

Abstract
Flow-induced production of nitric oxide (NO) by endothelial cells plays a fundamental role in vascular homeostasis. However, the mechanisms by which shear stress activates NO production remain unclear due in part to limitations in measuring NO, especially under flow conditions. Shear stress elicits the release of ATP, but the relative contribution of autocrine stimulation by ATP to flow-induced NO production has not been established. Furthermore, the importance of calcium in shear stress-induced NO production remains controversial, and in particular the role of capacitive calcium entry (CCE) has yet to be determined. We have utilized our unique NO measurement device to investigate the role of ATP autocrine signaling and CCE in shear stress-induced NO production. We found that endogenously released ATP and downstream activation of purinergic receptors and CCE plays a significant role in shear stress-induced NO production. ATP-induced eNOS phophorylation under static conditions is also dependent on CCE. Inhibition of protein kinase C significantly inhibited eNOS phosphorylation and the calcium response. To our knowledge, we are the first to report on the role of CCE in the mechanism of acute shear stress-induced NO response. In addition, our work highlights the importance of ATP autocrine signaling in shear stress-induced NO production.
Keywords: endothelial cells, nitric oxide, shear stress, capacitative calcium entry, ATP
Introduction
It is well established that endothelial cells (ECs) are involved in the regulation of vascular tone and are able to sense and response to shear stress caused by blood flowing across their surface. One important physiological response to the mechanical forces of shear stress is the release of nitric oxide (NO). NO is a major vasodilator that causes relaxation of the smooth muscle but is also involved in the inhibition of platelet aggregation (1, 2), adhesion of leukocytes to the endothelium (3, 4) and smooth muscle cell proliferation (5). Despite its importance, our understanding of the mechanisms of shear stress-induced NO production remains incomplete, due in part to difficulties in measuring NO in response to acute changes in flow.
Ample evidence exists for calcium dependent NO production in static cultures (6-10) and that shear stress stimulates a calcium response (11-13). Yet the role of calcium, its source (extra- vs intracellular, plasma membrane vs. endoplasmic reticulum) and mechanism for release under flow in NO production has been unclear and sometimes controversial. One mechanism for the flow-induced calcium response is through the release of ATP. A number of studies have shown that shear stress causes the release of ATP in blood vessels (14, 15) and endothelial cells (16, 17) and its release precedes and spatially coincides with the initiation of a calcium wave (13). In static cultures, the mechanism by which ATP stimulates a calcium response is well characterized and has been shown to increase endothelial nitric oxide synthase (eNOS) phosphorylation and NO production (14, 18, 19). Despite this evidence, some studies have discredited or diminished the contribution of ATP and calcium in shear stress-induced NO production using NO measurements (20, 21) and mathematical modeling (22).
ATP stimulates a calcium response through both the release from endoplasmic reticulum (ER) stores as well as the influx through store-operated channels (SOCs). This influx from the extracellular space has been termed capacitiatve calcium entry (CCE) and in static cultures maximally activates NO production as compared to the ER calcium release (6-8, 23). The shear stress-induced ATP release and the clear connection between calcium, ATP and SOCs under static conditions suggests a role for ATP autocrine stimulation in shear stress-induced NO production but the relative contribution and CCE dependence has yet to be confirmed or fully explored.
In this study, we used our novel technique for measuring NO directly and in real-time from endothelial cells under flow conditions to study the mechanism for shear stress-induced NO production. To accomplish this, we inhibited aspects of the ATP and SOC pathway to determine the relative contribution of each in the flow-induced NO response. Our data show that ATP autocrine signaling and the subsequent activation of CCE is in part responsible for shear stress-induced NO production. In addition, activation of CCE and its PKC dependency leads to an increase in eNOS phosphorylation as well as NO production under flow and static cultures.
Materials and Methods
Cell Culture
Primary culture Bovine Aortic Endothelial Cells (BAECs) were obtained from Dr. Peter Davies' Laboratory (University of Pennsylvania). BAECs were cultured in Dulbeccos modified Eagle's medium (Mediatech Cellgro), supplemented with 10% fetal bovine serum (Sigma), 2 mmol/l L-glutamine (Mediatech Cellgro), and penicillin-streptomycin (Mediatech Cellgro) as described previously (24).
Chemicals and Reagents
Suramin, apyrase, SKF-96365 (SKF), chelerythrine (Cheler), and Dulbecco's phosphate buffered saline (PBS) with calcium and magnesium were purchased from Sigma. PBS without calcium was purchased from Mediatech, Inc. BCA protein assay was purchased from Pierce.
Protein Assay
Total protein content was determined using bicinchoninic acid for colorimetric detection and quantification of total protein (BCA Protein Assay Kit). Standards were created using Albumin (BSA) diluted in lysis buffer (20 mM Tris, 1% Deoxycholate, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 2 mM PMSF, 0.1% SDS, 1 μg/mL leupeptin, 50 mM mM NaF, 10% glycerol, Pierce complete protease inhibitor, pH 7.4). Concentrations of BSA ranged from 2000 μg/ml to 0 μg/ml. 25 μL of each sample was used in each microplate well in triplicates. BCA working reagent was mixed in 1:50 ratio of BCA Reagent A to BCA Reagent B and 200 μL added to each well. Microplate was covered with aluminum foil and incubated at 37°C prior and read at 562 nm in a Tecan Infinite M200 Series. Blank absorbances were subtracted from all standard and experimental sample values. Standards were then plotted and a best-fit curve was used to calculate total protein content of experimental samples.
Nitric Oxide Measurements Under Flow
Our device consists of a parallel plate flow chamber in which a NO sensitive electrode is located in a stagnant upper compartment separated from the ECs and the flow field by a porous membrane, the details of which have been described previously (25). In typical NO responses, an increase in flow (shear stress) first causes the NO concentration to decrease due to increased convection. Then the NO concentration increases due to an increase in production, which overcomes these convection effects until a higher NO steady-state concentration is reached. Previously, we have shown that this time-dependent response represents the actual kinetics of NO production and not a transport lag of the device (25).
Cells were grown on the underside of transwell™ membranes until confluent. Membranes were washed in PBS with calcium/magnesium supplemented with 70 μM L-arginine 3× and then inserted in the flow chamber. In experiments to degrade endogenous ATP using apyrase, cells were exposed to multiple step changes in shear stress from a basal level of 0.1 dyn/cm2 to 1, 6,10 or 20 dyn/cm2 with 3-minute interval between step changes. The experimental solution was then exchanged with PBS with calcium/magnesium and 1U/mL apyrase, and the step change sequence was repeated.
In experiments involving the inhibition of SOCs or purinergic receptors, cells were pretreated with SOC blocker, SKF-96365 (50 μM, 10 min) or purinergic receptor antagonist, suramin (200 μM 10 min). Each membrane of cells was exposed to 3-5 step changes in shear stress from 0.1 to 10 dyn/cm2. Each cycle, the shear stress was held at 10 dyn/cm2 for 3 min then returned to 0.1 dyn/cm2 for 3 min before the next step increase to 10 dyn/cm2. The NO responses to each of the 3-5 step increases to 10 dyn/cm2 were of similar magnitude and were averaged to give a single value representing the response of that membrane and group of cells.
Each experimental condition was repeated 3 times (i.e. with 3 different membranes of cells) for SKF and suramin, and 4 times for treatment with apyrase. The Δ[NO] response to each step change was calculated using the average concentration of NO over a 10 s interval prior to the step increase and the average concentration at the new steady state over a 10 s interval.
eNOS Phosphorylation and Shear Stress
Cells grown on the underside of transwell membranes were washed 3× with experimental fluid and then inserted into the flow chamber. The chamber was placed in a water bath for 10 min prior to exposure to 10 dyn/cm2 for 0, 1 or 3 min. In experiments with apyrase, the circulating experimental fluid contained 1U/mL apyrase. The membrane was removed, and cells were harvested for western blots. The increase in phosphorylation after 3 min of shear stress was normalized by the untreated response. Each condition was repeated 5 times.
Western Blot
Samples were prepared for Western blot by scraping cells in lysis buffer at 4°C for 20 min. Samples were then centrifuged for 10 min at 10,000g, 4 °C to remove insoluble material. Cell lysates were normalized for protein content, separated by SDS-PAGE on a 4–20% Bis-Tris gel (Lonza), and transferred to a nitrocellulose membrane (Invitrogen, Iblot). After blocking in 5% Blotto (Bioexpress), membranes were incubated with primary peNOS (1179) antibody (Invitrogen) overnight at 4 °C followed by a secondary horseradish peroxidase-conjugated antibody for 2 h at room temperature. Following protein band detection, the membrane was stripped (Western blot stripping buffer, Pierce) for 8 min and then incubated overnight with primary eNOS antibody (BD biosciences) at 4 °C and subsequently by a secondary horseradish peroxidase-conjugated antibody at room temperature. Protein bands were detected using an enhanced chemiluminescence kit (Western Lightning, PerkinElmer), and visualized with a Fluorchem digital imager (Alpha Innotech). Band intensity was quantified using AlphaEase FC software.
Calcium Fluorescence Imaging
Cells were grown on 25 mm2 round glass coverslips for 2 days prior to experiments. Cells were incubated with 1.5 μmol/L fluo-3AM (Molecular Probes) for 40 minutes at room temperature. SKF-96365 (SKF) treated cells were incubated with 50 μM for 10 min and chelerythrine (cheler) treated cells were treated with 30 μM for 2 min prior to incubation with fluorescent dye. Following treatment, cells were stimulated with 500 μL of 100 μM ATP. Each experimental condition was repeated the following number of times: Untreated n=8, SKF n=5, Cher n=4. Cells were viewed with an inverted light microscope 20× (Nikon TE300 Eclipse microscope). All fluorescence measurements were expressed as a ratio (R) of fluorescence intensity (F) to the basal fluorescence intensity (F0): R= F/F0 which has been described previously (24).
ATP stimulation and eNOS Phosphorylation
Cells were grown on 25 mm2 round glass coverslips for 3 days prior to experiments. Experimental treatments include incubation with 50 μM SKF-96365 (SKF) for 10 min, 30 μM cheler for 2 min, and 30 μM BAPTA for 30 min. Stimulation in the presence of extracellular calcium: cells were washed 3× with PBS with calcium, stimulated with 100 μM ATP with calcium and then harvested at time intervals 0, 1, 3, 5, 10 min. In experiments without calcium, cells were washed 3× with calcium-free PBS then stimulated with calcium-free agonist, and cells were harvested at time intervals 0, 1, 3, 5, 10 min. After 10 min, 500 μL of PBS with calcium or 500 μL of calcium-free was added, and cells were harvested at time intervals (1, 3, and 10 min). Each experimental condition was repeated the following number of times: Untreated n=4, BAPTA n=4, ATP wo Ca n=5, addition of PBS with Ca+2 n=3, No Ca n=3, SKF n=5, Cheler n=4.
Results
The Shear Stress-Induced NO Response is due in part to ATP autocrine signaling
In order to determine the relative contribution of ATP autocrine stimulation on shear stress-induced NO production, we utilized direct real-time measurements NO concentrationas well as eNOS phosphorylation in response to changes in shear stress. First, we used apyrase to degrade endogenously released ATP. Steady-state levels of the NO response to shear stress were compared before and after apyrase treatment. Following stimulation at multiple shear stresses, the experimental fluid was exchanged with 1U/mL apyrase. Measurements with apyrase were made under the same protocol as prior to treatment with apyrase, with the shear stress starting at 0.1 dyn/cm2 or 1 dyn/cm2 and then increased to 1, 6, 10 or 20 dyn/cm2. In the sample traces (Figure 1A-D) this step change occurs at 50s. The steady-state concentration was offset to zero in order to show the comparison between individual NO responses due to the step changes. Whether the baseline shear stress was 0.1 or 1 dyn/cm2, the response to step changes to higher flow rates were not statistically different and were averaged within a single experiment for untreated and apyrase treatment. Comparison of the Δ[NO] response between untreated and apryase treated responses were found to be statistically significant for step changes to 6, 10 and 20 dyn/cm2 but not for 0.1 to 1 dyn/cm2 (Figure 1E). The steady-state NO response in the presence of apyrase were reduced to 57%, 77%, and 80% of untreated controls for shear stress steps of 6, 10, and 20 dyn/cm2, respectively. We also examined the effect of degradation of ATP on shear stress-induced eNOS phosphorylation. Cells were exposed to a step change to 10 dyn/cm2 in untreated cells for 1 min and 3 min, and the ratio of phosphorylated eNOS to total eNOS was determined and normalized by the sham control (no flow). Under normal conditions (no apyrase), shear stress caused a statistically significant increase in phosphorylation at 3 min but not at 1 min (data not shown). Treatment with apyrase to degrade endogenously-released ATP prevented the shear stress-induced phosphorylation of eNOS at 3 mins (Figure 2). We also interfered with ATP autocrine signaling by inhibiting downstream purinergic receptors using the non-specific purinergic receptor blocker suramin. Blocking purinergic receptors attenuated the Δ[NO] response to a step change from 0.1 to 10 dyn/cm2 to 40% of the untreated responses (Figure 3A,B).
Figure 1.
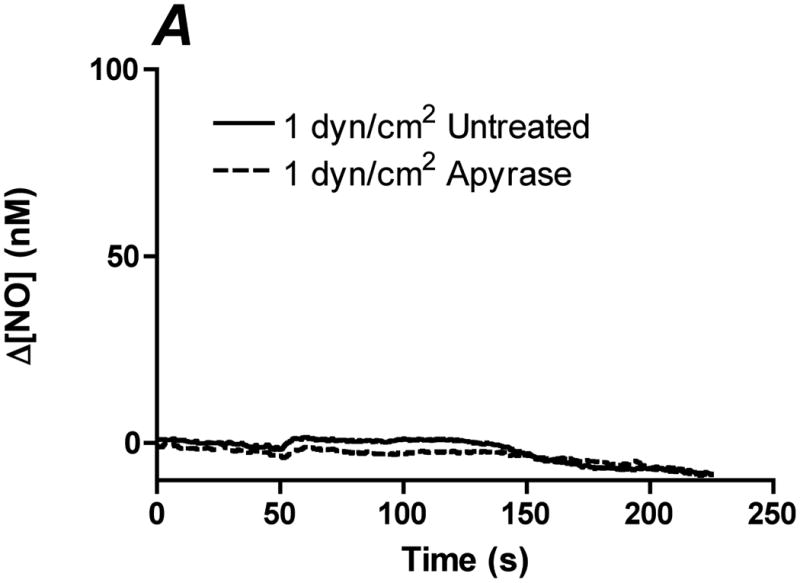
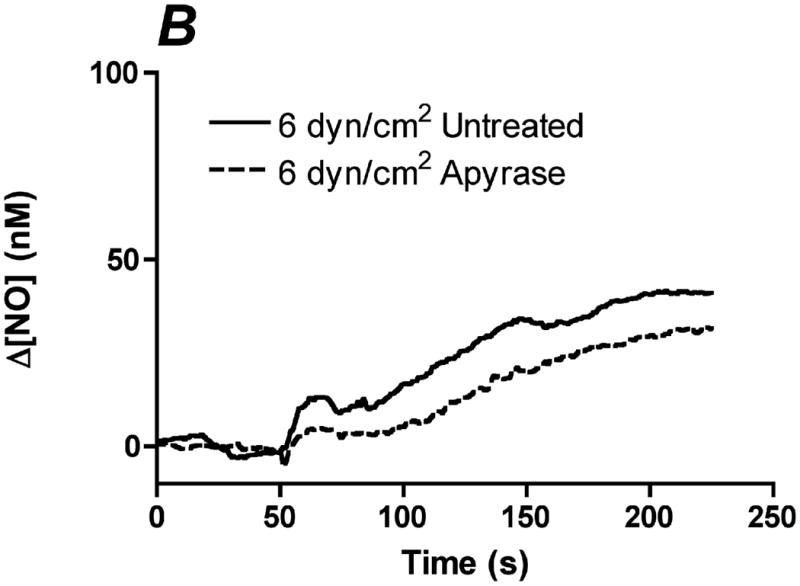
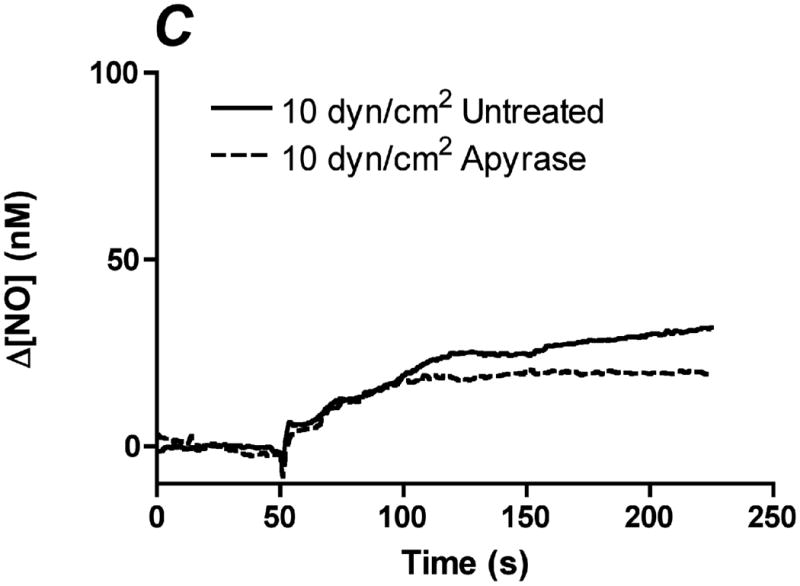
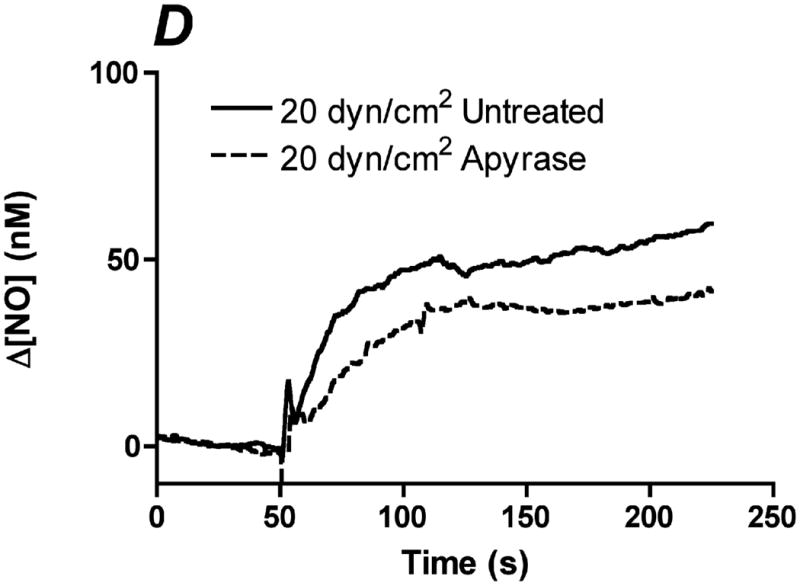
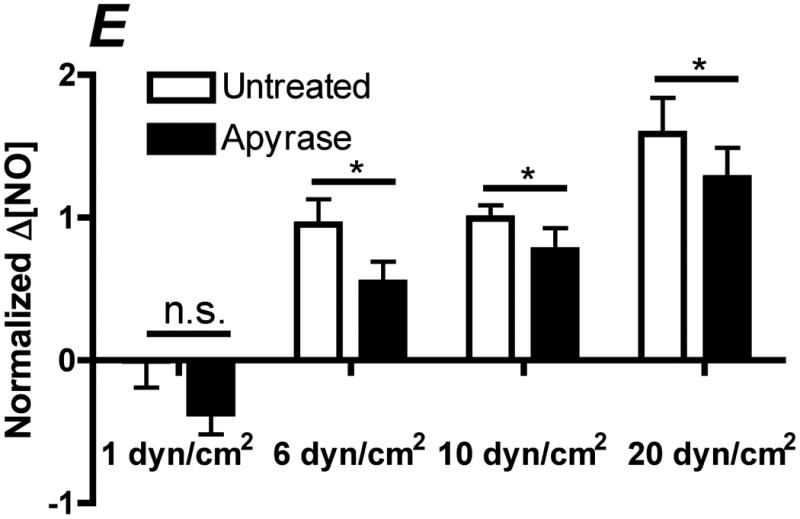
Degradation of endogenous ATP attenuates shear stress-induced NO production. A-D. Sample traces of the NO response before (solid line) and after (dotted line) treatment with 1U/mL of apyrase. The steady-state concentration was offset to zero in order to show the individual NO response due to the step change (at 50s). Sample responses from a step change to A. 1 dyn/cm2 B. 6 dyn/cm2 C. 10 dyn/cm2. D. 20 dyn/cm2. E. Within an experiment, apyrase showed an inhibition of the shear stress-induced NO response; however, due to the variability between experiments, we normalized the responses for an untreated step change to 10 dyn/cm2 within each experiment. Comparisons of the Δ[NO] response before and after treatment with apyrase. Δ[NO] between untreated and apryase treated cells were found to be statistically significant for step changes to 6, 10 and 20 dyn/cm2 but not to 1 dyn/cm2. Δ[NO] for apyrase treated cells averaged 57% for 6 dyn/cm2, 77% for 10 dyn/cm2 and 80% for 20 dyn/cm2 of the untreated responses. (Mean and SEM, one-tailed t-test, *p<0.05, n=4)
Figure 2.
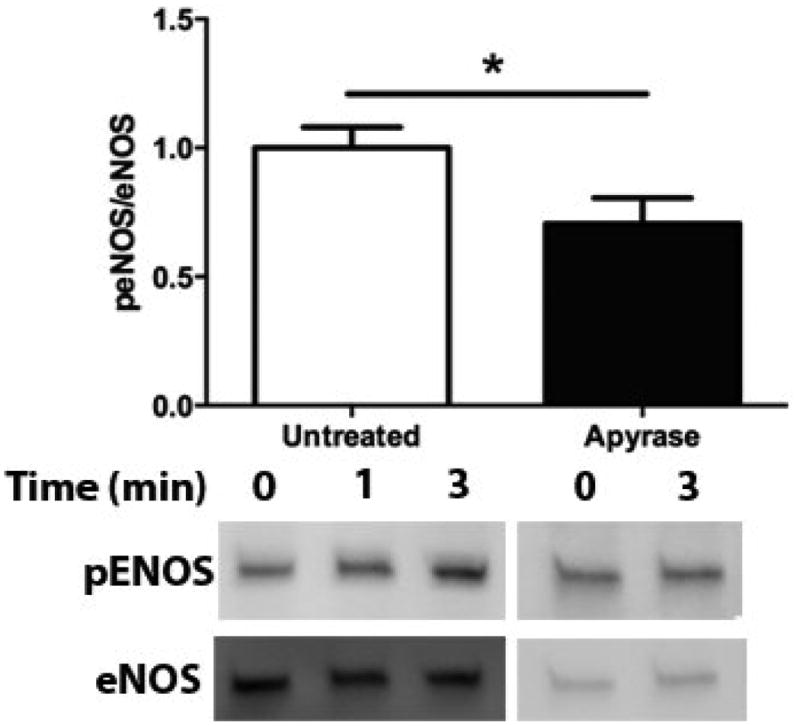
Apyrase attenuates shear stress-induced eNOS phosphorylation. Bar graph comparing shear stress-induced eNOS phosphorylation after 3 min in untreated and apyrase treated cells. Phosphorylation increased in untreated cells after 3 mins and was attenuated when endogenous ATP was degraded using apyrase as compared to the no flow condition. Responses are normalized by the untreated response (Mean and SEM were plotted, *p<0.05 one-tailed t-test n=5 each condition).
Figure 3.
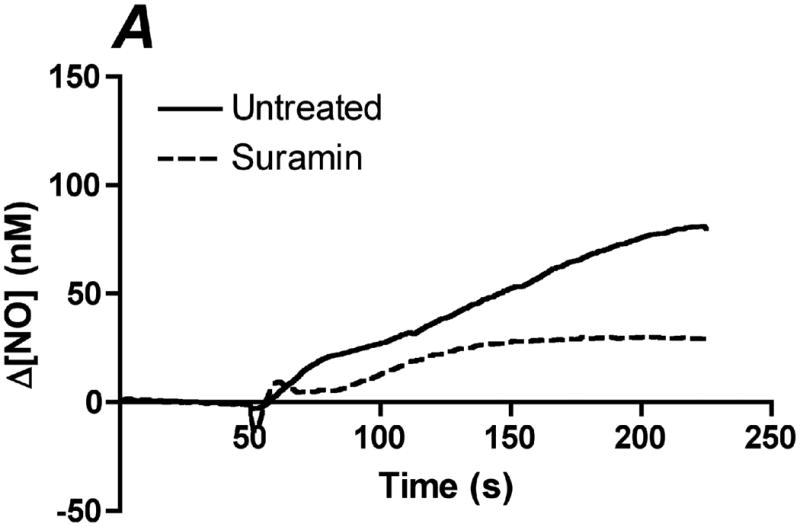
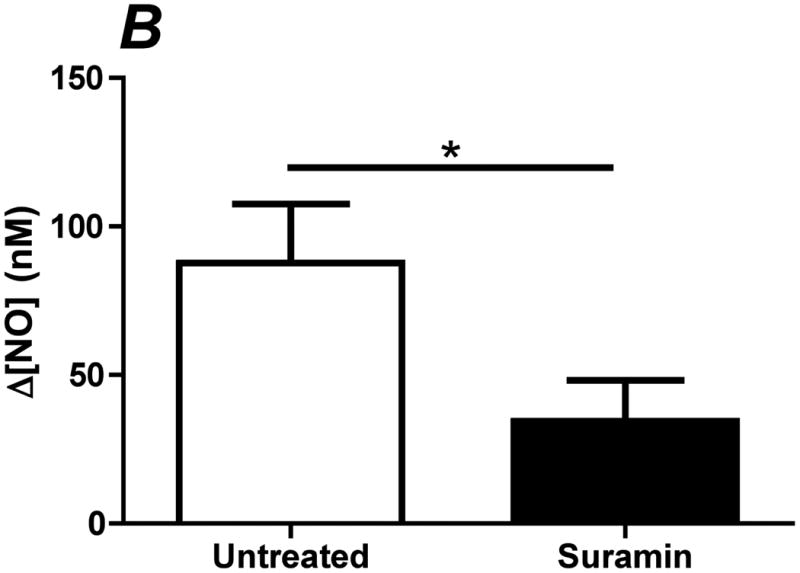
Inhibition of purinergic receptors attenuates shear stress-induced NO production. Purinergic receptors were blocked with using the non-specific blocker suramin. Cells were incubated with 200 μM of suramin for 10 min prior to insertion into the chamber. Each membrane was exposed to a series of 4 step changes from 0.1 to 10 dyn/cm2. A. Sample traces of the response for untreated (solid line) and suramin (dotted line) treated cells. The steady-state concentration was offset to zero in order to show the individual NO response due to the step change occurring at 50s. B. Bar graph comparing the Δ[NO] response to shear stress in untreated and suramin treated cells. The Δ[NO] response in cells treated with suramin averaged 40% of the untreated responses and were statistically significant. (Mean and SEM were plotted, one-tailed t-test. n=3 each, *p<0.05).
The Shear Stress-Induced NO Response is Dependent on CCE
Since a significant portion of the shear stress-induced NO response is dependent on endogenously released ATP and subsequent activation of purinergic receptors, we hypothesized that shear stress-induced NO production was mediated by the activation of SOCs. We and others have shown that eNOS is preferentially activated by CCE when stimulated with agonists under static cultures (6-8). This occurs because of the colocalization of eNOS with SOCs, which create high local calcium concentration gradients leading to maximal eNOS activity (23). To determine the role of CCE in shear stress-induced NO responses, we inhibited SOCs using SKF-96365. Inhibition of SOCs caused a 40% reduction in the Δ[NO] responses to 10 dyn/cm2 (Figure 4A,B).
Figure 4.
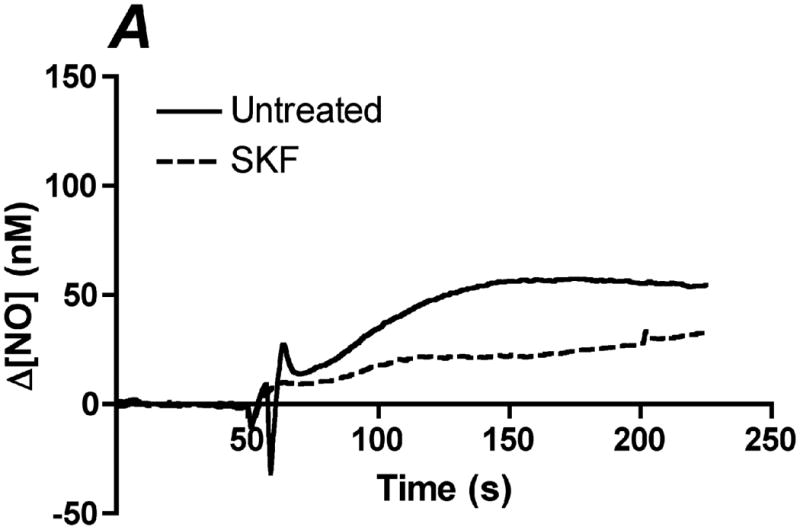
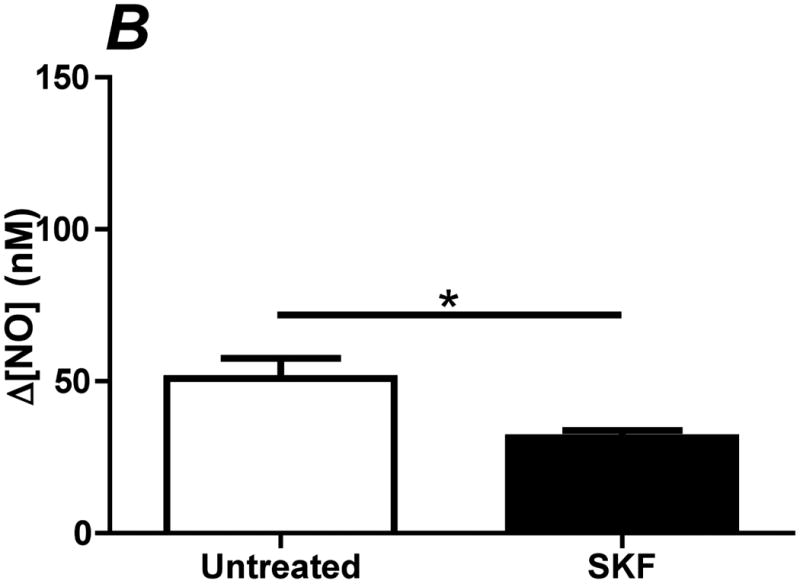
Inhibition of store-operated channels (SOC) attenuates shear stress-induced NO production. SOCs were inhibited using the blocker SKF-96365 (SKF). Cells were treated with 50 μM of SKF for 10 min prior to insertion into the chamber. Each membrane was exposed to a series of 5 step changes of 0.1 to 10 dyn/cm2. A. Sample traces of the response for untreated (solid line) and SKF treated (dotted line) cells. The steady-state concentration was offset to zero in order to show the individual NO response due to the step change occurring at 50s. B. Bar graph representing the Δ[NO] response to shear stress in untreated and SKF treated cells. The Δ[NO] response for SKF treated cells averaged 60% of the untreated responses and were found to be statistically significant (Mean and SEM, one-tailed t-test. n=3 each, *p<0.05).
In order to further elucidate the mechanism of CCE-dependent autocrine stimulation of NO production, we next focused on the mechanism of ATP induced eNOS phosphoration under static conditions. ATP-induced eNOS phosphorylation has been reported to be dependent on calcium (19); however, the exact nature of that relationship is still unclear. To determine the importance of increases in intracellular calcium in ATP-induced eNOS phosphorylation, we first chelated intracellular calcium using BAPTA. Treatment with BAPTA completely abolished the increase in eNOS phosphorylation in response to ATP stimulation (Figure 5).
Figure 5.
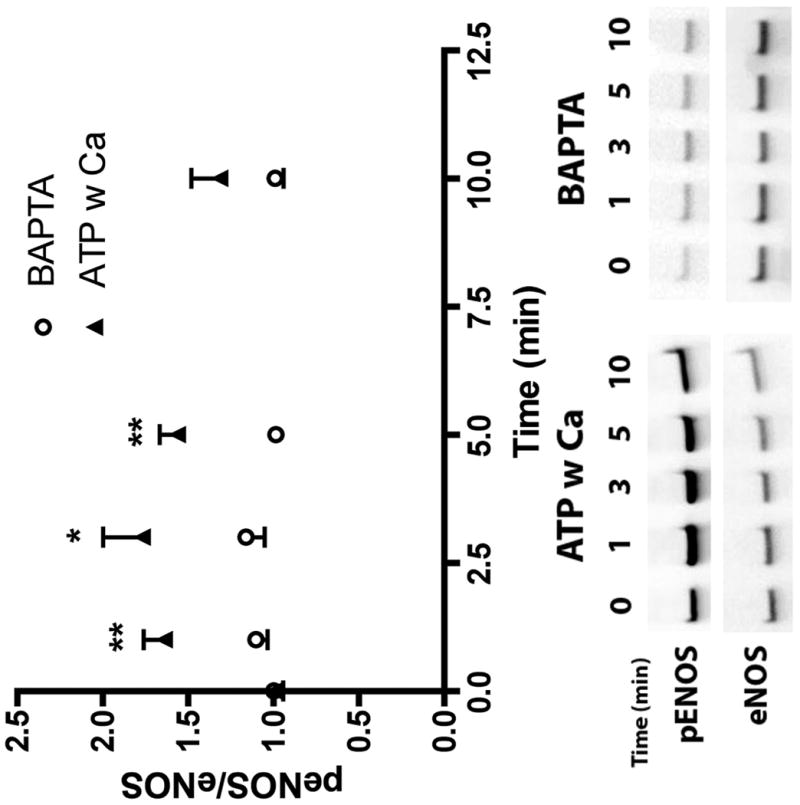
ATP induced eNOS phosphorylation is dependent on increases in intracellular calcium. Cells were simulated with 100 μM ATP with calcium. Prior to stimulation, cells were treated with 30 μM of BAPTA for 30 min. Cells were harvested before stimulation (t=0) and at time points 1, 3, 5, and 10 min after stimulation. All peNOS/eNOS ratios were normalized by t=0. Treatment with BAPTA abolished the eNOS phosphorylation response. (p<0.05 *; p<0.01 ** one-tailed t-test, n=4 each)
ATP stimulation elicits the release of calcium from the ER and concomitant influx via SOCs. To evaluate the relative effect on eNOS phosphorylation of calcium from the two sources, we separated them temporally by stimulating with ATP in the absence of extracellular calcium for 10 min followed by the replacement of extracellular calcium (Figure 6, open squares). This type of experiment is frequently used to separate ER calcium release and calcium influx from the extracellular space through SOCs (23). ATP stimulation in the presence of extracellular calcium (closed triangles) showed a rapid increase in phosphorylation that peaked at 3 min followed by a sustained elevation through 10 min. In contrast, ATP stimulation in the absence of extracellular calcium (open squares), elicited a transient peak in eNOS phosphorylation at 1 min that returned to baseline by 5 min. Addition of extracellular calcium at 10 min after stimulation (open squares) caused a sustained increase in eNOS phosphorylation over 10 min (representing time point 20 min) which was not seen with the addition of PBS without calcium (Eight Spoked Asterisks). To test our hypothesis that the influx of extracellular calcium specifically through SOCs was responsible for the large, sustained phosphorylation response, we treated cells with the SOC channel inhibitor SKF-96365 prior to stimulation with ATP. Typical calcium responses to ATP in untreated cells (Figure 7 A, B) showed a transient peak occurring within 30s followed by a sustained response, which continues past 3 min. When SOCs were inhibited, the peak calcium was reduced by 25% and the sustained response was strongly attenuated, returning to baseline by 150s. Inhibiting calcium influx via SOCs significantly attenuated ATP-stimulated eNOS phosphorylation at 1 and 3 min (Figure 8).
Figure 6.
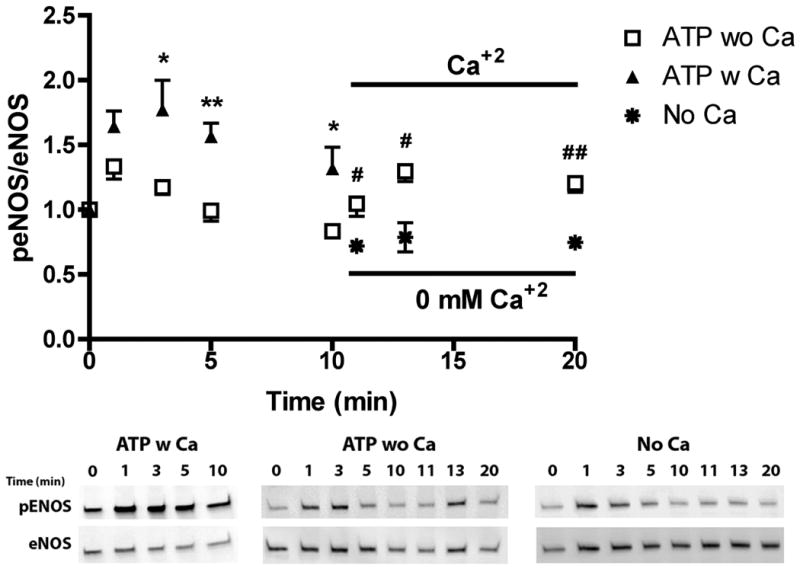
ATP induced eNOS phosphorylation is dependent on the calcium influx from the extracellular space. Cells were simulated with 100 μM ATP in the presence (ATP w CA) or absence of extracellular calcium (ATP wo Ca). Cells were harvested before stimulation (t=0) and at time points 1, 3, 5, and 10 min after stimulation. All peNOS/eNOS ratios were normalized by t=0. eNOS phosphorylation remained elevated after 1 min, which was not seen in the absence of extraceullular calcium. After the initial response to stimulation in the absence of calcium, PBS with calcium was added (t=10) which caused an increase in eNOS phosphorylation that was not observed when PBS without calcium was added. (p<0.05 *,#; p<0.01 **, ## two-tailed t-test; ATP wo Ca+2 n=5, ATP w Ca+2 n=4, addition of PBS with Ca+2 n=3, No Ca n=3)
Figure 7.
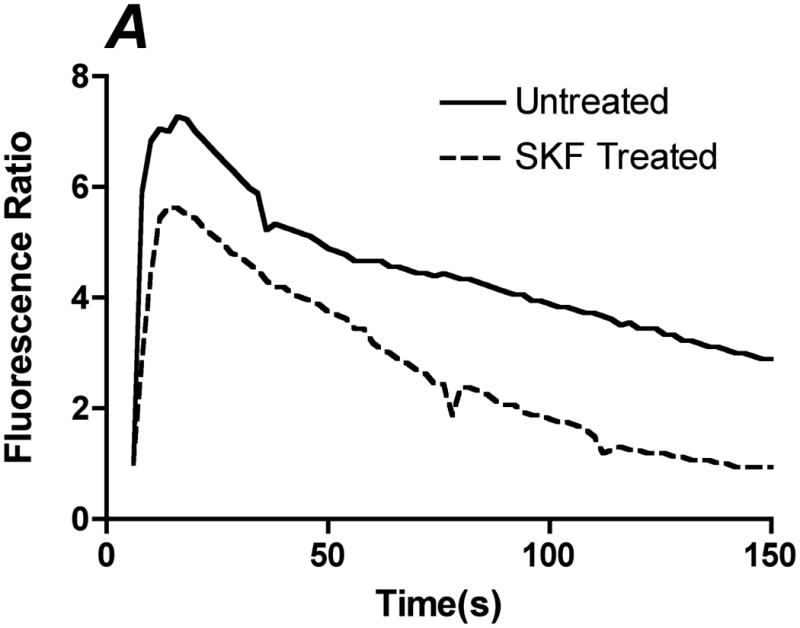
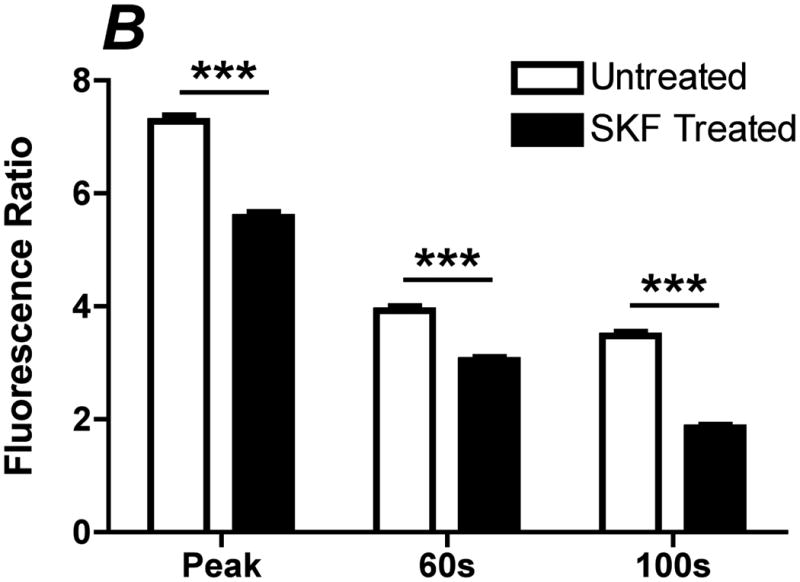
Inhibition of ATP-induced CCE by SKF-96365. Prior to the experiment, cells were incubated with PBS with Ca+2 or 50 μM of SOC inhibitor SKF-96365 for 10 min. A. Representative traces of the calcium response in untreated (solid line) and SKF (dotted line) treated cells to 500 μL of 100 μM ATP with calcium. B. Bar graph representing average responses for each condition at the peak response, 60s and 100s. SKF reduced transient and sustained calcium response, which was statically significant from untreated and SKF treated cells. Average responses represent multiple coverslips (cs) and total cell count among coverslips (n) Untreated: #cs= 8, n=512; SKF: #cs=5, n=298. (Mean and SEM, Two-tailed t-test p<0.0001)
Figure 8.
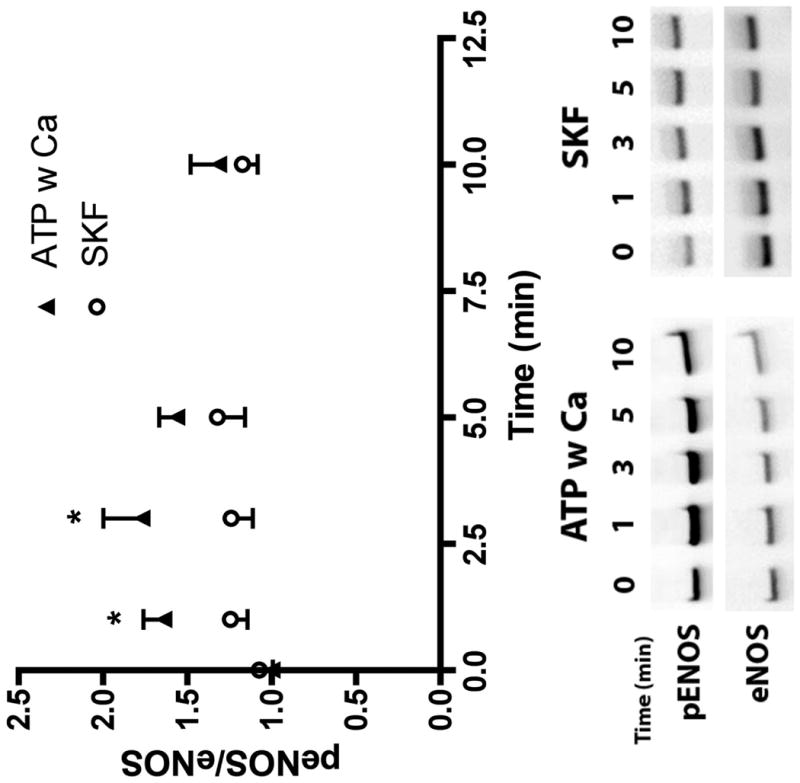
ATP induced eNOS phosphorylation is dependent on CCE. Cells were simulated with 100 μM ATP with calcium. Prior to stimulation, cells were treated with 50 μM of SKF-96365 for 10 min. Cells were harvested before stimulation (t=0) and at time points 1, 3, 5, and 10 min after stimulation. All peNOS/eNOS ratios were normalized by t=0. SKF-96365 treated cells (SKF) show an attenuated eNOS phosphorylation at 1 and 3 min, which was statistically significant. (p<0.05 * one-tailed t-test, ATP w Ca+2 n=4, SKF n=5)
Recently, PKC has been shown to be important for eNOS phosphorylation and CCE separately (26-28). Thus, we sought to investigate the role of PKC in CCE-dependent eNOS phosphorylation. Inhibiting PKC with the general PKC inhibitor chelerythrine reduced the transient calcium peak by 40% and attenuated the sustained response (Figure 9 A,B). Furthermore, inhibition of PKC significantly attenuated eNOS phosphorylation at 1 and 3 min (Figure 10), to an extent similar to inhibition of SOCs.
Figure 9.
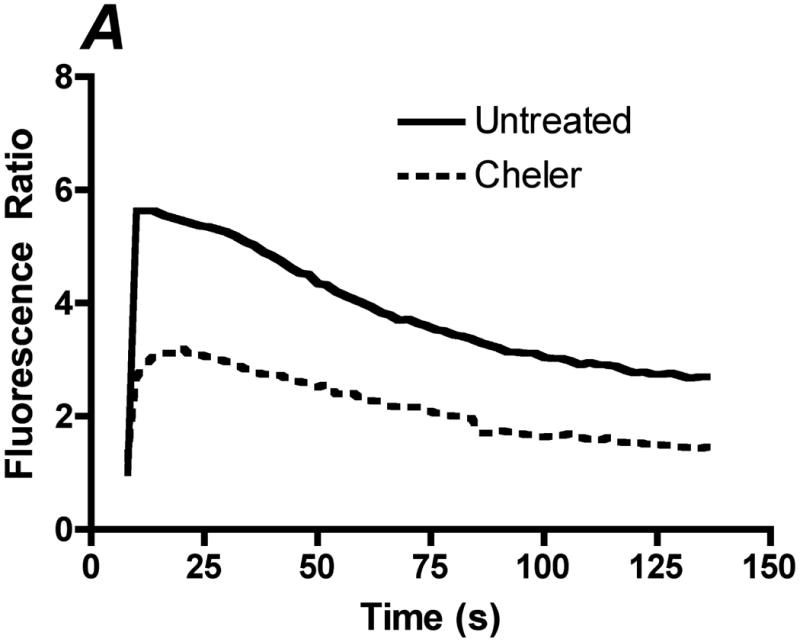
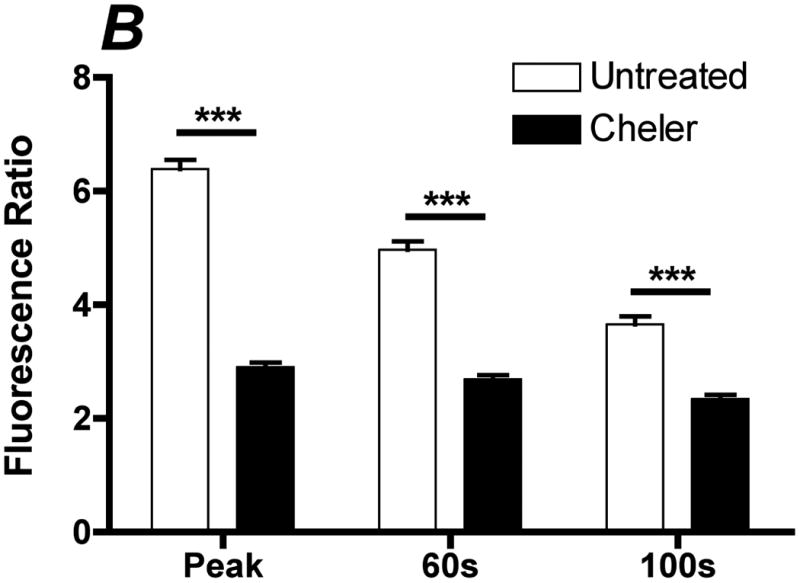
Inhibition of PKC attenuates the ATP stimulated sustained calcium response. Prior to stimulation with 500 μL of 100 μM ATP with calcium, cells were incubated with 30 μM of the general PKC inhibitor cheler for 2 min. A. Representative traces of the calcium response in untreated (solid line) and cheler (dotted line) treated cells to 500 μL of 100 μM ATP with calcium. B. Bar graph representing average responses for each condition at the peak response, 60(s) and 100(s). Cheler treated cells showed attenuation of the peak and sustained calcium response. Average responses represent multiple coverslips (cs) and total cell count among coverslips (n) Untreated: #cs= 3, n=120, Cheler: #cs=4, n=160, two-tailed t-test ***p<0.0001.
Figure 10.
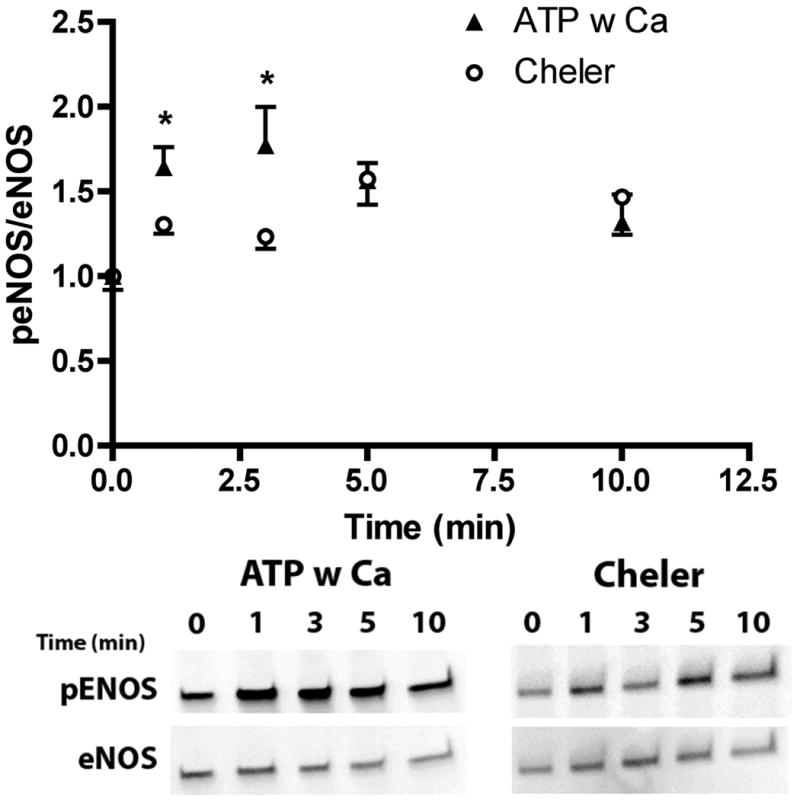
Inhibition of PKC attenuates ATP induced phosphorylation of eNOS. Prior to stimulation with 100 μM ATP with calcium, cells were treated with 30 μM of cheler for 2 min prior to stimulation. Cells were harvested before stimulation (t=0) and at time points 1, 3, 5, and 10 min after stimulation. All peNOS/eNOS ratios were normalized by t=0. Cheler treated cells showed an attenuated agonist stimulated phosphorylation of eNOS, which was statistically significant at 1 and 3 min (p<0.05 * one-tailed t-test, ATP w Ca+2 n=4, Cheler n=5).
Discussion
We have made use of our group's unique ability to measure NO directly and in real-time from endothelial cells under flow conditions to study not only the importance of the ATP autocrine pathway in shear stress-induced NO production but also how calcium dynamics, particularly CCE, contributes to the kinetics of the acute NO response.
Shear stress has been shown to cause the release of ATP, with higher flow rates releasing larger concentrations of ATP (17, 13). We utilized two approaches to interfere with autocrine stimulation in the flow induced NO response. Degradation of endogenously release ATP using apyrase inhibited NO production in response to a change in shear stress between 5-20 dyn/cm2 (Figure 1A-E). The degree of inhibition was less at higher shear stresses, which can be explained by the greater release rates at higher shear stresses and the diffusion limited reaction of apyrase with ATP. Further, ATP release is spatially dependent with maximal concentrations localized in membrane invaginations called caveolae where eNOS preferentially resides (13) and may further imped degradation by apryase. Inhibiting downstream purinergic receptors with suramin, which would not be diffusion limited, showed greater attenuation of the NO response at 10 dyn/cm2 than seen with apyrase (Figure 3A,B). The degree of inhibition is similar to in vivo studies by Silva et al. (18).
ATP stimulates purinergic receptors P2Y (G protein-coupled receptors (GPCRs)) and P2X (calcium channels). Both have been shown to be activated in response to shear stress (29, 30); however, a recent study showed that luminal P2Y purinergic receptors were responsible for flow-induced NO production (30). Activation of P2Y receptors stimulates ER store depletion and subsequent activation of SOCs responsible for CCE. We and others have shown that CCE preferentially activates NO production under static conditions (6-8, 23) and that ATP activates CCE, which led us to hypothesize that the increase in calcium specifically through SOCs caused by flow-induced autocrine activation of purinergic receptors was responsible for NO production. Using an SOC blocker, SKF-96365, we were able to inhibit the shear stress-induced NO response (Figure 4A,B). Previous studies have shown that under static conditions, SOC activation is predominantly responsible for the agonist stimulated NO response (7, 8, 31). In contrast, inhibition of SOCs caused a 40 % decrease in the flow-induced Δ[NO] whereas blocking purinergic receptors with suramin caused a 60% inhibition of the flow-induced Δ[NO]. Thus, SOC inhibition can only account for a portion of the ATP-dependent NO response to flow. This may be due to incomplete inhibition of SOCs by SKF or to the contributions of P2X purinergic receptors or other calcium sources (ER, L-type channels etc.). The identities of the SOCs inhibited by SKF in our experiments are beyond the scope of this paper. However, recent developments in calcium signaling point to members of the transient receptor potential (TRP) (see review (32)) and the Orai channels (33) as SOCs. To our knowledge, we are the first to report on the importance of CCE in the acute shear stress-induced NO production. Our experiments have demonstrated that the mechanism by which endogenously released ATP activates purinergic receptors and consequently SOCs plays a significant role in shear stress-induced NO production.
A number of researchers have reported that shear stress (34-37) and ATP (38, 39, 19, 10) increase eNOS phosphorylation at the serine residue. We detected a rise in eNOS phosphorylation in response to an increase in shear stress at 3 min (Figure 2), which is consistent with our static experiments showing maximal ATP-induced phosphorylation occurring around 3 min (Figure 6), noting that shear-induced ATP release occurs rapidly within a few seconds of the onset of flow (13). Flow induced eNOS phosphorylation was inhibited when endogenous ATP was degraded suggesting a role for ATP in shear stress-induced eNOS phosphorylation. ATP induces eNOS phosphorylation in static culture (18, 19); however, little is known about the role of calcium, and specifically CCE, in ATP-induced eNOS phosphorylation. Gonçalves da Silva et al.(19) reported that chelating intracellular calcium with BAPTA abolished the phosphorylation response 1 min after ATP stimulation (18, 19). Our data expand those results to demonstrate the full time course of the phosphorylation response to ATP as dependent on intracellular calcium changes (Figure 5). ATP mobilizes intracellular calcium from both the ER and the extracellular space. Inhibiting SOCs attenuated ATP-induced phosphorylation (Figure 8). By stimulating in the absence of extracellular calcium we were then able to separate those two events to show the importance of extracellular calcium specifically. Furthermore, phosphorylation could be recovered upon the re-addition of calcium corresponding with calcium influx through SOCs (Figure 6). Inhibition of SOCs decreased eNOS phosphorylation at t=1 and t=3 min (Figure 8). This is in agreement with the typical calcium response time course to ATP which peaks within 30 s and returns to baseline around 3 min (Figure 7). In addition, the NO response to flow also follows this timeline with a rapid response reaching a steady state concentration by 3 min (Figure 1,3 & 4). Inhibition of the magnitude of eNOS phosphorylation at early time points (via inhibition of SOCs and PKC) would therefore affect the magnitude of the steady state concentration, which is in agreement with our direct measurements under flow.
A number of studies have shown the importance of PKC in eNOS phosphorylation and CCE separately (26-28). Gonçalves da Silva et al.(19) showed an attenuation in ATP induced eNOS phosphorylation when PKC is inhibited which they have proposed is due to direct phosphorylation of eNOS by PKC (19). Our results indicated that inhibition of PKC attenuated both the ATP induced calcium response and eNOS phosphorylation (Figure 9 and 10). These responses are virtually identical to inhibition of SOCs (Figure 7 and 8) and suggest that PKC is upstream of CCE. Based on our data we have proposed the following mechanism for shear stress-induce NO production (Figure 11). Shear stress causes the release of ATP in nM-to-μM concentrations. ATP activates purinergic receptors in the plasma membrane and the release of inositol tri-phosphate (IP3). IP3 causes the depletion of ER calcium stores and activation of SOCs, which is PKC dependent. The influx of calcium through SOCs then preferentially activates eNOS to produce NO.
Figure 11.
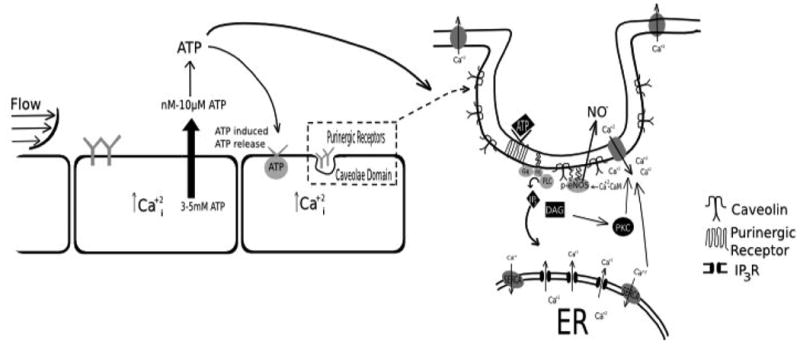
Proposed pathway for the mechanism of shear stress-induced NO. Flow causes the release of ATP in nM-μM levels, which activate purnergic receptors causing the release of IP3 and DAG. IP3 then causes the ER to deplete intracellular calcium stores. Depletion of ER calcium stores leads to the activation of store-operated channels (SOCs) (known as capacitative calcium entry (CCE)), which is PKC-dependent. CCE then activates calcium-dependent calmodulin (Ca+2-CaM), which leads to phosphorylation of eNOS and production of NO.
Our results demonstrate the importance of ATP autocrine signaling and activation of CCE in the shear stress-induced NO response. We showed that degrading ATP using apyrase led to decreased NO and eNOS phosphorylation in response to shear stress. In addition, purinergic receptor blocker suramin reduced shear stress-induced NO responses, further supporting the role of ATP. We also demonstrated the importance of CCE in the shear stress-induced NO response using the SOC inhibitor SKF-96365, which attenuated the NO response. Furthermore, we found that ATP-induced eNOS phosphorylation is dependent on both the activation of CCE and upstream PKC signaling. While our quantification of the shear stress-induced NO response focused on the change in the steady state [NO], our direct NO measurements show a more complex picture of the kinetics of the NO response and open the door to the development of a mathematical model which can take into account multiple components in the mechanism of NO production.
Acknowledgments
Funding Sources: NIH/HL068164, NSF/BES0301446, NSF/CBET0730547, NIH U01HL116256
Non-standard Abbreviations and Acronyms
- NO
Nitric oxide
- EC
endothelial cells
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N, N, N, N-tetraacetic acid
- ER
endoplasmic reticulum
- BAECs
bovine aortic endothelial cells
- IP3
inositol triphosphate
- PKC
protein kinase C
- DAG
diacylglycerol
- CCE
capacitative calcium entry
- SOCs
store operated channels
- SKF
SKF-96365
- Cheler
chelerythrine
- PBS
Dulbecco's phosphate buffered saline
- L-NAME
Nω-nitro-L-arginine methyl ester
- eNOS
endothelial nitric oxide synthase
Footnotes
Conflicts of Interest: Dr. Andrews has nothing to disclose. Dr. Jaron reports grants from NIH, from NSF, during the conduct of the study. Dr. Buerk has nothing to disclose. Dr. Barbee reports grants from NIH, grants from NSF, during the conduct of the study.
References
- 1.Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057–1058. doi: 10.1016/s0140-6736(87)91481-4. [DOI] [PubMed] [Google Scholar]
- 2.Radomski MW, Vallance P, Whitley G, Foxwell N, Moncada S. Platelet adhesion to human vascular endothelium is modulated by constitutive and cytokine induced nitric oxide. Cardiovasc Res. 1993;27:1380–1382. doi: 10.1093/cvr/27.7.1380. [DOI] [PubMed] [Google Scholar]
- 3.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsao PS, Buitrago R, Chan JR, Cooke JP. Fluid flow inhibits endothelial adhesiveness. Nitric oxide and transcriptional regulation of VCAM-1. Circulation. 1996;94:1682–1689. doi: 10.1161/01.cir.94.7.1682. [DOI] [PubMed] [Google Scholar]
- 5.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang YP, Shin WS, Kawaguchi H, Inukai M, Kato M, Sakamoto A, Uehara Y, Miyamoto M, Shimamoto N, Korenaga R, Ando J, Toyooka T. Contribution of sustained Ca2+ elevation for nitric oxide production in endothelial cells and subsequent modulation of Ca2+ transient in vascular smooth muscle cells in coculture. Journal of Biological Chemistry. 1996;271:5647–5655. doi: 10.1074/jbc.271.10.5647. [DOI] [PubMed] [Google Scholar]
- 7.Lin S, Fagan KA, Li KX, Shaul PW, Cooper DMF, Rodman DM. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. Journal of Biological Chemistry. 2000;275:17979–17985. doi: 10.1074/jbc.275.24.17979. [DOI] [PubMed] [Google Scholar]
- 8.Dedkova EN, Blatter LA. Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. Journal of Physiology-London. 2002;539:77–91. doi: 10.1113/jphysiol.2001.013258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi FX, Magness RR, Bird IM. Simultaneous imaging of [Ca2+]i and intracellular NO production in freshly isolated uterine artery endothelial cells: effects of ovarian cycle and pregnancy. Am J Physiol Regul Integr Comp Physiol. 2005;288:R140–148. doi: 10.1152/ajpregu.00302.2004. [DOI] [PubMed] [Google Scholar]
- 10.Xiao Z, Wang T, Qin H, Huang C, Feng Y, Xia Y. Endoplasmic reticulum Ca2+ release modulates endothelial nitric-oxide synthase via extracellular signal-regulated kinase (ERK) 1/2-mediated serine 635 phosphorylation. J Biol Chem. 2011;286:20100–20108. doi: 10.1074/jbc.M111.220236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen J, Luscinskas FW, Connolly A, Dewey CF, Jr, Gimbrone MA., Jr Fluid shear stress modulates cytosolic free calcium in vascular endothelial cells. Am J Physiol. 1992;262:C384–390. doi: 10.1152/ajpcell.1992.262.2.C384. [DOI] [PubMed] [Google Scholar]
- 12.Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow-induced nitric-oxide production in endothelial-cells. American Journal of Physiology. 1994;266:C628–C636. doi: 10.1152/ajpcell.1994.266.3.C628. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M, Ando J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci. 2011;124:3477–3483. doi: 10.1242/jcs.087221. [DOI] [PubMed] [Google Scholar]
- 14.Buxton IL, Kaiser RA, Oxhorn BC, Cheek DJ. Evidence supporting the Nucleotide Axis Hypothesis: ATP release and metabolism by coronary endothelium. Am J Physiol Heart Circ Physiol. 2001;281:H1657–1666. doi: 10.1152/ajpheart.2001.281.4.H1657. [DOI] [PubMed] [Google Scholar]
- 15.Mortensen SP, Thaning P, Nyberg M, Saltin B, Hellsten Y. Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol. 2011;589:1847–1857. doi: 10.1113/jphysiol.2010.203034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodin P, Bailey D, Burnstock G. Increased flow-induced ATP release from isolated vascular endothelial cells but not smooth muscle cells. Br J Pharmacol. 1991;103:1203–1205. doi: 10.1111/j.1476-5381.1991.tb12324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodin P, Burnstock G. Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J Cardiovasc Pharmacol. 2001;38:900–908. doi: 10.1097/00005344-200112000-00012. [DOI] [PubMed] [Google Scholar]
- 18.Silva G, Beierwaltes WH, Garvin JL. Extracellular ATP stimulates NO production in rat thick ascending limb. Hypertension. 2006;47:563–567. doi: 10.1161/01.HYP.0000197954.93874.ef. [DOI] [PubMed] [Google Scholar]
- 19.Gonçalves da Silva CG, Specht A, Wegiel B, Ferran C, Kaczmarek E. Mechanism of purinergic activation of endothelial nitric oxide synthase in endothelial cells. Circulation. 2009;119:871–879. doi: 10.1161/CIRCULATIONAHA.108.764571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanai AJ, Strauss HC, Truskey GA, Crews AL, Grunfeld S, Malinski T. Shear-stress induces ATP-independent transient nitric-oxide release from vascular endothelial-cells, measured directly with a porphyrinic microsensor. Circulation Research. 1995;77:284–293. doi: 10.1161/01.res.77.2.284. [DOI] [PubMed] [Google Scholar]
- 21.Ayajiki K, Kindermann M, Hecker M, Fleming I, Busse R. Intracellular pH and tyrosine phosphorylation but not calcium determine shear stress-induced nitric oxide production in native endothelial cells. Circ Res. 1996;78:750–758. doi: 10.1161/01.res.78.5.750. [DOI] [PubMed] [Google Scholar]
- 22.Comerford A, Plank MJ, David T. Endothelial nitric oxide synthase and calcium production in arterial geometries: an integrated fluid mechanics/cell model. J Biomech Eng. 2008;130:011010. doi: 10.1115/1.2838026. [DOI] [PubMed] [Google Scholar]
- 23.Hong D, Jaron D, Buerk DG, Barbee KA. Transport-dependent calcium signaling in spatially segregated cellular caveolar domains. American Journal of Physiology-Cell Physiology. 2008;294:C856–C866. doi: 10.1152/ajpcell.00278.2007. [DOI] [PubMed] [Google Scholar]
- 24.Hong D, Jaron D, Buerk DG, Barbee KA. Heterogeneous response of microvascular endothelial cells to shear stress. American Journal of Physiology-Heart and Circulatory Physiology. 2006;290:H2498–H2508. doi: 10.1152/ajpheart.00828.2005. [DOI] [PubMed] [Google Scholar]
- 25.Andrews AM, Jaron D, Buerk DG, Kirby PL, Barbee KA. Direct, real-time measurement of shear stress-induced nitric oxide produced from endothelial cells in vitro. Nitric Oxide. 2010;23:335–342. doi: 10.1016/j.niox.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma R, Kudlacek PE, Sansom SC. Protein kinase Calpha participates in activation of store-operated Ca2+ channels in human glomerular mesangial cells. Am J Physiol Cell Physiol. 2002;283:C1390–1398. doi: 10.1152/ajpcell.00141.2002. [DOI] [PubMed] [Google Scholar]
- 27.Zhang F, Wen Q, Mergler S, Yang H, Wang Z, Bildin VN, Reinach PS. PKC isoform-specific enhancement of capacitative calcium entry in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:3989–4000. doi: 10.1167/iovs.06-0253. [DOI] [PubMed] [Google Scholar]
- 28.Smani T, Patel T, Bolotina VM. Complex regulation of store-operated Ca2+ entry pathway by PKC-epsilon in vascular SMCs. Am J Physiol Cell Physiol. 2008;294:C1499–1508. doi: 10.1152/ajpcell.00365.2007. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto K, Korenaga R, Kamiya A, Qi Z, Sokabe M, Ando J. P2X(4) receptors mediate ATP-induced calcium influx in human vascular endothelial cells. American Journal of Physiology-Heart and Circulatory Physiology. 2000;279:H285–H292. doi: 10.1152/ajpheart.2000.279.1.H285. [DOI] [PubMed] [Google Scholar]
- 30.Cabral PD, Hong NJ, Garvin JL. ATP mediates flow-induced NO production in thick ascending limbs. Am J Physiol Renal Physiol. 2012;303:F194–200. doi: 10.1152/ajprenal.00504.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong D, Buerk DG, Barbee KA, Jaron D. Microelectrode measurements of NO release from endothelial cells in response to shear stress and ATP. BMES Meeting. 2005 Fall; [Google Scholar]
- 32.Kwan HY, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochim Biophys Acta. 2007;1772:907–914. doi: 10.1016/j.bbadis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Frischauf I, Schindl R, Derler I, Bergsmann J, Fahrner M, Romanin C. The STIM/Orai coupling machinery. Channels (Austin) 2008;2:261–268. doi: 10.4161/chan.2.4.6705. [DOI] [PubMed] [Google Scholar]
- 34.Corson MA, James NL, Latta SE, Nerem RM, Berk BC, Harrison DG. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circulation Research. 1996;79:984–991. doi: 10.1161/01.res.79.5.984. [DOI] [PubMed] [Google Scholar]
- 35.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 36.Kemeny SF, Figueroa DS, Andrews AM, Barbee KA, Clyne AM. Glycated collagen alters endothelial cell actin alignment and nitric oxide release in response to fluid shear stress. J Biomech. 2011;44:1927–1935. doi: 10.1016/j.jbiomech.2011.04.026. [DOI] [PubMed] [Google Scholar]
- 37.Yang B, Rizzo V. Shear Stress Activates eNOS at the Endothelial Apical Surface Through 1 Containing Integrins and Caveolae. Cell Mol Bioeng. 2013;6:346–354. doi: 10.1007/s12195-013-0276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cale JM, Bird IM. Dissociation of endothelial nitric oxide synthase phosphorylation and activity in uterine artery endothelial cells. Am J Physiol Heart Circ Physiol. 2006;290:H1433–1445. doi: 10.1152/ajpheart.00942.2005. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan JA, Grummer MA, Yi FX, Bird IM. Pregnancy-enhanced endothelial nitric oxide synthase (eNOS) activation in uterine artery endothelial cells shows altered sensitivity to Ca2+, U0126, and wortmannin but not LY294002--evidence that pregnancy adaptation of eNOS activation occurs at multiple levels of cell signaling. Endocrinology. 2006;147:2442–2457. doi: 10.1210/en.2005-0399. [DOI] [PubMed] [Google Scholar]