

Abstract
N-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-6-fluoronicotinamide ([18F]FNEM), a novel prosthetic agent that is thiol-specific, was synthesized using a one-pot two-step strategy: (1) 18F incorporation by a nucleophilic displacement of trimethylammonium substrate under mild conditions; (2) amidation of the resulting 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester with N-(2-aminoethyl)maleimide trifluoroacetate salt. The radiosynthesis of the maleimide tracer was completed in 75 min from [18F]fluoride with 26 ± 5% decay uncorrected radiochemical yield, and specific activity of 19–88 GBq/μmol (decay uncorrected). The in vitro cell uptake, in vivo biodistribution, and positron emission tomography (PET) imaging properties of its conjugation product with [Cys40]-exendin-4 were described. [18F]FNEM-Cys40-exendin-4 showed specific targeting of glucagon-like peptide 1 receptor (GLP-1R) positive insulinomas and comparable imaging results to our recently reported [18F]FPenM-Cys40-exendin-4.
Keywords: fluorine-18, thiol reactive prosthetic group, insulinoma imaging
Introduction
18F is the most clinically relevant positron emitting radioisotope1 because of its favorable nuclear decay properties (β+ 0.635 MeV, 97% abundance, half-life 109.8 min). 18F-labeled peptides have been more widely used for diagnostic imaging in cancer and other diseases due to their nonimmunogenic behavior, intrinsic pharmacokinetic properties, high affinity, and readily available solid-phase chemistry to allow tuning of these properties.
One consideration for the development of radiotracers is the ability to achieve selective radiolabeling. Peptides are challenging in this regard, as the most common labeling methods employ species that react at amines due to their facile reaction with activated carboxylic acid groups (N-succinimidyl-4-[18F]fluorobenzoate, [18F]SFB, for example)2,3 or with aldehydes.4,5 Peptides of sufficient length may have several amines all with slightly different reaction rates. Regioselective radiolabeling may be important to retain binding affinity and selectivity for a given peptide. The ability to easily modify peptides to reduce the number of reactive sites to one is an enticing property of peptides. Furthermore, labeling with [18F]SFB often requires large excess of peptide or protein to achieve reasonably high radiochemical yield, which compromises specific activity and/or requires HPLC purification strategies.6,7
On the basis of the above considerations, a more selective surrogate instead of amino reactive functional group is necessary. A free thiol group from a cysteine residue is able to meet these requirements and is extensively studied in radiolabeling of peptides. Peptides can be engineered to contain a free thiol group to allow specific labeling. Thiols react selectively with maleimides,8−11a-halogenketones,12−15 and phosphorothionate agents16,17 under mild conditions. The selectivity of prosthetic groups toward free thiol can be exemplified by bovine serum albumin (BSA) conjugation. BSA is a protein with 55 free amino groups and 35 thiol groups, 34 of which form a disulfide bond and with just one free thiol group for site-specific conjugation (Cys34).18,19 Selective labeling of Cys34 with 19F units that may be used as a potential 19F MRI agent has recently been developed.20
Glucagon-like peptide-1 (GLP-1) is an important glucose-dependent hormone released mainly from the small intestine during the ingestion of food.21 Its receptor (GLP-1R) is a G protein-coupled receptor mainly expressed in the pancreatic islet cells. GLP-1R is highly expressed in insulinomas.22 Detection of insulinomas can be difficult by CT, echography, or MRI, due to their relatively small size in the pancreas. Thus, GLP-1R provides a very promising target for receptor-targeted imaging and therapy of insulinomas. However, the native GLP-1 is very unstable in vivo and can be degraded by dipeptidyl-peptidase-IV via cleavage of two N-terminal residues, with a half-life less than 2 min, which limits its biomedical application.23 We have been interested in the development of a peptide based imaging agent for GLP-1R based on radiometal24 or fluorine-1825−27 labeling to overcome the limitations with promising results. However, radiometal labeled peptides are thought to metabolize to radiometal-chelated amino acids that are able to be trapped in the tubular lysosomes, thereby delivering high radiation doses to the kidneys with potential nephrotoxicity.28
Our group developed a series of 18F-radiolabeled prosthetic groups for the purpose of labeling cysteine-engineered GLP-1 analogues for tumor targeting with considerable success. Two thiol site-specific prosthetic groups containing maleimide units, N-[2-(4-[18F]fluorobenzamido)ethyl]-maleimide ([18F]FBEM)29 and N-5-[18F]fluoropentylmaleimide ([18F]FPenM),27 were developed for the peptide conjugation. The synthesis of these two prosthetic maleimides required three chemical steps and two reaction vessels. [18F]FBEM was synthesized with 17.3 ± 7.1% yield (decay uncorrected) using an Eckert and Ziegler module; the total synthetic time is approximately 100 min, and the measured specific activity was 91–176 GBq/μmol (end of synthesis). The most recently developed [18F]FPenM has comparable radiolabeling yield (14 ± 3% decay uncorrected yield in 110 min).
Both of our previous maleimide prosthetic groups displayed good imaging properties when conjugated to GLP-1 analogue, [Cys40]-exendin-4 with low kidney uptake or rapid kidney clearance. However, neither was synthesized rapidly nor easily. Herein, we describe a new thiol-specific prosthetic agent, N-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-6-fluoronicotinamide ([18F]FNEM) by a one-pot two-step strategy: (1) 18F incorporation by a nucleophilic displacement of trimethylammonium substrate under mild conditions; (2) amidation of the resulting 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester with N-(2-aminoethyl)maleimide trifluoroacetate salt. The synthesis begins with a previously reported, high yielding synthesis of tetrafluorophenyl 2-[18F]fluoronicotinamide.30 The maleimide tracer was completed in 75 min from [18F]fluoride with 26 ± 5% decay uncorrected yield, and specific activity 19–88 GBq/μmol (decay uncorrected). The in vitro cell uptake, in vivo biodistribution, and PET imaging properties of its conjugation product with [Cys40]-exendin-4 are described.
Materials and Methods
Reagents and Instrumentation
Analytical thin layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates (Merck) with visualization by ultraviolet (UV) irradiation at 254 nm or staining with KMnO4. The synthesized compounds were purified by silica gel chromatography. [Cys40]-exendin-4 was prepared by solid-phase peptide synthesis (CS Bio, Menlo Park, CA). 1H, 19F, and 13C NMR spectra were carried out on a Bruker 300 MHz NMR spectrometer, equipped with a 1H/19F/13C 5 mm multinuclear probe. LC/MS analysis was conducted on a Waters LC–MS system (Waters, Milford, MA) that included an Acquity UPLC unit coupled to the Waters Q-Tof Premier high-resolution mass spectrometer.27
Chemistry
N,N,N-Trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium Chloride 1
Compound 1 was synthesized by modifying a literature method.30 Briefly, to a solution of 6-chloronicotinic acid (4.4 g, 27.9 mmol) and 2,3,5,6-tetrafluorophenol (TFP) (4.8 g, 28.9 mmol) in dioxane (150 mL) was added N,N′-dicyclohexylcarbodiimide (DCC) (5.7 g, 27.6 mmol); the mixture was stirred overnight at room temperature. Dicyclohexylurea (DCU) was removed by filtration, and the filtrate was evaporated in vacuum. The residue was purified by silica gel flash chromatography using hexane/CH2Cl2 (5/1, v/v) as the eluent to afford compound 1 as a white solid (7.2 g, 84%). 1H NMR (300 MHz, CDCl3) δ 9.21–9.20 (m, 1H), 8.44–8.41 (m, 1H), 7.58–7.55 (m, 1H), 7.17–7.05 (m, 1H); 13C NMR (75.5 MHz, CDCl3) δ 160.8, 157.6, 152.2, 148.2–147.8 (m), 144.8–144.5 (m), 142.7–142.4 (m), 140.6, 139.3–139.2 (m), 139.1–139.0 (m), 125.0, 122.6, 104.1 (t, J = 22.7 Hz); 19F NMR (282 MHz, CDCl3) δ −138.21 to −138.31 (m, 2F), −152.40 to −152.56 (m, 2F); mass (ESI) m/z 305.9 [M + H]+.
N,N,N-Trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium Trifluoromethanesulfonate 3
Compound 3 was synthesized by modifying a literature method with improved yield.30 Briefly, to a solution of compound 1 (1.0 g, 3.3 mmol) in dry THF (15 mL) was added 1 M trimethylamine solution in THF (9.0 mL). A white precipitate was found 10 min after the reaction started, which was allowed to proceed overnight. The precipitate was collected and washed with cold Et2O and cold CH2Cl2 successively. The solid residue was suspended in CH2Cl2, and TMSOTf (1.7 mL, 9 mmol) was added over 10 min. The mixture was concentrated, and the residue was recrystallized from EtOAc to afford compound 3 as a white solid (0.9 g, 57% yield). 1H NMR (300 MHz, CD3OD) δ 9.42–9.41 (m, 1H), 8.95–8.92 (m, 1H), 8.28–8.25 (m, 1H), 7.63–7.51 (m, 1H), 3.74 (s, 9H); 13C NMR (75.5 MHz, CD3SOCD3) δ 164.8, 159.1, 149.3, 147.6–147.3 (m), 144.4–144.0 (m), 141.8, 139.6–139.3 (m), 136.9–136.5 (m), 136.3–136.1 (m), 128.9, 120.7 (q, J = 322.5 Hz), 115.6, 95.4 (t, J = 23.9 Hz), 54.6; 19F NMR (282 MHz, CD3OD) δ – 81.66 (s, 3F), –142.36 to –142.52 (m, 2F), –156.81 to –156.95 (m, 2F); mass (ESI) m/z 329.5 [M – CF3SO3]+.
2,3,5,6-Tetrafluorophenyl 6-(2,3,5,6-Tetrafluorophenoxy)nicotinate
To a solution of triflate 3 (86 mg, 0.30 mmol) and TFP (60 mg, 0.36 mmol) in acetonitrile (0.5 mL) was added DIPEA (57 μL, 0.33 mmol); the mixture was stirred at room temperature for 2 h. The residue was concentrated and purified by silica gel flash chromatography using hexane/CH2Cl2 as the eluent to afford the compound as a white solid (70 mg, 90%). 1H NMR (300 MHz, CDCl3) δ 8.96 (d, J = 2.1 Hz, 1H), 8.57–8.53 (m, 1H), 7.33–7.30 (m, 1H), 7.15–7.02 (m, 2H); 13C NMR (75.5 MHz, CDCl3) δ 164.9, 160.9, 151.3, 148.2–147.8 (m), 144.9–144.5 (m), 143.2–142.3 (m), 139.9–139.0 (m), 131.8, 129.5, 120.2, 111.3, 104.2–102.9 (m); 19F NMR (282 MHz, CDCl3) δ –138.48 to –139.16 (m, 2H), –152.52 to –152.95 (m, 2H); mass (ESI) m/z 435.9 [M + H]+.
General Procedure for the Condensation of Aromatic Carboxylic Acid with N-(2-aminoethyl)maleimide Trifluoroacetate Salt
To a solution of N-(2-aminoethyl)maleimide trifluoroacetate salt (1.0 equiv) in anhydrous DMF at 0 °C was added aromatic carboxylic acid (1.5 equiv), HOBt (1.5 equiv), HBTU (1.5 equiv), 3 Å molecular sieves, and DIPEA (2.5 equiv) successively. The reaction proceeded at 0 °C for 0.5 h and continued at room temperature overnight. After confirmation from TLC that the starting material was consumed completely, the mixture was quenched with ice water and extracted with CH2Cl2. The organic extracts were washed with water and brine, respectively, then dried and the solvent rotary evaporated. The residue was purified by silica gel column chromatography with CH2Cl2/MeOH as the eluent to afford the amide compound.
N-(2-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-6-fluoronicotinamide 5 (FNEM)
Compound 5 was prepared according to the general procedure as a white solid (83 mg, yield 79%). 1H NMR (300 MHz, CDCl3) δ 8.61 (d, J = 2.4 Hz, 1H), 8.24–8.17 (m, 1H), 7.01–6.97 (m, 1H), 6.95 (br, 1H), 6.74 (s, 2H), 3.85–3.82 (m, 2H), 3.68–3.63 (m, 2H); 13C NMR (75.5 MHz, CDCl3) δ 171.3, 164.9, 163.6, 147.2 (d, J = 15.9 Hz), 140.8 (d, J = 9.1 Hz), 134.5, 128.3 (d, J = 4.5 Hz), 109.9 (d, J = 37.0 Hz), 40.3, 37.5; 19F NMR (282 MHz, CDCl3) δ – 63.37 (d, J = 5.6 Hz); mass (ESI) m/z 264.0 [M + H]+.
N-(2-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-3,3,3-triphenylpropanamide 6
Compound 6 was prepared according to the general procedure as a light yellow solid (112 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ 7.32–7.26 (m, 12H), 7.25–7.18 (m, 3H), 6.67 (s, 2H), 5.14 (t, J = 5.1 Hz, 1H), 3.54 (s, 2H), 3.41–3.37 (m, 2H), 3.12–3.06 (m, 2H); 13C NMR (75.5 MHz, CDCl3) δ 171.1, 170.8, 146.5, 134.3, 129.4, 128.2, 126.6, 56.4, 48.6, 38.8, 37.3; mass (ESI) m/z 425.1 [M + H]+.
N-(2-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-2-naphthamide 7
Compound 7 was prepared according to the general procedure as a light yellow solid (60 mg, yield 49%). 1H NMR (300 MHz, CDCl3) δ 8.28 (s, 1H), 7.89–7.78 (m, 4H), 7.57–7.48 (m, 2H), 7.06 (br, 1H), 6.69 (s, 2H), 3.86–3.83 (m, 2H), 3.73–3.68 (m, 2H); 13C NMR (75.5 MHz, CDCl3) δ 171.3, 168.0, 134.9, 134.4, 132.8, 131.5, 129.2, 128.6, 127.9, 127.82, 127.77, 126.9, 123.7, 39.9, 37.7; mass (ESI) m/z 295.0 [M + H]+, 589.1 [2 M + H]+.
1-Hexyl-1H-pyrrole-2,5-dione 8
Hexylamine (50 mg, 0.5 mmol) was dissolved in a saturated aqueous solution of NaHCO3 (2 mL). The solution was put on ice-bath, and after 5 min, N-(methoxycarbonyl)maleimide (93 mg, 0.6 mmol) was added. The resulting solution was stirred on ice-bath for 30 min and then at room temperature for an additional 30 min until all the starting material was consumed completely as confirmed by TLC. The mixture was purified through silica gel flash chromatography using CH2Cl2/MeOH as the eluent to afford compound 8 as a colorless liquid (68 mg, yield 76%). 1H NMR (300 MHz, CDCl3) δ 6.68 (s, 2H), 3.50 (t, J = 7.2 Hz, 2H), 1.59–1.54 (m, 2H), 1.28–1.24 (m, 6H), 0.89–0.84 (m, 3H); 13C NMR (75.5 MHz, CDCl3) δ 171.1, 134.2, 38.1, 31.5, 28.7, 26.6, 22.7, 14.2; mass (EI) m/z 110.0 [M – n-C5H11]+, 181.1 M+.
General Procedure for the Synthesis of FNEM-[Cys40]-exendin-4 and N-Hexylmaleimido-[Cys40]-exendin-4
To a solution of [Cys40]-exendin-4 in degassed PBS, FNEM or N-hexylmaleimide in acetonitrile was added and incubated for 1 h; then the mixture was subjected to semipreparative HPLC (Vydac C18 protein column, 9.4 × 250 mm, flow rate 5.0 mL/min, solvent A, 0.1% TFA in water, solvent B, 0.1% TFA in CH3CN). The elution profile was isocratic at 25% solvent B for 5 min, then a gradient to 55% solvent B over 25 min, and finally to 90% B over the next 5 min)27 to give FNEM-[Cys40]-exendin-4 and N-hexylmaleimido-[Cys40]-exendin-4, respectively. The peak at about 21 or 23 min was collected for FNEM-[Cys40]-exendin-4 and N-hexylmaleimido-[Cys40]-exendin-4, respectively. The fractions were lyophilized for further use.
FNEM-[Cys40]-exendin-4
White solid, 2.91 mg, 78% yield. Analytical HPLC tR = 19.3 min; HPLC-MS 1518.5 [M + 3H]3+, 1138.9 [M + 4H]4+; deconvolves to 4552.0. Elemental composition C199H297FN54O64S2: exact mass, 4550.1071; molecular weight, 4552.9974.
N-Hexylmaleimide-[Cys40]-exendin-4
White solid, 0.84 mg, 77% yield. Analytical HPLC tR = 22.3 min; HPLC-MS 1491.5 [M + 3H]3+, 1118.6 [M + 4H]4+, 895.3 [M + 5H]5+; deconvolves to 4471.5. Elemental composition C197H302N52O63S2: exact mass, 4468.1468; molecular weight, 4471.0040.
Radiochemical Synthesis of [18F]FNEM 5
TBAHCO3 (0.8 M in H2O, 30 μL), acetonitrile (200 μL), and [18F]fluoride (23–86 mCi) were added to a test tube, and the solvent was evaporated under a stream of argon while being heated at 100 °C. The fluoride was dried by adding acetonitrile (200 μL) three times and each evaporated. Then triflate 9 (9 mg, 18.8 μmol) in acetonitrile/tBuOH (300 μL/100 μL) was added. The reaction mixture was heated at 40 °C for 10 min, cooled to room temperature, and then N-(2-aminoethyl)maleimide trifluoroacetate salt (12 mg, 47.2 μmol) and pyridine (7.6 μL) in acetonitrile (170 μL) was added to the mixture. The reaction solution was further heated at 60 °C for 15 min. The solvent was evaporated by argon flow and diluted with 1 mL of 10% aqueous CH3CN; the mixture was centrifuged and subjected to semipreparative HPLC purification with Phenomenex Luna 5 μm C18 column (250 × 10 mm, flow rate 4.0 mL/min). The collected fraction was diluted with 10 mL of water, and the product was trapped on two stacked Sep-Pak C18 plus cartridges. The cartridge was washed with H2O (3 mL) and hexane (2 mL) successively, and the product was eluted with 10% EtOH in CH2Cl2 (1.5 mL). The solvent was removed under argon flow. A typical one-pot two-step radiolabeling would require 75 min. The purity of compound 5 was confirmed by analytical HPLC (Phenomenex Luna 3 μm C18 column, 150 × 4.6 mm, flow rate 1.0 mL/min, isocratic elution with 15% CH3CN in 0.1% TFA/H2O). Rt = 8.9 min.
Radiochemical Synthesis of [18F]FNEM-[Cys40]-exendin-4
[18F]FNEM (5, 7–20 mCi) was dissolved in ethanol (10 μL), and [Cys40]-exendin-4 (100–200 μg) in 100 μL 0.1% sodium ascorbate in degassed PBS was added, and the reaction mixture was incubated at room temperature for 30 min. Then to the mixture was added N-hexylmaleimide 8 (160 μg) in acetonitrile (150 μL), and the reaction stood for another 20 min. Then 0.1% TFA (100 μL) was added, and the mixture was subjected to semipreparative HPLC purification. The collected fractions were diluted with water and passed through a C18 BondElut cartridge. The product was eluted with 1.5 mL of 10 mM HCl in ethanol, and the volume was reduced to about 100–200 μL on a rotary evaporator. The residue was diluted by PBS for further studies. MS 1138.7 [M + 4H]4+, 1518.3 [M + 3H]3+; deconvolves to 4552.0. Elemental composition C199H297FN54O64S2: exact mass, 4550.1071; molecular weight, 4552.9974.
Mouse Serum Stability Study
To study the stability of [18F]FNEM-[Cys40]-exendin-4 in serum, the radiotracer (142 μCi) was mixed with freshly harvested mouse serum (200 μL). A 50 μL aliquot was removed at 0 min, and the remaining sample was incubated at 37 °C. Additional aliquots of 50 μL were removed at 30, 60, and 90 min. Each aliquot was mixed with 50 μL of CH3CN and centrifuged. A portion of the supernatant was taken for radioHPLC analysis using an online radioactivity detector.
Cell Culture and Animal Model
The animal study protocol was in accordance with the principles and procedures outlined in the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Clinical Center, National Institutes of Health (Animal protocol NIBIB 13-01). Rat insulinoma cell line INS-1 was obtained from the American Type Culture Collection (ATCC, Manassas, VA). INS-1 cells were grown in RPMI-1640 culture medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (FBS, Invitrogen), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Invitrogen) at 37 °C in a humidified atmosphere containing 5% CO2. INS-1 tumors were developed in 5–6 week-old female Balb/c mice (n = 10). Each mouse underwent inoculation of about 5 × 106 INS-1 cells in the right shoulder. The tumor growth was monitored by caliper measurement.
Cell Experiments
The GLP-1R binding assay was performed according to a reported procedure27 to determine binding affinities of FNEM-[Cys40]-exendin-4 and exendin-4. The IC50 values were calculated using a GraphPad Prism software. The INS-1 cell uptake and efflux of [18F]FNEM-[Cys40]-exendin-4 were also conducted as previously reported.27
PET Imaging
When the INS-1 tumor reached 8–10 mm in size (18–24 days after inoculation), PET imaging studies were performed using an Inveon small animal PET scanner (Siemens Preclinical Solutions). Tumor mice were randomly divided into the control group and the blocking group (n = 5/group). For the control group, about 1.11 MBq (30 μCi) of [18F]FNEM-Cys40-exendin-4 was injected through tail vein under isoflurane anesthesia. For exendin-4 blocking group, unlabeled exendin-4 (100 μg) was injected (i.v. tail vein) 15 min before the injection of 1.11 MBq (30 μCi) [18F]FNEM-Cys40-exendin-4. For both groups, a 5 min acquisition was performed at 1 and 2 h after tracer injection. The images were reconstructed using a 2D OSEM algorithm without correction for attenuation or scattering. The mean pixel values within the three-dimensional regions of interest (3D-ROIs) were converted to MBq/mL/min using a predetermined calibration factor. By assuming a tissue density of 1 g/mL, imaging ROI-derived % ID/g was obtained.
Ex Vivo Biodistribution
Immediately after the 2 h microPET imaging, tumor model mice in both groups were sacrificed, and INS-1 tumor, blood, major organs, or tissues were harvested and wet weighed. The radioactivity of each organ or tissue was measured using a γ-counter, and the results were expressed as percentages of the injected dose per gram of tissue (%ID/g).
Statistical Analysis
Quantitative data were expressed as mean ± SD, and the results were compared using Student’s t test. P value of <0.05 is considered statistically significant.
Results
Chemistry and Radiochemistry
Following literature procedures,30 we first synthesized compound 1 from 6-chloronicotinic acid and 2,3,5,6-tetrafluorophenol by N,N′-dicyclohexylcarbodiimide condensation. With slight modification of the literature procedure, we found that chloride 2 was obtained in good yield using 1 M trimethylamine solution in THF instead of using trimethylamine gas. The chloride salt 2 with poor solubility in acetonitrile was converted to the trifluoromethanesulfonate salt by adding trimethylsilyl trifluoromethanesulfonate (TMSOTf) to a suspension of 2 in dichloromethane. Purified needle-shaped 3 was conveniently produced by recrystallization of the concentrated organic phase from ethyl acetate. This triflate salt 3 had excellent solubility in the commonly used radiolabeling solvent acetonitrile (Scheme 1).
Scheme 1. Synthesis of Precursor 3 and One-Pot Two-Step Radiosynthesis of [18F]FNEM 5.
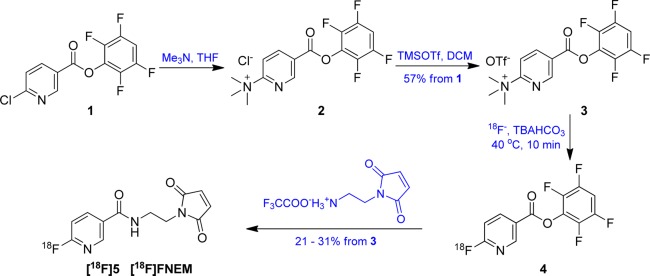
Next we evaluated radiolabeling of the triflate salt 3. The radiolabeling proceeded well when using the optimized conditions developed by Olberg.30 Olberg used an Oasis MCX Plus Sep-Pak (Waters) to purify the resulting radiolabeled product 4, eliminating the time-consuming HPLC purification step. However, in the subsequent peptide conjugation, 2 mg of peptide in 3 mL of buffer was required. It was our observation, that the product 4, purified by solid-phase extraction, contained a significant amount of 2,3,5,6-tetrafluorophenyl 6-(2,3,5,6-tetrafluorophenoxy)nicotinate, resulting from 2,3,5,6-tetrafluorophenol substitution of the trimethylammonium leaving group. Indeed we found that 3 efficiently reacted with 2,3,5,6-tetrafluorophenol in the presence of base to provide 95% isolated yield of the side product. Although the incorporation of [18F]fluoride was very good, we observed HPLC purification to be adversely affected by the tBuOH in the reaction (see discussion below). Subsequently, we found that with smaller volume (0.4 mL instead of 1 mL) of solvent and changing the tBuOH/MeCN ratio from 4/1 to 1/3 still gave 67–82% radiochemical yields.
Next we focused on maleimide incorporation without requiring a solvent change. We evaluated the amount of N-(2-aminoethyl)maleimide and different bases to achieve the desired coupling. Base with higher pKa typically gave lower yield or completely degraded polar stuff (Table 1, entry 1, 9); diisopropylethylamine proved to be an appropriate base for the coupling while higher temperature with excess amount of the base afforded no product (Table 1, compare entry 2, 3). We also unsuccessfully tried the coupling in aqueous buffer, which gave very low yield (Table 1, entry 4). Finally, we found pyridine (pKa 5.25) formed a stable solution with N-(2-aminoethyl)maleimide and afforded good yield for the condensation; further increasing the amount of maleimide and elevating the reaction temperature gave improved results (Table 1, entry 5–8). The desired product was verified by coinjection of authentic FNEM, which was prepared by the condensation of 6-fluoronicotinic acid and N-(2-aminoethyl)maleimide. The radiotracer was further confirmed by LC–MS.
Table 1. Condition Screening for the Radiosynthesis of [18F]FNEM 5.
| entry | –OTf substrate | aminoethyl maleimide | temperature | base | yielda |
|---|---|---|---|---|---|
| 1 | 9 mg, 18.8 μmol (1 equiv) | 7.2 mg (1.5 equiv) | 40 °C | DMAP 3 equiv | 14% |
| 2 | 9 mg, 18.8 μmol (1 equiv) | 7.2 mg (1.5 equiv) | 40 °C | DIPEA 3 equiv | 47% |
| 3 | 9 mg, 18.8 μmol (1 equiv) | 7.2 mg (1.5 equiv) | 60 °C | DIPEA 5 equiv | none |
| 4 | 9 mg, 18.8 μmol (1 equiv) | 12 mg (2.5 equiv) | 40 °C | PBS pH 9.0 | 10% |
| 5 | 9 mg, 18.8 μmol (1 equiv) | 7.2 mg (1.5 equiv) | 40 °C | pyridine 3 equiv | 54% |
| 6 | 9 mg, 18.8 μmol (1 equiv) | 7.2 mg (1.5 equiv) | 60 °C | pyridine 5 equiv | 58% |
| 7 | 9 mg, 18.8 μmol (1 equiv) | 12 mg (2.5 equiv) | 60 °C | pyridine 5 equiv | 73% |
| 8 | 9 mg, 18.8 μmol (1 equiv) | 12 mg (2.5 equiv) | 60 °C | pyridine 8 equiv | 61% |
| 9 | 9 mg, 18.8 μmol (1 equiv) | 12 mg (2.5 equiv) | 60 °C | 2,4,6-collidine 5 equiv | none |
The yield for the second radiolabeling step based on integrated radioactivity of individual peaks relative to the total radioactivity peak areas.
We knew we needed to remove the side product (product 3 + tetrafluorophenol). First we tried fluorous cartridge to separate the two components based on the fluorophilicity interaction instead of eluent polarity but were unsuccessful. We returned our focus to HPLC purification because we had good analytical conditions. At the end of the reaction, we diluted the reaction solution to 0.4, 1, or 4 mL and injected onto a semipreperative column. Our initial semipreparative conditions (isocratic with 0.1% TFA in 15% water, 85% acetonitrile) for HPLC resulted in a product peak with a 4 min peak width (baseline to baseline) at 18 min. The amount of aqueous dilution of the reaction mixture (up to 0.6, 1, or 4 mL with water) did not improve the peak shape. Even on an analytical system, optimal peak shape was not obtained unless the tert-butanol was completely evaporated. Because of the large volume of collected fraction from the semipreparative column, we were unable to trap the desired product on a solid phase extraction column.
Attempts to modulate the peak width using more basic HPLC eluents, such as ammonium acetate buffer (50 mM, pH 6.4) or PBS buffer (pH 7.2), provided no improvement in peak shape. The retention time of [18F]FNEM was around 8.9 min with 15% isocratic acetonitrile for analytical HPLC in all tested buffers (0.1% TFA, pH 6.4, pH 7.4). We hypothesized that the tert-butanol in the reaction solvent was causing the unfavorable peak shape. Evaporation of the reaction solvent prior to HPLC injection provided better resolution. Typically the tracer was collected in 26 ± 5% (n = 8) uncorrected yield from EOB, and the total radiochemical synthesis time was around 75 min with specific activity 19–88 GBq/μmol (n = 8). Moreover, the trapping efficiency from the HPLC eluate was around 70% when the HPLC fraction (approximately 6 mL) was diluted to 21 mL with pure water. Up to 90% of the activity was trapped when a stack of two Waters C18 plus cartridges was used. Following washes with H2O and hexane, the tracer was efficiently eluted (80 ± 5% recovery) with 10% ethanol in dichloromethane.
In order to demonstrate the application of this novel nicotinic maleimide prosthetic group, we applied the methods previously used for [18F]FBEM-[Cys40]-exendin-4.26 The solvent was evaporated under inert gas flow, and the remaining radioactivity was subjected to coupling with [Cys40]-exendin-4 using 100 μL degassed PBS as the buffer. After a 30 min incubation, the reaction was quenched with 100 μL of 0.1% TFA and subjected to preparative HPLC. Typically the [18F]FNEM-[Cys40]-exendin-4 was collected in around 40% decay uncorrected yield based on starting [18F]FNEM. In contrast to our recently developed [18F]FPenM-[Cys40]-exendin-4, where the target tracer could be chromatographically separated due to slightly longer retention time from parent excess [Cys40]-exendin-4, the [18F]FNEM-[Cys40]-exendin-4 could not be separated from excess starting material. This was consistent with the fact that [18F]FNEM was more polar than [18F]FBEM and [18F]PenM. Efforts to improve the separation with changes in speed of gradient or pH of buffer were unsuccessful.
Our next approach to achieve higher chemical purity was to consume excess [Cys40]-exendin-4 with a much more lipophilic maleimide that could be added following reaction time with [18F]FNEM. Two lipophilic maleimide prosthetic agents 6 and 7 were prepared through condensation of 3,3,3-triphenylpropionic acid and 2-naphthoic acid with N-(2-aminoethyl)maleimide), respectively (Scheme 2). After the radiolabeling reaction, we added 10 equiv of 6 or 7 in acetonitrile to consume excess free thiol compound, but the trial failed probably due to the high hydrophobicity of 6 and 7 that precludes dispersion into aqueous phase.
Scheme 2. Structures of Maleimido-Containing Prosthetic Groups Used for the Capture of Excess Free Thiol Reagent.
Inspired by our previous work, we synthesized another maleimide prosthetic agent 8 with a hexyl chain, which would have similar lipophilicity to fluoropentyl group and may capture excess [Cys40]-exendin-4 in aqueous medium. Indeed, following the 30 min radiochemical incorporation, a 20 min incubation at room temperature with excess N-hexylmaleimide produced a new nonradioactive peak with 2 min later retention time from [18F]FNEM-[Cys40]-exendin-4. The new peak was collected and its identity confirmed as a hexylmaleimde conjugate by LC–MS. About 60% of the starting material was consumed by the added N-hexylmaleimide, and the resulting specific activity of [18]FNEM-[Cys40]-exendin-4 was 0.2–0.6 Ci/μmol, 2 to 4 times higher than achievable without the N-hexylmaleimide addition. The trapped and eluted [18F]FNEM-[Cys40]-exendin-4 was concentrated and delivered for animal study.
Mouse Serum Stability Study
The stability of [18F]FNEM-[Cys40]-exendin-4 was studied at 37 °C in mouse serum and was shown to be stable up to 90 min. Trace amount of a polar component was produced, which may be due to oxidation of methionine group on the peptide (Figure 1).31,32 The tracer is stable enough to acquire appropriate PET imaging results. The extraction efficiency from the mouse serum was around 80%, as determined by the ratio of radioactivity in the supernatant compared to the pellet.
Figure 1.

Stability of [18F]FNEM-[Cys40]-exendin-4 incubated with mouse serum at 0, 0.5, 1, and 1.5 h, respectively.
Cell Binding Assay
The IC50 values of exendin-4 and FNEM-[Cys40]-exendin-4, using 125I-GLP-1 as radioligand, in INS-1 cells are displayed in Figure 2. The developed FNEM-[Cys40]-exendin-4 showed high binding affinity (0.44 nM) although exhibited slightly lower binding affinity than parent exendin-4 (0.18 nM), suggesting that labeling [Cys40]-exendin-4 with FNEM did not significantly change the binding affinity toward GLP-1R expressed on INS-1 cells.
Figure 2.
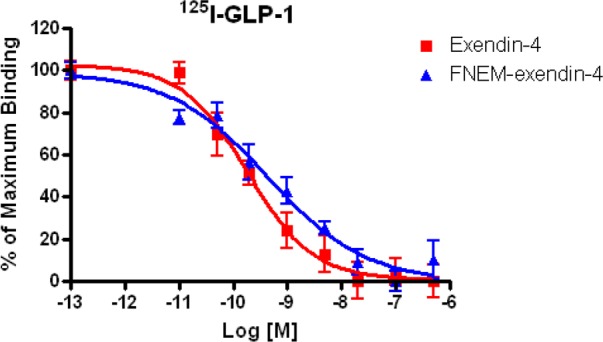
Inhibition curves of exendin-4 (red ■) and FNEM-[Cys40]-exendin-4 (blue ▲) derived from competitive GLP-1R binding assay using 125I-GLP-1 as radioligand.
Cellular Uptake and Efflux Assay
Cellular uptake and efflux of [18F]FNEM-[Cys40]-exendin-4 was evaluated using INS-1 tumor cells (Figure 3). Uptake was apparent (0.34 ± 0.04%) at 15 min, and there was sustained increase until 60 min (0.63 ± 0.08%), then the uptake decreased at 2 h (0.38 ± 0.02%). The uptake was effectively inhibited in the presence of a blocking dose of exendin-4. The efflux appeared to be biphasic with an early rapid washout, reflecting the loss of surface receptor binding, followed by a slow loss of radioactivity from the cells, representing the clearance of internalized radioactivity, about 42% of the activity effluxed from the cells by 60 min.
Figure 3.

INS-1 cell uptake (a, ■), block (a, ▲), and efflux (b, ■) of [18F]FNEM-[Cys40]-exendin-4.
PET Imaging and ex Vivo Biodistribution
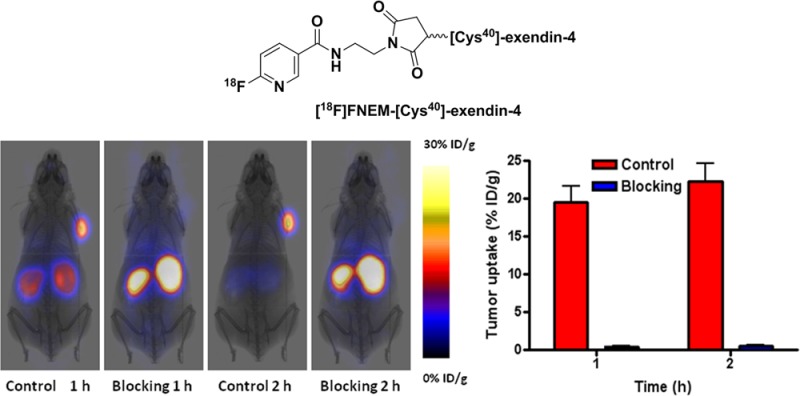
The PET images clearly showed high uptake of [18F]FNEM-[Cys40]-exendin-4 in the INS-1 tumor at 60 min post injection (19.56 ± 4.57 %ID/g), which remained high at 2 h time point (22.28 ± 5.17 %ID/g). This tracer is stable against defluorination as negligible tracer uptake was found in the bone; this was consistent with the mouse serum stability study results prior to in vivo imaging. Kidney uptake of [18F]FNEM-[Cys40]-exendin-4 was modest (8.28 ± 3.43%ID/g) at 1 h and with most of the tracer cleared from the kidneys by 2 h p.i. (2.49 ± 0.40 %ID/g). Liver uptake was low (1.64 ± 0.10 %ID/g at 1 h; 1.69 ± 0.24 %ID/g at 2 h p.i.).
GLP-1R specific of [18F]FNEM-[Cys40]-exendin-4 in vivo was evaluated by injecting a blocking dose (100 μg) of [Cys40]-exendin-4 15 min prior to the administration of [18F]FNEM-[Cys40]-exendin-4 (Figure 4d). The presence of a blocking dose of [Cys40]-exendin-4 significantly reduced the tumor uptake (0.46 ± 0.22%ID/g at 60 min p.i.). The specific tumor uptake of [18F]FNEM-[Cys40]-exendin-4 was further confirmed by biodistribution using dissected tissues. The biodistribution of [18F]FNEM-[Cys40]-exendin-4 in tumors, conducted following microPET imaging, showed similar values, compared with the PET imaging results at 2 h post-tracer injection. In the control group, INS-1 tumor uptake was 23.06 ± 3.87 %ID/g, while in blocking group the tumor showed only 0.35 ± 0.23 %ID/g (P < 0.01). Tumor-to-kidney ratio was 11, and tumor-to-liver ratio was 63.8 (Figure 4). The blocking dose led to reduced uptake in the GLP-1R positive organs such as pancreas, stomach, and lung.
Figure 4.

Representative PET images of INS-1 tumor mice at 1 (a) and 2 h (b) postinjection of [18F]FNEM-[Cys40]-exendin-4 (30 μCi) for the control and blocking groups (n = 5/group). (c) Quantification of tumor uptake at 1 and 2 h postinjection. (d) Direct tissue sampling measurement of the biodistribution of [18F]FNEM-[Cys40]-exendin-4 right after the PET acquisition at 2 h time point.
Discussion
Direct radiolabeling of peptides/proteins usually requires high temperature and strong basic medium, with which most peptides are incompatible.33−39 Labeling strategies for the introduction of 18F into peptides or proteins most often utilize radiolabeled prosthetic groups.40−43 The small 18F-labeled prosthetic groups often require multiple synthetic steps to construct before final conjugation to the ligand of interest under mild conditions.44 Several factors are essential to the successful application of 18F-labeled prosthetic groups: speed of synthesis, selectivity of reactivity, and specific activity.
The speed of a radiochemical synthesis is an important factor in the utility and translation of any radiotracer employing a short-lived radionuclide. The application of rapid synthetic reactions and solid-phase extraction methods, instead of HPLC, generally reduce preparation time at the potential expense of lower purity. In the case of 18F, the development of labeling procedures that can be conducted in aqueous media avoids the time required to render fluoride anhydrous for traditional labeling methods. These methods include the use of silicon33,45−47 or boron48−51 based fluorine acceptor groups for 18F incorporation, oxime formation under aqueous conditions by conjugation of [18F]FDG or its derivatives with oxy-amine functionalized peptides,4,52−54 and chelation of the prelabeled Al18F complex for direct [18F]radiolabeling of macromolecules.55−57 Gouverneur et al.58 reported the radiolabeling of fluorous-tagged precursors by nucleophilic fluorination and subsequent purification by fluorous solid phase extraction based on the different affinities of the unreacted substrate and the radiolabeled product for the stationary phase.
High specific activity of the final radiolabeled peptide is often desirable since the receptors or enzymes, recognized by the peptides, would be competitively bound with nonradioactive ligands. Sufficiently low concentration (<0.1 × Kd) of the tracer bound to the target is usually required to avoid pharmacological effects for regulatory approval.59 With our ongoing interest in the development of efficient radiolabeling strategies, we developed three thiol site-specific 18F-radiolabeling prosthetic groups recently. The efficiency in terms of radiochemical yield, preparation time, and specific activity was outlined in Table 2. The newly developed [18F]FNEM was achieved with higher yield in shorter time using a one-pot two-step strategy compared to our previously described [18F]FBEM27 and [18F]FPenM,26 which employ a three-step synthesis and two reaction vessels. We attempted to obtain a 18F radiolabeled maleimide prosthetic group using one-step radiolabeling method that was unsuccessful, due to the lability of maleimide group under harsh conditions. All three thiol-site specific groups showed high specific activity and good radiochemical purity (Table 2).
Table 2. Three Thiol-Site Prosthetic Groups Compared in Terms of Radiolabeling Efficiency and Tumor Uptake.
| tracers | mode | method | time (min) | yielda | SA (GBq/μmol) | tumor uptake (%ID/g)b |
|---|---|---|---|---|---|---|
| [18F]FBEM | automated | two-pot three-step | 95 | 17 ± 7% | 91–176c | 30.27 ± 5.44% |
| [18F]FPenM | manual | two-pot three-step | 110 | 14 ± 3% | 20–49c | 33.21 ± 4.79% |
| [18F]FNEM | manual | one-pot two-step | 75 | 26 ± 5% | 19–88c | 23.06 ± 3.87% |
Decay uncorrected yield.
Tumor uptake is based on biodistribution of corresponding [Cys40]-exendin-4 conjugates.
Specific activity of the prosthetic agents after purification.
The conjugation with [Cys40]-exendin-4 using the developed thiol site-specific prosthetic agents proceeded smoothly with 30–40% decay uncorrected yield. The images for insulinoma targeting showed similar results for [18F]FBEM-[Cys40]-exendin-4 and [18F]FPenM-[Cys40]-exendin-4, but slightly lower tumor uptake was observed for [18F]FNEM-[Cys40]-exendin-4. We ascribed the difference to the following possibilities: (a) because of the lack of chromatographic separation between [Cys40]-exendin-4 and [18F]FNEM-[Cys40]-exendin-4, relatively low specific activity was achieved, and the excess free [Cys40]-exendin-4 was unable to be consumed completely although additional maleimide substrate was added to the mixtures to capture the remaining free thiol group after radiolabeling; (b) the nitrogen of the nicotinamide in the [18F]FNEM may be protonated and affect the tumor binding affinity as compared to the [18F]FPenM-[Cys40]-exendin-4 version. There is a literature report that replacement of a phenyl group with a pyridine moiety will affect the binding affinities of somatostatin receptors in terms of electrostatic potentials through tuning the water solubility and hydrogen bonding capacity, apparently it would change the pKa and configuration after modification.60
Conclusions
A new prosthetic group, [18F]FNEM, synthesized via an one-pot two-step strategy was developed with shorter reaction time and higher radiolabeling yield than our earlier developed thiol-site specific prosthetic groups [18F]FBEM and [18F]FPenM. The application of [18F]FNEM was demonstrated in the synthesis and evaluation of [18F]FENM-[Cys40]-exendin-4 for imaging GLP-1R positive INS-1 insulinoma xenografted mice. The tracer has similar tumor uptake compared with [18F]FBEM-[Cys40]-exendin-4 and [18F]FPenM-[Cys40]-exendin-4, previously developed by our group, and shows high tumor-to-normal tissue ratios for insulinoma imaging. The tracer has rapid renal clearance and low accumulation in the liver. [18F]FNEM may be used to site-specifically radiolabel thiol-containing proteins, antibodies, aptamers, and oligonucleotides.
Acknowledgments
This work was supported by the intramural research program of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. The authors acknowledge the NIH Warren Grant Magnuson Clinical Center’s PET Department for radioisotope production.
Author Contributions
∥ X. Yue and X. Yan contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science 2011, 3346056639–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäding P.; Füchtner F.; Wüst F. Module-assisted synthesis of the bifunctional labelling agent N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB). Appl. Radiat. Isot. 2005, 633329–32. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan G.; Zalutsky M. R. Synthesis of N-succinimidyl 4-[18F]fluorobenzoate, an agent for labeling proteins and peptides with 18F. Nat. Protoc. 2006, 141655–61. [DOI] [PubMed] [Google Scholar]
- Glaser M.; Morrison M.; Solbakken M.; Arukwe J.; Karlsen H.; Wiggen U.; Champion S.; Kindberg G. M.; Cuthbertson A. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjugate Chem. 2008, 194951–7. [DOI] [PubMed] [Google Scholar]
- Namavari M.; Cheng Z.; Zhang R.; De A.; Levi J.; Hoerner J. K.; Yaghoubi S. S.; Syud F. A.; Gambhir S. S. A novel method for direct site-specific radiolabeling of peptides using [18F]FDG. Bioconjugate Chem. 2009, 203432–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchar M.; Pretze M.; Kniess T.; Steinbach J.; Pietzsch J.; Loser R. Site-selective radiolabeling of peptides by 18F-fluorobenzoylation with [18F]SFB in solution and on solid phase: a comparative study. Amino Acids 2012, 4341431–43. [DOI] [PubMed] [Google Scholar]
- Jahan M.; Nag S.; Krasikova R.; Weber U.; Muhs A.; Pfeifer A.; Spenger C.; Willbold D.; Gulyas B.; Halldin C. Fluorine-18 labeling of three novel d-peptides by conjugation with N-succinimidyl-4-[18F]fluorobenzoate and preliminary examination by postmortem whole-hemisphere human brain autoradiography. Nucl. Med. Biol. 2012, 393315–23. [DOI] [PubMed] [Google Scholar]
- Shiue C. Y.; Wolf A. P.; Hainfeld J. F. Synthesis of 18F-labelled N-(p-[18F]fluorophenyl)maleimide and its derivatives for labelling monoclonal antibody with 18F. J. Labelled Compd. Radiopharm. 1989, 261–12287–289. [Google Scholar]
- Toyokuni T.; Walsh J. C.; Dominguez A.; Phelps M. E.; Barrio J. R.; Gambhir S. S.; Satyamurthy N. Synthesis of a new heterobifunctional linker, N-[4-(aminooxy)butyl]maleimide, for facile access to a thiol-reactive 18F-labeling agent. Bioconjugate Chem. 2003, 1461253–9. [DOI] [PubMed] [Google Scholar]
- de Bruin B.; Kuhnast B.; Hinnen F.; Yaouancq L.; Amessou M.; Johannes L.; Samson A.; Boisgard R.; Tavitian B.; Dolle F. 1-[3-(2-[18F]Fluoropyridin-3-yloxy)propyl]pyrrole-2,5-dione: design, synthesis, and radiosynthesis of a new [18F]fluoropyridine-based maleimide reagent for the labeling of peptides and proteins. Bioconjugate Chem. 2005, 162406–20. [DOI] [PubMed] [Google Scholar]
- Dollé F.; Hinnen F.; Lagnel B.; Boisgard R.; Sanson A.; Russo-Marie F.; Tavitian B. Session 3: Radiopharmaceutical Chemistry. J. Labelled Compd. Radiopharm. 2003, 46S1S15–S20. [Google Scholar]
- Kilbourn M. R.; Dence C. S.; Welch M. J.; Mathias C. J. F-18 labeling of proteins. J. Nucl. Med. 1987, 284462–470. [PubMed] [Google Scholar]
- Dence C. S.; McCarthy T. J.; Welch M. J. Improved synthesis of [18F]4-fluorophenacyl bromide: the use of polymer supported perbromide. Appl. Radiat. Isot. 1993, 446981–3. [DOI] [PubMed] [Google Scholar]
- Dolle F.; Hinnen F.; Vaufrey F.; Tavitian B.; Crouzel C. A general method for labeling oligodeoxynucleotides with 18F for in vivo PET imaging. J. Labelled Compd. Radiopharm. 1997, 394319–330. [Google Scholar]
- Kuhnast B.; de Bruin B.; Hinnen F.; Tavitian B.; Dolle F. Design and synthesis of a new [18F]fluoropyridine-based haloacetamide reagent for the labeling of oligonucleotides: 2-bromo-N-[3-(2-[18F]fluoropyridin-3-yloxy)propyl]acetamide. Bioconjugate Chem. 2004, 153617–27. [DOI] [PubMed] [Google Scholar]
- Kuhnast B.; Dolle F.; Vaufrey F.; Hinnen F.; Crouzel C.; Tavitian B. Fluorine-18 labeling of oligonucleotides bearing chemically-modified ribose–phosphate backbones. J. Labelled Compd. Radiopharm. 2000, 438837–848. [Google Scholar]
- Kuhnast B.; Hinnen F.; Boisgard R.; Tavitian B.; Dolle F. Fluorine-18 labelling of oligonucleotides: Prosthetic labelling at the 5 ′-end using the N-(4-18F[fluorobenzyl)-2-bromoacetamide reagent. J. Labelled Compd. Radiopharm. 2003, 46121093–1103. [Google Scholar]
- Habeeb A. F. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal. Biochem. 1966, 143328–36. [DOI] [PubMed] [Google Scholar]
- Markus G.; Karush F. The disulfide bonds of human serum albumin and bovine γ-globulin. J. Am. Chem. Soc. 1957, 79, 134–139. [Google Scholar]
- Yue X. Y.; Feng Y.; Yu Y. B. Synthesis and characterization of fluorinated conjugates of albumin. J. Fluorine Chem. 2013, 152, 173–181. [Google Scholar]
- Christ E.; Wild D.; Reubi J. C. Glucagonlike peptide-1 receptor: an example of translational research in insulinomas: a review. Endocrinol. Metab. Clin. North Am. 2010, 394791–800. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Chen W. Radiolabeled glucagon-like peptide-1 analogues: a new pancreatic beta-cell imaging agent. Nucl. Med. Commun. 2012, 333223–7. [DOI] [PubMed] [Google Scholar]
- Deacon C. F.; Johnsen A. H.; Holst J. J. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J. Clin. Endocrinol. Metab. 1995, 803952–7. [DOI] [PubMed] [Google Scholar]
- Kiesewetter D. O.; Guo N.; Guo J.; Gao H.; Zhu L.; Ma Y.; Niu G.; Chen X. Evaluation of an [(18)F]AlF-NOTA analog of exendin-4 for imaging of GLP-1 receptor in insulinoma. Theranostics 2012, 210999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Niu G.; Yang M.; Quan Q.; Ma Y.; Murage E. N.; Ahn J. M.; Kiesewetter D. O.; Chen X. PET of insulinoma using 18F-FBEM-EM3106B, a new GLP-1 analogue. Mol. Pharmaceutics 2011, 851775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesewetter D. O.; Gao H.; Ma Y.; Niu G.; Quan Q.; Guo N.; Chen X. 18F-radiolabeled analogs of exendin-4 for PET imaging of GLP-1 in insulinoma. Eur. J. Nucl. Med. Mol. Imaging 2012, 393463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X.; Kiesewetter D. O.; Guo J.; Sun Z.; Zhang X.; Zhu L.; Niu G.; Ma Y.; Lang L.; Chen X. Development of a new thiol site-specific prosthetic group and its conjugation with [cys40]-exendin-4 for in vivo targeting of insulinomas. Bioconjugate Chem. 2013, 2471191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr T. M.; Goldenberg D. M.; Becker W. Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: present status, future prospects and limitations. Eur. J. Nucl. Med. 1998, 252201–12. [DOI] [PubMed] [Google Scholar]
- Wang H.; Gao H.; Guo N.; Niu G.; Ma Y.; Kiesewetter D. O.; Chen X. Site-specific labeling of scVEGF with fluorine-18 for positron emission tomography imaging. Theranostics 2012, 26607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olberg D. E.; Arukwe J. M.; Grace D.; Hjelstuen O. K.; Solbakken M.; Kindberg G. M.; Cuthbertson A. One step radiosynthesis of 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J. Med. Chem. 2010, 5341732–40. [DOI] [PubMed] [Google Scholar]
- Shimada K.; Mitamura K. Derivatization of thiol-containing compounds. J. Chromatogr., Biomed. Appl. 1994, 6591–2227–41. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J.; Lin Y. A.; Davis B. G. Chemical modification of proteins at cysteine: opportunities in chemistry and biology. Chem.—Asian J. 2009, 45630–40. [DOI] [PubMed] [Google Scholar]
- Ting R.; Adam M. J.; Ruth T. J.; Perrin D. M. Arylfluoroborates and alkylfluorosilicates as potential PET imaging agents: High-yielding aqueous biomolecular 18F-labeling. J. Am. Chem. Soc. 2005, 1273813094–13095. [DOI] [PubMed] [Google Scholar]
- Schirrmacher R.; Bradtmoller G.; Schirrmacher E.; Thews O.; Tillmanns J.; Siessmeier T.; Buchholz H. G.; Bartenstein P.; Wangler B.; Niemeyer C. M.; Jurkschat K. 18F-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew. Chem., Int. Ed. 2006, 45366047–50. [DOI] [PubMed] [Google Scholar]
- Mu L. J.; Hohne A.; Schubiger R. A.; Ametamey S. M.; Graham K.; Cyr J. E.; Dinkelborg L.; Stellfeld T.; Srinivasan A.; Voigtmann U.; Klar U. Silicon-based building blocks for one-step 18F-radiolabeling of peptides for PET imaging. Angew. Chem., Int. Ed. 2008, 47264922–4925. [DOI] [PubMed] [Google Scholar]
- Becaud J.; Mu L. J.; Karramkam M.; Schubiger P. A.; Ametamey S. M.; Graham K.; Stellfeld T.; Lehmann L.; Borkowski S.; Berndorff D.; Dinkelborg L.; Srinivasan A.; Smits R.; Koksch B. Direct one-step 18F-labeling of peptides via nucleophilic aromatic substitution. Bioconjugate Chem. 2009, 20122254–2261. [DOI] [PubMed] [Google Scholar]
- Jacobson O.; Zhu L.; Ma Y.; Weiss I. D.; Sun X. L.; Niu G.; Kiesewetter D. O.; Chen X. Y. Rapid and simple one-step 18F labeling of peptides. Bioconjugate Chem. 2011, 223422–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Lang L.; Hu S.; Guo N.; Zhu L.; Sun Z.; Ma Y.; Kiesewetter D. O.; Niu G.; Xie Q.; Chen X. Comparison of three dimeric 18F-AlF-NOTA-RGD tracers. Mol. Imaging Biol. 2014, 16, 274–283. [DOI] [PubMed] [Google Scholar]
- Dialer L. O.; Selivanova S. V.; Muller C. J.; Muller A.; Stellfeld T.; Graham K.; Dinkelborg L. M.; Kramer S. D.; Schibli R.; Reiher M.; Ametamey S. M. Studies toward the development of new silicon-containing building blocks for the direct 18F-labeling of peptides. J. Med. Chem. 2013, 56197552–63. [DOI] [PubMed] [Google Scholar]
- Li Z. B.; Conti P. S. Radiopharmaceutical chemistry for positron emission tomography. Adv. Drug Delivery Rev. 2010, 62111031–1051. [DOI] [PubMed] [Google Scholar]
- Li X. G.; Haaparanta M.; Solin O. Oxime formation for fluorine-18 labeling of peptides and proteins for positron emission tomography (PET) imaging: A review. J. Fluorine Chem. 2012, 143, 49–56. [Google Scholar]
- Fani M.; Maecke H. R.; Okarvi S. M. Radiolabeled peptides: valuable tools for the detection and treatment of cancer. Theranostics 2012, 25481–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamous M.; Haberkorn U.; Mier W. Synthesis of peptide radiopharmaceuticals for the therapy and diagnosis of tumor diseases. Molecules 2013, 1833379–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirrmacher R.; Wangler C.; Schirrmacher E. Recent developments and trends in 18F-radiochemistry: syntheses and applications. Mini-Rev. Org. Chem. 2007, 44317–429. [Google Scholar]
- Wängler C.; Kostikov A.; Zhu J.; Chin J.; Wängler B.; Schirrmacher R. Silicon-[18F]fluorine radiochemistry: basics, applications and challenges. Appl. Sci. 2012, 2, 277–302. [Google Scholar]
- Glaser M.; Iveson P.; Hoppmann S.; Indrevoll B.; Wilson A.; Arukwe J.; Danikas A.; Bhalla R.; Hiscock D. Three methods for 18F labeling of the HER2-binding affibody molecule ZHER2:2891 including preclinical assessment. J. Nucl. Med. 2013, 54111981–8. [DOI] [PubMed] [Google Scholar]
- Wangler B.; Kostikov A. P.; Niedermoser S.; Chin J. S.; Orchowski K.; Schirrmacher E.; Iovkova-Berends L.; Jurkschat K.; Wangler C.; Schirrmacher R. Protein labeling with the labeling precursor [18F] SiFA-SH for positron emission tomography. Nat. Protoc. 2012, 7111964–1969. [DOI] [PubMed] [Google Scholar]
- Li Z. B.; Lin T. P.; Liu S. L.; Huang C. W.; Hudnall T. W.; Gabbai F. P.; Conti P. S. Rapid aqueous [18F]-labeling of a bodipy dye for positron emission tomography/fluorescence dual modality imaging. Chem. Commun. 2011, 47339324–9326. [DOI] [PubMed] [Google Scholar]
- Liu S.; Park R.; Conti P. S.; Li Z. ″Kit like″ 18F labeling method for synthesis of RGD peptide-based PET probes. Am. J. Nucl. Med. Mol. Imaging 2013, 3197–101. [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Li Y.; Lozada J.; Wong M. Q.; Greene J.; Lin K. S.; Yapp D.; Perrin D. M. Kit-like 18F-labeling of RGD-19F-arytrifluroborate in high yield and at extraordinarily high specific activity with preliminary in vivo tumor imaging. Nucl. Med. Biol. 2013, 406841–9. [DOI] [PubMed] [Google Scholar]
- Liu Z. B.; Li Y.; Lozada J.; Schaffer P.; Adam M. J.; Ruth T. J.; Perrin D. M. Stoichiometric leverage: rapid 18F-aryltrifluoroborate radiosynthesis at high specific activity for click conjugation. Angew. Chem., Int. Ed. 2013, 5282303–2307. [DOI] [PubMed] [Google Scholar]
- Hultsch C.; Schottelius M.; Auernheimer J.; Alke A.; Wester H. J. 18F-Fluoroglucosylation of peptides, exemplified on cyclo(RGDfK). Eur. J. Nucl. Med. Mol. Imaging 2009, 3691469–1474. [DOI] [PubMed] [Google Scholar]
- Wuest F.; Hultsch C.; Berndt M.; Bergmann R. Direct labelling of peptides with 2-[18]fluoro-2-deoxy-d-glucose ([18]FDG). Bioorg. Med. Chem. Lett. 2009, 19185426–5428. [DOI] [PubMed] [Google Scholar]
- Wuest F.; Berndt M.; Bergmann R.; van den Hoff J.; Pietzsch J. Synthesis and application of [18F]FDG-maleimidehexyloxime ([18F]FDG-MHO): A [18F]FDG-based prosthetic group for the chemoselective 18F-labeling of peptides and proteins. Bioconjugate Chem. 2008, 1961202–1210. [DOI] [PubMed] [Google Scholar]
- Smith T. A. D. [18]Fluoride labelling of macromolecules in aqueous conditions: silicon and boroaryl-based [18]fluorine acceptors, [18]FDG conjugation and Al18F chelation. J. Labelled Compd. Radiopharm. 2012, 558281–288. [Google Scholar]
- D’Souza C. A.; McBride W. J.; Sharkey R. M.; Todaro L. J.; Goldenberg D. M. High-yielding aqueous 18F-labeling of peptides via Al18F chelation. Bioconjugate Chem. 2011, 2291793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride W. J.; Sharkey R. M.; Goldenberg D. M. Radiofluorination using aluminum-fluoride (Al18F). EJNMMI Res. 2013, 3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejot R.; Fowler T.; Carroll L.; Boldon S.; Moore J. E.; Declerck J.; Gouverneur V. Fluorous synthesis of 18F radiotracers with the [18F]fluoride ion: nucleophilic fluorination as the detagging process. Angew. Chem., Int. Ed. 2009, 483586–589. [DOI] [PubMed] [Google Scholar]
- Cai L. S.; Lu S. Y.; Pike V. W. Chemistry with [18F]fluoride ion. Eur. J. Org. Chem. 2008, 172853–2873. [Google Scholar]
- Prasad V.; Birzin E. T.; McVaugh C. T.; Van Rijn R. D.; Rohrer S. P.; Chicchi G.; Underwood D. J.; Thornton E. R.; Smith A. B. III; Hirschmann R. Effects of heterocyclic aromatic substituents on binding affinities at two distinct sites of somatostatin receptors. Correlation with the electrostatic potential of the substituents. J. Med. Chem. 2003, 46101858–69. [DOI] [PubMed] [Google Scholar]