, eq 2 can be simplified to
| (6) |
This equation indicates that it is possible to determine Keq and r from the FRET signals if independent measurements can provide information about how the quantum yield of the acceptor changes as a function of urea concentration. Fortunately, this can be done easily in the current case because even when PheCN is present, the Trp (acceptor) fluorescence can be selectively excited by using an excitation wavelength of 290 nm. As shown (Figure 6b), the Trp fluorescence of LysM-P obtained by directly exciting the Trp fluorophore is different from that obtained via FRET excitation. It is apparent that the unfolding transition reported by Trp fluorescence is less pronounced than that reported by FRET, due to the fact that the Trp residue in the LysM domain is largely exposed to solvent. Nonetheless, it has been used as a probe for monitoring the unfolding of this peptide (35). To determine the thermodynamics of the cooperative folding–unfolding transition of the LysM domain, we globally fit those FRET data and Trp fluorescence intensities, presented in Figure 6b, to eq 6 and the following equation:
| (7) |
where Furea represents the fluorescence intensity of Trp obtained in urea solutions while F0 represents that obtained in buffer solution. In addition, the donor–acceptor distance r and the quantum yield of the acceptor (QA) are assumed to be linearly dependent on the concentration of urea:
| (8) |
| (9) |
During the fitting, ΔG°(0), m, rf, , p, and q are treated as global parameters. As shown (Figure 6b), the best fits yield the following thermodynamic and structural parameters: ΔG°(0) = 1.4 ± 0.1 kcal/mol, m = 0.42 ± 0.05 kcal mol−1 M−1, and rf = 13 ± 2 Å. The fact that the value of ΔG°(0) determined from the FRET method is comparable to that (1.2 ± 0.2 kcal/mol) determined from CD spectroscopy (Figure 7) validates the FRET method. In addition, this result is also consistent with the study of Nickson et al. (35), which showed that the ΔG° for the unfolding of the LysM domain is 3.0 kcal/mol at 10 °C and pH 7. Since the LysM domain is more stable at neutral pH and lower temperatures (35), under the current experimental conditions (pH 5.8 and 20 °C) the LysM domain is expected to exhibit a smaller ΔG°(0). In addition, the m value recovered from the fit, 0.42 kcal mol−1 M−1, is also very close to that calculated from a commonly used model, 0.5 kcal mol−1 M−1 (48). Finally, the obtained rf value, 13 Å, matches the expected value estimated from the NMR structure of the LysM domain. Taken together, these results indicate that the above FRET model provides a reliable estimate of the folding–unfolding thermodynamics of the LysM domain, supporting the general applicability of this approach to protein conformational studies. It should be noted, however, that the FAD used in this analysis contains Trp fluorescence arising from direct excitation of the fluorophore at 240 nm (39), which ranges from 3 to 10% depending on the urea concentration. This additional contribution certainly skews the FDA/FAD ratios, thus introducing errors to the resultant thermodynamic and structural parameters. To achieve a more rigorous analysis, it is desirable to subtract out such a contribution. The latter can be done simply by using either a reference protein that does not contain the PheCN donor or the Trp fluorescence data obtained with selective excitation of Trp at 290 nm in conjunction with a scaling factor (determined by the difference in Trp absorbances at 290 and 240 nm as well as the difference in the excitation intensities at these two wavelengths).
Figure 7.

Mean residue ellipticity of LysM-P at 222 nm vs temperature. The solid line is the best fit of these data to a two-state model discussed in the text.
CONCLUSIONS
We have studied the urea-induced unfolding of two small proteins, the villin headpiece subdomain and the LysM domain, using a FRET method, aiming to demonstrate the utility of the PheCN–Trp FRET pair in the study of protein folding–unfolding transitions. Our results show that this FRET pair is capable of monitoring unfolding transitions with site-specific resolution. Because PheCN can be incorporated into peptides via a standard solid-phase peptide synthesis method or into proteins via genetic manipulation, this FRET pair is expected to be generally applicable to many systems. While we present a FRET method which can be used to quantitatively analyze two-state unfolding scenarios, the PheCN–Trp FRET pair perhaps is more useful and informative in the study of non-two-state proteins or those that unfold in discrete steps. Via systematic variation of the position of the PheCN (donor) or Trp residue (acceptor), it is possible to dissect the sequence or order of the conformational events involved in the folding and unfolding of the protein in question. In addition, it is also possible to use PheCN and Trp to perform more complicated FRET experiments, such as one donor and two acceptors or two donors and one acceptor. Such experiments are particularly useful in that they allow one to probe how different segments in a protein move in response to a conformational perturbation.
Footnotes
Supported by the National Institutes of Health (GM-065978). J.M.G. is a trainee of the NIH Structural Biology Training Program at the University of Pennsylvania.
Abbreviations: CD, circular dichroism; FRET, fluorescence resonance energy transfer; HP35; villin headpiece subdomain; LysM, lysin motif; PheCN, p-cyanophenylalanine.
References
- 1.Sapsford KE, Berti L, Igor L, Medintz IL. Materials for fluorescence resonance energy transfer analysis: Beyond traditional donor-acceptor combinations. Angew Chem, Int Ed. 2006;45:4562–4588. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 2.Royer CA. Probing protein folding and conformational transitions with fluorescence. Chem Rev. 2006;106:1769–1784. doi: 10.1021/cr0404390. [DOI] [PubMed] [Google Scholar]
- 3.Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karolin J, Fa M, Wilczynska M, Ny T, Johansson LBA. Donor-donor energy migration for determining intramolecular distances in proteins: I. Application of a model to the latent plasminogen activator inhibitor-1 (PAV-1) Biophys J. 1998;74:11–21. doi: 10.1016/S0006-3495(98)77762-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tucker MJ, Oyola R, Gai F. Conformational distribution of a 14 residue peptide in solution: A fluorescence resonance energy transfer study. J Phys Chem B. 2005;109:4788–4795. doi: 10.1021/jp044347q. [DOI] [PubMed] [Google Scholar]
- 6.Tucker MJ, Tang J, Gai F. Probing the kinetics of membrane-mediated helix folding. J Phys Chem B. 2006;110:8105–8109. doi: 10.1021/jp060900n. [DOI] [PubMed] [Google Scholar]
- 7.Tang J, Signarvic RS, DeGrado WF, Gai F. Role of helix nucleation in the kinetics of binding of mastoparan X to phospholipid bilayers. Biochemistry. 2007;46:13856–13863. doi: 10.1021/bi7018404. [DOI] [PubMed] [Google Scholar]
- 8.Schultz KC, Supekova L, Ryu Y, Xie J, Perera R, Schultz PG. A genetically engineered infrared probe. J Am Chem Soc. 2006;128:13984–13985. doi: 10.1021/ja0636690. [DOI] [PubMed] [Google Scholar]
- 9.Aprilakis KN, Taskent H, Raleigh DP. Use of the novel fluorescent amino acid p-cyanophenylalanine offers a direct probe of hydrophobic core formation during the folding of the N-terminal domain of the ribosomal protein L9 and provides evidence for two-state folding. Biochemistry. 2007;46:12308–12313. doi: 10.1021/bi7010674. [DOI] [PubMed] [Google Scholar]
- 10.Getahun Z, Huang CY, Wang T, De Leon B, DeGrado WF, Gai F. Using nitrile-derivatized amino acids as infrared probes of local environment. J Am Chem Soc. 2003;125:405–411. doi: 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee S, Chowdhury P, DeGrado WF, Gai F. Site-specific hydration status of an amphipathic peptide in AOT reverse micelles. Langmuir. 2007;23:11174–11179. doi: 10.1021/la701686g. [DOI] [PubMed] [Google Scholar]
- 12.McKnight CJ, Doering DS, Matsudaira PT, Kim PS. A thermostable 35 residue subdomain within villin headpiece. J Mol Biol. 1996;260:126–134. doi: 10.1006/jmbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 13.McKnight CJ, Matsudiara PT, Kim PS. NMR structure of the 35 residue villin headpiece subdomain. Nat Struct Biol. 1997;4:180–184. doi: 10.1038/nsb0397-180. [DOI] [PubMed] [Google Scholar]
- 14.Bateman A, Bycroft M. The structure of the LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD) J Mol Biol. 2000;299:1113–1119. doi: 10.1006/jmbi.2000.3778. [DOI] [PubMed] [Google Scholar]
- 15.Hansen KC, Rock RS, Larsen RW, Chan SI. A method for photoinitiating protein folding in a nondenaturing environment. J Am Chem Soc. 2000;122:11567–11568. [Google Scholar]
- 16.Kubelka J, Eaton WA, Hofrichter J. Experimental tests of villin subdomain folding simulations. J Mol Biol. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Tang Y, Sato S, Vugmeyster L, McKnight CJ, Raleigh DP. Dynamic NMR line-shape analysis demonstrates that the villin headpiece subdomain folds in the microsecond time scale. J Am Chem Soc. 2003;125:6032–6033. doi: 10.1021/ja028752b. [DOI] [PubMed] [Google Scholar]
- 18.Brewer SH, Vu DM, Tang Y, Li Y, Franzen S, Raleigh DP, Dyer RB. Effect of modulating unfolded state structure on the folding kinetics of the villin headpiece subdomain. Proc Natl Acad Sci USA. 2005;102:16662–16667. doi: 10.1073/pnas.0505432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brewer SH, Song B, Raleigh DP, Dyer RB. Residue specific resolution of protein folding dynamics using isotope-edited infrared temperature jump spectroscopy. Biochemistry. 2007;46:3279–3285. doi: 10.1021/bi602372y. [DOI] [PubMed] [Google Scholar]
- 20.Gao JM, Kelly JW. Toward quantification of protein backbone-backbone hydrogen bonding energies: An energetic analysis of an amide-to-ester mutation in an α helix within a protein. Protein Sci. 2008;17:1096–1101. doi: 10.1110/ps.083439708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan Y, Wang L, Kollman PA. The early stage of folding of villin headpiece subdomain observed in 200 ns fully solvated molecular dynamics simulation. Proc Natl Acad Sci USA. 1998;95:9897–9902. doi: 10.1073/pnas.95.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan DC, Kuntz DI. Protein folding as biased conformational diffusion. J Phys Chem B. 2002;106:3255–3262. [Google Scholar]
- 23.Islam SA, Karplus M, Weaver DL. Application of the diffusion-collision model to the folding of three-helix bundle proteins. J Mol Biol. 2002;318:199–215. doi: 10.1016/S0022-2836(02)00029-3. [DOI] [PubMed] [Google Scholar]
- 24.Shen MY, Freed KF. All-atom fast protein folding simulations: The villin headpiece. Proteins. 2002;49:439–445. doi: 10.1002/prot.10230. [DOI] [PubMed] [Google Scholar]
- 25.Jang SM, Kim E, Shin S, Pak Y. Ab initio folding of helix bundle proteins using molecular dynamics simulations. J Am Chem Soc. 2003;125:14841–14846. doi: 10.1021/ja034701i. [DOI] [PubMed] [Google Scholar]
- 26.Lin CY, Hu CK, Hansmann UH. Parallel tempering simulations of HP-36. Proteins. 2003;52:436–445. doi: 10.1002/prot.10351. [DOI] [PubMed] [Google Scholar]
- 27.Ripoll DR, Vila JA, Scheraga HA. Folding of the villin headpiece subdomain from random structures. Analysis of the charge distribution as a function of pH. J Mol Biol. 2004;339:915–925. doi: 10.1016/j.jmb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 28.Herges T, Wenzel W. Free-energy landscape of the villin headpiece in an all-tom force field. Structure. 2005;13:661–668. doi: 10.1016/j.str.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Carr JM, Wales DJ. Global optimization and folding pathways of selected α-helical proteins. J Chem Phys. 2005;123:234901. doi: 10.1063/1.2135783. [DOI] [PubMed] [Google Scholar]
- 30.Ensign DL, Kasson PM, Pande VS. Heterogeneity even at the speed limit of folding: Large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J Mol Biol. 2006;374:806–816. doi: 10.1016/j.jmb.2007.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandyopadhyay S, Chakraborty S, Bagchi B. Coupling between hydration layer dynamics and unfolding kinetics of HP-36. J Chem Phys. 2006;125:084912. doi: 10.1063/1.2335451. [DOI] [PubMed] [Google Scholar]
- 32.Wickstrom L, Okur A, Song K, Hornak V, Raleigh DP, Simmerling CL. The unfolded state of the villin headpiece helical subdomain: Computational studies of the role of locally stabilized structure. J Mol Biol. 2006;360:1094–1107. doi: 10.1016/j.jmb.2006.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei H, Duan Y. Two-stage folding of HP-35 from Ab initio simulations. J Mol Biol. 2007;370:196–206. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buist G, Steen A, Kok J, Kuipers OP. LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol Microbiol. 2008;68:838–847. doi: 10.1111/j.1365-2958.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- 35.Nickson AA, Stoll KE, Clark J. Folding of a LysM domain: Entropy-enthalpy compensation in the transition state of an ideal two-state folder. J Mol Biol. 2008;380:557–569. doi: 10.1016/j.jmb.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank BS, Vardar D, Buckley DA, McKnight CJ. The role of aromatic residues in the hydrophobic core of the villin headpiece subdomain. Protein Sci. 2002;11:680–687. doi: 10.1110/ps.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lei H, Wu C, Liu H, Duan Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc Natl Acad Sci USA. 2007;104:4925–4930. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vermeulen W, Van Troys M, Bourry D, Dewitte D, Rossenu S, Goethals M, Borremans FAM, Vanderkerckhove J, Martins JC, Ampe C. Identification of the PXW sequence as a structural gatekeeper of the headpiece C-terminal subdomain fold. J Mol Biol. 2006;359:1277–1292. doi: 10.1016/j.jmb.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 39.Tucker MJ, Oyola R, Gai F. A novel fluorescent probe for protein binding and folding studies: p-Cyano-phenylalanine. Biopolymers. 2006;83:571–576. doi: 10.1002/bip.20587. [DOI] [PubMed] [Google Scholar]
- 40.Tang Y, Rigotti DJ, Fairman R, Raleigh DP. Peptide models provide evidence for significant structure in the denatured state of a rapidly folding protein: The villin headpiece subdomain. Biochemistry. 2004;43:3264–3272. doi: 10.1021/bi035652p. [DOI] [PubMed] [Google Scholar]
- 41.Du D, Gai F. Understanding the folding mechanism of an α-helical hairpin. Biochemistry. 2006;45:13131–13139. doi: 10.1021/bi0615745. [DOI] [PubMed] [Google Scholar]
- 42.Vermeulen W, Vanhaesebrouck P, Van Troys M, Verschueren M, Fant F, Goethals M, Ampe C, Martins JC, Borremans FAM. Solution structures of the C-terminal headpeice subdomains of human villin and advillin, evaluation of headpiece F-actin-binding requirements. Protein Sci. 2004;13:1276–1287. doi: 10.1110/ps.03518104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Havlin RH, Tycko R. Probing site-specific conformational distributions in protein folding with solid-state NMR. Proc Natl Acad Sci USA. 2005;102:3284–3289. doi: 10.1073/pnas.0406130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du D, Tucker MJ, Gai F. Understanding the mechanism of β-hairpin folding via ϕ-value analysis. Biochemistry. 2006;45:2668–2678. doi: 10.1021/bi052039s. [DOI] [PubMed] [Google Scholar]
- 45.Förster T. Ann Phys. 1948;2:55–75. [Google Scholar]; Förster T. In: Biological Physics. Mielczarek EV, Greenbaum E, Knox RS, editors. American Institute of Physics; New York: 1993. pp. 148–160. (English translation) [Google Scholar]
- 46.Greene RF, Jr, Pace CN. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, α-chymotrytsin, and β-lactoglobulin. J Biol Chem. 1974;249:5388–5393. [PubMed] [Google Scholar]
- 47.Santoro MM, Bolen DW. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 48.Myers JK, Pace N, Scholtz JM. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]