

Abstract
We detail an investigation of a peptide-based catalyst 6 that is effective for the site- (>100:1:1) and enantioselective epoxidation (86% ee) of farnesol. Studies of the substrate scope exhibited by the catalyst are included, along with an exploration of optimized reaction conditions. Mechanistic studies are reported, including relative rate determinations for the catalyst and propionic acid, a historical perspective, truncation studies, and modeling using NMR data. Our compiled data advances our understanding of the inner workings of a catalyst that was identified through combinatorial means.
Introduction
The recorded study of catalytic asymmetric epoxidation reads like an epic.1 Epoxidation methods populate a large swath of the chemical literature because of the synthetic utility of epoxide products.2 Methods based on transition metals that allow for the use of directing groups (e.g., allylic alcohol bearing substrates) have achieved high levels of enantioselectivity and have served as benchmarks as the field has strived for expanded substrate scope. More remote directing groups,3 and metal-free based catalysts4–11 have also proven effective for various classes of olefins. So, too, have directing group-free processes proven to be of great importance.12
As part of an effort to explore the limits of effective catalysts based on “minimal peptides,”13 we have been studying an unusual catalytic cycle for epoxidation based on the proteinogenic amino acid aspartic acid (1, Figure 1). While this role is not known for aspartate in enzymatic catalysis, which instead most often employs co-factors for biosynthetic epoxidations,14 we have been intrigued about the possible intermediacy of aspartyl peracids (2) – co-factor-free intermediates – for peptide-based epoxidation catalysis. Our initial studies into the epoxidation of allylic carbamates8a,b were based on a catalytic cycle employing an aspartyl peptide catalyst, and hydrogen peroxide as the oxidant (Figure 1). The plausible catalytic cycle involves carboxyl activation with a carbodiimide reagent and capture of the derived active ester either as the carbodiimide adduct (3) or its nucleophile-derived adduct (e.g., 4) with hydrogen peroxide. The aspartyl peracid then delivers an O-atom to the olefin, regenerating the catalyst. This cycle proved to be robust and portable to other substrate types.15
Figure 1.
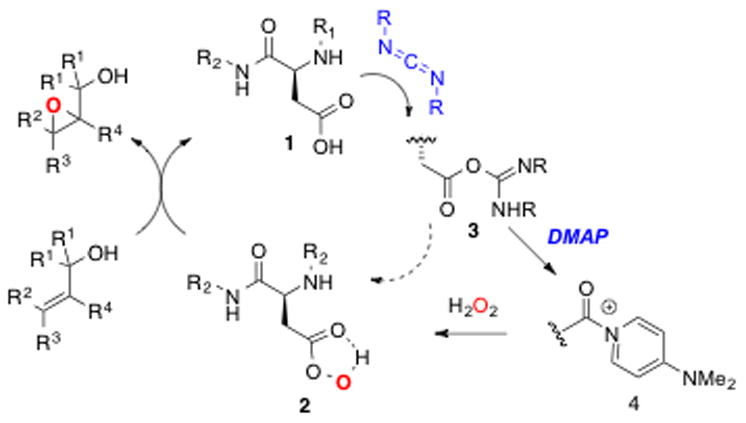
Aspartic acid-based peptide catalytic cycle.
A particular challenge for all types of asymmetric reactions is presented by substrates that possess multiple copies of the same functional group.16 In situations of this type, exemplified by farnesol (5, Scheme 1), the challenge of site selectivity is introduced alongside the challenge of stereoselectivity.17 We recently wondered if the aspartyl peptides might be adapted to meet the challenge of polyene epoxidation, given that this type of comprehensive selectivity challenge is often encountered in natural systems, and surmounted by enzymes.18 The challenge is particularly difficult in substrates like farnesol since, intrinsic to the substrate, there are reactivity hierarchies that render olefinic sites of differing reactivity. Thus, for a catalyst to be found that is selective for each site, some may have to overcome intrinsically low reactivity at a given position.
Scheme 1.

Catalytic epoxidations of farnesol.
We recently approached this challenge with farnesol employing a combinatorial split-and-pool approach.8d As shown in Scheme 1, we found a peptide-based catalyst 6 that is highly selective in forming the 2,3-epoxide of farnesol (8) over the 6,7-epoxide (9) or the 10,11-epoxide (10) with 1:1:>100 site selectivity (10:9:8) and 86% ee in forming 8. In addition, we found another catalyst (7) that exhibits highly unique site selectivity for the 6,7-position (1:8:1, 10:9:8) of farnesol, albeit with low enantioselectivity (10% ee). Notably, both out-comes are entirely different from the result one observes with either stoichiometric use of m-CPBA as the oxidant or when propionic acid is used as a catalyst under conditions analogous to those employed for catalysts 6 and 7. The unique behaviors of catalysts 6 and 7, along with the fact that each was discovered through a combinatorial screening that was relatively unencumbered by mechanistic hypotheses, have spawned studies of their catalytic mechanisms. We have recently reported studies targeted at understanding the mechanism of action of the 6,7-selective catalyst, 7.19 Herein, we describe studies targeted at elucidating the basis of the selectivity exhibited by the 2,3-selective catalyst (6), including an assessment of its substrate scope. Notably, while substantial insight may be gleaned about possible transition state ensembles, a conclusive single-transition state model does not fit the available data uniquely.
Results and discussion
Optimization of Reaction Conditions
We initially wished to explore substrate scope as part of a mechanistic inquiry that might shed light on the topological requirements of 6 in the transition states for various substrates. As part of this analysis, we revisited the reaction conditions, as the catalyst was projected to be less enantioselective for some substrates compared to farnesol.
Interestingly, in some cases the addition of Et3N led to higher enantioselectivity, but at the expense of isolated yield. The origin of this effect is not fully understood. However, it is possible that the presence of base disrupts carboxyl activation of the Asp side chain, which occurs under neutral conditions. Under neutral conditions, the acid and the carbodiimide are presumed to form the O-acyl urea as shown in Figure 2, Path A (purple). In the presence of base, however, it has been observed that O-acylurea formation is slower as the carboxylate salt (Figure 2, Path B, green).20 In either scenario, it is possible that catalyst deactivation through a base-assisted O-to N-acyl transfer from the activated carboxylate (Figure 2, red) might also conspire to reduce the efficiency of the catalysis in the presence of additional base.
Figure 2.

Catalyst deactivation in the presence of Et3N.
Substrate Scope
Encouraging results for enantioselective allylic epoxidation from our earlier report prompted us to further investigate the substrate scope of the reaction (Table 1). Previously reported results for prenol and nerol, which were epoxidized using previously reported conditions (a), indicated that (Z)-olefins were best suited for the system.8d Epoxynerol (11) can be synthesized with 93% ee and 79% yield and epoxyprenol (12) is formed in 92% ee and 75% gas chromatography (GC) yield (Table 1, entries 1 and 2, respectively). The optimal pattern for the substitution, in terms of steric bulk of substituents, remained an intriguing avenue for investigation.
Table 1.

| ||||
|---|---|---|---|---|
| Entry | Product | Conditions | Yield (%)f | ee (%)f |
| 18d |
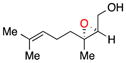 11 |
a | 79 | 93 |
|
| ||||
| 28d,d |
 12 |
a | 75 | 92 |
|
| ||||
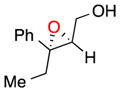
| 3 |
 13 |
b | 60 | 96 |
| c | 73 | 96 | ||
|
| ||||
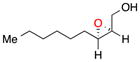
| 421a |
 14 |
b | 48 | 97 |
| c | 74 | 92 | ||
|
| ||||
| 521d |
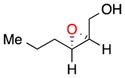 15 |
b | 46 | 91 |
| c | 29 | 91 | ||
|
| ||||
| 621a |
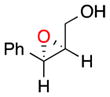 16 |
b | 54 | 83 |
|
| ||||
| 721e |
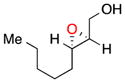 17 |
b | 55 | 75 |
| c | 47 | 74 | ||
|
| ||||
| 8b |
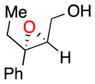 18 |
b | 36 | 67 |
|
| ||||
| 921f,b |
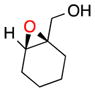 19 |
b | 29 | 63 |
| c | 43 | 61 | ||
|
| ||||
| 103c |
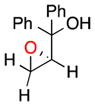 20 |
b | 9 | 86 |
| c | 10 | 84 | ||
|
| ||||
| 1121c |
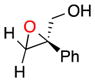 21 |
b | 13 | 13 |
Conditions a: Reaction run in CH2CI2. Conditions b: Reaction run with Et3N (0.1 equiv) in CHCI3. Conditions c: Reaction run in CHCI3.
Epoxidation was run over 2 days with 2 equivalents of DIC
Due to the volatity of the substrate, all of the crude material was derivatized with a benzoyl group. The yield reported is over two steps: epoxidation and derivatization.
Reported yieds and ee values are the average of triplicate expoxidation.
GC Yield
Average of duplicate epoxidations unless otherwise noted.
With these observations in mind, we proceeded to assess the substrate scope under two sets of conditions: (b) those that included the addition of triethylamine, and (c) those that lacked triethylamine. The scope reveals the effectiveness of peptide 6 with (Z)-trisubstituted alkenes. With base, (Z)-3-phenyl-pent-2-en-1-ol, gives its epoxide (13) in 60% yield and 96% ee (entry 3b). Without base, 13 can be isolated in 73% yield and 96% ee (entry 3c). (Z)-Disubstituted epoxides (entries 4–6) like 14 can also be synthesized with excellent selectivity (97% ee, condition b; 92% ee, condition c), and are perhaps the best suited for catalyst 6. Other cis alkenes like cis-hex-2-en-1-ol and cis-cinnamyl alcohol are also epoxidized in high enantioselectively. Compounds 15 and 16 can be isolated with 91% ee (46% yield) and 83% ee (54% yield) with base, (entries 5b and 6b, respectively). Without base, 15 is afforded in 29% yield and 91% ee (entry 5c).
(E)-Alkenes like trans-oct-2-en-1-ol can be epoxidized with somewhat lower ee. For example, 17 is afforded in 55% yield with 75% ee (entry 7b) or in 47% yield with 74% ee without a base additive (entry 7c). The improved performance of the peptide for (Z)- over (E)-alkenes is also apparent in the treatment of the two isomers of 3-phenyl-pent-2-en-1-ol. Whereas the (Z)-isomer is epoxidized in high yield and ee (entry 3), the corresponding (E)-3-phenyl-pent-2-en-1-ol is converted to 18 in 36% yield and 67% ee (entry 8). Analysis of cyclohex-1-enyl-1-methanol reveals that the substrate also exhibits lower selectivity. With base, 19 can be isolated in 29% yield and 63% ee (entry 9b). Without base, the yield increases to 43% and its ee goes down to 61% (entry 9c). We note that the stereochemical configuration of 19 is alternate to the other products we examined. The parent olefin leading to this outcome is unique in that it is cyclic and disubstituted at the 2-position. The nuances associated with catalyst-substrate interactions are addressed globally below.
The terminal, sterically encumbered 1,1-diphenylprop-2-en-1-ol gives epoxide 20 in low yields (9% and 10%, respectively), but with high ee (86% and 84%) under either set of reaction conditions (entries 10b and 10c, respectively). Catalyst 6 is particularly ineffective in discriminating the prochiral faces of 2-phenylprop-2-en-1-ol, producing 21 in 13% yield with 12% ee (entry 11). Our results with various olefins, especially those with Z-geometry, reveal a number of effective asymmetric epoxidations with good to excellent enantiomeric excesses.
Rate Studies
Investigations of the rate of the peptide-catalyzed process show epoxidation of cis-hex-2-en-1-ol is faster in the presence of peptide than it is the presence of propionic acid (Figure 3). The reaction was monitored by GC, tracking the disappearance of cis-hex-2-en-1-ol in aliquots of the reaction mixture relative to a dodecane internal standard. When the reaction is examined in the early stages of the reaction (the first 240 minutes), and treated as if possessing a profile that is first order in substrate, kobs for the catalyst 6-mediated reaction (1×10−2 M/min) is approaching two orders of magnitude faster than the propionic acid-catalyzed reaction (2×10−4 M/min; See Supporting Information for details). Furthermore, the selectivity achieved by catalyst 6 for the allylic position with farnesol serves as a competition experiment (i.e., the epoxidation of the allylic olefin is faster than other positions). These data show that the epoxidation event is accelerated by the peptide relative to propionic acid, suggesting hydrogen bonding contacts between catalyst 6 and the hydroxyl-bearing substrate.22
Figure 3.
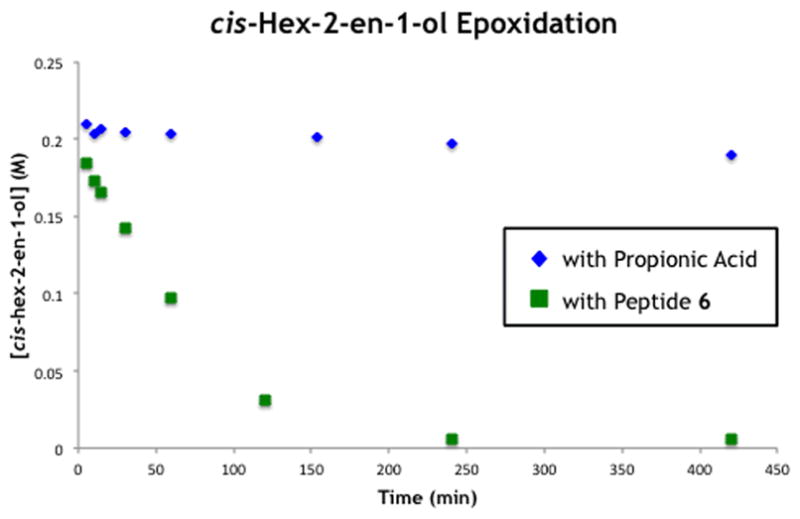
Plot of catalyst 6- and propionic acid-catalyzed epoxidation of cis-hex-2-en-1-ol. The experimental conditions match those employed for the reactions in Table 1. (See Supporting Information for details).
Analogue Studies
Our group has often found value in examining truncated peptide catalysts to understand how remote sequence elements influence selectivity.8e,23 While we had examined truncated versions of the catalyst 6 on-bead,8d we endeavored to perform this type of analysis on farnesol with peptides in solution (Table 2). While catalyst 6 delivers excellent selectivity in solution (1:1:>100, 10:9:8; 86% ee for 8), each of the further truncated analogues of 6 delivers incrementally worse selectivity. The steepest decline seems to occur with 22, a pentamer in which the C-terminal Asn(Trt)-OCH3 has been replaced with a methyl ester. Catalyst 22 delivers about a quarter of the enantiomeric ratio of 6, with site selectivity of 1.1:1.0:18 (10:9:8). It is important to note that the changes in site selectivity and enantioselectivity can be magnified by differences in conversion.24
Table 2.
Incremental truncation of catalyst 6 and the effect on catalysts’ selectivity.
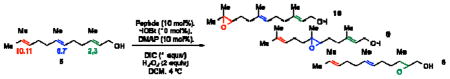
| |||||
|---|---|---|---|---|---|
| Catalyst | Entry | Site-Selective | Enantioselectivity | ||
| 10, 11-(10) | 6, 7-(9) | 2, 3-(8) | ee (or) of 8 | ||
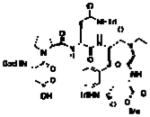 6 |
18d | 1.0 | 1.0 | >100 | 86% (13:1) |
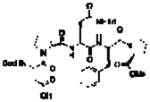 22 |
2 | 1.0 | 1.0 | 18 | 62% (4.2:1) |
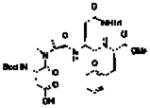 23 |
3 | 1.0 | 1.0 | 14 | 55% (3.5:1) |
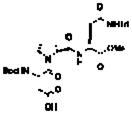 24 |
4 | 1.0 | 1.0 | 5.2 | 16% (1.4:1) |
The further truncated tetramer 23 exhibits another loss in selectivity, furnishing 1.0:1.0:14 (10:9:8) site selectivity and 8 in 55% ee. Removing the i + 3 residue to give the trimer 24 leads to further reduced site selectivity (1.0:1.0:5.2, 10:9:8) and enantioselectivity in forming 8 (16% ee). Interestingly, while the full catalyst 6 is clearly the most selective, none of the truncated peptides leads to a complete loss in selectivity for epoxide 8. We note parenthetically that losses of site selectivity correlate with reductions in the observed enantioselectivity within this series of truncated catalysts. It is interesting to speculate about the significance of the correlation. The drops in both metrics of selectivity relate to energetic separation of competing transition states, and perhaps also their multiplicity. The study of site selectivity, in this manner, may provide a window into the transition state ensemble that could be less visible when the binary read-out enantioselectivity alone is assessed.
In any case, these studies prompt two fundamental questions: What are the loci of peptide-substrate interactions and how does the C-terminal region of catalyst 6 promote selective epoxidation?
One interpretation of these truncation data is that catalyst 6’s essential points of interaction with substrate are localized to the N-terminal residues. Alternatively, the means by which catalyst 6 interacts with the substrates may change with each truncation. Henbest and others have demonstrated that hydroxyl-groups are capable of directing peracids with stereoselectivity by themselves.5,25 The influence of the Henbest-type mechanism in the present case is unclear, but catalyst 6 (or its analogues) may adopt conformations that favor this mode of epoxidation (Figure 4, A). It is also possible that the peptide directs the epoxidation exclusively through other interactions (C) or using a combination of these interactions (B).
Figure 4.
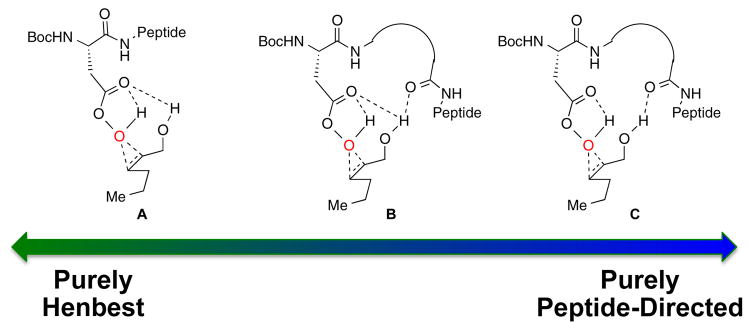
Gradient scale of possible transition states with generic peptide catalysts that range from a purely Henbest-like epoxidation to one in which enantioinduction is exclusively the result of interaction with the peptide.
The important role of the C-terminal region of catalyst 6 is also of interest. One possibility is that the disposition of this region helps the catalyst 6 adopt a more optimal conformation to achieve selectivity, perhaps stabilizing other interactions taking place between catalyst and substrate. In support of this hypothesis, the 1D 1H NMR of 22 indicates that there are at least two species present. It is likely that catalyst 22 exists as at least two conformational isomers.26 If removal of the C-terminal Asn(Trt) results in a number of conformations, it is possible that the i + 5 Asn(Trt) exists to bias the conformation of 6 toward a number of conformations that lead to favorable selectivity. Another scenario that is not supported by experiment is that the C-terminal region of catalyst 6 may interact directly with the substrate.
We may now also reflect on information that we garnered from previous catalysts from our screening libraries, which led to the development of catalyst 6. First, we note that a number of catalysts that we screened displayed favorable selectivity in the production of epoxide 8. Studies of the most site-selective library for farnesol epoxidation suggest that the identity of the i + 4 side chain (a DPhe in catalyst 6) may not be important, as a number of catalysts were identified with variability in this position. Given the broad sequence tolerance8d we decided to investigate the conformation of catalyst 6 in solution using 2D NMR techniques.
NMR Structure Elucidation
In pursuit of the nature of stereochemical information transfer, we investigated the solution structure of catalyst 6 employing NMR. A ROESY experiment yielded numerous data that allowed assignment of through-space interactions (Figure 5a). Crystallography and NMR Systems (CNS)27 was used to calculate plausible structures that satisfied NMR-derived distance restraints.28
Figure 5.

a. Unabridged ROESY-observed, through-space 1H-1H interactions. (See SI for rOe input classifications for CNS calculations.) b. Ensemble of the ten lowest scored structures of 6 produced by CNS to satisfy distances calculated from its ROESY spectrum with a proposed γ-turn. c. Three subgroupings of ensembles, within the ten structures shown in b, that are most closely related.
A variety of techniques may be brought to bear on the study of peptide-based catalyst structures29 Among these, modelling of NMR data using CNS required several decisions about data treatment. For instance, we found that the ranges (bins) to which the distance restraints were assigned influenced the subpopulations within the structure ensembles, so we have focused on the most consistent set of structures for presentation herein (see supporting information for details).
While the structures show some variations, there are several generalizations that may be found in the lowest energy structures as calculated by CNS. For example, these structures exhibit an internal hydrogen bond between i + 3 and i + 5 that is consistent with a γ-turn30 motif (Figure 5b) from the methyl-capped C-terminus to Asn(Trt) 6, Pro 5, and through the DPhe 4 residue (Figure 5b). When the ten structures that best fit our data are parsed out into three sub-ensembles (Figure 5c), it becomes clear that, despite the common γ-turn across the whole of the ensemble, our data accommodate multiple conformations of catalyst 6.
However, with the perceived importance of the C-terminal region of 6 on both structure and selectivity, we looked for other areas of homogeneity within the peptide (Figure 6a). Pairwise alignment of the non-hydrogen backbone atoms of the N-terminal portion of the sequence (Figure 6b, i), the central Asn(Trt) residue (Figure 6b, ii), and the C-terminal region (Figure 6b, iii) was carried out in Pymol31 using the NMR ensemble average structure as an arbitrary reference. The higher RMSD values of catalyst 6 for a given region of the sequence suggest more conformational possibilities, while low values imply fewer conformational possibilities. The i, i + 1, i + 2 residues of catalyst 6 in the NMR ensemble overlay with an RMSD value of 0.10 ± 0.05 (Figure 6b, portion i). Similarly, the i + 3, i + 4, i + 5 residues overlay with an RMSD value of 0.11 ± 0.06 (Figure 6b, portion iii). However, when same treatment is applied to the central portion of 6 (the i + 1, i + 2, and i + 3 residues), the structures within the ensemble diverge (Figure 6b, portion ii). The & backbone atoms of the i + 2 Asn(Trt) residue and the preceding and trailing atoms necessary to define its ϕ, ψ torsion angles overlay with an RMSD of 0.6 ± 0.6. This analysis suggests that both the proline-containing N- (Figure 6b, portion i) and C-termini (Figure 6b, portion iii) are more conformationally homogeneous relative to a more conformationally heterogeneous central portion of catalyst 6. Our CNS and truncation data indicate the C-terminus of catalyst 6 is essential to its selectivity. Yet, it is less clear whether this influence is through a direct interaction with the substrate, or a ‘buttressing’ effect that stabilizes the selective catalyst conformation.
Figure 6.

a. Ensemble of the ten lowest scored peptide conformations with portions highlighted for further analysis. b. Images of i and i + 1 (i); i + 1, i + 2, and i + 3 (ii); and i+3, i+4, and i + 5 (iii) with pair-wise fitting of all non-hydrogen backbone atoms in Pymol.
Our studies do not culminate in a unique model for asymmetric induction by catalyst 6. Instead, we are able to envision several structural arrangements that may accommodate an allylic alcohol, and in particular a Z-allylic alcohol, in a manner that is consistent with both the asymmetric reactions themselves and the ground state calculated NMR structures. With the oft-quoted caveat that the ground state conformations may not resemble transition state structures,32 we consider the three ensembles shown in Figure 7. Of particular note is that if the two better defined catalyst sectors (N-terminal region and separated C-terminal region) observed by NMR are preserved in the transition state for O-atom transfer, then it is plausible that an H-bonding array harnesses the directing hydroxyl group through several interactions.8e As shown in Figure 7a and 7c, the carbonyl oxygen atoms of i Asp and i + 1 Pro seem strong candidates for providing these contacts. An alternative arrangement is shown in Figure 7b, wherein the carbonyls of i Asp and i + 4 DPhe may present greater proximity to the i Asp side chain, housing the peracid moiety and the O-atom that is transferred. In each of the ensembles, the anchoring of the hydroxyl group of the Z-allylic alcohol to the peptide presents the face of the double bond that is oxidized proximal to the Asp side chain. For each of the other substrates, it is plausible that analogous hydrogen bonds may be formed, but that differential steric interactions may arrange the substrate such that the most accessible face of the olefin is oxidized. Simultaneously, the contribution of “Henbest-type” interactions (cf., Figure 4) may be superimposed upon the ensembles of Figure 7. These details seem beyond the resolution of our experimental interrogation of the system. However, a limited ensemble of possible transition states may be considered as a result of the analysis.
Figure 7.

Possible models for peptide-substrate interaction with catalyst 6. In each drawing, the aspartic acid side chain is drawn as the carboxylic acid itself (as opposed to the activated peracid species) since our NMR studies are of the acid. Conformational changes upon peracid formation are possible, but not readily observed under the conditions of our experiments.
Conclusions
In this study, we have probed the mechanistic basis of a peptide-based catalyst that is quite effective for the asymmetric epoxidation of certain types of allylic alcohols. The origin of the catalyst for these reactions was a combinatorial study that was minimally biased in terms of the nature of the peptide sequences that were originally introduced into the library. Of note, when peptides of shorter sequences were employed, particular drops in efficiency were observed – suggesting that essentially every piece of catalyst 6 contributes to its interesting performance.
On the other hand, examination of other substrates revealed that the substrate of the screen – farnesol – was not the “best” substrate for peptide catalyst 6. Some Z-configured allylic alcohols provide a higher level of enantioselectivity when subjected to oxidation with catalyst 6. It seems appropriate to wonder about the result of performing the entire combinatorial screening study anew, with Z-allylic alcohols as the test substrate. Would catalysts like 6 emerge as the best ones? Or could we design new catalysts for alternative substrates based on our mechanistic studies?
An analysis of the solution structures that may be populated by peptide 6 offers perspectives that are only partially guiding. We are able to significantly limit the field of possible catalyst conformations through our studies. Plausible loci of interactions between catalyst and substrate may be proposed. At the same time, kinetic experiments show unambiguously that catalyst 6 operates through a mechanism defined by a specific rate acceleration relative to that observed with an aliphatic carboxylic acid catalyst. This observation is consistent with our models and complementary to a very different sequence that delivers site selectivity of a different type (7, 6,7-selectivity within farnesol).8e While a unique transition state model that accounts for observed selectivity is not derived, features of members of an ensemble of structures that may contribute to the observed selectivity may be derived. Taken together, these studies establish further that combinatorial screens of peptide-based catalysts can often lead to catalysts of substantial prowess. The generality of the catalysts can vary, and their modes of operation can be difficult to establish definitively. In the end, it may be that these same challenges persist with catalysts that emanate from studies aptly called “design.” It remains a theme of some discovery-based research agendas that “how to do something” can be an easier question to address than “why does it work?”
Supplementary Material
Figure 8.
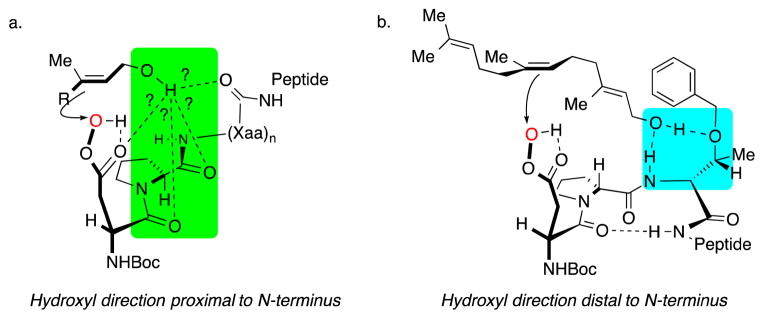
Composite of possible catalyst-substrate interactions that rationalize observed selectivity with: a. catalyst 6 for the epoxidation of allylic alchols and b. a single possibility for catalyst 7 in the epoxidation of the 6,7-olefin of farnesol.8e
Acknowledgments
The authors wish to thank Dr. Eric Paulson for discussions about NMR structure elucidation. This work was supported by National Institutes of Health (NIH R01-GM096403) to S.J.M. Additionally, P.A.L. was partially supported by NIH CBI-TB-GM-067543.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.(a) Sharpless KB. Angew Chem Int Ed. 2002;41:2024–2032. [PubMed] [Google Scholar]; (b) Katsuki T, Martin VS. Org React. 1996;48:1–299. [Google Scholar]; (c) Wong OA, Shi Y. Chem Rev. 2008;108:3958–3987. doi: 10.1021/cr068367v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Breuer M, Ditrich K, Habicher T, Hauer B, Keβeler M, Stürmer R, Zelinski T. Angew Chem, Int Ed. 2004;43:788–824. doi: 10.1002/anie.200300599. [DOI] [PubMed] [Google Scholar]; (b) Xi N, Alemany LB, Ciufolini MA. J Am Chem Soc. 1998;120:80–86. [Google Scholar]; (c) Chenault HK, Dahmer J, Whitesides GM. J Am Chem Soc. 1989;111:6354–6364. [Google Scholar]
- 3.(a) Zhang W, Yamamoto H. J Am Chem Soc. 2007;129:286–287. doi: 10.1021/ja067495y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhi L, Wei Z, Yamamoto H. Angew Chem Int Ed. 2008;47:7520–7522. doi: 10.1002/anie.200802523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Olivares-Romero JL, Zhi L, Yamamoto H. J Am Chem Soc. 2013;135:3411–3413. doi: 10.1021/ja401182a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li Z, Yamamoto H. J Am Chem Soc. 2010;132:7878–7880. doi: 10.1021/ja100951u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Zhu Y, Wang Q, Cornwall RG, Shi Y. Chem Rev [Online] 2014 doi: 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]; (b) Davis RL, Still J, Naicker T, Jiang H, Jorgensen KA. Angew Chem Int Ed. 2014;53 doi: 10.1002/anie.201400241. In Press. [DOI] [PubMed] [Google Scholar]
- 5.Henbest HB, Wilson RAL. J Chem Soc. 1957:1958–1965. [Google Scholar]
- 6.Tian H, She X, Shu L, Hongwu Y, Shi Y. J Am Chem Rev. 2000;122:11551–1552. [Google Scholar]
- 7.(a) Julia SN, Masana J, Vega JC. Angew Chem Int Ed. 1980;19:929–931. [Google Scholar]; (b) Julia S, Guizer J, Masana J, Rocas J, Colonna S, Annuziata R, Molinan H. J Chem Soc Perkins Trans 1. 1982:1317–1324. [Google Scholar]
- 8.(a) Peris G, Jakobsche CE, Miller SJ. J Am Chem Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jakobsche CE, Peris G, Miller SJ. Angew Chem Int Ed. 2008;47:6707–6711. doi: 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Romney DK, Miller SJ. Org Lett. 2012;14:1138–1141. doi: 10.1021/ol3000712. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lichtor PA, Miller SJ. Nature Chem. 2012;4:990–995. doi: 10.1038/nchem.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lichtor PA, Miller SJ. J Am Chem Soc. 2014;136:5301–5308. doi: 10.1021/ja410567a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang D, Yip Y-C, Tang M-W, Wong M-K, Zheng J-H, Cheng KK. J Am Chem Soc. 1996;118:491–492. [Google Scholar]
- 10.Davis FA, Havakal ME, Awad SB. J Am Chem Soc. 1983;105:3123–3126. [Google Scholar]
- 11.Aggarwal VK, Wang MF. Chem Comm. 1996:191–192. [Google Scholar]
- 12.(a) Zhang W, Loebach JL, Wilson SR, Jacobsen EN. J Am Chem Soc. 1990;112:2801–2803. [Google Scholar]; (b) Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J Am Chem Soc. 1997;119:11224–11235. [Google Scholar]
- 13.Jarvo ER, Miller SJ. Tetrahedron. 2002;58:2481–2495.Miller SJ. Acc Chem Res. 2004;37:601–610. doi: 10.1021/ar030061c.Colby-Davie E, Mennen SM, Xu Y, Miller SJ. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w.Wennemers H. Chem Commun. 2011;47:12036–12041. doi: 10.1039/c1cc15237h.For recent examples see: Mbofana CT, Miller SJ. J Am Chem Soc. 2014;136:3285–3292. doi: 10.1021/ja412996f.Metrano AJ, Miller SJ. J Org Chem. 2014;79:1542–1554. doi: 10.1021/jo402828f.
- 14.Thibodeaux CJ, Chang W-C, Liu H-W. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolundzic F, Noshi MN, Tjandra M, Movassagi M, Miller SJ. J Am Chem Soc. 2011;133:9104–9111. doi: 10.1021/ja202706g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robles O, Romo D. Nat Prod Rep. 2014;31:318–334. doi: 10.1039/c3np70087a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahatthananchai J, Dumas AM, Bode JM. Angew Chem Int Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- 18.Koeller KM, Wong C-H. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- 19.Lichtor PA, Miller SJ. ACS Comb Sci. 2011;13:321–326. doi: 10.1021/co200010v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan LC, Cox BC. J Org Chem. 2007;72:8863–8869. doi: 10.1021/jo701558y. [DOI] [PubMed] [Google Scholar]
- 21.Stereochemical assignments made my comparisons to the same epoxides in the following publications: Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780.Wang B, Wu XY, Wong OA, Nettles B, Zhao MX, Chen D, Shi Y. J Org Chem. 2009;74:3986–3989. doi: 10.1021/jo900330n.Wang B, Wong OA, Zhao M-X, Shi Y. J Org Chem. 2008;73:9539–9543. doi: 10.1021/jo801576k.Katsuki T, Oguma T, Egami H. J Am Chem Soc. 2010;132:5886–5895. doi: 10.1021/ja100795k.Sarabia F, Vivar-Garcia C, Carcia-Castro M, Martin-Ortiz J. J Org Chem. 2011;76:3129–3150. doi: 10.1021/jo1025964.Lifchits O, Mahlau M, Reisinger CM, Lee A, Fares C, Polyak I, Gopakumar G, Theil W, List B. J Am Chem Soc. 2013;135:6677–6693. doi: 10.1021/ja402058v.
- 22.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;4:1307–1370. [Google Scholar]
- 23.Fierman MB, O’Leary DJ, Steinmetz WE, Miller SJ. J Am Chem Soc. 2004;126:6967–6971. doi: 10.1021/ja049661c. [DOI] [PubMed] [Google Scholar]
- 24.While we do not observe large quantities of diepoxide or triepoxides in these reactions, their formation, even in small quantities, can modulate site selectivity and/or enantioselectivity to a corresponding extent. This general phenomenon has been described previously. For example, see: Schreiber SL, Schreiber TS, Smith DB. J Am Chem Soc. 1987;109:1525–1529.
- 25.(a) Bach RD, Estevez CM, Winter JE, Glukhoutsev MN. J Am Chem Soc. 1998;120:680–685. [Google Scholar]; (b) Adams W, Bach RD, Dmitrenko O, Saha-Moller C. J Org Chem. 2000;65:6715–6728. doi: 10.1021/jo0009548. [DOI] [PubMed] [Google Scholar]
- 26.See Supporting Information for details.
- 27.(a) Brunger AT, Adams PD, Clore GM, Gros P, Kunstleve-Grosse RW, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]; (b) Brunger AT. Nature Protocols. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 28.All details and decisions that went into the CNS calculations are detailed in the Supporting Information.
- 29.For example, see: Kamrath MZ, Garand E, Jordan PA, Leavitt CM, Wolk AB, Van Stipdonk MJ, Miller SJ, Johnson MA. J Am Chem Soc. 2011;133:6440–6448. doi: 10.1021/ja200849g.Garand E, Kamrath MZ, Jordan PA, Wolk AB, McCoy AB, Miller SJ, Johnson MA. Science. 2012;335:694–698. doi: 10.1126/science.1214948.
- 30.Némethy G, Printz MP. Macromolecules. 1972;5:755–758. [Google Scholar]
- 31.The Pymol Molecular Graphics System, Version 1.5.0.4, Schrodinger, LLC.
- 32.Truhlar DG, Garrett BC, Klippenstein SJ. J Phys Chem. 1996;100:12771–12800. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.