

Abstract
BACKGROUND
Multiple studies show that reactive oxygen species (ROS) play a major role in prostate cancer (PCa) development and progression. Previously, we reported an induction of Spermidine/Spermine N1-Acetyl Transferase (SSAT) by androgen-activated androgen receptor (AR)-JunD protein complex that leads to over-production of ROS in PCa cells. In our current research, we identify small molecules that specifically block AR-JunD in this ROS-generating metabolic pathway.
METHODS
A high throughput assay based on Gaussia Luciferase reconstitution was used to identify inhibitors of the AR-JunD interaction. Selected hits were further screened using a fluorescence polarization competitor assay to eliminate those that bind to the AR Ligand Binding Domain (LBD), in order to identify molecules that specifically target events downstream to androgen activation of AR. Eleven molecules were selected for studies on their efficacy against ROS generation and growth of cultured human PCa cells by DCFH dye-oxidation assay and DNA fluorescence assay, respectively. In situ Proximity Ligation Assay (PLA), SSAT promoter-luciferase reporter assay, and western blotting of apoptosis and cell cycle markers were used to study mechanism of action of the lead compound.
RESULTS
Selected lead compound GWARJD10 with EC50 10 μM against ROS production was shown to block AR-JunD interaction in situ as well as block androgen-induced SSAT gene expression at IC50 5 μM. This compound had no effect on apoptosis markers, but reduced cyclin D1 protein level.
CONCLUSIONS
Inhibitor of AR-JunD interaction, GWARJD10 shows promise for prevention of progression of PCa at an early stage of the disease by blocking growth and ROS production.
Keywords: AR-JunD protein-protein interaction, small molecule inhibitors, cellular reactive oxygen species, cyclin D1
INTRODUCTION
Advanced hormone refractory metastatic prostate cancer (PCa) is the second leading cause of cancer deaths of US men [1]. PCa recurs in approximately 30% of patients after their first-line of therapy of either radical prostatectomy or ionizing radiation [2]. Although Androgen-Deprivation Therapy (ADT) initially causes the regression of the early stage recurrent PCa, unfortunately over 80% of the patients fail ADT and progress to androgen-independent castration-resistant tumor (CRPCa) [2]. Therefore, identification of an effective agent to prevent PCa progression at an early stage of recurrence is a major focus for PCa research.
Cellular Reactive Oxygen Species (ROS), such as hydroxyl radical, superoxide, hydrogen peroxide, and nitric oxide, are naturally occurring carcinogens in prostate epithelial cells [3]. When cellular ROS production overwhelms detoxification capacity of cells, oxidative stress occurs [4]. It has been established that oxidative stress causes DNA, RNA, and phospholipid damages [5]. ROS also act as a signal for promoting cell proliferation that can contribute to cancer development and progression, including prostate cancer [3,6,7]. Data published from our and other laboratories have shown that androgen signaling is a major source of ROS generation in prostatic epithelial cells [8–11]. We have further shown that one pathway of androgen-induced oxidative stress generation involves activation of AP-1 transcription factor JunD [12,13] followed by androgen receptor (AR)-JunD association [14] that induces an enzyme Spermidine/Spermine N1-Acetyl Transferase (SSAT). Induction of SSAT initiates a major polyamine oxidation pathway that generates excess hydrogen peroxide (H2O2) production in polyamine-rich prostatic epithelial cells [15]. We established that JunD is required for androgen-induced SSAT gene expression and ROS generation [14]. We hypothesize that specific inhibitors of AR-JunD interaction that act downstream of AR activation will specifically inhibit androgen induced SSAT gene expression and consequently prevent the excess ROS production, and should therefore prevent the initiation and progression of PCa.
In our previously published study we used a Gaussia luciferase enzyme reconstitution assay to demonstrate a direct interaction of activated AR with JunD [14]. Here, we performed a high throughput screen of NCI Diversity Set [16] and Life Chemicals [17] small molecule libraries to identify potential candidates that may inhibit this interaction without anti-androgenic activity due to binding to the AR-ligand binding domain (LBD). Selected compounds have been further characterized for ROS and cell growth inhibitory effects. One of the lead compounds, GWARJD10, significantly reduced androgen-induced ROS production in LNCaP cells, as well as proliferation of androgen-dependent LNCaP and castrate-resistant C4-2 cells. GWARJD10 was studied further to confirm its proposed mechanism of action in blocking PCa progression.
Studies on the mechanism of action of GWARJD10 showed that this compound significantly reduces the interaction between AR and JunD and also reduces the transcriptional activity of SSAT promoter in LNCaP human PCa cells cultured in the presence of androgen. Further studies showed that this compound significantly reduces cyclin D1 expression in the presence of androgen in both LNCaP and C4-2 cells. No significant effect on apoptosis markers (e.g., cleaved PARP) at this concentration of the compound was observed in either of the two cell lines.
MATERIALS AND METHODS
Cell Culture
Androgen-dependent LNCaP human prostate carcinoma cells were obtained from the American Type Culture Collection. Castrate-resistant LNCaP C4-2 cells [18] were a kind gift from Ajit Verma (Department of Human Oncology, UW-Madison), with permission from George Thalmann (Department of Urology, Inselspital, Bern, Switzerland). LNCaP cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and grown in the same medium (F5 medium) for androgen-dependent growth studies. For studies on androgen-induced ROS, LNCaP cells were grown in DMEM supplemented with 1% FBS and 4% charcoal stripped serum (F1C4 medium) as described before [8]. This combination of stripped and non-stripped serum was previously shown to sufficiently deplete androgen content while limiting the adverse growth effects not related to hormone depletion that occur with the use of 5% stripped serum [8]. LNCaP C4-2 cells were maintained and grown for androgen-independent growth studies in the F1C4 medium.
Hep3B human hepatoma cells with no endogenous AR were obtained from the Small Molecule Screening and Synthesis Facility at the University of Wisconsin Carbone Cancer Center (UWCCC SMSSF) and maintained in RPMI 1640 supplemented with 10% FBS and antibiotics, as explained previously [14]. Synthetic androgen R1881 (methyltrienolone, NEN) was used as an androgen analog in cell culture studies at 1–2 nmol/l for maximal induction of JunD and ROS as described before [12]. For Hep3B transfected cells, 2 nmol/l R1881 was used to induce the activity of AR after transfection.
For direct cDNA synthesis from cells for qrtPCR, 1.7 × 105 LNCaP cells were cultured in each well of a 12 well plate in F1C4 medium, treated with or without 2 nM R1881 and 5 μM GWARJD10, and incubated for 24, 48, and 72 hr. After incubation, cells from each time point were lysed by a lysis solution provided in a kit by Life Technologies™ following the manufacturer instructions (see below).
Antibodies Used for Western Blot and In Situ Proximity Ligation Assay (PLA)
Mouse monoclonal antibody for cyclin D1 (Santa Cruz, sc-8396), mouse monoclonal antibody for cleaved PARP (Cell Signaling #9546), mouse monoclonal antibody for Androgen Receptor (Santa Cruz, sc-7305), and rabbit polyclonal antibody for JunD (Santa Cruz, sc-74) were used. β-actin mouse monoclonal antibody (AC-15) for loading and normalization control was obtained from Sigma-Aldrich.
Transfection of Constructs cmv-Gluc1-AR and cmv-JunD-Gluc2 Into Hep3B Cells and Generation of Large Scale Cell Lysate for High Throughput Screening
Transfection of cells was done using Lipofectamine 2000 reagent (Invitrogen) per manufacturer’s instructions as explained previously [14]. Briefly, Hep3B cells were seeded and 1 day later co-transfected with 3 μg each of cmv-Gluc1-AR and cmv-JunD-Gluc2 constructs. Transfections with cmv-Gluc1-AR or cmv-JunD-Gluc2 alone were used as negative controls, and cmv-Gluc1-smad3 co-transfected with cmv-PKB-Gluc2 [19] was used as a positive control. Two to 3 hr after transfection, cells were washed and refed with DMEM without serum and either treated with 2 nmol/l R1881 (+R) or left untreated (−R). The cells were incubated after transfection at 37°C under 5% CO2 for 48 hr, then lysed using a lysis buffer provided in a Gaussia luciferase assay kit from New England Biolabs (Ipswich, MA) following the manufacturer’s protocol.
Initial High Throughput Screening (HTS) of NCI Diversity Set Library Containing ~2000 Small Molecules and Life Chemicals Library Containing 25,000 Drug-Like Small Molecules
Five microliter of cell lysate per well was plated in 384 well plates using MicroFlo Select (BioTek Instruments, Inc., Winooski, VT). Two columns in each plate were used for negative controls (for DMSO and for lysates from cells that were transfected with both constructs but were not treated with R1881). One hundred nanoliters of 1 mmol/l of each potential inhibitor (final concentration of 20 μmol/l for each compound) and control DMSO from the NCI Diversity Set and Life Chemicals Library were added to each well using a robotic pin-based liquid handler (Biomek FX, Beckman Coulter, Inc.) following a procedure published by the UWCCC SMSSF [20]. The plates were sealed by MicroSeal B Film (BioRad) and incubated overnight at 37°C in dark. Ten microliter of Renilla Luciferase substrate (Promega) was added to each well, and the bioluminescence activity of Gaussia luciferase in lysates was measured using a Synergy 4 plate scanner (BioTek, Winooski, VT). Compounds that blocked greater than 30% of the luciferase activity were considered as “hits” in this and all subsequent screening assays. Z′ factor values of above 0.5 are generally accepted as significant for identification of “hits” in a HTS assay [21]. For all screens, our Z′-factor was above 0.56.
Secondary Screen of the Identified Hits of the Initial HTS
For eliminating false positives, including non-specific inhibitors and toxins, 5 μl lysate/well from cells that were co-transfected with vectors containing cmv-Gluc1/cmv-Gluc2 (negative control) or with cmv-smad3-Gluc1/cmv-Gluc2-PKB (positive control) [19] were plated in a 384 well plate and 0.1 μl of each identified inhibitor from 1 mmol/l stock (final concentration of 20 μmol/l) was added to each well. Plates were sealed as before and incubated for an overnight at 37°C in dark, then the bioluminescence activity was measured as described earlier. Only the “hits” that inhibit Gaussia Luciferase reconstitution in an AR-JunD system, but fail to inhibit GL-reconstitution in the secondary screen with the positive control were considered as “true” inhibitors of the AR-JunD complex (Fig. 1, “Confirmed Hits“).
Fig. 1.

Schematic presentation of high throughput screen and selection of small molecule inhibitors of AR-JunD interaction. NCI Diversity Set library of ~2,000 small molecules and a Life Chemical Library of 25,000 small molecules were assayed in a high throughput screen (HTS) based on a Gaussia Luciferase (GL) reconstitution due to AR and JunD interaction (see Methods). Z′ factor for all screens was >0.56. Eleven hundred small molecules that showed >30% inhibition of GL in the first screen were further screened with cmv-smad3-Gluc1/cmv-Gluc2-PKB control to eliminate false positives. Twenty-two confirmed hits were considered as compounds of interest for further testing and screened for their ability to compete with androgen for binding to androgen receptor ligand-binding domain (AR-LBD; see Fig. 2), to eliminate antiandrogens since our focus is on inhibiting AR post androgen activation. Eleven of the non-antiandrogens were selected for further invitro testing based on their structure and/or greater than 50% block of luciferase activity in the first HTS.
Fluorescence Polarization AR Ligand Binding Competition Assay
Since inhibition of AR activity and its interaction with JunD might have been due to the competition of these inhibitors with androgen for binding to the Ligand Binding Domain of AR (AR-LBD) and thus blocking androgen activation of AR, we used a commercially available assay to determine the anti-androgenic AR-LBD binding activity of these inhibitors. Selected hits from the dual HTS of the NCI Diversity Set and Life Chemical libraries were tested by PolarScreen™ Androgen Receptor Competitor Assay kit (Invitrogen) to determine their abilities for binding to the AR-LBD. Fluorescence polarization assay with graded concentrations of the compounds of interest was performed according to the manufacturer’s supplied protocol, in comparison to clinical antiandrogen Casodex® (bicalutamide).
Growth and ROS Assays
Cells collected for experiments were counted and seeded in 96-well tissue culture plates at a density of 4,000 cells per well in 200 μl medium described above 1 day prior to treatment with compounds in varying doses. Androgen treatments for ROS studies were done on the same day. Treatments were carried out over 96 hr at 37°C under 5% CO2. Measurements of ROS were used as an indicator of redox status and DNA levels were measured as an indicator of growth. The 96-well culture plates were assayed for estimation of ROS levels in intact cells using the fluorescent dye 2′, 7′-dichlorofluorescein di-acetate (DCF; Molecular Probes, Inc.) following a published procedure [22]. In brief, cell cultures were washed with 200 μl Kreb’s Ringer buffer prewarmed to 37°C, incubated under 5% CO2 at 37°C in 100 μl Kreb’s Ringer buffer containing 10 μg/ml DCF dye for 45 min. Each 96-well culture plate was scanned on a Synergy 4 plate scanner (Biotek) using the 485/530 nm filter excitation and emission set and then frozen at −70°C for the subsequent analysis of DNA content. For DNA analysis, each culture plate frozen to −70°C was equilibrated to room temperature protected from light. Hoechst dye was then added to each well at a final concentration of 6.7 μg/ml in 200 μl of high salt TNE buffer (10 mmol/l Tris, 1 mmol/L EDTA, 2 mol/L NaCl [pH 7.4]). After further incubation at room temperature for over 2 hr under protection from light, culture plates were scanned on a Synergy 4 plate scanner using the 360/460 nm filter excitation and emission set. The DCF fluorescence units were normalized to the Hoechst-DNA fluorescence units for each well and used as a measure of the level of ROS being generated. The DNA fluorescence units were also used as a measure of cell growth.
Quantitative In Situ Proximity Ligation Assay (PLA)
For quantifying the level of inhibition of AR-JunD complex formation by the lead compound GWARJD10, we performed a quantitative in situ Proximity Ligation Assay (PLA) [23] in intact cells using a kit from OLINK Bioscience (Uppsala, Sweden). LNCaP cells (2 × 104 cells/well) were seeded in eight-well polylysine coated chamber slides in F1C4 media [8] and treated with 2 nmol/l R1881 (positive control) or without R1881 (negative control) in control channels, or treated with or without 2 nmol/l R881 with 5 μmol/l of GWARJD10 inhibitor. In brief, after 72 hr incubation at 37°C under 5% CO2, cells were washed and then fixed in 4% Paraformaldehyde in PBS for 10 min at room temperature. Cells were then permeabilized with 0.5% Triton-X100 in PBS for 10 min at room temperature. Subsequently cells were washed with 0.05% Tween 20 in PBS (PBS-T). The instructions provided by manufacturer were followed for PLA assay. Briefly, after the cells were permeabilized and washed with PBS-T, one drop of Duolink II Blocking Solution (1×) was added to each well after complete removal of wash buffer, slides were incubated for 1 hr at 37°C in a 5% CO2 incubator. After 1 hr, the blocking solution was completely removed and 1/100 dilution of rabbit polyclonal antibody for JunD and 1/100 dilution of mouse monoclonal antibody for AR were added to each well and in one well only antibody diluent was added as a negative control. Slides were incubated for 1 hr at 37°C under 5% CO2 and then washed with supplied wash buffer. Forty microliter of two PLA probes (Duolink II anti-Mouse MINUS and Duolink II anti-Rabbit PLUS) at 1:5 in antibody diluent was added to each well. After 1 hr incubation at 37°C under 5% CO2, slides were washed and 40 μl Duolink II Ligation stock (×5) was added to each well and incubated for 30 min at 37°C under 5% CO2. After incubation, slides were washed and 40 μl of diluted Duolink II Amplification Mix was added to each well and incubated for 100 min at 37°C under 5% CO2. Finally, slides were washed and mounted using one drop of solution containing DAPI. A Nikon TI-U inverted fluorescence microscope with excitation/emission 594/624 filters was used to acquire images of PLA-processed slides at ×20 magnification. The red fluorescent spots were counted and normalized against nuclei blue DAPI for a quantitative measure of AR-JunD interaction.
SSAT Full Length Promoter-Luciferase (ssatP-luc) Reporter Assay
LNCaP (1 × 105) cells were seeded in each well of 12-well polylysine coated plates. The day after seeding, cells were co-transfected with 2 μg of the previously published [14] ssatP-luc vector and 0.1 μg of cmv-betagalactosidase (internal control for transfection) (Promega) using Lipofectamine 2000 (Invitrogen) and following the manufacturer’s instructions. After transfection, cells were treated with or without 2 nmol/LR1881 (±R) with 1 or 5 μmol/l of GWARJD10 or zero dose vehicle control.
Luciferase assay was performed using a kit from Promega as instructed by the company and described previously [14], for quantitation of androgen-induced SSAT promoter activity.
PSAc DNA Synthesis and Quantitative Real Time PCR (qrtpcr)
Cells-to-cDNA™ II, reverse transcription without RNA isolation kit from Ambion® by Life Technologies™ (AM 1723) was used to synthesize cDNA directly from cells following the instructions provided by the manufacturer. Briefly, 100 μl of lysis solution was added to cells in each well of a 12 well plate, and the lysed cells were incubated at 75°C for 10 min. The genomic DNA was lysed by adding 2 μl of DNaseI per 100 μl lysed cells and incubated for 15 min at 37°C. The DNaseI was inactivated by heating the solution to 75°C and the reverse transcription of released RNA was achieved by incubating the lyses at 42°C for 1 hr using reverse transcription mix provided in the kit. The reverse transcription was stopped by heating the samples to 95°C for 10 min. The cDNAs were subjected to real time PCR using IQ™ Syber® Green supermix (Bio-Rad). Each reaction was normalized by coamplification of 18srRNA. Triplicates of samples were run on a Bio-Rad CFX-96 real-time cycler. The sequences of primers used for amplification of PSA and 18srRNA cDNAs were as follows:
PSA: forward: 5′-GACCACCTGCTACGCCTCA and reverse: 5′-GGAGGTCCACACTGAAGTTTC
18srRNA: forward 5′-CGCCGCTAGAGGTGAAATCT and reverse 5′-CGAACCTCCGACTTTCGTT.
Western Blot
Cell lysates were prepared using modified Radio-immunoprecipitation (RIPA) buffer (50 mmol/l Tris-HCl, PH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mmol/l NaCl and 1 mmol/l EDTA) containing a tablet of complete protease inhibitors from Roche (Indianapolis, IN). Total protein from each sample was separated on a 4–12% Bis-Tris gel NuPAGE Novex from Life Technologies. The gel was transferred onto a PVDF membrane, blocked and then incubated with antibody as explained before [14]. The membrane was developed with Enhanced Chemiluminescent (ECL) substrate from Thermo Scientific after incubation with the appropriate HRP-conjugated secondary antibodies.
Statistical Analyses
An unpaired two-tailed heteroscedastic Student’s t-test with a confidence level of 0.05 was performed for data comparisons and significance determination.
RESULTS
Identification of Specific Inhibitors of AR-JunD Interaction
For identification of specific inhibitors of the AR-JunD interaction, a NCI Diversity Set library containing 2,000 small molecules and Life Chemicals Library containing 25,000 drug-like small molecules were screened using Gaussia luciferase reconstitution assay as explained in the Materials and Methods (see Methods). The screening scheme is shown (Fig. 1). The assay used conditions where both AR and JunD proteins were overexpressed in a non-prostate cancer cell line (Hep3B cells) without AR background. This cell line was selected to avoid the competition from binding of the endogenous AR. Eleven hundred molecules were identified as inhibitors in this first screen, based on greater than 30% block of luciferase activity. The second screen was performed using the cell lysates from the positive smad3/PKB control [19] (see Methods). Molecules that inhibited the interaction of smad3/PKB were considered to be non-specific inhibitors and were discarded. Only the “confirmed hits” from the second screen were considered as specific inhibitors of AR-JunD interaction. Twenty-two such molecules were considered as compounds of interest and selected for further testing and analysis.
Lack of AR-LBD Binding Activity of Specific Inhibitors of AR-JunD Interaction
Using the PolarScreen™ Androgen Receptor Competitor Assay kit from Invitrogen, all 22 selected confirmed specific inhibitors of AR-JunD interaction were tested for their ability to bind to the AR Ligand Binding Domain (AR-LBD). Data for select compounds are shown (Fig. 2). Antiandrogens, such as the positive control bicalutamide and compound GWARJD22 shown (Fig. 2), exhibited a dose dependent decrease in polarization value. As our goal is to specifically target and block steps downstream to AR activation by androgen, the five compounds found to be antiandrogens by this assay were set aside and not studied further at this time. Seventeen compounds that did not show a decrease in polarization value, which indicates their inability to compete with androgen for binding to AR-LBD, were considered for further studies.
Fig. 2.

Screening of compounds for anti-androgenic properties. The PolarScreen™ Androgen Receptor Competitor Assay was used to determine if the confirmed hits have anti-androgenic properties. Bicalutamide (Casodex®), a clinical antiandrogen, was used as a control. A classic dose response of bicalutamide competition with fluorescence tagged androgen (fluoromone) for binding to the ligand binding domain (LBD) of AR was observed. In contrast, the GWARJD inhibitors numbers 7, 10, and 14, which are the focus of this paper, did not compete with androgen for binding to LBD of AR in the same dose range. All 22 selected confirmed hits were tested in this manner, and based on this assay 17 were classified as non-anti-androgens while 5 were classified as antiandrogens, as represented by GWARJD22 showing a curve similar to bicalutamide. N = 3 per data point, nonlinear regression fit curves, representative from two assays performed for each compound.
AR-JunD Inhibitors Prevent Cell Growth in Both LNCaP and Its Castrate-Resistant Variant C4-2 Cells in Culture
Of the 17 specific inhibitors of AR-JunD interaction selected from the dual HTS and AR-LBD assay data described above, 11 compounds were selected for further studies based on their structures that allow for ease in chemical synthesis and/or greater than 50% block of luciferase activity in the first HTS. These 11 compounds were tested in cell culture for efficacy against the androgen-induced ROS production in which AR-JunD interaction plays a key role [14], as well as efficacy against androgen-dependent and -independent PCa cell growth.
Data obtained from the ROS and cell growth studies for the three most effective compounds, GWARJD07 (NSC693573), GWARJD10 (F1174-3266), and GWARJD14 (F3382-1718), are shown (Fig. 3), with their structures shown (Fig. 3a). Results from GWARJD07 (CPC507) showed that at 0.8 μmol/l, it causes a marked inhibition of androgen-induced ROS production in androgen-dependent LNCaP cells (Fig. 3b) and inhibits growth by 50% at less than 500 nmol/l (IC50 < 500 nmol/l; Fig. 3c). In castrate-resistant C4-2 cells, GWARJD07 showed a marked effect at 30 nmol/l on cell growth and its IC50 is less than 100 nmol/l (Fig. 3c). This compound, however, has an imidazole ring and thus, has anti-oxidant properties. As chemical anti-oxidants such as alpha-tocopherol failed in PCa therapy/prevention [24], we focused on GWARJD10 and GWARJD14 that have no chemical anti-oxidant properties. GWARJD10 also showed inhibitory effects on both ROS production and PCa cell growth, with inhibition of androgen-induced ROS production by 50% at less than 10 μmol/l in LNCaP cells (EC50 < 10 μmol/l; Fig. 3b) and growth inhibition in both LNCaP and C4-2 cells by 50% at less than 5 μmol/l (IC50 < 5 μmol/l; Fig. 3c and d), respectively. GWARJD14 was found to inhibit androgen-induced ROS production by 50% at approximately 25 μM (EC50 ~25 μmol/l; Fig. 3b), and inhibit both androgen-dependent LNCaP and castrate-resistant C4-2 cell growth by 50% at less than 10 μmol/l (IC50 < 10 μmol/l; Fig. 3c and d), respectively. A cell viability assay showed no cytotoxicity at the growth inhibitory IC50 concentrations for each of these compounds, and the concentrations at which 50% cytotoxicity was observed for each was well above their IC50 concentration (data not shown), thus the observed growth inhibition was likely not due to cytotoxic properties of the compounds. We selected the more effective compound GWARJD10 for confirming its proposed mechanism of action and thereby, establishing the proof-of-principle.
Fig. 3.

ROS and growth analysis: compounds (structures shown in a) were tested for their ability to block androgen-induced ROS production (b) and inhibit androgen-dependent (c) and androgen-independent (d) prostate cancer cell growth. (b) Dose response effect of compounds on ROS in LNCaP treated with 1 nmol/l R1881 androgen that induces AR-JunD interaction and subsequent ROS over-production. (c) Dose responses effect on growth of androgen-dependent LNCaP cells in growth stimulatory androgen complete medium (F5). (d) Dose response effect on growth of castrate-resistant variant LNCaP C4-2 cells in androgen-deprived medium (F1C4). All treatments were carried out over 96 hr. Data points are the average from three experiments with N = 6 per condition in each experiment (#s 10 and 14) or N = 6 in one experiment (#7). n.e., not evaluable due to extremely low DNA values.
Inhibition of Interaction of AR-JunD by GWARJD10 in Androgen-Dependent LNCaP Cells
Results from the quantitative in situ Proximity Ligation Assay (PLA; see Methods) demonstrate the interaction of AR-JunD in R1881 treated LNCaP cells (Fig. 4a). In androgen-dependent LNCaP cells, R1881 activates Androgen Receptor (AR) and JunD that initiates their interaction and translocation to the nucleus (Fig. 4a), where they may bind to the promoter of genes that are involved in growth and ROS production. In LNCaP cells that were treated with 2 nmol/l R1881 and 1 or 5 μmol/l GWARJD10, AR and JunD interaction was significantly abrogated (P <0.003; Fig. 4b and c).
Fig. 4.

Inhibition of androgen-induced AR-JunD interaction by GWARJD10 in LNCaP cells. The DuoLink Proximity Ligation Assay (PLA) technique was used to analyze the effect of lead compound GWARJD10 on androgen-induced AR-JunD interaction. Androgen-dependent LNCaP cells were treated for 72 hr either with 2 nmol/l androgen R1881, positive control (a) or with 2 nmol/l R1881 and 5 μmol/l of GWARJD10 (b). In (a), androgen-induced interactions of AR with JunD are represented by red spots in the cytoplasm and by pink spots in the nuclei which are the merge of red spots with blue DAPI. With the addition of GWARJD10 (b), the interactions of the two proteins were inhibited and few AR-JunD interaction spots were observed (magnification 20×). In (c), LNCaP cells treated with (+) or without (−) 2 nmol/l androgen R1881 (R) and either 0.25% DMSO drug vehicle control (Veh.C.) or 1 or 5 μmol/l GWARJD10 for 72 hr were analyzed by PLA for AR-JunD interaction. Fluorescent spots resulting from AR-JunD interaction (red) were counted and normalized to the number of cells (blue, DAPI) per field using an inverted fluorescent microscope. The three fold induction of AR-JunD interaction by androgen (P <0.001) was significantly reduced by both 1 and 5 μmol/l GWARJD10 (P <0.003). The level of AR-JunD interaction in +R, 5 μmol/L GWARJD10 was not significantly different from androgen-deprived control −R, Veh. C.N = 12 fields counted percondition across two experiments(6 fields per experiment).
GWARJD10 Significantly Reduces Androgen-Induced Transcriptional Activity of ssat Promoter
Previously, we showed that JunD binds to the SSAT promoter [14] and also showed that the expression of SSAT enzyme is ~30-fold higher in LNCaP cells treated with R1881 compared to the control [15]. Because there is no AR Response Element (ARE) in the SSAT Promoter sequence, an increase in the SSAT gene expression is likely due to the complex formation between AR and JunD, where JunD binds to the AP1 consensus sequence in ssat Promoter [14]. Here, we show in (Fig. 5) that 5 μmol/l GWARJD10 causes a significant threefold reduction in the androgen-induced ssat Promoter activity (P = 0.003).
Fig. 5.

Effect of GWARJD10 on androgen receptor activity. (a) Inhibition of androgen-induced SSAT promoter transcriptional activity by GWARJD10 in LNCaP cells: LNCaP cells were transiently transfected with an SSAT promoter-luciferase reporter vector and treated with 2 nmol/L androgen R1881 or vehicle control with or without 1 or 5μmol/lGWARJD10. Cell lysates were collected after 72hr, the relative light units (RLU) of firefly luciferase activity was measured by a luminometer at 480nm, and the ratio of RLU for androgen treated (+R1881) versus vehicle control (−R1881) cells was determined. Similarly, the ratios of RLU for the LNCaP cells treated with androgen (+R1881) versus vehicle control (−R1881) with 1 and 5μmol/l GWARJD10 were determined. SSAT transcriptional activity was significantly reduced with 5μmol/lof GWARJD10 in the androgen treated cells. N≥3 percondition. *P = 0.003 compared to zero dose control. (b) Inhibition of induction of PSA: LNCaP cells in low androgen F1C4 medium were treated with drug GWARJD10 at 5μmol/l (D) and/or androgen R1881 at 2 nmol/l (R). The conditions included: vehicle control (−R, −D), 5μmol/l of GWARJD10 drug (−R, +D), androgen-stimulated (+R, −D), and androgen plus 5μmol/l GWARJD10 (+R, +D). GWARJD10 significantly reduced PSA mRNA under the androgen-deprived condition (P <0.01 for −R, −D compared to −R, +D at all timepoints). As expected, 2nM androgen stimulation led to a significant increase in PSA mRNA (P <0.001 for +R, −D compared to −R, −D at all timepoints). However, GWARJD10 did not affect PSA mRNA levels under the 2nM androgen-stimulated condition, as no difference was observed at any timepoint for +R, −D compared to +R,+D. N= 6 percondition.
GWARJD10 Significantly Reduces AR Transcriptional Activity as Measured by PSA Expression in LNCaP Cells Under Low Androgen (F1C4) Condition But Does Not Affect Androgen Induction of AR Transcriptional Activity
As shown in Figure 5b, GWARJD10 significantly reduced the transcriptional activity of AR, as measured by PSA mRNA expression, in low androgen (−R) environment (P <0.01 for all timepoints for drug treated cells compared to control in −R condition). Stimulation with 2 nM androgen R1881 (+R) significantly increased AR transcriptional activity with respect to PSA expression as expected (P <0.001 for all timepoints for androgen treated cells compared to low androgen without drug), but GWARJD10 had no effect on this induction of AR transcriptional activity by androgen, as levels of PSA mRNA were the same in cells treated with androgen without or with drug (+R, −D compared to +R, +D).
GWARJD10 Reduces the Expression of Cyclin D1 in the Presence of 2 nmol/l R1881 But Does Not Reduce the Expression of cPARP in the Presence or Absence of Androgen
LNCaP and C4-2 cells were treated with vehicle control or with 2 nmol/l R1881 with or without 1 or 5 μmol/l GWARJD10. We observed a significant decrease in cyclin D1 expression in 2 nmol/l R1881 and 5 μmol/l GWARJD10-treated LNCaP and C4-2 cells after 72 hr compared to controls. Representative western blots are shown in Figure 6, panels c and d, respectively. Quantitation of the bands showed that cyclin D1 level was significantly reduced on average across independent experiments to 65 ± 6% in LNCaP cells (P = 0.001, N = 3) and 71 ± 13% in C4-2 cells (P = 0.05, N = 4) as compared to control. We, however, did not observe any significant change in cleaved PARP after treatment with 5 μmol/l of GWARJD10 when compared to the controls (Fig. 6, panels a–d)
Fig. 6.
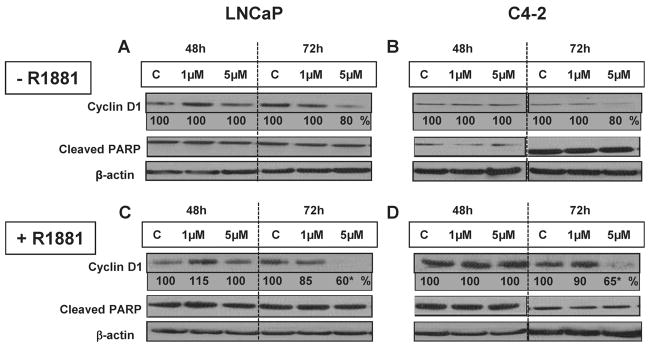
Effect of GWARJD10 on apoptosis marker (Cleaved PARP) and cell cycle marker (Cyclin D1) in androgen-dependent LNCaP cells and its castrate-resistant variant C4-2 cells. LNCaP or C4-2 cells were treated with (+) or without (−) 2 nmol/l androgen R1881 and zero (C), 1 or 5 μmol/l of lead compound GWARJD10 for 48 and 72 hr. Cells were then harvested and protein levels were determined by Western blot analysis. β-actin was used as loading control and normalization. Percentage of each protein band compared to C (%) at each timepoint was calculated after normalizing each band against β-actin. Representative blots are shown. The level of Cleaved PARP (89 kDa) did not change under treatment conditions. Cyclin D1 (37 kDa) level was markedly reduced in R1881 treated LNCaP and C4-2 cells after 72 hr. The western blots were run three times for cell lysates from three to four different experiments per condition with similar results. Statistical analysis of bands across the experiments showed significantly lower (*) average cyclin D1 of 65% in LNCaP cells (P = 0.001, N = 3) and 71% in C4-2 cells (P = 0.05, N = 4) compared to control.
DISCUSSION
In the present study, we searched for small molecule inhibitors in two chemical libraries, a NCI Diversity Set [16] and a Life Chemicals Library [17] containing approximately 2,000 and 25,000 molecules, respectively, that can specifically block the interaction of activated AR with JunD. Twenty-two such inhibitors were selected from the screen and analyzed for their AR-LBD binding property. Eleven compounds that do not compete with androgen for binding to AR-LBD and have structures that are amenable to chemical synthesis were studied further in ROS and growth assays in prostate cancer cells. Three lead compounds, GWARJD07, GWARJD10, and GWARJD14, were markedly effective in blocking androgen-induced cellular ROS, as well as inhibiting growth of androgen-dependent LNCaP and its castrate-resistant variant C4-2 cells (Fig. 3). GWARJD10 was selected as the lead compound, based on its structure, lack of chemical anti-oxidant properties, and effect against ROS and growth as explained in the Results section, for further studies of the mechanism of action.
The rationale for seeking the agents that do not inhibit AR ligand-binding was based on the fact that current antiandrogens used in the clinic that generally target AR LBD, that is, compete with androgen for binding to AR-LBD, are only partially effective. Furthermore, there are reports that suggest an agonist potential of a major clinical anti-androgen that targets the AR-LBD [25–28], and/or the development of resistance of PCa to a more recent anti-androgen such as Enzalutamide through induction of mutations in the AR-LBD or activation of alternative pathways that leads to castrate-resistant PCa [29,30]. Recent evidence also shows the importance of inhibiting AR activation by a small molecule that binds to its N-terminus Domain (NTD) instead of the LBD at the C-terminus zone [31]. These inhibitors may block AR activation by inhibiting its interaction with other co-activators [31,32]. Mutational analysis have revealed that NTD is involved in transcriptional activity of AR and mutations in this region do not affect androgen-binding but abrogate AR transcriptional activity [33], therefore targeting AR NTD by small molecules may inhibit AR activity without affecting androgen-binding to AR [31].
Although we did not map the binding sites of these inhibitors in AR-JunD complex structure, the results from the AR ligand-binding competition assay showed that none of the selected inhibitors of AR-JunD interaction can effectively compete with androgen-binding to the AR-LBD domain. Furthermore, the results from quantitative analysis of PSA expression, where lead compound GWARJD10 had no effect on androgen activation of AR to induce PSA, yet did reduce AR transcriptional activity under androgen deprived conditions, indicate that the inhibition of AR transcriptional activity by this compound is likely through its binding to other sites of AR, for example, NTD or DBD, and not to the LBD. The reduction in PSA expression as well as the lower transcriptional activity of SSAT promoter by this compound are not due to lower expression or degradation of AR or JunD proteins, since we did not observe any changes in AR or JunD protein levels or their stability in western blots under the conditions that the studies were performed (data not shown). Therefore, we concluded that these inhibitors bind to other domains in AR and likely at the JunD-AR interface. We are currently investigating the binding location of GWARJD10 in AR-JunD complex using structural biology approaches.
After identification of GWARJD10 as the lead compound, we used the IC50 concentration (5 μmol/l) of this compound to study its effect on the disruption of AR-JunD interaction in LNCaP cells using a Proximity Ligation Assay (PLA). The results from the PLA studies demonstrate a significant disruption of AR and JunD interaction in androgen treated LNCaP cells by GWARJD10 (Fig. 4c). We have shown before that JunD is activated and translocated to the nucleus in androgen treated LNCaP cells [14]. Concerted activation and translocation of both transcription factors to the nucleus may lead to the activation of genes regulated by both AR and JunD, or the AR-JunD complex may transcribe a new set of genes that is not activated by either of them alone. For example, we have shown before that activated AR with JunD is involved in the induction of the ssat gene expression, yielding over-expression of SSAT enzyme that causes significant generation of ROS in the presence of androgen in LNCaP cells [14,15]. The ability of 5 μmol/l GWARJD10 to block the androgen-induced ssat promoter transcriptional activity (Fig. 5) provides further evidence of the ability of this compound to inhibit AR-JunD interaction. Thus, blocking the interaction between the AR and JunD may down regulate the expression of genes that are involved in growth and ROS generation in PCa cells.
Studies on the mechanism of GWARJD10 in growth inhibition of LNCaP and C4-2 cells show that this compound at 5 μmol/l is not cytotoxic and probably does not affect apoptosis since the level of cleaved PARP, a marker of apoptosis, did not change under different conditions (Fig. 6; panels a–d). We also looked at other apoptosis markers such as Caspases 3 and 7, and likewise did not observe changes in the level of expression of these markers (data not shown). Since cell kill or apoptosis did not appear to be a factor in growth inhibitory effects of GWARJD10 under our conditions, we looked at a cell cycle marker cyclin D1. Cyclin D1 is a cell cycle regulator protein. Its over-expression has been shown to be associated with prostate cancer cell metastasis to the bone [34]. Additional support for potential role of cyclin D1 in androgen-independent PCa was derived from both in vitro and in vivo data [35]. Our data indicate that the growth inhibitory effect of this compound is at least in part due to its effect on cyclin D1 and related cell cycle regulatory gene expression. GWARJD10 at IC50 5 μmol/l caused approximately 20% decrease in cyclin D1 in androgen-dependent LNCaP and castrate-resistant C4-2 cells (Fig. 6a, b). Interestingly, the effect of GWARJD10 on cyclin D1 was more pronounced under high ROS conditions. There are reports that explored the role of ROS on cyclin D1 in different conditions [36,37]. These reports showed that ROS degrade cyclin D1 and thereby prevent cell proliferation. We have observed that at 2 nmol/l R1881, which is the growth inhibitory concentration of R1881 and maximum ROS production [8], the expression of cyclin D1 is dramatically reduced (compare Fig. 6a with Fig. 6c in control lanes). GWARJD10 caused an additional 40% decrease in cyclin D1 under these conditions (Fig. 6c, d), and also blocked ROS production by approximately 40% (Fig. 3b) at 5 μmol/l. The results from this study suggest that GWARJD10 at 5 μmol/l inhibits cell growth by reducing cyclin D1 expression in prostate cancer cells and also reduces ROS production in these cells by blocking AR-JunD interaction and thus, preventing ssat gene expression. Therefore, we believe this class of compounds will inhibit PCa growth, and could prevent progression to CRPCa that may be due to accumulation of ROS through this pathway. Further studies are needed to address how GWARJD10 concurrently causes the reduction in cyclin D1 and ROS.
In vivo studies with GWARJD10 are currently underway to elucidate the effectiveness of this compound in inhibition of growth and progression of prostate cancer.
Acknowledgments
Grant sponsor: Department of Defense; Grant number: W81XWH-10-1-0169; Grant sponsor: National Institutes of Health grant; Grant number: P30 CA014520 (UWCCC CCSG); Grant sponsor: Prostate Cancer Foundation.
The authors thank the UWCCC Small Molecule Screening and Synthesis Facility for assistance with HTS assays and analysis, and the UWCCC Experimental Pathology Laboratory for assistance with the Nikon TI-U inverted fluorescence microscope for PLA quantitation. Additionally, the authors thank Colby Pharmaceutical Company, San Jose, CA for sharing CPC507.
Footnotes
Conflict of interest: Farideh Mehraein-Ghomi, Dawn R. Church, Hirak S. Basu, and George Wilding have potential conflict of interest.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Tindall DJ, Scardino PT. Recent advances in prostate cancer. Singapore: World Scientific Publishing Co. Pte.Ltd; 2011. [Google Scholar]
- 3.Jorgenson TC, Zhang W, Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013;73(20):6118–6123. doi: 10.1158/0008-5472.CAN-13-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newmeyer DD, Ferguson-Miller S. Mitochondira: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 5.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Ann Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 6.Shulze-Osthoff K, Bauer M, Vogt M, Wesselborg S, Baeuerle PA. Reactive oxygen species as primary and second messengers in the activation of transcription factors. In: Forman HJ, Cadenas E, editors. Oxidative stress and signal transduction. New York: Chapman and Hall; 1997. pp. 239–259. [Google Scholar]
- 7.Shiota M, Yokomizo A, Naito S. Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic Biol Med. 2011;51(7):1320–1328. doi: 10.1016/j.freeradbiomed.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Ripple MO, Henry WF, Rago RP, Wilding G. Prooxidant-antioxidant shift induced by androgen treatment of human prostate carcinoma cells. J Natl Cancer Inst. 1997;89:40–48. doi: 10.1093/jnci/89.1.40. [DOI] [PubMed] [Google Scholar]
- 9.Ripple MO, Hagopian K, Oberley TD, Schatten H, Weindruch R. Androgen-induced oxidative stress in human LNCaP prostate cancer cells is associated with multiple mitochondrial modifications. Antiox Redox Signal. 1999;1:71–81. doi: 10.1089/ars.1999.1.1-71. [DOI] [PubMed] [Google Scholar]
- 10.Seddiqui IA, Raisuddin S, Shukla Y. Protective effects of black tea extract on testosterone induced oxidative damage in prostate. Cancer Lett. 2005;227(2):125–132. doi: 10.1016/j.canlet.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 11.Miyake H, Hara I, Gleave ME, Eto H. Protection of androgen-dependent human prostate cancer cells from oxidative stress-induced DNA damage by overexpression of clusterin and its modulation by androgen. Prostate. 2004;61(4):318–323. doi: 10.1002/pros.20087. [DOI] [PubMed] [Google Scholar]
- 12.Church DR, Lee E, Thompson TA, Basu HS, Ripple MO, Ariazi EA, Wilding G. Induction of AP-1 activity by androgen activation of the androgen receptor in LNCaP human prostate carcinoma cells. Prostate. 2005;63:155–168. doi: 10.1002/pros.20172. [DOI] [PubMed] [Google Scholar]
- 13.Mehraein-Ghomi F, Lee E, Church DR, Thompson TA, Basu HS, Wilding G. JunD mediates androgen-induced oxidative stress in androgen dependent LNCaP human prostate cancer cells. Prostate. 2008;68:924–934. doi: 10.1002/pros.20737. [DOI] [PubMed] [Google Scholar]
- 14.Mehraein-Ghomi F, Basu HS, Church DR, Hoffmann FM, Wilding G. Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells. Cancer Res. 2010;70:4560–4568. doi: 10.1158/0008-5472.CAN-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basu HS, Thompson TA, Church DR, Clower CC, Mehraein-Ghomi F, Amlong CA, Martin CT, Woster PM, Lindstrom MJ, Wilding G. A small molecule polyamine oxidase inhibitor androgen-induced oxidative stress and delays prostate cancer progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2009;69(19):7689–7695. doi: 10.1158/0008-5472.CAN-08-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.NCI Diversity set is a small library, ideal for beginning a screening campaign. [accessed in 2010];It consists of a collection of 2,000 synthetic molecules selected from the full NCI screening collection to allow users to focus their cancer screening efforts on a small scale. http://dtp.nci.nih.gov/dtpstandard/ChemData.
- 17. [accessed the library in 2011];Life Chemicals diversity libraries (Life CHEM1 and Life CHEM2) consists of 25000 new diverse, drug-like compounds selected from Life Chemicals, Inc. repository each, available for HTS through the UWCCC Small Molecule Screening Facility. http://hts.wisc.edu/htslibraries.php. Life Chemicals: http://www.lifechemicals.com.
- 18.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LWK. Derivation of androgen independent human LNCaP prostatic cancer cell subline: Role of bone stromal cells. Int J Cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 19.Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3(12):977–979. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- 20.Goel SA, Guo LW, Wang B, Guo S, Roenneburg D, Ananiev GE, Hoffman FM, Kent KC. High-throughput screening identifies Idarubicin as a preferential inhibitor of smooth muscle versus endothelial cell proliferation. PLOS One. 2014;9(2):e89349. doi: 10.1371/journal.pone.00899349. also see http://hts.wisc.edu and the references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 22.Rago RP, Mitchen J, Wilding G. DNA fluorometric assay in 96-well tissue culture plates using Hoechst 33258 after cell lysis by freezing in distilled water. Anal Biochem. 1990;191:31–34. doi: 10.1016/0003-2697(90)90382-j. [DOI] [PubMed] [Google Scholar]
- 23.Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues. Methods. 2008;45(3):227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 24.Klein EA. Selenium and vitamin E. Interesting biology and dashed hope. J Natl Cancer Inst. 2009;101:283–285. doi: 10.1093/jnci/djp009. [DOI] [PubMed] [Google Scholar]
- 25.Guerrero J, Alfaro IE, Gomez F, Protter AA, Bernales S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate. 2013;73:1291–1305. doi: 10.1002/pros.22674. [DOI] [PubMed] [Google Scholar]
- 26.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63(1):149–153. [PubMed] [Google Scholar]
- 27.Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K, Klocker H. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumor progression in a new model system. Br J Cancer. 1999;81(2):242–251. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawata H, Ishikura N, Watanabe M, Nishminoto A, Tsunenari T, Aoki Y. Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate. 2010;70(7):745–754. doi: 10.1002/pros.21107. [DOI] [PubMed] [Google Scholar]
- 29.Joseph JD, Lu N, Qian J, Sensintaffar J. A clinically relevant androgen receptor mutation confers resistance to second-generation anti-androgens Enzalutamide and ARN-509. Cancer Discovery. 2013;3(9):1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 30.Nadiminty N, Tummala R, Liu C, Yang J, Lou W, Evans CP, Gao AC. NF-κB2/p52 induces resistant to Enzalutamide in prostate cancer: Role of androgen receptor and its variants. Mol Cancer Ther. 2013;12(8):1629–1637. doi: 10.1158/1535-7163.MCT-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 32.Sadar MD, Williams DE, Mawji NR, Patrick BO, Wikanta T, Chasanah E, Irianto HE, Soest RV, Andersen RJ. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org Lett. 2008;10(21):4947–4950. doi: 10.1021/ol802021w. [DOI] [PubMed] [Google Scholar]
- 33.Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5:1396–1404. doi: 10.1210/mend-5-10-1396. [DOI] [PubMed] [Google Scholar]
- 34.Drobnjak M, Osman I, Scher HI, Fazzari M, Cordon-Cardo C. Overexpression of Cyclin D1 is associated with metastatic prostate cancer to bone. Clinical Cancer Research. 2000;6:1891–1895. [PubMed] [Google Scholar]
- 35.Chen Y, Martinez LA, LaCava M, Coghlan L, Conti CJ. Increased cell growth and tumorigenicity in human prostate LNCaP cells by overexpression of cyclin D1. Oncogene. 1998;16:1913–1920. doi: 10.1038/sj.onc.1201719. [DOI] [PubMed] [Google Scholar]
- 36.Pyo CW, Choi JH, Oh SM, Choi SY. Oxidative stress induced cyclinD1 depletion and its role in cell cycle processing. Biochim Biophys Acta. 2013;1830(11):5316–5325. doi: 10.1016/j.bbagen.2013.07.030. [DOI] [PubMed] [Google Scholar]
- 37.Lim JH, Lee YM, Chun YS, Park JW. Reactive oxygen species-mediated cyclin D1 degradation mediates tumor growth retardation in hypoxia, independently of p21cip1 and hypoxia-inducible factor. Cancer Sci. 2008;99(9):1798–1805. doi: 10.1111/j.1349-7006.2008.00892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]