

Abstract
Most new mutations are deleterious and are eventually eliminated by natural selection. But in an adapting population, the rapid amplification of beneficial mutations can hinder the removal of deleterious variants in nearby regions of the genome, altering the patterns of sequence evolution. Here, we analyze the interactions between beneficial “driver” mutations and linked deleterious “passengers” during the course of adaptation. We derive analytical expressions for the substitution rate of a deleterious mutation as a function of its fitness cost, as well as the reduction in the beneficial substitution rate due to the genetic load of the passengers. We find that the fate of each deleterious mutation varies dramatically with the rate and spectrum of beneficial mutations and the deleterious substitution rate depends nonmonotonically on the population size and the rate of adaptation. By quantifying this dependence, our results allow us to estimate which deleterious mutations will be likely to fix and how many of these mutations must arise before the progress of adaptation is significantly reduced.
Keywords: adaptation, genetic load, hitchhiking
RECENT years have witnessed an increased interest in the evolutionary dynamics of rapid adaptation. Once regarded as an obscure limit of population genetics, this regime has since been observed in a variety of empirical settings, from laboratory evolution experiments (Barrick et al. 2009; Lang et al. 2013) to natural populations of pathogenic viruses (Strelkowa and Lässig 2012), bacteria (Lieberman et al. 2014), and certain cancers (Nik-Zinal et al. 2012). In these populations, natural selection plays a central role in driving beneficial variants to fixation, and significant theoretical and empirical effort has been devoted to the study of these beneficial mutations and the dynamics by which they spread through the population (see Sniegowski and Gerrish 2010 for a review). Yet even in the most rapidly adapting populations, the vast majority of new mutations are neutral or deleterious, and much less is known about how these variants influence (or are influenced by) adaptation in nearby regions of the genome. As a result, even the most basic questions about this process remain unanswered. How deleterious must a mutation be before it is effectively purged by selection? Which deleterious mutations have the largest influence on the spread of the adaptive mutations? And how do the answers to these questions depend on the size of the population and the spectrum of beneficial mutations? These questions are the focus of the present study.
In the absence of other mutations, the fate of a deleterious variant is determined by the interplay between natural selection and genetic drift. Selection purges harmful variants from the population on a timescale inversely proportional to the fitness cost, sd, of the deleterious mutation. Meanwhile, random fluctuations from genetic drift can drive these variants to fixation, which requires a time proportional to the effective population size, Ne. Deleterious mutations with will be purged long before they can fluctuate to high frequency, while mutations with will barely feel the effects of selection before they fix. The presence of this “drift barrier” at has long been recognized (Kimura 1968; King and Jukes 1969; Ohta 1973). It suggests that fewer deleterious mutations will accumulate in larger populations and that those that do fix will have a smaller effect on fitness. However, even a small number of beneficial mutations can change this picture considerably.
When beneficial mutations are available, natural selection must purge deleterious variants and amplify beneficial mutations simultaneously. These forces can conflict with each other in closely linked regions of the genome, leading to a second source of stochasticity known as genetic draft (Gillespie 2000). Thus, provided that the cost of a deleterious mutation is not too high, it can hitchhike to high frequency with a beneficial “driver” mutation that happens to arise on the same genetic background (Maynard Smith and Haigh 1974). The fixation of these deleterious “passengers” imposes a direct cost on the fitness of the population, which can be ameliorated only by future compensatory mutations. Deleterious mutants also impose an opportunity cost on the fitness of the population when they hinder the fixation of driver mutations that arise on poor genetic backgrounds (Charlesworth 1994; Peck 1994). Thus, even when they are not destined to fix, segregating deleterious variants still contribute to an overall mutation load, which reduces the fraction of available genetic backgrounds where adaptation can proceed unhindered.
Together, deleterious passengers and the mutation load can dramatically reduce the rate of adaptation and, in extreme cases, can even lead to fitness decline (Silander et al. 2007) and mutational meltdown (Gabriel et al. 1993). Conversely, even a small number of beneficial mutations can bias the spectrum of deleterious mutations that accumulate during the course of evolution (Schiffels et al. 2011). These interactions between adaptation and constraint have been the subject of extensive theoretical study (Charlesworth 1994; Peck 1994; Barton 1995; Orr 2000; Johnson and Barton 2002; Bachtrog and Gordo 2004; Desai et al. 2007; Hartfield and Otto 2011; Jiang et al. 2011; Schiffels et al. 2011; Goyal et al. 2012; McFarland et al. 2014), but many aspects of this process remain poorly characterized. In particular, theory still struggles to account for observed variation in the fitness effects of new mutations and how these disparate mutations combine to determine overall levels of hitchhiking and the genetic load. This gap in our understanding is especially problematic for the largest and most rapidly adapting populations, where multiple beneficial driver mutations compete for fixation at the same time. As we will see, these populations actually accumulate more deleterious mutations, even as they become more efficient at finding and fixing adaptive variants. It is therefore unsurprising that deleterious passengers are thought to play an important role in the adaptive process (Pybus et al. 2007; Covert et al. 2013; McFarland et al. 2013; Łuksza and Lässig 2014).
In this article, we study the effects of deleterious passengers in a simple model of widespread adaptation. We employ a perturbative approach, leveraging a recent mathematical description of adaptation in the absence of deleterious mutations (Good et al. 2012; Fisher 2013). This enables us to obtain simple analytical predictions for genomic substitution rates across a broad range of beneficial and deleterious fitness effects. In particular, we find that the maximum cost of a passenger is inversely proportional to the coalescence timescale, Tc, over which the fates of new common ancestors are determined. This constitutes a natural generalization of the traditional drift barrier in the presence of widespread genetic draft, where the population-size dependence of Tc can be dramatically altered. We end by discussing the relevance of these findings for recent microbial evolution experiments and comment on directions for future work.
Model
We consider a population of N nonrecombining haploid individuals that accumulate mutations at a per genome rate U. We assume an infinite-sites model, in which the fitness effect of each mutation is drawn from a distribution of fitness effects, ρ(s), that remains constant over the relevant time interval. We further partition the distribution of fitness effects (DFE) into its beneficial and deleterious components,
| (1) |
where Ub and Ud denote the per-genome rates of beneficial and deleterious mutations, respectively. For concreteness, we primarily focus on a simplified “two-effect” DFE,
| (2) |
where beneficial and deleterious mutations each have a characteristic fitness effect. In a later section, we show how our analysis can be extended to more general distributions, provided that Ubρb(s) and Udρd(s) satisfy certain technical conditions.
These assumptions define a simple model of sequence evolution with a straightforward computational implementation. We wish to use this model to study the impact of deleterious mutations on the long-term genetic composition of the population, which is determined by the average substitution rate, R(s), of new mutations as a function of their fitness effect. In particular, we wish to quantify the relative contributions from beneficial and deleterious mutations. For the simple two-effect DFE in Equation 2, this is uniquely determined by the total beneficial and deleterious substitution rates, Rb = R(sb) and Rd = R(−sd). However, for more general DFEs, there is some ambiguity in how we define the net contribution from beneficial and deleterious mutations. For example, the raw substitution rates and tend to be dominated by neutral or nearly neutral mutations, which have a negligible impact on the fitness of the population. To avoid this bias, we focus on the weighted substitution rates,
| (3) |
where and represent the average fitness effects of the underlying DFE. For the two-effect DFE in Equation 2, Rb and Rd coincide with the raw rates of sequence evolution, as desired. For more general DFEs, the substitution rate of each mutation is weighted by its contribution to the total fitness of the population, so that the total rate of adaptation is simply
Heuristic Analysis and Intuition
Before we perform any explicit calculations, it is useful to consider the dynamics of deleterious mutations from a heuristic perspective. This allows us to identify many of the relevant fitness scales and helps build intuition for the more detailed calculations below. Our discussion resembles the traditional drift-barrier argument from the Introduction, but it applies for a much broader range of populations where drift is no longer the dominant evolutionary force.
Deleterious mutations can be classified into two fundamental regimes, depending on the substitution rates of the mutations involved. Sufficiently weakly selected mutations accumulate nearly neutrally (Rd ≈ Ud), while sufficiently strongly selected mutations rarely fix (Rd ≈ 0). The transition between these two regimes occurs for some characteristic cost which can be estimated from the fundamental timescales of the system. When a deleterious mutation arises, it originates on a particular genetic background, and it competes with this background lineage until one of them is driven to extinction. If the fitness of the background is sufficiently high, then the deleterious mutation may increase in frequency in the short term. Yet on average, the frequency of the mutant relative to its background decays exponentially at rate sd (see Figure 1). Thus, deleterious mutations are typically purged by selection on a characteristic timescale
Figure 1.

A schematic depiction of the fundamental evolutionary timescales. A deleterious variant (del) is purged relative to its background on a characteristic timescale Td. The fate of a future common ancestor is determined on a characteristic timescale Tc, and the fate of a beneficial mutation is determined over a characteristic timescale Tb.
The fixation of these mutations is governed by the underlying coalescent process. At each site in the genome, exactly one of the present-day individuals will grow to become an ancestor to the entire population. We let Tc denote the characteristic timescale over which the fates of new common ancestors are determined. This does not imply that new common ancestors have fixed within Tc generations, but only that their chances of extinction are negligible beyond this point (see Figure 1). As we demonstrate below, Tc is closely related to the coalescent timescale that determines the levels of neutral diversity in the population. When the genealogy of the population is dominated by drift, Tc is simply proportional to the population size (Tc ≈ 2N), but in general Tc can vary in an arbitrary way with the underlying parameters. A deleterious mutation can fix only if it arises in a future common ancestor and evades natural selection for Tc generations. If Td ≪ Tc, selection will typically purge the deleterious variant before its descendants reach fixation, while mutations with Td ≫ Tc will barely feel the effects of selection before they fix. This implies that the crossover between effectively neutral (Rd ≈ Ud) and effectively lethal (Rd ≈ 0) substitution rates must occur for where c is an (1) constant.
So far, we have considered two classes of deleterious mutations: those that fix and those that do not, with a transition between the two regimes at In an adapting population, there is also a third class of deleterious mutations: those that are most likely to hinder the spread of the beneficial variants, regardless of whether or not they fix. The typical cost of these maximally interfering mutations can be estimated from a similar timescale argument. Here, the relevant timescale is not Tc but rather the characteristic time Tb over which the fate of a beneficial mutation is determined (see Figure 1). When the fates of beneficial mutations are controlled by genetic drift, Tb is simply the drift time of the beneficial mutation (Tb ∼ 1/sb), but in general Tb can be an arbitrary function of the underlying parameters. For a deleterious mutation to hinder the fixation of a beneficial variant, it must fix (or nearly fix) within the beneficial lineage and begin to feel the effects of selection within Tb generations. These conditions are jointly satisfied when Tb ∼ Td, so the maximally interfering mutations have where b is another (1) constant.
Thus, without performing any explicit calculations, we see that a simple heuristic argument is sufficient to determine the relevant deleterious fitness effects in terms of the fundamental timescales of the system. In the following sections, we rederive these results more rigorously with explicit calculations of Rb and Rd in several different parameter regimes. Although these calculations are somewhat less general than the heuristic argument above, they allow us to predict the quantitative nature of the transitions near and in addition to the location of the transitions themselves. Perhaps more importantly, these calculations provide explicit expressions for Tc and Tb in terms of the underlying parameters N and Uρ(s), which enables us to estimate when deleterious passengers are likely to be important in practice.
Analysis
Although our model is simple, it can be difficult to analyze the evolution of a tightly linked genome directly at the sequence level. The fate of any particular variant is strongly influenced by additional mutations (particularly beneficial driver mutations) that segregate in the same genetic background, as well as by competing mutations that arise elsewhere in the population. Keeping track of the arrival times and haplotype structure of these mutations can rapidly become unwieldy when the genome contains more than a handful of selected sites.
Fortunately, previous work has shown that many of these difficulties can be avoided by utilizing an intermediate level of description, where the distribution of fitnesses within the population plays a central role (Haigh 1978; Tsimring et al. 1996). Instead of tracking individual genotypes, we focus on the total fraction of the population, f(X, t), with (log) fitness X. We can then write down a consistent set of equations governing the evolution of the fitness distribution without reference to the underlying genotypes. Similarly, the fate of a new mutation can be recast as a competition between the fitness distribution of its descendants and that of the background population (see Appendix A). This leads to a dramatic simplification in large populations, since the distribution of fitnesses can be highly predictable even when sequence evolution is highly stochastic.
The dynamics of the fitness distribution and the fates of individual mutations have been well characterized in the absence of deleterious mutations (Neher et al. 2010; Hallatschek 2011; Neher and Shraiman 2011; Good et al. 2012; Fisher 2013). Thus, rather than looking for an exact solution in the deleterious case, we utilize a perturbative approach, focusing only on the leading-order corrections to the substitution rate in the limit that Ud → 0. This strategy allows us to exploit our existing knowledge of the evolutionary dynamics in the absence of deleterious mutations. In particular, it implies that the fundamental fitness scales and can be estimated from previously derived formulas in the Ud → 0 limit. Although this limit may seem unrealistic (given that deleterious mutations are at least as common as their beneficial counterparts), these leading-order expressions will turn out to be surprisingly accurate in large populations, even when Ud ≫ Ub. This accuracy is due to the fact that the coupling constant in the perturbative expansion is controlled by the fitness differences in the population, rather than by Ub. These fitness differences are often much larger than Ub for many biologically relevant parameters (Desai and Fisher 2007), so the total perturbative correction can be small even when Ud ≫ Ub. In the following analysis, we distinguish between different adaptive regimes, depending on the frequency of strongly beneficial driver mutations, which can dramatically influence the timescales Tc and Tb.
No driver mutations
We start by reviewing the simplest case, where beneficial driver mutations can be neglected. This assumption applies not only when Ub = 0, but also in small populations where drivers fix less frequently than neutral coalescence events (N Rb ≪ 1). Both conditions are sufficient to ensure that the coalescent timescale is dominated by genetic drift (Tc ∼ N). In the absence of deleterious mutations, there is no fitness diversity within the population [i.e., f0(X, t) ≈ δ(X)]. To leading order, the fate of a particular individual is determined solely by the balance between selection and drift, and the fixation probability is given by the standard formula
| (4) |
where X denotes the fitness of the individual (Fisher 1930; Wright 1931). The deleterious substitution rate trivially follows by averaging over the potential fitness backgrounds of new deleterious mutations:
| (5) |
Thus, we recover the well-known result that deleterious mutations accumulate neutrally when sd ≪ 1/2N and are exponentially suppressed when sd ≫ 1/2N. As we argued on heuristic grounds above, the transition between these two extremes occurs at when the lifetime of a deleterious mutation (Td ∼ 1/sd) is on the order of the neutral coalescence time (Tc ∼ N).
Of course, the substitution rate in Equation 5 eventually breaks down for large values of Ud, when many deleterious mutations segregate in the population simultaneously. Interference between these mutations can drive additional mutants to fixation via Muller’s ratchet (Muller 1964), thereby accelerating the rate of sequence evolution. Yet while the ratchet can alter both the functional form of Equation 5 and the location of the transition to neutrality, it preserves the monotone dependence of these quantities on the population size (see Appendix C). Thus, in the absence of drivers, both the number of deleterious substitutions and the fitness effects of these mutations tend to decrease in larger populations (Figure 2A).
Figure 2.

The deleterious substitution rate in the limit that beneficial driver mutations are rare. Symbols represent simulations of a two-effect DFE for fixed Ub = 10−9, sb = 10−2, and Ud = 10−4, with sd tuned between 10−5 and 10−2. In the top panel, the red lines give the drift-dominated predictions from Equation 5, while the bottom panel shows the sweep-dominated predictions from Equation 9.
In the following sections, we assume for simplicity that the population size is large enough that none of the deleterious mutations would fix due to drift alone (Nsd ≫ 1). This is a reasonable assumption for most microbial evolution experiments, where the effective population sizes are on the order of 105 or greater (Kawecki et al. 2012). This also allows us to focus on true passenger mutations, which could never fix without the help of a beneficial driver. (For an analysis of the opposite regime, see McFarland et al. 2014.) As we demonstrate below, these passenger mutations display a qualitatively different dependence on the population size than Equation 5 would predict.
Rare driver mutations
In larger populations, beneficial drivers substitute sufficiently often that the coalescence timescale is dominated by the sweep time of a beneficial mutation. In this case, we still expect deleterious mutations to accumulate neutrally when but this threshold is no longer tied to the inverse population size. Again, it is useful to begin with the simplest case, where beneficial mutations are sufficiently rare that they fix independently. This requires that the waiting time for a successful driver [Twait ≈ 1/(2NUbsb)] is much longer than its fixation time so that the coalescent timescale is given by Tc ∼ Twait. The effects of deleterious mutations in this “rare driver” regime have been studied by a number of previous authors (Charlesworth 1994; Peck 1994; Orr 2000; Johnson and Barton 2002). This earlier work primarily focuses on the reduction in the beneficial substitution rate, with analytical results available in the limiting case where sd > sb. Here, we generalize these results and derive simple analytical formulas for Rb and Rd that are valid across the full range of deleterious fitness costs. These formulas can then be contrasted with their counterparts in the multiple-driver regime below.
In the absence of deleterious mutations, a population in the rare driver regime is typically fixed for a single genotype (i.e., the last successful driver) whose fitness can be taken to be X = 0. Deleterious mutations create variation around this genotype, which is temporarily depleted when the next driver sweeps to fixation (Figure 3). In the time period between sweeps, the distribution of fitness is governed by the deterministic dynamics,
| (6) |
where denotes the mean fitness of the population (Haigh 1978). The solution to this equation, subject to the initial condition can be obtained via standard methods (Johnson 1999; Etheridge et al. 2007; Desai and Fisher 2011). The leading-order contribution is given by
| (7) |
where we have neglected terms of order At long times (t ≫ 1/sd), this distribution approaches the standard mutation–selection balance, f(−sd) ≈ Ud/sd, but it is possible that the next driver will occur before this equilibrium is reached (Johnson and Barton 2002). The waiting time for the next successful driver is exponentially distributed with mean Tc ≈ 1/(2NUbsb), so the average fraction of deleterious individuals at the time of the next sweep is
| (8) |
This reduces to the standard mutation–selection balance when Previous studies often neglect this relaxation phase, since they focus on deleterious fitness effects with But based on our heuristic discussion, we expect that most of the successful passenger mutations will have where the deviations from mutation–selection balance in Equation 8 start to become important. Indeed, as we will see below, it is exactly this time-dependent behavior that drives most of the deleterious hitchhiking in these populations.
Figure 3.

A schematic illustration of the fitness distribution. (A) In the rare driver regime, the population is predominantly composed of the last successful driver, with a deleterious subpopulation given by Equation 7. (B) In the multiple-driver regime, the fitness distribution approaches a steady-state shape, f(x), which translates toward higher fitness at rate v. The red line depicts the fixation probability, w(x), which increases rapidly with x before transitioning to the standard Haldane result at x ≈ xc. Future common ancestors are located in a narrow region of width near the nose. Effectively lethal mutations remove an individual from this region, thereby reducing the pool of potential ancestors. Effectively neutral mutations leave an individual within this region, with a small fitness cost relative to its immediate ancestor.
When the next driver mutation does arise, it can drag a deleterious passenger to fixation in one of two ways. The deleterious mutation can arise before the driver mutation, so that the driver originates directly in a deleterious background. These passenger-first events occur at rate NUbf(−sd)pfix(sb − sd). Alternatively, the deleterious mutation can arise after the driver and fix within the driver lineage while it is still rare. In Appendix C, we show that these driver-first events occur at rate When this rate is much smaller than the passenger-first scenario above, but it becomes comparable in magnitude when sd ∼ sb. Combining these two expressions, we find that the total deleterious substitution rate is given by
| (9) |
A similar expression was derived by Schiffels et al. (2011), using a different method of analysis. The substitution rate in Equation 9 approaches the neutral limit when sd ≪ 2NUbsb and decays as a power law when sd ≫ 2NUbsb. This power-law decay is significantly slower than the exponential falloff in Equation 5 (Figure 2) and cannot be recast as a simple reduction in effective population size. Thus, even when deleterious mutations would never drift to fixation on their own, frequent drivers can still cause these variants to accumulate like neutral mutations. Note that the time-dependent fitness distribution in Equation 7 played a crucial role in the emergence of this effectively neutral regime. Once the fitness distribution has reached mutation–selection balance, hitchhiking is already reduced by a factor of NUb ≪ 1. In agreement with our heuristic argument, the border of the effectively neutral regime is located at when the lifetime of a deleterious mutation (Td ∼ 1/sd) is on the order of the coalescence time (Tc ∼ Twait). However, in contrast to the nonadapting case, is now an increasing function of the population size. This implies that more deleterious mutations will accumulate in larger and more rapidly adapting populations and that the average cost of each passenger will increase as well (Figure 2B).
To calculate the total rate of sequence evolution and the rate of adaptation, we must also understand how deleterious mutations influence the fixation of the drivers. In the absence of deleterious mutations, drivers substitute at rate Rb = 2NUbsb, and deleterious mutations will reduce this rate by decreasing the effective fitness advantage of the drivers. Part of the reduction in driver fitness arises from the fixation of deleterious passengers within the driver lineage before it completes its sweep. This can occur via either of the two hitchhiking scenarios (passenger-first hitchhiking or driver-first hitchhiking) described above. Deleterious mutations can also influence the fates of unloaded drivers through the time-varying mean fitness in Equation 7. The corresponding reduction in Rb is somewhat more difficult to obtain compared to the deleterious substitution rate, since Rb depends on nonequilibrium properties of f(X, t) that are not captured by the simple average in Equation 8. Nevertheless, since the fate of a driver mutation is determined while it is at low frequency, the reduction in Rb can still be obtained using standard branching-process techniques (Johnson and Barton 2002). We carry out this calculation in Appendix C and find that the leading-order reduction in Rb is given by
| (10) |
Most of the interesting reduction occurs for when the fractional change in Rb becomes independent of Tc. In other words, these deleterious mutations have all reached mutation–selection balance by the time that the next driver arises. When sd < sb, Equation 10 has a simple interpretation as a reduction in the establishment probability of each driver due to the deleterious passengers that accumulate during the establishment time, These “tunneling” events are crucial for obtaining the proper sd dependence in Equation 10; traditional arguments based on the mutation load (Kimura and Maruyama 1966) would otherwise suggest that Rb is independent of sd. In the opposite case where sd > sb, we recover the well-known “background selection” (Charlesworth 1994) or “ruby in the rough” (Peck 1994) behavior observed in previous studies. In this case, loaded drivers can never fix, and Equation 10 can be interpreted as a reduction in the effective population size equal to the fraction of mutation-free individuals in the population.
These results show that the largest reduction in Rb occurs for which is much larger than the size of a typical passenger mutation In other words, the maximally interfering mutations rarely hitchhike to fixation (Rd ≈ 0). The disparity between these two scales can be explained in terms of the heuristic argument above. When drivers are rare, the fates of beneficial mutations are primarily influenced by drift. The driver fates are therefore determined during the drift time, which is much shorter than both the fixation time of a driver and the waiting time between sweeps [Twait ∼ 1/(2NUbsb)]. This will change dramatically in the multiple-driver regime below.
Multiple-driver mutations
As the population size increases, the waiting time between drivers is eventually dwarfed by the time that it takes each driver to fix. Multiple drivers will segregate in the population at the same time, and these mutations will interfere with each other as they compete for fixation. In this clonal interference regime, drivers and passengers are both dominated by the effects of genetic draft, and coalitions of multiple drivers are often required to drive a lineage to fixation (Rouzine et al. 2003; Desai and Fisher 2007). Recent empirical work in microbial populations suggests that this regime is likely to be the rule rather than the exception (De Visser et al. 1999; Miralles et al. 1999; Perfeito et al. 2007; Batorsky et al. 2011; Miller et al. 2011; Strelkowa and Lässig 2012; Kvitek and Sherlock 2013; Lang et al. 2013; Lee and Marx 2013; Barroso-Batista et al. 2014), so it is important that we extend our previous analysis to this potentially more realistic scenario.
At long times, a population in the multiple-driver regime reaches a steady state in which the continuous production of new mutations is balanced by the depletion of this diversity due to natural selection (Tsimring et al. 1996; Rouzine et al. 2003; Desai and Fisher 2007). The fitness distribution f(X, t) behaves like a “traveling wave,” with a characteristic shape f(x) that translates toward higher fitness at a constant rate v (see Figure 3). In some respects, this multidriver equilibrium is simpler to analyze than the rare driver regime above, where the punctuated nature of adaptation required us to explicitly account for departures from steady state. Averaged over short fitness scales and timescales, the steady-state shape of the fitness distribution is described by the deterministic dynamics,
| (11) |
where x denotes the relative fitness, This deterministic equation holds throughout the bulk of the fitness distribution, but starts to break down near the high-fitness “nose” (see Figure 3) where most successful drivers originate. In large populations, the nose constitutes just a small fraction of the total population, so we can approximate the effects of genetic drift in this regime with a suitable linear branching process (Neher et al. 2010; Good et al. 2012; Fisher 2013). The fixation probability, w(x), for a lineage with relative fitness x satisfies a related differential equation,
| (12) |
where the nonlinear term gives the contribution from genetic drift (see Appendix A). The marginal fixation probability, pfix(s), of a mutation with fitness effect s can be calculated from w(x) by averaging over the distribution of background fitnesses,
| (13) |
where the consistency condition pfix(0) ≈ 1/N serves to uniquely determine v as a function of the underlying parameters N and Uρ(s) (Hallatschek 2011; Good et al. 2012; Fisher 2013).
In the absence of deleterious mutations, we have previously derived an approximate solution to Equations 11–13 in the strong selection regime where (Good et al. 2012). Unfortunately, this solution contains several pathologies that render it unsuitable for the perturbative analysis below (Fisher 2013). In Appendix B, we derive a modified version of this solution that corrects these issues. The resulting fixation probability is characterized by a narrow boundary layer near a critical fitness value, xc ≥ sb. Above this point, lineages fix without the need for additional driver mutations, so w0(x) ≈ 2x. Below xc, the fixation probability rapidly declines as
| (14) |
The fitness distribution displays a similar transition near xc, taking a simple Gaussian form below xc,
| (15) |
and vanishing above this threshold (Fisher 2013). The location of the boundary can be obtained from an integral transform of Equation 12, which yields an auxiliary condition,
| (16) |
which uniquely determines xc as a function of Ub, sb, and v0 (Good et al. 2012).
The solution in Equations 14–16 requires that and The first of these conditions places a lower bound on the amount of clonal interference in the population: there must be sufficient fitness variation that the parents of successful drivers have abnormally high fitness (i.e., greater than one standard deviation from the mean). This distinguishes the clonal interference regime from the rare driver limit above. The second condition places an upper bound on the amount of fitness diversity in the population: individuals that compose the bulk of the population (i.e., within one standard deviation from the mean) typically harbor the same number of driver mutations. In terms of the underlying parameters, these two conditions require that Nsb ≫ 1 and Ub ≪ sb (see Appendix B). We focus on this regime because it is thought to apply to a broad range of microbial evolution experiments, at least in the initial phases of adaptation (Desai et al. 2007; Parfeito et al. 2007; Wiser et al. 2013; Barroso-Batista et al. 2014).
Deleterious mutations lead to a reduction in the fixation probability and deviations from the Gaussian fitness distribution, which can alter the rate of adaptation and the location of the nose in potentially complex ways. However, we can still investigate the leading-order effects of deleterious passengers, using the same perturbative strategy that we employed above. We rewrite the substitution rates in the suggestive form
| (17a) |
| (17b) |
with corresponding expansions for w(x), f(x), and v,
| (17c) |
| (17d) |
| (17e) |
Note that we have not included an expansion for xc, since this is simply a property of w0(x) rather than a measurable quantity like v. Thus, with a slight abuse of notation, we continue to use xc to denote the zeroth-order value With these definitions in hand, we can substitute Equation 17 into Equations 11–13 and equate like powers of Ud to obtain at a corresponding set of equations for Δrb, Δrd, g(x), and h(x) (see Appendix C). The correction to the deleterious substitution rate is particularly easy to calculate, since the zeroth order (Ud = 0) contribution vanishes. To leading order, we find that
| (18) |
which interpolates between the effectively neutral limit (Rd ≈ Ud) and the effectively lethal limit (Rd ≈ 0) illustrated in Figure 4. In sufficiently large populations where xc ≫ sb, Equation 18 reduces to
| (19) |
which shows that the border of the effectively neutral regime occurs at This crossover has a natural interpretation in terms of the fundamental timescales of the system. The “nose-to-mean” time Tsweep ∼ xc/v0 is the time required for the current fitness of the nose to become the mean fitness of the population. When xc ≫ sb, we have previously shown that this is also the timescale over which the fates of new common ancestors and successful drivers are decided, so that Tc ∼ Tb ∼ Tsweep (Desai et al. 2013). Solving for v0 and xc, one can show that
| (20) |
which is much greater than 1/sb (i.e., Tcsb ≫ 1) and is approximately independent of the population size (Desai et al. 2013). Thus, like the rare driver regime above, the number of deleterious passengers does not necessarily decrease in larger and more rapidly adapting populations. Yet in this case, the cost of a typical passenger does not increase as rapidly with N or Ub, which reflects the fact that adaptation is not limited by the supply of beneficial mutations.
Figure 4.

Corrections to the substitution rate in the limit that driver mutations are common. Symbols denote the results of forward-time simulations for various combinations of Ud and sd, with the remaining parameters fixed at N = 107, Ub = 10−5, sb = 10−2. The quantities Δrb and Δrd are calculated from Equation 17, with v0 measured from simulations and xc estimated from Equation 16. For comparison, the solid red lines show the first-order predictions from Equations 18 and 21, using the same estimated values of v0 and xc.
Corrections to the beneficial substitution rate can be obtained in a similar way, although the algebra is more involved because Δrb, g(x), and h(x) are not independent. We carry out this calculation in Appendix C, and we find that the leading-order correction to Rb is given by
| (21) |
We compare this formula with simulations in Figure 4. In sufficiently large populations where xc ≫ sb, Equation 21 reduces to the simple form
| (22) |
which depends only on the compound parameter In the limit that which is consistent with the equal accumulation of nearly neutral passengers in the nose and in the bulk population (Desai and Fisher 2007). On the other hand, when the reduction in Rb is consistent with a simple reduction in population size, Ne = N(1 − Ud/sd), similar to the ruby in the rough limit of the rare driver regime above. In between these two extremes, Equation 21 predicts a maximum reduction in Rb at an intermediate value of This is consistent with our heuristic argument that passengers should influence the fates of drivers only if they are purged on the same timescale that determines the fate of a driver mutation, Tb ∼ Tsweep. Unlike the rare driver regime, the maximally interfering mutations in this case are approximately the same size as a typical passenger mutation and are much smaller than the size of a typical driver
Distributions of fitness effects
Our analysis has so far assumed that the DFE is given by the simple two-effect form in Equation 2. On the surface, such an assumption seems to conflict with empirical measurements of the DFE, which typically involve at least severalfold variation in the magnitudes of beneficial and deleterious mutations (Eyre-Walker and Keightley 2007). In this section, we discuss how our analysis can be extended to this more biologically realistic scenario.
At the level of approximation considered here, distributions of deleterious fitness effects do not pose any major problems for our analysis. Since we have focused only on the leading-order contributions in Ud, the net effects of a deleterious DFE can be obtained simply by averaging over the deleterious effect sizes,
| (23a) |
| (23b) |
where Δrd(sd) and Δrb(sd) are given by Equations 18 and 21. The outcome of this average can depend rather sensitively on both the supply of beneficial mutations [which controls the quantitative dependence of Δrd(sd) and Δrb(sd)] and the relative variation in ρd(sd). In the broad distribution limit where ρd(sd) is approximately uniform, the reduction in the beneficial substitution rate will be dominated by the density of deleterious mutations near The effects on the deleterious substitution rate are more subtle. When Δrd(sd) decays exponentially with the fitness cost, Rd will be dominated by mutations near as expected. However, when Δrd(sd) decays as an inverse power of sd, the weighting factor in Equation 23a will remove much of the Tc dependence in the integrand. In this case, significant contributions to Rd will arise from all deleterious mutations in the range
In the rare driver regime, a similar averaging scheme can be applied for a distribution of beneficial fitness effects, so that However, this average breaks down in the multiple-driver regime, since the beneficial substitution rate is not a simple linear function of Ub. Instead, previous work has shown that a large class of beneficial DFEs can be approximated by a single-effect DFE, provided that the effective selection coefficient and the mutation rate are chosen appropriately (Hegreness et al. 2006; Desai and Fisher 2007; Good et al. 2012). In agreement with this earlier work, we find that the single-sb approximation holds for Δrb and Δrd as long as the effective parameters are associated with the expressions in Good et al. (2012):
| (24a) |
| (24b) |
Thus, we can extend our results to a distribution of fitness effects simply by replacing and in all of our previous expressions. The only difference is that and now vary as a function of the population size and mutation rate, so the scaling of the various quantities (e.g., Tc and Tb) can change.
As an example, we consider the case where beneficial mutations follow an exponential distribution, In this case, we have previously shown that the effective selection strength and mutation rate are given by
| (25) |
where for all but the largest population sizes (Good et al. 2012). Thus, the fittest individuals in the population typically contain only one more driver mutation than the mean, and the dynamics of adaptation bear some resemblance to the selective sweeps pictured above (Schiffels et al. 2011). In this “quasi-sweep” regime, the size of a typical driver increases with the population size in such a way that the nose-to-mean time,
| (26) |
remains a decreasing function of N. This is similar to the rare driver regime above, although the dependence on N is much weaker in this case. Substituting our expressions for and into Equations 18 and 21, we find that the substitution rates are given by
| (27a) |
| (27b) |
We compare these predictions to simulations in Figure 5. Similar to the single-sb case, we again observe a transition from a regime of effective neutrality (Rd ≈ Ud) to a regime of effective lethality (Rd ≈ 0) at a characteristic effect size This threshold is much smaller than the size of an average beneficial mutation as well as the size of a typical driver Since these results are also consistent with earlier work by Schiffels et al. (2011), which found that Writing the typical cost of a passenger in this way emphasizes the quasi-sweep nature of the exponential DFE. Like the rare driver regime, the deleterious substitution rate in Equation 27a decays as a power law when as opposed to the exponential dependence in Equation 19. Similarly, the reduction in the beneficial substitution rate in Equation 27b is maximized when where hitchhiking is already unlikely (Rd ≈ 0). This illustrates a subtle feature of the single-s equivalence principle above. If we condition on the size of a successful driver our results are insensitive to the shape of the DFE. However, if we condition on the size of an average potential mutation then the relevant deleterious mutations can depend rather sensitively on the shape of ρb(s).
Figure 5.

Corrections to the substitution rate for an exponential distribution of beneficial fitness effects. Symbols denote the results of forward-time simulations for N = 105 (triangles) and N = 107 (circles), with the remaining parameters the same as in Figure 4. Once again, Δrb and Δrd are calculated using the simulated value of v0, with xc estimated from Equation 16 and and estimated from Equation 25. For comparison, the solid red lines show the first-order predictions from Equation 27, using the same estimated values of v0, xc, and
Discussion
In any sufficiently complex organism, spontaneous mutations will have a broad range of effects on reproductive fitness. This leads to a natural question: Which (if any) of these mutations will influence the evolutionary dynamics of the population? If certain mutations are more important than others, is it possible to focus only on a subset of potential mutations? In the general case, these questions can be difficult to answer. Adaptive changes account for just a small fraction of all possible mutations, but when they do arise, beneficial variants are rapidly amplified by selection and dramatically alter the evolution of the population. Deleterious mutations, in contrast, have a negligible impact individually, but their greater numbers can nevertheless lead to a large collective influence. Given this competition between the scales of mutation and selection, it is possible that beneficial and deleterious mutations both play an important role in certain populations.
Here, we have introduced a quantitative mathematical framework for characterizing the effects of deleterious passengers in rapidly evolving populations. By leveraging previous results in the absence of deleterious mutations, we derived simple formulas for the rates of sequence evolution when beneficial and deleterious mutations possess a broad range of fitness effects. These results provide important qualitative intuition about the effects of deleterious passengers, since they allow us to estimate which deleterious mutations are most likely to hitchhike to fixation and which ones will hinder the fixation of the drivers.
In the case of hitchhiking, we found that the maximum cost of a passenger is determined by the inverse of the coalescent timescale, which reduces to the traditional drift-barrier in the absence of other mutations. When drivers are more common, the location of this neutral threshold can grow to be much larger than the inverse population size, with a qualitatively different dependence on N (Table 1). Thus, as observed in previous studies (Schiffels et al. 2011), larger and more rapidly adapting populations will often accumulate a larger number of more strongly deleterious mutations, although the total fraction of deleterious substitutions must still decline. This increased deleterious load can have important implications for the stability of rapidly adapting proteins [e.g., in influenza (Strelkowa and Lässig 2012)] and is likewise relevant when inferring the prevalence of adaptive mutations from changes in dN/dS ratios (Ostrow et al. 2014). It is important to note, however, that the deleterious load can increase with N only in the presence of strongly beneficial driver mutations (Tcsb ≫ 1) and not as a general consequence of linkage. Indeed, we find that decreases (weakly) with N in “infinitesimal” models of linked selection (Tcsb ≪ 1) (Tsimring et al. 1996; Cohen et al. 2005; Hallatschek 2011; Neher and Hallatschek 2013; Neher et al. 2013; Good et al. 2014) and in the presence of Muller’s ratchet (Söderberg and Berg 2007).
Table 1. Summary of the deleterious substitution rates in the different adaptive regimes.
| Adaptive regime | Deleterious substitution rate | Typical passenger cost | Related work |
|---|---|---|---|
| No drivers | r(x) ≈ 2x/(e2x − 1) | Fisher (1930); Wright (1931) | |
| Rare drivers | r(x) ≈ 1/(1 + x) | Johnson and Barton (2002) | |
| Multiple drivers, | r(x) ≈ (1 − e−2x)/(2x) | Schiffels et al. (2011) | |
| Multiple drivers, | r(x) ≈ e−x | Desai and Fisher (2007) | |
| Infinitesimal limit | r(x) ≈ e−x | Hallatschek (2011) |
To compare functional forms, we show the scaled substitution rate, r(x) = c1R(−c2x), where the scaling parameters c1 and c2 are chosen so that r(0) = −r′(0) = 1.
In addition to the size of a successful passenger, our framework also allows us to identify mutations that are most likely to hinder fixation of the drivers. We saw that this influence is maximized for deleterious mutations of an intermediate effect set by the stochastic phase of a successful driver mutation. When drivers are rare, this is simply the drift time and deleterious mutations above this threshold limit the rate of adaptation through a well-known reduction in effective population size (Charlesworth 1994; Peck 1994; Orr 2000; Wilke 2004). However, when driver mutations interfere with each other (RbTc ≫ 1), this stochastic phase becomes much longer than since multiple beneficial mutations are required for fixation. Thus, the relevant deleterious fitness effects are usually much smaller than the size of a typical driver, which emphasizes the importance of the two-effect DFE that we used throughout our analysis. Previous work has often focused on a simpler “single-effect” DFE, where the fitness effects of beneficial and deleterious mutations are identical (sb = sd) (Woodcock and Higgs 1996; Rouzine et al. 2003, 2008; Goyal et al. 2012). Our present results suggest that these models may underestimate the importance of deleterious mutations, since they implicitly neglect mutations with the largest potential influence.
These findings suggest that the cumulative influence of deleterious passengers depends rather sensitively on the distribution of beneficial and deleterious fitness effects, in addition to the population size and mutation rate. This makes it difficult to estimate the relevance of deleterious passengers in practice, since these distributions are known only to a very rough degree of approximation (Eyre-Walker and Keightley 2007). The notable exception is in experimental microbial populations, where high-throughput screens have enabled a much more detailed characterization of the fitness effects of new mutations (Elena et al. 1998; Wloch et al. 2001; Sanjuán et al. 2004; Kassen and Bataillon 2006; Qian et al. 2012; Frenkel et al. 2014). As a concrete example, we have recently estimated ρb(s) for a strain of Saccharomyces cerevisiae in rich media to be an exponential distribution with Ub = 10−4 and (Frenkel et al. 2014), which is also consistent with recent measurements of the yeast deletion collection (Figure 6A). If we assume that ρd(s) can also be estimated from these deletion measurements, then our model implies that deleterious mutations will start to influence the rate of adaptation in these populations only when Ud ∼ 10−2 (Figure 6B), which is already larger than the total per-genome mutation rate in yeast (Zhu et al. 2014). Deleterious passengers are therefore unlikely to impede the rate of adaptation in these populations. Recent work has shown that may decrease by as much as a factor of 4 over ∼1000 generations of evolution due to the effects of diminishing-returns epistasis (Chou et al. 2011; Khan et al. 2011; Wiser et al. 2013; Kryazhimskiy et al. 2014). This lowers our estimate of only by a factor of 2, which suggests that deleterious mutations are also unlikely to contribute to the long-term differences in evolvability of these strains in the absence of more complicated epistatic interactions. Of course, it is not surprising that deleterious mutations play a small role here, since these populations have been studied precisely because they repeatedly adapt to their laboratory environment. In more well-adapted populations, mutator strains have been observed to evolve toward lower mutation rates (McDonald et al. 2012; Maharjan et al. 2013; Wielgoss et al. 2013), suggesting that the deleterious load may eventually become more of a burden. However, without a more detailed estimate of the DFE, this hypothesis is merely speculative.
Figure 6.

Breakdown of the linear approximation at high Ud. (A) Fitness effects of all single-gene deletions in yeast, measured by Qian et al. (2012) in rich media. The shape of the beneficial DFE is approximated by an exponential distribution with mean sb = 0.01, while the deleterious DFE is approximated by a Weibull distribution with mean and shape k = 0.64 (red lines). (B) Corrections to the rate of adaptation as a function of the deleterious substitution rate, assuming that the beneficial and deleterious DFE shapes are given by the fits in A. The remaining parameters are N = 105 and Ub = 10−4 (Frenkel et al. 2014). Symbols denote the results of forward-time simulations, while the predicted scaling relation Δv ∝ Ud is shown in solid green. For comparison, the dashed lines indicate the point where the accumulation of deleterious mutations overwhelms the rate of adaptation (v ≈ 0). (C) Analogous corrections for the beneficial and deleterious substitution rates. In this case, the predicted scaling relation is Δr ∝ 1.
In the present article, we have considered only the most basic effects of deleterious passengers on the long-term patterns of sequence evolution, leaving many potential avenues for future work. Most notably, we have omitted any discussion about the patterns of sequence diversity within the population, which provide a snapshot of the selective forces operating in the recent past. Deleterious passengers are potentially even more relevant for these contemporary data, since mutations that are too deleterious to fix can still generate appreciable fitness diversity if they are sufficiently common (Haigh 1978). Our perturbative corrections to the fitness distribution can potentially capture some of this deleterious diversity, but the effects on neutral polymorphism and the implications for recombining chromosomes will require additional analysis. Recent work by Weissman and Hallatschek (2014) could potentially be used to address these questions. In this case, the relevant parameters would be the beneficial and deleterious mutation rates within an appropriately defined linkage block, the length of which must be self-consistently determined by the resulting coalescence timescale (Neher et al. 2013; Good et al. 2014; Weissman and Hallatschek 2014).
A second limitation of our analysis is that it is fundamentally perturbative in nature. By focusing only on the leading-order corrections to the dynamics in powers of Ud/xc, our results are primarily applicable when the net effect of deleterious mutations is small (i.e., v0 − v ≪ v0). A similar approximation is implicit in earlier work by Schiffels et al. (2011). Thus, these studies have explicitly neglected cases where many deleterious mutations fix cooperatively due to Muller’s ratchet (Söderberg and Berg 2007; McFarland et al. 2013) or the long-term “fixed point” where the accumulation of beneficial and deleterious mutations balances (Goyal et al. 2012). In practice, our expressions are often quite accurate, even permitting estimates of the v ≈ 0 fixed point in certain cases (Figure 6B). However, a more thorough characterization of this fixed point [and the shapes of ρb(s) and ρd(s) that are attained there] remains an important avenue for future work.
Acknowledgments
We thank Daniel Balick, Ivana Cvijović, and Karri DiPetrillo for useful discussions. This work was supported in part by a National Science Foundation (NSF) Graduate Research Fellowship, the James S. McDonnell Foundation, the Alfred P. Sloan Foundation, the Harvard Milton Fund, grant PHY 1313638 from the NSF, and grant GM104239 from the National Institutes of Health. Simulations in this article were performed on the Odyssey cluster supported by the Research Computing Group at Harvard University.
Appendix A: Stochastic Model of the Fitness Distribution and the Mean-Field Approximation
As described in the text, the dynamics of our evolutionary model can be recast in terms of the population fitness distribution, f(X, t), which tracks the fraction of individuals in each “fitness class” X. In the diffusion limit, these fitness classes obey the Langevin dynamics,
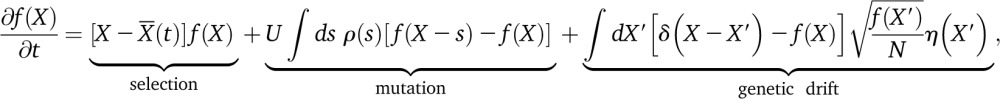 |
(A1) |
where is the mean fitness of the population and η(X) is a Brownian noise term of the Ito form (see Good and Desai 2013 for further discussion). Equation A1 represents a natural generalization of the single-locus diffusion equation for a genome with a large number of selected sites. To track the fate of a lineage founded by an individual with fitness X0, we introduce the labeled fitness classes g(X, t) and f(X, t) corresponding to the focal lineage and the background population, respectively. These fitness classes obey a generalized version of Equation A1,
| (A2a) |
| (A2b) |
with the initial condition For general N and Uρ(s), there is no closed-form solution of Equation A3. However, in sufficiently large populations we can employ a “mean-field” approximation that has been used in several previous studies (Neher et al. 2010; Neher and Shraiman 2011; Good et al. 2012; Fisher 2013).
The basic idea behind this approximation is that there is a separation of frequency scales between the regime where genetic drift is important and the regime where population saturation is important (Desai and Fisher 2007). In other words, the fate of a lineage is determined while it is still rare while most of the remaining population evolves deterministically. From these assumptions, we can rewrite Equation A3 in the simpler form,
| (A3a) |
| (A3b) |
where we have approximated the dynamics of the focal lineage by a simple linear branching process. The fixation probability of this lineage can then be deduced using standard techniques from the theory of branching processes. We introduce the generating functional which satisfies the partial differential equation
| (A4) |
subject to the initial condition H[φ(X), 0] = exp[−φ(X0)] = 1 − φ(X0). [The last equality follows from the standard diffusion limit used to derive Equation A1. Here, φ(X) has “units” of fitness or inverse population size and thus becomes infinitesimally small in the limit that N → ∞ and s → 0. Since we neglect saturation effects, we know that at long times the focal lineage must either go extinct or diverge to infinity. This yields a relation between the generating function and the fixation probability,
| (A5) |
where θ(x) is the Heaviside step function. Thus, we must simply solve Equation A4 to obtain the fixation probability for any subset of the focal lineage. We can achieve this via the method of characteristics. Letting τ denote backward time, the characteristic equation for φ(X) is given by
| (A6) |
with the backward-time initial condition φ(X, τ = 0) = φ(X). Meanwhile, the characteristic for H is simply ∂τH = 0, so we can immediately conclude that H[φ(X), t] = 1 − φ(X0, τ = t) and therefore that the fixation probability for a subset of the focal lineage is given by
| (A7) |
Finally, we let w(X) be the unique long-time limit of φ(X, τ) when φ(X) > 0, or
| (A8) |
In other words, w(X) is simply the fixation probability of a new lineage founded by a single individual with fitness X at time t = 0. Similar derivations of Equation A8 can be found in Good et al. (2012) and Fisher (2013), although we will sometimes require the more general expression in Equation A7.
Within our mean-field approximation, the fixation probability, pfix(s), for a new mutation with fitness effect s can be obtained by averaging over the fitness backgrounds that the mutation could have arisen on. By construction, this distribution of backgrounds is simply f(X), so that
| (A9) |
We must then match the “microscopic” dynamics of pfix(s) with the deterministic solution for f(X, t) to obtain a self-consistent description of the evolutionary dynamics (Hallatschek 2011; Neher et al. 2010; Neher and Shraiman 2011; Good et al. 2012; Fisher 2013). We carry out this procedure for several simple cases below.
Appendix B: Zeroth-Order Solution Without Deleterious Mutations
In this section, we review the solution of the mean-field model in the absence of deleterious mutations (Ud = 0). This “zeroth-order” solution forms the basis of the perturbative analysis described in the text. Mirroring our discussion in the text, we distinguish between the rare driver regime and the multiple-driver regime.
Rare Driver Mutations
When driver mutations are rare [also known as the successional mutations or strong selection–weak mutation (SSWM) regime (Desai and Fisher 2007)], multiple beneficial mutations rarely segregate in the population at the same time. We can therefore neglect the mutation terms in the mean-field approximation in Equations A3a and A6. When a beneficial mutation arises, the population is fixed for a single genotype, which (without loss of generality) can be assumed to have fitness X = 0. In other words, the deterministic solution for the fitness distribution is simply f(X, t) ≈ δ(X). The differential equation for ϕ(X, τ) in Equation A6 reduces to
| (B1) |
from which we can obtain the long-term solution for the fixation probability,
| (B2) |
Given w(X), the fixation probability of a new mutation trivially follows from Equation A9,
| (B3) |
which we immediately recognize as the large-N limit of the single-locus fixation probability, pfix(s) = 2s/(1 − e−2Ns). The beneficial substitution rate is therefore given by Rb = 2NUbsb. For this solution to be self-consistent, we require that no new beneficial mutations establish during the fixation time of the driver, or
| (B4) |
This also implies that Rb ≪ sb.
Multiple-Driver Mutations
In large populations, the rare driver condition in Equation B4 breaks down and multiple beneficial mutations will segregate in the population at the same time. In this case, the mean-field fitness distribution is no longer a δ-function (with discrete jumps when drivers fix), but rather an extended traveling wave f(X, t) = f(X − vt) that steadily increases in fitness at rate v. Without loss of generality, we can assume that the mean fitness of the population is zero when the new mutation arises. Then we can change variables to the relative fitness, so that Equations A3a and A6 become
| (B5a) |
| (B5b) |
with as described in the text. For this solution to be self-consistent, we require that pfix(0) ≈ 1/N; i.e., exactly one individual from the current population will become a future common ancestor (Hallatschek 2011; Good et al. 2012; Fisher 2013). This serves to completely determine v, f(x) and w(x) as a function of N and Ubρb(s). There are many different regimes to consider (see Fisher 2013 for additional discussion), but we focus on a particular case that is relevant for many microbial evolution experiments. We assume that the typical background fitness of a successful driver is much larger than the standard deviation of the fitness distribution (xbg ≫ σ). This ensures that there is a substantial amount of clonal interference in the population, which is consistent with the empirical observation that the fates of drivers strongly depend on background fitness (Lang et al. 2013). Second, we assume that the fitness effect of a typical driver is also much larger than the standard deviation of the fitness distribution This ensures that there is not too much clonal interference in the population and is consistent with direct measurements of the fitness distribution (Desai et al. 2007) and forward-time simulations, using parameters inferred from marker divergence experiments (Frenkel et al. 2014). In terms of the underlying parameters, these two conditions apply whenever (Desai and Fisher 2007; Fisher 2013).
Assuming that these two conditions are met, we can use the approximate solution to Equation B5 derived in Good et al. (2012). This earlier work focused on the rate of adaptation and the distribution of fixed beneficial mutations, which were relatively insensitive to the approximate forms of f(x) and w(x) that we employed. In contrast, the perturbative analysis described in the text is much more sensitive to the precise details of our approximation scheme. Fortunately, we can obtain a suitable generalization of the analysis in Good et al. (2012) by enforcing a basic symmetry constraint: we demand that the approximate expression for w(x) varies self-consistently under infinitesimal boosts (i.e., changes in v) and translations (changes in ). The resulting solution will still be incorrect in several key ways (see Fisher 2013 for additional discussion), but it will be sufficient to calculate the leading-order corrections from deleterious mutations at the level of approximation required here.
When the contributions from the mutation terms in Equation B5 are small for most of the relevant fitnesses. For sufficiently large x, the fixation probability satisfies the reduced equation
| (B6) |
which has the solution
| (B7) |
Here, is a constant of integration that must be set to ensure that w(x) matches onto the correct branch of the solution for smaller x. The solution in Equation B7 has a characteristic “shoulder” shape. For the fixation probability saturates to the Haldane limit w(x) ≈ 2x, which reflects a dominant balance between the selection [xw(x)] and drift [w(x)2/2] terms in Equation B6. In this regime, a new mutation will fix provided that it survives genetic drift (Good et al. 2012). For the fixation probability is rapidly reduced due to clonal interference, which reflects a dominant balance between the selection [xw(x)] and mean fitness [v∂xw(x)] terms in Equation B6. In this regime, a new mutation will fix only if it can generate further beneficial mutations to outrun the steady increase in the mean fitness. The width of the crossover between these two regimes is of order v/xc, which becomes increasingly sharp in the limit that
Since the fixation probability must vanish when x → −∞, it is clear that the shoulder solution breaks down for smaller values of x where the effects of “lucky” beneficial mutations become important. In this regime, w(x) will depend rather sensitively on the precise shape of the DFE, since a mutation will survive only if it is rescued by an usually large driver mutation. Yet by definition, successful mutations that land in this regime are atypical and represent a small fraction of all substitutions. We therefore introduce a negligible amount of error by assuming that w(x) vanishes below a certain threshold, so that
| (B8) |
Note that the functional form of Equation 14 is independent of Ubρb(s), which enters only through location of xmin and xc. In Good et al. (2012), we had previously assumed that xmin ≈ 0, but if taken literally, this leads to the pathological behavior pointed out by Fisher (2013). For the present analysis, we assume only that which is sufficient to ensure that xmin drops out of the analysis in Good et al. (2012). The precise value of xmin is set by the symmetry considerations below. As described in Good et al. (2012), the location of xc can be obtained from an integral transform of Equation B5b, which reduces to
in the limit that and To satisfy the symmetry constraints below, we need to introduce an (1) “fudge factor” F, so that this relation is instead given by
| (B9) |
Then we can substitute our approximate expression for w(x) from Equation B8 and evaluate the integrals to obtain a condition for xc,
| (B10) |
where the effective selection coefficient and mutation rate are given by Equation 24 in the main text. Together, these expressions completely determine w(x) as a function of v and Ubρb(s).
An approximate expression for the fitness distribution can be obtained in a similar manner. Ignoring the mutation term, f(x) satisfies the reduced equation −∂xf(x) = xf(x), which has a simple Gaussian solution with mean zero and variance σ2 = v. This solution is valid for fitnesses up to x ≈ xc, after which the input from the mutation term causes f(x) to rapidly approach zero and eventually turn negative (Rouzine et al. 2008; Goyal et al. 2012; Fisher 2013). This is an artifact of our mean-field approximation, which neglects the increasingly important effects of drift near x ≈ xc. However, like the behavior of the fixation probability for x ≪ xmin, mutations that originate from x ≫ xc are highly atypical (since they rely on a chance fluctuation of the fitness distribution) and therefore constitute a relatively small fraction of all substitutions. We therefore introduce a negligible amount of error by assuming that f(x) vanishes above xc, so that
| (B11) |
Then we can substitute our approximate expressions for f(x) and w(x) into the consistency condition and obtain a second relation,
| (B12) |
which uniquely determines v as a function of N and Ubρ(s).
To determine xmin and F, we enforce a symmetry constraint on our approximate solution for w(x). Specifically, we assume that under infinitesimal boosts and translations, we should get the same expression for w(x) whether we expand our approximate solution or solve Equation B5b perturbatively. Although this may seem like an abstract requirement, these symmetry transformations turn out to be intimately related to the sd → 0 and sd → ∞ limits of the full deleterious model.
First, suppose that we perform an infinitesimal boost by perturbing v = v0(1 + ε), with ε ≪ 1. This is equivalent to adding an additional term εv0∂xw(x) to the left-hand side of Equation B5b. According to our analysis above, the nonperturbative solution is
| (B13) |
where δx and (and the corresponding δs and δU) are defined by
| (B14a) |
| (B14b) |
| (B14c) |
From the definition of there are no contributions to δx from δs and δU to lowest order in ε, which shows that
| (B15) |
and
| (B16) |
where we have defined Then for any test function ζ(x), we have
| (B17) |
and we see that the contribution from the nonperturbative boundary layer between xc and vanishes to lowest order in ε. Thus, by solving Equation B5b and then expanding, we find that
| (B18) |
Now we try to obtain this solution by expanding Equation B5b in powers of ε and solving perturbatively. To that end, we rewrite w(x) in the form w(x) = w0(x)[1 + εg(x)], which yields a related equation for g(x):
| (B19) |
We can solve this equation using the same approximation methods that we used for Equation B5b above. For x ≫ xc, the mutation and mean fitness terms can be ignored, and we have
| (B20) |
Meanwhile, for x < xc, the drift and mutation terms can be ignored, and we find that
| (B21) |
where C is a constant of integration. Since w(x) is continuous at x = xc, it is tempting to fix C by demanding that g(x) is also continuous at xc, but this would not be correct. We saw in Equation B16 that w(x) develops a nonperturbative boundary layer between xc and In this region (which does not contribute to any integrals at leading order), w(x) changes rapidly to ensure continuity, but the perturbative correction g(x) is not continuous. Instead, we fix C using the integral transform above that we used to determine xc. This yields the relation
| (B22) |
which reduces to
| (B23) |
and hence
| (B24) |
Thus, we see that the perturbative solution matches our previous expression in Equation B18 provided that
| (B25) |
This provides one relation between xmin and F and is sufficient to show that
Now we repeat this analysis for an infinitesimal translation, x → x − εxc, which is equivalent to adding a −εxcw(x) term in Equation B5b. In this case, the nonperturbative solution is
| (B26) |
which, when expanded to first order in ε, yields
| (B27) |
Again, the boundary layer does not contribute to any integrals of w(x) at leading order, so we can simply drop it from our expansion to obtain
| (B28) |
We now try to obtain this solution in the opposite order by expanding Equation B5b and solving perturbatively. Defining w(x) = w0(x)[1 + εg(x)], we see that g(x) satisfies
| (B29) |
whose solution is simply
| (B30) |
The constant C is again determined by the integral condition
| (B31) |
which shows that
| (B32) |
This perturbative solution matches our previous expression in Equation B28 if
| (B33) |
which provides a second relation between xmin and F. Combined with the first relation in Equation B25, this shows that
| (B34) |
This completes our analysis of the zeroth-order solution in the multiple-mutations regime.
The Infinitesimal Limit
In addition to the “strong driver” limit we can also analyze the behavior of the multiple-driver regime in the “weak driver” limit A particularly simple case is the infinitesimal limit, where Ub → ∞ and sb → 0 in such a way that the product remains fixed (Tsimring et al. 1996; Cohen et al. 2005; Neher et al. 2010; Hallatschek 2011; Neher and Shraiman 2011; Neher and Hallatschek 2013; Neher et al. 2013; Good et al. 2014). Since sb → 0, the driver mutations behave as if they were effectively neutral, so the substitution rate is simply
| (B35) |
However, the overall evolutionary dynamics are far from neutral. Since we are simultaneously taking Ub → ∞, there are enough infinitesimal drivers segregating within the population that the total variance in fitness remains finite. It is straightforward to solve for the distribution of fitnesses and the fixation probability in this limit, since the mutation terms in Equation B5 can be expanded in a Taylor series,
| (B36a) |
| (B36b) |
where σ2 = v − Us is the variance in fitness within the population. In large populations, w(x) again develops a sharp boundary layer near x ≈ xc, above which it approaches the Haldane form w(x) ≈ 2x. Below xc, f(x) and w(x) both satisfy a modified Airy equation, so that
| (B37) |
| (B38) |
where Ai(z) is the solution to the Airy equation that converges for large z (Hallatschek 2011). The full solution is obtained by matching w(x) and its derivative at x = xc. For large N, the argument of the Airy function will be close to the first zero, z0 ≈ −2.33 (Fisher 2013). Expanding around this point, we find that
| (B39) |
where the relationship between xc and σ2 (i.e., the analogue of Equation 16) is given by
| (B40) |
Finally, we can solve for σ2 by substituting these expressions into the self-consistency condition π(0) ≈ 1/N, which yields
| (B41) |
in the limit of large N (Tsimring et al. 1996; Cohen et al. 2005; Hallatschek 2011).
Appendix C: Leading-Order Corrections from Deleterious Mutations
In this section, we derive the leading-order corrections to Rb and Rd in the presence of deleterious mutations. As with any perturbative calculation, this will lean heavily on the zeroth-order (Ud = 0) solutions derived in Appendix B, which we denote using subscripts/superscripts [e.g., , v0, f0(x)].
Rare Driver Mutations
In the rare driver regime, adaptation is a highly nonequilibrium process, since drivers arise and fix on very different timescales. Johnson and Barton (2002) have previously investigated the effects of deleterious mutations in this regime numerically, using the same branching process approximations employed in the present work. Our analysis therefore constitutes a special case of their method, valid in the limit that Ud → 0. By restricting the parameter regime in this way, we obtain simple analytical formulas for the beneficial and deleterious substitution rates that can be compared to the multiple-driver regime below.
If we assume that the last successful driver fixed t generations ago, then the leading-order deleterious corrections to f(X, t) are given by Equation 7 in the main text, which has mean fitness When a new driver arises, it creates a new subpopulation g(X) that sweeps through the population, provided that it survives genetic drift (see Appendix A). There are three different types of successful drivers:
-
1.
The driver occurs on an f(0) background and g(sb) survives drift. This is a classic “unburdened” sweep, since the g(sb) lineage will come to dominate the population. We denote the probability of this event by f(0, t)pfix(0, 0, t).
-
2.
The driver occurs on an f(0) background and g(sb) goes extinct, but not before it creates an additional deleterious mutation in g(sb − sd) that survives drift. The driver mutation is successful, but it carries a deleterious passenger. We denote the probability of this event by f(0, t)pfix(0, 1, t).
-
3.
The driver occurs on an f(−sd) background and g(sb − sd) survives drift, so the deleterious background hitchhikes to fixation. We denote the probability of this event by f(−sd, t)pfix(1, 1, t).
From our analysis in Appendix A, the fixation probabilities are given by the long-time behavior of φ(X, τ),
| (C1a) |
| (C1b) |
where φ(X, τ) satisfies
| (C2a) |
| (C2b) |
This system of equations is difficult to solve in general (Johnson and Barton 2002), but they are straightforward to solve perturbatively in the limit that Ud is small. To that end, we rewrite φ(x) in the form As described in Appendix B, the zeroth-order stationary solutions are given by
| (C3) |
and the first-order correction φ1(sb, τ) satisfies the linearized equation
| (C4) |
with the initial condition φ1(sb, 0) = 0. This equation can be solved using elementary methods, and we find that
| (C5a) |
| (C5b) |
| (C5c) |
Given these fixation probabilities, successful sweeps arise as an inhomogeneous Poisson process with rate
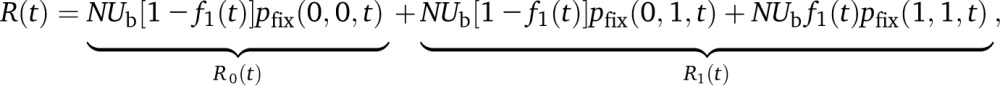 |
(C6) |
where R1(t) and R0(t) are the rates of burdened and unburdened sweeps, respectively. The average time between sweeps is given by
| (C7) |
and the probability of a deleterious mutation hitchhiking on any given sweep is
| (C8) |
Thus, the beneficial and deleterious substitution rates are given by
| (C9a) |
| (C9b) |
in agreement with Equations 9 and 10 in the text.
Multiple-driver mutations
The deleterious corrections in the multiple-driver regime are more straightforward to calculate, since the time dependence of f(X, t) is already accounted for in the steady-state traveling wave. Substituting Equation 17 into Equations 11 and 12 and equating powers of Ud/xc, we obtain a set of linearized equations for g(x) and h(x),
| (C10a) |
| (C10b) |
where we have defined A similar expansion of the consistency condition, yields a third relation,
| (C11) |
which uniquely determines g(x), h(x), and η. To carry out this calculation, it is useful to separate g(x) and h(x) into parts that depend on η and parts that depend on sd:
| (C12) |
where the individual components satisfy
| (C13a) |
| (C13b) |
| (C13c) |
| (C13d) |
Substituting these definitions into the consistency condition in Equation C11, we transform the implicit equation for η into an explicit formula
| (C14) |
We must then simply solve for the g’s and h’s and evaluate the required integrals.
We begin by focusing on gη(x). Here, the solution is identical to the “boost” transformation discussed in Appendix B, with Thus, we can immediately conclude that
| (C15) |
Similarly for hη(x), we find that and after renormalizing,
| (C16) |
Combining these expressions for gη(x) and hη(x), we can simplify the denominator of Equation C14, so that
| (C17) |
Now we turn our attention to gs(x) and hs(x). For the latter, we find that
| (C18) |
and hence after renormalization
| (C19) |
For gs(x), we can neglect the first-derivative term when x > xc, so that
| (C20) |
Meanwhile, for x < xc we can neglect the w0(x)gs(x) term and obtain gs(x) by direct integration:
| (C21) |
To solve for C, we return to the same integral transform that we used for the symmetry transformations in Appendix B. For infinitesimal translations, we already saw that the terms do not contribute to C in this regime. The remaining terms yield
| (C22) |
and hence
| (C23) |
Due to the separation of fitness scales and we can keep only the zeroth-order terms in which yields
| (C24) |
Putting all of this together in our expression for η, we see that the xxc/v0 terms from the gs(x) and hs(x) integrals cancel, and we obtain
| (C25) |
| (C26) |
The last term can be neglected since and we find that
| (C27) |
The Infinitesimal Limit
The leading-order corrections in the infinitesimal limit can be obtained in a similar manner. In this case, however, it makes more sense to partition the fitness effects into deleterious and infinitesimal components,
| (C28) |
rather than the beneficial/deleterious division in Equation 1. Here, ρi(s) can also contain deleterious fitness effects as long as they are sufficiently close to the infinitesimal limit (Tc|s| ≪ 1). Recall that the mutational diffusion constant does not depend on the sign of s, so these results will also apply when the fitness of the population is declining due to Muller’s ratchet. Provided that Ud ≪ xc, the substitution rate for the remaining deleterious mutations is given by
| (C29) |
where the last approximation captures the interesting dependence in the large-N limit where σ2 ≫ D2/3. Neher and Hallatschek (2013) have recently shown that the coalescent timescale is given by in this regime, so for fixed Tc, Equation C29 is identical to Equation 19 derived in the text. In this case, however, we see that
| (C30) |
which increases weakly with population size. This implies that larger populations will tend to fix fewer and more weakly deleterious mutations, similar to the behavior in the single-locus case.
To calculate the change in the infinitesimal substitution rate, we must solve for the corrections to w(x) and f(x) in the same manner as the previous section. In this case, however, we note that the infinitesimal substitution rate formally diverges in the infinitesimal limit (Ui → ∞), so the fractional change in Rb is simply
| (C31) |
Footnotes
Communicating editor: N. H. Barton
Literature Cited
- Bachtrog D., Gordo I., 2004. Adaptive evolution of asexual populations under Muller’s ratchet. Evolution 58: 1403–1413. [DOI] [PubMed] [Google Scholar]
- Barrick J. E., Yu D. S., Yoon S. H., Jeong H., T. K. Oh et al. , 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461: 1243–1247. [DOI] [PubMed] [Google Scholar]
- Barroso-Batista, J., A. Sousa, M. Lourenco, M.-L. Bergman, J. Demengeot et al., 2014 The first steps of adaptation of Escherichia coli to the gut are dominated by soft sweeps. arXiv:1311.2435. [DOI] [PMC free article] [PubMed]
- Barton N. H., 1995. Linkage and the limits to natural selection. Genetics 140: 821–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batorsky R., Kearney M. F., Palmer S., Maldarelli F., Rouzine I., et al. , 2011. Estimate of effective recombination rate and average selection coefficient for HIV in chronic infection. Proc. Natl. Acad. Sci. USA 108: 5661–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., 1994. The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet. Res. 63: 213–227. [DOI] [PubMed] [Google Scholar]
- Chou H.-H., Chiu H.-C., Delaney N. F., Segre D., Marx C. J., 2011. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332: 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E., Kessler D. A., Levine H., 2005. Front propagation up a reaction rate gradient. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 72: 066126. [DOI] [PubMed] [Google Scholar]
- Covert A. W., III, Lenski R. E., Wilke C. O., Ofria C., 2013. Experiments on the role of deleterious mutations as stepping stones in adaptive evolution. Proc. Natl. Acad. Sci. USA 110: E3171–E3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser J. A. G. M., Zeyl C. W., Gerrish P. J., Blanchard J. L., Lenski R. E., 1999. Diminishing returns from mutation supply rate in asexual populations. Science 283: 404–406. [DOI] [PubMed] [Google Scholar]
- Desai M. M., Fisher D. S., 2007. Beneficial mutation selection balance and the effect of genetic linkage on positive selection. Genetics 176: 1759–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M. M., Fisher D. S., 2011. The balance between mutators and nonmutators in asexual populations. Genetics 188: 997–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M. M., Fisher D. S., Murray A. W., 2007. The speed of evolution and the maintenance of variation in asexual populations. Curr. Biol. 17: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M. M., Walczak A. M., Fisher D. S., 2013. Genetic diversity and the structure of genealogies in rapidly adapting populations. Genetics 193: 565–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena S. F., Ekunwe L., Hajela N., Oden S. A., Lenski R. E., 1998. Distribution of fitness effects caused by random insertion mutations in Escherichia coli. Genetica 102/103: 349–358. [PubMed] [Google Scholar]
- Etheridge, A., P. Pfaffelhuber, and A. Wakolbinger, 2007 How often does the ratchet click? Facts, heuristics, and asymptotics, pp. 365–390 in Trends in Stochastic Analysis, edited by P. M. J. Blath and M. Scheutzow. Cambridge University Press, Cambridge, UK/London/New York. [Google Scholar]
- Eyre-Walker A., Keightley P. D., 2007. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8: 610–618. [DOI] [PubMed] [Google Scholar]
- Fisher D. S., 2013. Asexual evolution waves: fluctuations and universality. J. Stat. Mech. 2013: P01011. [Google Scholar]
- Fisher R. A., 1930. The distribution of gene ratios for rare mutations. Proc. R. Soc. Edinb. 50: 204–219. [Google Scholar]
- Frenkel E. M., Good B. H., Desai M. M., 2014. The fates of mutant lineages and the distribution of fitness effects of beneficial mutations in laboratory budding yeast populations. Genetics 196: 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel W., Lynch M., Burger R., 1993. Muller’s ratchet and mutational meltdowns. Evolution 47: 1744–1757. [DOI] [PubMed] [Google Scholar]
- Gillespie J. H., 2000. Genetic drift in an infinite population: the pseudohitchhiking model. Genetics 155: 909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good B. H., Desai M. M., 2013. Fluctuations in fitness distributions and the effects of weak linked selection on sequence evolution. Theor. Popul. Biol. 85: 86–102. [DOI] [PubMed] [Google Scholar]
- Good B. H., Rouzine I. M., Balick D. J., Hallatschek O., Desai M. M., 2012. Distribution of fixed beneficial mutations and the rate of adaptation in asexual populations. Proc. Natl. Acad. Sci. USA 109: 4950–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good B. H., Walczak A. M., Neher R. A., Desai M. M., 2014. Genetic diversity in the interference selection limit. PLoS Genet. 10: e1004222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal S., Balick D. J., Jerison E. R., Neher R. A., Shraiman B. I., et al. , 2012. Dynamic mutation-selection balance as an evolutionary attractor. Genetics 191: 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigh J., 1978. The accumulation of deleterious genes in a population. Theor. Popul. Biol. 14: 251–267. [DOI] [PubMed] [Google Scholar]
- Hallatschek O., 2011. The noisy edge of traveling waves. Proc. Natl. Acad. Sci. USA 108: 1783–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfield M., Otto S. P., 2011. Recombination and hitchhiking of deleterious alleles. Evolution 65: 2421–2434. [DOI] [PubMed] [Google Scholar]
- Hegreness M., Shoresh N., Hartl D., Kishony R., 2006. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science 311: 1615–1617. [DOI] [PubMed] [Google Scholar]
- Jiang X., Xu Z., Li J., Shi Y., Wu W., et al. , 2011. The influence of deleterious mutations on adaptation in asexual populations. PLoS ONE 6: e27757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T., 1999. The approach to mutation-selection balance in an infinite asexual population, and the evolution of mutation rates. Proc. Biol. Sci. 266: 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T., Barton N. H., 2002. The effect of deleterious alleles on adaptation in asexual populations. Genetics 162: 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen R., Bataillon T., 2006. Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria. Nat. Genet. 38: 484–488. [DOI] [PubMed] [Google Scholar]
- Kawecki T. J., Lenski R. E., Ebert D., Holis B., Olivieri I., et al. , 2012. Experimental evolution. Trends Ecol. Evol. 27: 547–560. [DOI] [PubMed] [Google Scholar]
- Khan A. I., Din D. M., Schneider D., Lenski R. E., Cooper T. F., 2011. Negative epistasis between beneficial mutations in an evolving bacterial population. Science 332: 1193–1196. [DOI] [PubMed] [Google Scholar]
- Kimura M., 1968. Evolutionary rate at the molecular level. Nature 217: 624–626. [DOI] [PubMed] [Google Scholar]
- Kimura M., Maruyama T., 1966. The mutational load with epistatic gene interactions in fitness. Genetics 54: 1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. L., Jukes T. H., 1969. Non-Darwinian evolution. Science 164: 788–798. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S. K., Rice D. P., Jerison E. R., Desai M. M., 2014. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science 344: 1519–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek D. J., Sherlock G., 2013. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 9: e1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang G. I., Rice D. P., Hickman M. J., Sodergren E., Weinstock G. M., et al. , 2013. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500: 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.-C., Marx C. J., 2013. Synchronous waves of failed soft sweeps in the laboratory: remarkably rampant clonal interference of alleles at a single locus. Genetics 193: 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman T. D., Flett K. B., Yelin I., Martin T. R., McAdam A. J., et al. , 2014. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet. 46: 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łuksza M., Lässig M., 2014. A predictive fitness model for influenza. Nature 507: 57–61. [DOI] [PubMed] [Google Scholar]
- Maharjan R. P., Liu B., Li Y., Reeves P. R., Wang L., et al. , 2013. Mutation accumulation and fitness in mutator subpopulations of Escherichia coli. Biol. Lett. 9: 20120961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J., Haigh J., 1974. Hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McDonald M., Hsieh Y., Yu Y., Chang S., Leu J., 2012. The evolution of low mutation rates in experimental mutator populations of Saccharomyces cerevisiae. Curr. Biol. 22: 1235–1240. [DOI] [PubMed] [Google Scholar]
- McFarland, C., L. Mirny, and K. S. Korolev, 2014 A tug-of-war between driver and passenger mutations in cancer and other adaptive processes. arXiv:1402.6354. [DOI] [PMC free article] [PubMed]
- McFarland C. D., Korolev K. S., Kryukovc G. V., Sunyaev S. R., Mirny L. A., 2013. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 110: 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. R., Joyce P., Wichman H. A., 2011. Mutational effects and population dynamics during viral adaptation challenge current models. Genetics 187: 185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles R., Gerrish P. J., Moya A., Elena S. F., 1999. Clonal interference and the evolution of RNA viruses. Science 285: 1745–1747. [DOI] [PubMed] [Google Scholar]
- Muller H. J., 1964. The relation of recombination to mutational advance. Mutat. Res. 1: 2–9. [DOI] [PubMed] [Google Scholar]
- Neher R., Shraiman B. I., 2011. Genetic draft and quasi-neutrality in large facultatively sexual populations. Genetics 188: 975–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher R., Shraiman B. I., Fisher D. S., 2010. Rate of adaptation in large sexual populations. Genetics 184: 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher R. A., Hallatschek O., 2013. Genealogies in rapidly adapting populations. Proc. Natl. Acad. Sci. USA 110: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher R. A., Kessinger T. A., Shraiman B. I., 2013. Coalescence and genetic diversity in sexual populations under selection. Proc. Natl. Acad. Sci. USA 110: 15836–15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zinal S., Loo P. V., Wedge D. C., L. B. Alexandrov, C. D. Greenman et al., 2012. The life history of 21 breast cancers. Cell 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T., 1973. Slightly deleterious mutant substitutions in evolution. Nature 246: 96–98. [DOI] [PubMed] [Google Scholar]
- Orr H. A., 2000. The rate of adaptation in asexuals. Genetics 155: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrow S. L., Barshir R., DeGregori J., Yeger-Lotem E., Hershberg R., 2014. Cancer evolution is associated with pervasive positive selection on globally expressed genes. PLoS Genet. 10: e1004239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito L., Fernandes L., Mota C., Gordo I., 2007. Adaptive mutations in bacteria: high rate and small effects. Science 317: 813. [DOI] [PubMed] [Google Scholar]
- Peck J. R., 1994. A ruby in the rubbish. Genetics 137: 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pybus O. G., Rambaut A., Belshaw R., Freckleton R. P., Drummond A. J., et al. , 2007. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 24: 845852. [DOI] [PubMed] [Google Scholar]
- Qian W., Ma D., Xiao C., Wang Z., Zhang J., 2012. The genomic landscape and evolutionary resolution of antagonistic pleiotropy in yeast. Cell Rep. 2: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine I., Brunet E., Wilke C., 2008. The traveling-wave approach to asexual evolution: Muller’s ratchet and the speed of adaptation. Theor. Popul. Biol. 73: 24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine I. M., Wakeley J., Coffin J. M., 2003. The solitary wave of asexual evolution. Proc. Natl. Acad. Sci. USA 100: 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Moya A., Elena S. F., 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101: 8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffels S., Szöllösi G., Mustonen V., Lässig M., 2011. Emergent neutrality in adaptive asexual evolution. Genetics 189: 1361–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silander O. K., Tenaillon O., Chao L., 2007. Understanding the evolutionary fate of finite populations: the dynamics of mutational effects. PLoS Biol. 5: e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski P. D., Gerrish P. J., 2010. Beneficial mutations and the dynamics of adaptation in asexual populations. Philos. Trans. R. Soc. B 365: 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg R. J., Berg O. G., 2007. Mutational interference and the progression of Muller’s ratchet when mutations have a broad range of deleterious fitness effects. Genetics 177: 971–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelkowa N., Lässig M., 2012. Clonal interference in the evolution of influenza. Genetics 192: 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimring L., Levine H., Kessler D., 1996. RNA virus evolution via a fitness-space model. Phys. Rev. Lett. 90: 088103. [DOI] [PubMed] [Google Scholar]
- Weissman D., Hallatschek O., 2014. The rate of adaptation in large sexual populations with linear chromosomes. Genetics 196: 1167–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgoss S., Barrick J. E., Tenaillon O., Wiser M. J., W. J. Ditmaret al. , 2013. Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc. Natl. Acad. Sci. USA 110: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke C. O., 2004. The speed of adaptation in large asexual populations. Genetics 167: 2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiser M. J., Ribeck N., Lenski R. E., 2013. Long-term dynamics of adaptation in asexual populations. Science 342: 1364–1367. [DOI] [PubMed] [Google Scholar]
- Wloch D. M., Szafraniec K., Borts R. H., Korona R., 2001. Direct estimate of the mutation rate and the distribution of fitness effects in the yeast Saccharomyces cerevisiae. Genetics 159: 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock G., Higgs P. G., 1996. Population evolution on a multiplicative single-peak fitness landscape. J. Theor. Biol. 179: 61–73. [DOI] [PubMed] [Google Scholar]
- Wright S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Siegal M. L., Hall D. W., Petrov D. A., 2014. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl. Acad. Sci. USA 111: E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]