

Abstract
Purpose.
Extensive death of uninfected bystander neuronal cells is an important component of the pathogenesis of cytomegalovirus retinitis. Our previous results have shown that caspase 3–dependent and –independent pathways are involved in death of uninfected bystander cells during murine cytomegalovirus (MCMV) retinitis and also that Bcl-2, an important inhibitor of apoptosis via the Bax-mediated mitochondrial pathway, is downregulated during this process. The purpose of this study was to determine whether Bax-mediated mitochondrial damage has a significant role in the death of uninfected retinal cells.
Methods.
BALB/c mice, Bax−/− mice, or Bax+/+ mice were immunosuppressed with methylprednisolone and infected with 5 × 103 plaque-forming units (PFU) of the K181 strain of MCMV via the supraciliary route. Injected eyes were analyzed by plaque assay, electron microscopy, hematoxylin and eosin (H&E) staining, TUNEL assay, Western blot (for caspase 3, caspase 12, Bax, receptor interacting protein-1 [RIP1] and receptor interacting protein-3 [RIP3]), as well as immunohistochemical staining for MCMV early antigen and cleaved caspase 3.
Results.
Significantly more Bax was detected in mitochondrial fractions of MCMV-infected eyes than in mitochondrial fractions of mock-infected control eyes. Furthermore, the level of cleaved caspase 3 was significantly lower in MCMV-infected Bax−/− eyes than in MCMV-infected Bax+/+ eyes. However, more caspase 3–independent cell death of uninfected bystander retinal cells and more cleaved RIP1 were observed in Bax−/− than in Bax+/+ eyes.
Conclusions.
During MCMV retinitis, Bax is activated and has an important role in death of uninfected bystander retinal cells by caspase 3–dependent apoptosis. Although the exact mechanism remains to be deciphered, active Bax might also prevent death of some types of uninfected retinal cells by a caspase 3–independent pathway.
Keywords: murine cytomegalovirus, retinitis, apoptosis, Bax
Bax is activated during murine cytomegalovirus retinitis and plays an important role in the death of uninfected bystander retinal cells by caspase 3–dependent apoptosis.
Introduction
Human cytomegalovirus (HCMV) retinitis is a serious ocular complication in patients immunosuppressed as a result of acquired immunodeficiency syndrome (AIDS), chemotherapy, or malignancy, and in newborns who are congenitally infected.1–5 Although highly active antiretroviral therapy (HAART) has resulted in a significant decrease in the number of new cases of AIDS-related HCMV retinitis, the disease continues to be a chronic sight-threatening ophthalmologic problem among AIDS patients who do not respond to HAART or who discontinue therapy.6–10 The HIV infection is now a chronic disease with survival after infection estimated at >14 years.11 However, because of late testing among many high-risk patients, approximately one-third of patients with newly-diagnosed HIV infection in the United States will still have progression to AIDS within one year of diagnosis,12 resulting in a population at ongoing risk for HCMV retinitis.
Although retinal necrosis is one of the hallmarks of HCMV retinitis, apoptotic cells have been observed during microscopic examination of biopsied eyes from HCMV retinitis patients.13–15 Our laboratory has used a mouse model in which injection of murine cytomegalovirus (MCMV) into the supraciliary space of immunosuppressed mice causes retinal infection with histopathologic features that mimic those observed in ocular specimens obtained from human patients.16–21 Although MCMV infects specific types of retinal neurons, such as horizontal cells and bipolar cells at an early stage of infection,21 other types of retinal neuronal cells, such as rod and cone photoreceptors, amacrine cells, and ganglion cells, are rarely infected with MCMV.21 Virus-infected cells are protected from premature death by several MCMV encoded proteins, which inhibit apoptosis and programmed necrosis (or necroptosis), allowing the virus to complete its replication program and produce abundant progeny.22–24 Paradoxically, uninfected bystander cells located throughout the neural retina undergo cell death, leading to marked disruption of retinal architecture and eventual blindness.18 Increasing evidence suggests that apoptosis of uninfected bystander neuronal cells appears to be an important component of the pathogenesis of CMV retinitis.13–15,18–21,25–28 Multiple apoptosis inducers, including TNF-α,25–28 Fas ligand,15 and iNOS,28 have been detected in HCMV- and MCMV-infected retinal tissue or retinal cells. Caspase 3–dependent and –independent apoptosis has been demonstrated during MCMV retinitis.25,28 Since Bcl-2, an important inhibitor of the mitochondrial pathway, is downregulated and tBid, a proapoptotic factor in the mitochondrial pathway, is upregulated during MCMV retinitis,25 we hypothesized that mitochondrial damage has a significant role in the apoptosis of uninfected retinal cells. Bax-induced mitochondrial outer membrane permeabilization (MOMP) is considered to be one of the key control switches of apoptosis.29 Therefore, the purpose of this study was to test this hypothesis by comparing MCMV-induced apoptosis in Bax−/− and Bax+/+ mice.
Methods
Virus and Virus Titration
The original stock of MCMV strain K181 was kindly provided by Edward Morcarski, PhD, Emory University, Atlanta, Georgia, United States. Virus was prepared from the salivary glands of MCMV-infected BALB/c mice as described previously.16 The titer of the virus stock was determined by plaque assay on mouse embryonic fibroblast (MEF) cells. Aliquots of stock virus were stored at −70°C, and a fresh aliquot was thawed and diluted to the appropriate concentration for each experiment.
Mice
The BALB/c mice and Bax+/− mice (strain name, B6.129X1-Baxtm1Sjk/J; stock number, 002994) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and Bax−/− and Bax+/+ mice were obtained by crossing two Bax+/− parental mice. Adult (6~8 weeks old) mice were used in all experiments. Mice were housed in accordance with National Institutes of Health (NIH) guidelines. Mice were maintained on a 12-hour light cycle alternating with a 12-hour dark cycle and were given unrestricted access to food and water. All ocular injections were performed following anesthesia with a mixture of 42.9 mg/mL ketamine, 8.57 mg/mL xylazine, at a dose of 0.5 to 0.7 mL/kg body weight. The treatment of animals in this study conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and was approved by the Institutional Animal Care and Use Committee of Georgia Regents University.
Antibodies
The antibodies used in this study were obtained from the following sources: anti-cleaved caspase 3, anti-caspase 12, and anti-receptor interacting protein 1 (RIP1) were from Cell Signaling Technology, Inc. (Danver, MA, USA). Anti-β-actin was from Sigma-Aldrich Corp. (St. Louis, MO, USA). Texas Red-labeled Avidin and Texas Red–labeled anti-rabbit IgG were from Vector Laboratories, Inc. (Burlingame, CA, USA). Anti-RIP3, goat anti-rabbit IgG-HRP, and goat anti-mouse IgG-HRP were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Experimental Plan
Mice were immunosuppressed (IS) by intramuscular injection of 2.0 mg sterile methylprednisolone acetate suspension every 3 days beginning on day −2. This treatment typically depletes 93% of the CD4+ and CD8+ T cells as well as macrophages from MCMV-infected mice as assayed by flow cytometry of splenocytes.17 Mice were injected with 1 × 104 plaque-forming units (PFU) of MCMV contained in a volume of 2 μL via the supraciliary route on day 0 and euthanized at various time points after infection. Enucleated eyes were homogenized in serum-free tissue culture medium using a handheld tissue homogenizer (Biospec Products, Inc., Racine, WI, USA) and plated on MEF cells for measurement of virus titer. Eyes of additional mice were removed and prepared for immunohistochemistry, electron microscopy (EM), or Western blot analysis.
Immunohistochemistry
Anti-MCMV early antigen (EA)21 was labeled with FITC (Sigma-Aldrich Corp.) or biotinylated (Sulfo-NHS-LC-Biotin; Pierce, Rockford, IL, USA) according to the manufacturer's instructions. Injected eyes were embedded in optimal cutting temperature (OCT) compound (Tissue-Tek; VWR Scientific, Houston, TX, USA), snap frozen, and sectioned on a cryostat. Frozen sections were fixed with 4% paraformaldehyde for 15 minutes. For double staining with TUNEL and MCMV EA, sections were first stained with TUNEL (In Situ Cell Death Detection Kit, Fluorescein; Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer's instructions. After washing and blocking, biotinylated anti-EA was applied to the section, followed by Texas Red-labeled Avidin. For double staining of cleaved caspase 3 and MCMV EA, sections were first stained with rabbit anti-mouse cleaved caspase 3 followed by Texas Red–labeled anti-rabbit IgG. After washing and blocking, sections were stained with FITC-labeled anti-MCMV EA. Slides were mounted with anti-fade medium containing 4′6-diamidino-2-phenylindole (DAPI, Vectashield; Vector Laboratories, Inc.) and examined on an Axioplan 2 microscope (Zeiss, Inc., Jena, Germany). Images were analyzed using Axiovision Rel. 4.7 software.
Western Blot Analysis
Proteins were extracted from normal eyes, MCMV-injected eyes, or medium-injected eyes as described previously.25,27,28 Mitochondrial and cytosolic fractions were collected as described by Wei et al.30 Briefly, fresh or frozen eye tissues were homogenized in a lysis buffer containing 0.27 M sucrose, 1 mM ethylene glycol tetraacetic acid (EGTA), 5 mM Tris-HCl (pH 7.4). Homogenates were centrifuged at 600g for 10 minutes at 4°C to remove cell debris, and nuclei and supernatants were collected and centrifuged at 10,000g for 5 minutes at 4°C. The pellet, containing the mitochondrial fraction, was washed three times and dissolved in lysis buffer. The supernatants from the 10,000g centrifugation were recentrifuged at 100,000g for 60 minutes to collect the cytosolic fraction. Western blot analysis was performed as previously described.25,27,28 Briefly, equal amounts of proteins were loaded for SDS-PAGE, followed by electroblotting onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Piscataway, NJ, USA). Following blocking with 5% nonfat dry milk for 1 hour at room temperature, membranes were incubated overnight at 4°C with primary antibody and for 1 hour at room temperature with horseradish peroxidase (HRP)–conjugated secondary antibody. Immune complexes were detected using chemiluminescence (ECL; GE Healthcare) and exposure to x-ray film. To verify equal loading among lanes, membranes were stained with anti-β-actin. The density of each band was analyzed using Image J software (NIH).
Electron Microscopy (EM)
Eyes of experimental and control mice were fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer overnight at 4°C, washed in cacodylate buffer, postfixed with 4% osmium tetroxide for 1 hour at room temperature, dehydrated in a graded ethanol series, and embedded in resin (Pure Embed 812 mixture; Electron Microscopy Science, Hatfield, PA, USA). Ultrathin sections were stained with uranyl acetate and lead citrate, and visualized in an electron microscope (Jem 1230; JEOL, Tokyo, Japan). Quantification of apoptotic cells was performed by counting the number of apoptotic cells in a 45 × 45 μm area of the electron micrographs and expressed as the mean number of apoptotic cells ± SEM.
Retinitis Scoring
To analyze the extent of retinopathy, a scoring system was used. This was modified from a previously published study.16 Frozen sections were fixed with 4% paraformaldehyde for 15 minutes, and stained with hematoxylin and eosin (H&E). Six sections were evaluated per eye, and changes in the posterior segment of each section were evaluated microscopically as follows: 0, normal or injection artifact; 1/2, mild atypical retinopathy defined as absence of cytomegaly plus retinal folds involving less than three-quarters of the retinal section; 1, moderate atypical retinopathy defined as absence of cytomegaly plus retinal folds involving more than three-quarters of the retinal section plus photoreceptor atrophy or retinal infiltration by leukocytes involving more than one-quarter of the retina; 2, retinal infection defined as cytomegaly of retinal cells plus partial-thickness retinal necrosis or full-thickness necrosis extending from the ciliary body, but not beyond one-quarter of the retinal section from the ciliary body; 3, necrotizing retinitis defined as cytomegaly plus full-thickness retinal necrosis existing further than one-quarter of a retinal section from the ciliary body or full-thickness retinal necrosis extending from the ciliary body through one quarter of the section; 4, severe necrotizing retinitis defined as cytomegaly with full-thickness necrosis involving the entire retinal section. The average score for all retinal sections of each eye was used to determine the mean retinal score. Data were analyzed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) to determine if significant differences existed between treatment groups.
Statistical Analysis
All data were expressed as means ± SEM, with n representing the number of mice used in each of the experimental groups. Statistical analyses were used to determine the significance of observed differences between treatment groups in all experiments. Statistical significance was calculated by means of either a 2-tailed t-test or 1-way ANOVA with Bonferroni's post hoc test using the GraphPad Prism 5 Analysis tool. P values < 0.05 were considered to be significant.
Results
Bax is Activated and Translocated to the Mitochondria During MCMV Infection
Bax is normally constrained in an inactive form in the cytosol through interactions between its groove and transmembrane domains. Upon reception of apoptotic stimuli, Bax is activated and translocated to the mitochondria, forming oligomeric pores with other molecules on the outer membrane, resulting in the release of apoptogenic factors, such as cytochrome c, and apoptosis-inducing factor (AIF).30–32 To determine if Bax is activated during MCMV infection, we analyzed Bax accumulation in mitochondria. To this end, mitochondrial fractions were isolated from MCMV-infected and mock-infected control eyes, and Bax expression was analyzed by Western blotting. As shown in Figure 1, more mitochondrial Bax was detected in MCMV-infected eyes than in control eyes, confirming that Bax is activated during MCMV retinitis.
Figure 1.
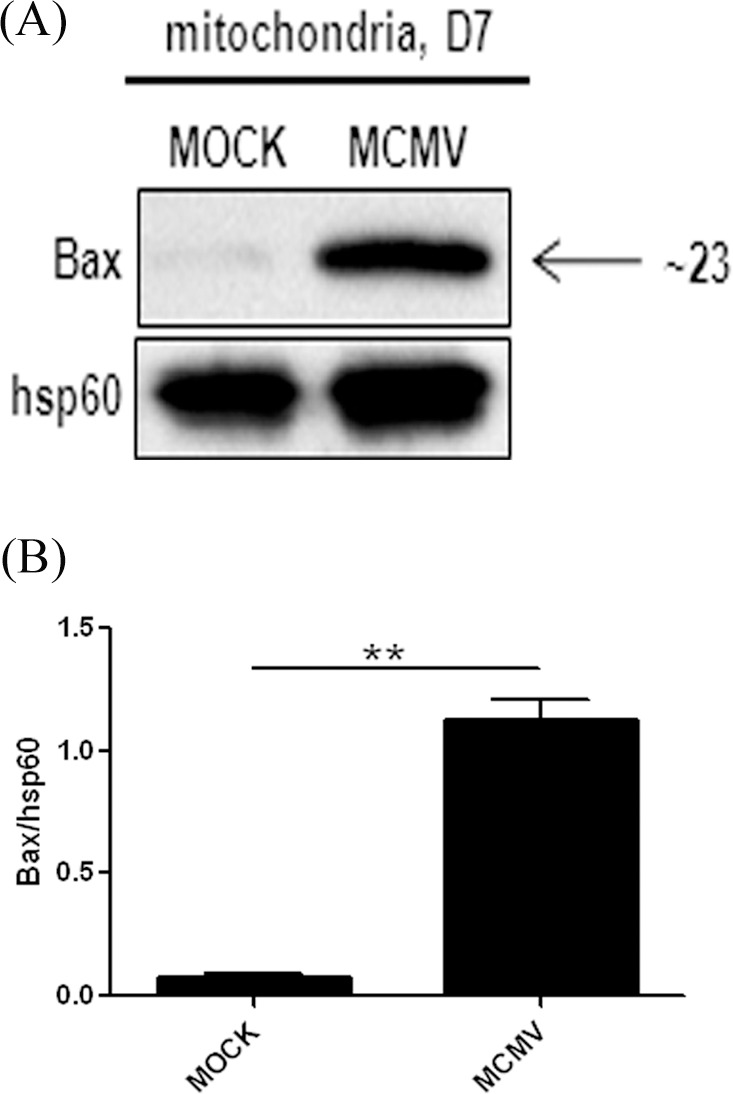
(A) Western blot showing Bax expression in the mitochondrial fraction from injected eyes of IS BALB/c mice, either mock-infected (MOCK) or MCMV-infected at day 7 after injection. (B) Ratio of Bax to Hsp60. Data are shown as mean ± SEM (n = 4) and compared by a 2-tailed t-test. **P < 0.01.
Viral Spread and Replication in Bax−/− Mice
To determine if Bax has a role in the death of bystander retinal cells during MCMV infection, Bax−/− and Bax+/+ mice were immunosuppressed and infected with the K181 strain of MCMV. Injected eyes were removed at day 7 after infection and virus titers were determined by plaque assay. As shown in Figure 2, the titer of virus in injected eyes of Bax−/− mice and Bax+/+ control mice was similar. The MCMV injected eyes and uninfected control eyes also were sectioned, fixed, stained with H&E, and scored for retinitis.16 As shown in Figures 3A and 3B, the retinal structure of uninfected Bax−/− mice was indistinguishable from that of control Bax+/+ mice. Cytomegalic cells were present in the inner retina, as well as in the retinal pigment epithelium in infected Bax+/+ (Fig. 3C) and infected Bax−/− (Fig. 3D) eyes. Additionally, the infected eyes in both groups exhibited shortening of the photoreceptors, atypical folding of the retinal layers, as well as leukocytes infiltration. However, the spread of MCMV in the inner retina was more extensive and retinitis was more severe in the injected eyes of Bax−/− mice (Fig. 3D) than in those of Bax+/+ control mice (Fig. 3C). The average section score of infected eyes of Bax−/− mice was significantly higher than those of control mice (Fig. 3E).
Figure 2.
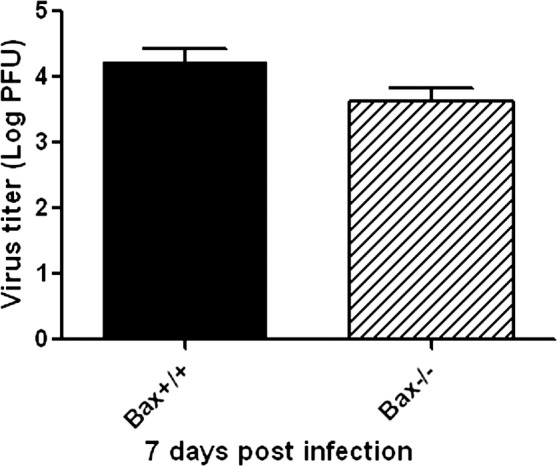
Titer of MCMV (Log10 ± SEM PFU/mL) in MCMV-injected eyes of IS Bax+/+ and Bax−/− mice at day 7 after injection. Data are shown as mean ± SEM (n = 4). Statistical analysis by 2-tailed t-test indicated no significant difference between the 2 groups.
Figure 3.

(A–D) Representative photomicrographs of H&E staining in uninjected and MCMV-injected eyes of IS Bax+/+ and Bax−/− mice at day 7 after injection. (E) Scoring of retinitis in MCMV-injected eyes of IS Bax+/+ and Bax−/− mice at day 7 after injection. Data are shown as mean ± SEM (n = 5) and compared by a 2-tailed t-test. *P < 0.05.
Caspase 3–Dependent Apoptosis in Bax−/− Mice
To investigate whether caspase 3–dependent apoptosis was affected by Bax deficiency, relative levels of cleaved caspase 3 were determined in injected eyes of Bax−/− and Bax+/+ mice by Western blot. Cleaved caspase 3 was not detected in mock-injected eyes of immunosuppressed Bax−/− and Bax+/+ mice. In contrast, cleaved caspase 3 was detected in MCMV-injected eyes of Bax−/− and Bax+/+ mice (Fig. 4A), and quantification of band density showed significantly lower levels of cleaved caspase 3 in injected eyes of Bax−/− mice than in those of Bax+/+ mice (Fig. 4B). To investigate whether decreased caspase 3–dependent apoptosis was localized to virus-infected cells or uninfected bystanders, sections of MCMV-injected eyes and mock-injected control eyes were stained with cleaved caspase 3 and MCMV EA. The MCMV EA–positive virus-infected cells were observed in the ciliary body (not shown), choroid, RPE layer, and inner retina in the injected eyes of Bax−/− and Bax+/+ mice (Figs. 4C, 4D). However, fewer cleaved caspase 3–positive cells were observed in the inner retinas of Bax−/− (Fig. 4D) than in the retinas of Bax+/+ (Fig. 4C) injected eyes. In injected eyes of Bax−/− and Bax+/+ mice, the majority of cleaved caspase 3–positive cells in the inner retina were MCMV-uninfected cells (Figs. 4C, 4D), suggesting that depletion of Bax resulted in decreased death of bystander cells by caspase 3–dependent apoptosis.
Figure 4.

(A) Western blot of cleaved caspase 3 in MOCK-injected and MCMV-injected eyes of IS Bax+/+ and Bax−/− mice at day 7 after injection. (B) Ratio of cleaved caspase 3 to β-actin. Data are shown as mean ± SEM (n = 4) and compared by ANOVA. *P < 0.05, Bax+/+, MCMV versus Bax×/×, MCMV. (C, D) Merged photomicrographs (×200) of staining for MCMV EA (green), cleaved caspase 3 (red), and DAPI (blue) in MCMV-injected eyes of an IS Bax+/+ and an IS Bax−/− mouse at day 7 after injection. (E, F) Merged photomicrographs (×200) of staining for MCMV EA (red), TUNEL (green), and DAPI (blue) in MCMV-injected eyes of an IS Bax+/+ and an IS Bax−/− mouse at day 7 after injection. (G) Quantification of the number of TUNEL-positive cells in retinas of MCMV-injected Bax+/+ eyes and MCMV-injected Bax−/− eyes at day 7 after injection. The average number of TUNEL-positive cells for all retinal sections of each eye was used to determine the mean number of TUNEL-positive cells. Data are shown as the mean ± SEM (n = 4) and compared by 2-tailed t-test. *P < 0.05.
Caspase 3–Independent Cell Death
Although depletion of Bax reduced caspase 3–dependent apoptosis, a more severe retinal disruption was observed in MCMV-infected Bax−/− mice. Since our previous results suggested that caspase3-dependent and –independent pathways are involved in the death of bystander retinal cells during MCMV retinitis,25,28 we determined whether Bax depletion affects caspase 3–independent death of bystander retinal cells during MCMV retinal infection. Using the TUNEL assay combined with MCMV EA staining, we observed that, in contrast to cleaved caspase 3 staining (Figs. 4C, 4D), significantly more TUNEL-positive cells were observed in the inner retinas of Bax−/− injected eyes than in the inner retinas of Bax+/+ injected eyes (Figs. 4E–G). Consistent with our previous observations, the majority of TUNEL-positive cells were uninfected (MCMV EA–negative) bystander retinal cells in both groups (Figs. 4E, 4F).
To augment the light microscopy studies, additional injected eyes were subjected to EM to determine the number of cells featuring hallmarks characteristic of apoptosis.21 As shown in Figure 5, significantly more apoptotic photoreceptor cells were observed in the outer nuclear layer of Bax−/− mice than in the outer nuclear layers of wild type Bax+/+ mice (6.67 ± 3.79 vs. 2.53 ± 1.80; n = 15, P < 0.001) . These apoptotic photoreceptor cells exhibited nuclear shrinkage and prominent chromatin condensation.21 In contrast, nuclear fragmentation was rarely seen in the retinas of MCMV-infected Bax−/− or wild type Bax+/+ mice. Although the majority of photoreceptors with an apoptotic nuclear morphology also had shrinkage and condensation of the cytoplasm (Fig. 5, arrows), a few also exhibited a marked loss of cytoplasm due to cell lysis (Fig. 5, arrowhead).
Figure 5.

(A, B) The EMs showing photoreceptors exhibiting typical features of apoptosis (nuclear shrinkage and strong chromatin condensation, arrows) in the outer nuclear layer of MCMV-injected eyes of an IS Bax+/+ and an IS Bax−/− mouse at day 7 after injection. A few photoreceptors with nuclear condensation also exhibited a marked loss of cytoplasm due to cell lysis (arrowhead). (C) Quantification of the number of photoreceptor cells characterized with apoptosis-like cell death in retinas of MCMV-injected Bax+/+ and Bax−/− eyes at day 7 after injection. Data are shown as mean ± SEM (n = 15 fields) and compared by a 2-tailed t-test. ***P < 0.001.
The TUNEL and EM results suggested that depletion of Bax increases caspase 3–independent apoptosis-like cell death. Since our previous results suggested that ER stress and caspase 12 activation are induced by ocular MCMV infection,25 activation of caspase 12 might induce release and translocation of AIF from the mitochondria to the nucleus33 and cause AIF-induced caspase-independent apoptosis-like cell death.34–36 To determine if increased cell death in Bax−/− injected eyes was related to caspase 12 activation, cleaved caspase 12 was measured by Western blot in MCMV-injected eyes and mock-injected control eyes. Consistent with previous observations,25 cleaved caspase 12 was increased in wild type mice after MCMV infection. However, lower levels of cleaved caspase 12 were detected in MCMV injected Bax−/− eyes (Figs. 6A, 6B), suggesting that increased caspase 3–independent apoptosis-like cell death in Bax−/− injected eyes was not related to caspase 12 activation.
Figure 6.

(A) Western blot of cleaved caspase 12 in MOCK-injected and MCMV-injected eyes of IS Bax+/+ and Bax−/− mice at day 7 after injection. (B) Ratio of cleaved caspase 12 to β-actin. Data are shown as mean ± SEM (n = 4) and compared by ANOVA. *P < 0.05, Bax+/+, MCMV versus Bax−/−, MCMV. (C) Western blot of RIP1 and RIP3 in MOCK-injected and MCMV-injected eyes of IS Bax+/+ and Bax−/− mice at day 7 after injection. The membrane was initially stained with anti-RIP1 and anti-β-actin and subsequently with anti-RIP3 after treatment with stripping buffer. (D) Ratio of cleaved RIP1 to β-actin. Data are shown as mean ± SEM (n = 4) and compared by ANOVA. *P < 0.05, Bax+/+, MCMV versus Bax−/−, MCMV. (E) Ratio of full length RIP1 to β-actin. Data are shown as mean ± SEM (n = 4) and compared by ANOVA. *P < 0.05, Bax+/+, MCMV versus Bax−/−, MCMV. (F) Ratio of full length RIP3 to β-actin. Data are shown as mean ± SEM (n = 4) and compared by ANOVA. *P < 0.05, Bax+/+, MCMV versus Bax−/−, MCMV.
Necroptosis, a form of programmed necrosis dependent on two related RIP homotypic interaction motif (RHIM)–containing signaling adaptors, RIP1 and RIP3,37 may also have a role in retinal cell death during MCMV retinitis. Although initially considered specific to apoptosis-like cell death, AIF also has been implicated in necroptotic death induced by DNA damage, such as that triggered by MMNG38–40 (AIF-mediated necroptotic death is also called parthanatos41). In this case, AIF is released from the mitochondrial intermembrane space following the overactivation of poly(ADP-ribose) polymerase 1 (PARP1). Chemical inhibition or genetic ablation of PARP1, as well as of AIF, prevents DNA-damage–induced death.38–40 The RIP1 and RIP3 are activated during MCMV ocular MCMV infection,42 suggesting that necroptosis also might be involved in death of bystander retinal cells. To determine if necroptosis is affected by Bax depletion, Western blots were performed to measure expression of RIP1 and RIP3. Although more cleaved RIP1 was detected after MCMV ocular infection in knockout and wild type mice compared to control, mock-infected mice, a significantly higher level of cleaved RIP1 was detected in MCMV-infected Bax−/− eyes than in MCMV-infected Bax+/+ eyes (Figs. 6C, 6D; P < 0.05, compared by ANOVA). Levels of full length RIP1 (Figs. 6C, 6E) and RIP3 (Figs. 6C, 6F) were lower in MCMV-infected Bax−/− eyes than in MCMV-infected Bax+/+ eyes.
Discussion
The experiments presented herein provided evidence that Bax is activated and has an important role in death of uninfected bystander retinal cells by caspase 3–dependent apoptosis during MCMV retinitis. Although the exact mechanism remains to be deciphered, depletion of Bax increases death of some types of uninfected retinal cells by a caspase 3–independent pathway.
Apoptosis is an actively controlled process of cell suicide characterized by distinctive morphological and biochemical changes that occur in response to a wide variety of stimuli43 and is important in eliminating cells whose survival otherwise might prove harmful to the organism as a whole, thereby providing a defense against viral infection and the development of cancer. Mitochondria have a critical role in apoptosis by releasing the apoptogenic factor cytochrome c from the intermembrane, which further triggers caspase 3 activation.44 This process, known as MOMP, is tightly regulated by the Bcl-2 family proteins. Proapoptotic Bcl-2 family members, Bax and Bak, change their conformation when activated by BH3 domain-only proteins and permeabilize the mitochondrial outer membrane (MOM), whereas prosurvival members inhibit permeabilization.45 Our results demonstrated that depletion of Bax significantly reduces death of uninfected bystander retinal cells by caspase 3–dependent apoptosis, supporting our hypothesis that Bax-mediated mitochondrial damage has an important role in caspase 3–dependent apoptosis in the retina.
Our results also suggested that Bax is important for preventing and/or limiting MCMV spread in the inner retina, since more extensive retinal infection and more severe retinitis were observed in Bax−/− eyes than in Bax+/+ eyes. Since virus-infected cells can be destroyed by apoptotic processes,46–48 the induction of early cell death would severely limit virus production and thereby reduce or eliminate spread of progeny virus in the host. Cytomegalovirus encodes the m38.5 protein, which binds cytosolic Bax; thus, preventing MOMP.49–52 The m38.5 should prevent Bax-mediated mitochondrial damage efficiently in most MCMV-infected ocular cells since similar amounts of MCMV were recovered from Bax−/− and Bax+/+ eyes and the majority of virus-infected cells were not TUNEL-positive, nor did they contain high levels of cleaved caspase 3. However, we cannot discount the possibility that the efficiency of binding of m38.5 might vary among cell types, and, therefore, active Bax might still have a role in inducing premature cell death of certain retinal cells and limit viral spread inside the retina. Our previous results showed that loss of photoreceptor cells correlated with spread of MCMV from the initial site of infection in the RPE layer to Müller cells during MCMV retinitis.21 Therefore, increased cell death of photoreceptors in Bax−/− mice should allow virus to spread more widely within the retina and increase retinal damage.
Depletion of Bax increases caspase 3–independent apoptosis-like cell death of photoreceptors as well as RIP1 activation during MCMV retinal infection. The RIP1 is an important upstream kinase that exerts strategic control over multiple cellular pathways involved in regulating inflammation and cell death.53 The RIP1 regulates inflammatory signaling in response to stimuli, such as TNF and ligands of the toll-like receptor (TLR) family in kinase-dependent and -independent manners.54,55 Furthermore, RIP1 kinase is a crucial upstream regulator of necroptosis.37–40,54 Although the exact mechanism of how and where RIP1 is activated, and the roles it has during MCMV retinitis remain to be determined, this unexpected result suggests that Bax might have an important role in prevention of cell death by necroptosis. The PARP1- and AIF-mediated necroptosis (parthanatos) has been shown convincingly to contribute to the death of neurons exposed to a variety of cytotoxic stimuli in vivo.41 Such neurotoxic triggers include, but presumably are not limited to methylnitronitrosoguanidine (MNNG),38–40 N-methyl-d-aspartate (NMDA),56,57 glutamate,58 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP),59 irradiation,60 trauma,61,62 retinal detachment,63 and perinatal64–66 as well as adult cerebral ischemia.67–69 Additional studies using RIP3−/−, and Bax−/−/RIP3−/− double knockout mice, and chemical inhibition or genetic ablation of PARP1 or AIF are needed to define the roles that PARP1- and AIF-mediated necroptosis might have in the death of uninfected bystander retinal cells, and how the processes of apoptosis and necroptosis interact during the evolution of cytomegalovirus retinitis. Supplementary Material includes TUNEL assay and H&E staining in uninfected control eyes (Supplementary Fig. S1) and Western blot of other BH3 domain proapoptotic proteins (Supplementary Fig. S2).
Acknowledgments
Supported by National Institutes of Health Grant EY009169 and the Retinal Research Foundation.
Disclosure: J. Mo, None; B. Marshall, None; J. Covar, None; N.Y. Zhang, None; S.B. Smith, None; S.S. Atherton, None; M. Zhang, None
References
- 1. Cohen J. AIDS therapy. New hope against blindness. Science. 1995; 268: 368–369. [DOI] [PubMed] [Google Scholar]
- 2. Drew WL. Cytomegalovirus infection in patients with AIDS. Clin Infect Dis. 1992; 14: 608–615. [DOI] [PubMed] [Google Scholar]
- 3. Istas AS, Demmler GJ, Dobbins JG, Stewart JA. Surveillance for congenital cytomegalovirus disease: a report from the National Congenital Cytomegalovirus Disease Registry. Clin Infect Dis. 1995; 20: 665–670. [DOI] [PubMed] [Google Scholar]
- 4. Jabs DA. Ocular manifestations of HIV infection. Trans Am Ophthalmol Soc. 1995; 93: 623–683. [PMC free article] [PubMed] [Google Scholar]
- 5. Gallant JE, Moore RD, Richman DD, Keruly J, Chaisson RE. Incidence and natural history of cytomegalovirus disease in patients with advanced human immunodeficiency virus disease treated with zidovudine. The Zidovudine Epidemiology Study Group. J Infect Dis. 1992; 166: 1223–1227. [DOI] [PubMed] [Google Scholar]
- 6. Palella FJ Jr, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998; 338: 853–860. [DOI] [PubMed] [Google Scholar]
- 7. Holtzer CD, Jacobson MA, Hadley WK, et al. Decline in the rate of specific opportunistic infections at San Francisco General Hospital, 1994–1997. Aids. 1998; 12: 1931–1933. [PubMed] [Google Scholar]
- 8. Jacobson MA, Stanley H, Holtzer C, Margolis TP, Cunningham ET. Natural history and outcome of new AIDS-related cytomegalovirus retinitis diagnosed in the era of highly active antiretroviral therapy. Clin Infect Dis. 2000; 30: 231–233. [DOI] [PubMed] [Google Scholar]
- 9. Jabs DA, Van Natta ML, Holbrook JT, et al. Longitudinal study of the ocular complications of AIDS: 1. Ocular diagnoses at enrollment. Ophthalmology. 2007; 114: 780–786. [DOI] [PubMed] [Google Scholar]
- 10. Jabs DA. AIDS and ophthalmology, 2008. Arch Ophthalmol. 2008; 126: 1143–1146. [DOI] [PubMed] [Google Scholar]
- 11. Braithwaite RS, Roberts MS, Chang CC, et al. Influence of alternative thresholds for initiating HIV treatment on quality-adjusted life expectancy: a decision model. Ann Intern Med. 2008; 148: 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Centers for Disease C, Prevention. Late HIV testing—34 states, 1996–2005. MMWR Morb Mortal Wkly Rep. 2009; 58: 661–665. [PubMed] [Google Scholar]
- 13. Chiou SH, Liu JH, Hsu WM, et al. Up-regulation of Fas ligand expression by human cytomegalovirus immediate-early gene product 2: a novel mechanism in cytomegalovirus-induced apoptosis in human retina. J Immunol. 2001; 167: 4098–4103. [DOI] [PubMed] [Google Scholar]
- 14. Buggage RR, Chan CC, Matteson DM, Reed GF, Whitcup SM. Apoptosis in cytomegalovirus retinitis associated with AIDS. Curr Eye Res. 2000; 21: 721–729. [PubMed] [Google Scholar]
- 15. Chiou SH, Liu JH, Chen SS, et al. Apoptosis of human retina and retinal pigment cells induced by human cytomegalovirus infection. Ophthal Res. 2002; 34: 77–82. [DOI] [PubMed] [Google Scholar]
- 16. Atherton SS, Newell CK, Kanter MY, Cousins SW. T cell depletion increases susceptibility to murine cytomegalovirus retinitis. Invest Ophthalmol Vis Sci. 1992; 33: 3353–3360. [PubMed] [Google Scholar]
- 17. Duan Y, Ji Z, Atherton SS. Dissemination and replication of MCMV after supraciliary inoculation in immunosuppressed BALB/c mice. Invest Ophthalmol Vis Sci. 1994; 35: 1124–1131. [PubMed] [Google Scholar]
- 18. Bigger JE, Tanigawa M, Zhang M, Atherton SS. Murine cytomegalovirus infection causes apoptosis of uninfected retinal cells. Invest Ophthalmol Vis Sci. 2000; 41: 2248–2254. [PubMed] [Google Scholar]
- 19. Zhang M, Atherton SS. Apoptosis in the retina during MCMV retinitis in immunosuppressed BALB/c mice. J Clin Virol. 2002; 25 (suppl 2): S137–S147. [DOI] [PubMed] [Google Scholar]
- 20. Zhang M, Zhou J, Marshall B, Xin H, Atherton SS. Lack of iNOS facilitates MCMV spread in the retina. Invest Ophthalmol Vis Sci. 2007; 48: 285–292. [DOI] [PubMed] [Google Scholar]
- 21. Zhang M, Xin H, Roon P, Atherton SS. Infection of retinal neurons during murine cytomegalovirus retinitis. Invest Ophthalmol Vis Sci. 2005; 46: 2047–2055. [DOI] [PubMed] [Google Scholar]
- 22. Handke W, Krause E, Brune W. Live or let die: manipulation of cellular suicide programs by murine cytomegalovirus. Med Microbiol Immunol. 2012; 201: 475–486. [DOI] [PubMed] [Google Scholar]
- 23. Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010; 7: 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ebermann L, Ruzsics Z, Guzman CA, et al. Block of death-receptor apoptosis protects mouse cytomegalovirus from macrophages and is a determinant of virulence in immunodeficient hosts. PLoS Pathog. 2012; 8: e1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang M, Covar J, Marshall B, Dong Z, Atherton SS. Lack of TNF-alpha promotes caspase-3-independent apoptosis during murine cytomegalovirus retinitis. Invest Ophthalmol Vis Sci. 2011; 52: 1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dix RD, Cousins SW. Susceptibility to murine cytomegalovirus retinitis during progression of MAIDS: correlation with intraocular levels of tumor necrosis factor-alpha and interferon-gamma. Curr Eye Res. 2004; 29: 173–180. [DOI] [PubMed] [Google Scholar]
- 27. Zhou J, Zhang M, Atherton SS. Tumor necrosis factor-alpha-induced apoptosis in murine cytomegalovirus retinitis. Invest Ophthalmol Vis Sci. 2007; 48: 1691–1700. [DOI] [PubMed] [Google Scholar]
- 28. Zhang M, Marshall B, Atherton SS. Murine cytomegalovirus infection and apoptosis in organotypic retinal cultures. Invest Ophthalmol Vis Sci. 2008; 49: 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Renault TT, Teijido O, Antonsson B, Dejean LM, Manon S. Regulation of Bax mitochondrial localization by Bcl-2 and Bcl-x(L): keep your friends close but your enemies closer. Int J Biochem Cell Biol. 2013; 45: 64–67. [DOI] [PubMed] [Google Scholar]
- 30. Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007; 72: 53–62. [DOI] [PubMed] [Google Scholar]
- 31. Brooks C, Dong Z. Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle. 2007; 6: 3043–3047. [DOI] [PubMed] [Google Scholar]
- 32. Zhou C, Pan W, Wang XP, Chen TS. Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. J Cell Path. 2012; 227: 3778–3786. [DOI] [PubMed] [Google Scholar]
- 33. Sanges D, Comitato A, Tammaro R, Marigo V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci U S A. 2006; 103: 17366–17371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Daugas E, Nochy D, Ravagnan L, et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000; 476: 118–123. [DOI] [PubMed] [Google Scholar]
- 35. Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002; 111: 147–150. [DOI] [PubMed] [Google Scholar]
- 36. Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002; 115: 4727–4734. [DOI] [PubMed] [Google Scholar]
- 37. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010; 22: 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu Y, Huang S, Liu ZG, Han J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem. 2006; 281: 8788–8795. [DOI] [PubMed] [Google Scholar]
- 39. Moubarak RS, Yuste VJ, Artus C, et al. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007; 27: 4844–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cregan SP, Fortin A, MacLaurin JG, et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002; 158: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014; 35C: 24–32. [DOI] [PubMed] [Google Scholar]
- 42. Chien H, Dix RD. Evidence for multiple cell death pathways during development of experimental cytomegalovirus retinitis in mice with retrovirus-induced immunosuppression: apoptosis, necroptosis, and pyroptosis. J Virol. 2012; 86: 10961–10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. White E. Life, death, and the pursuit of apoptosis. Genes Devel. 1996; 10: 1–15. [DOI] [PubMed] [Google Scholar]
- 44. Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005; 58: 495–505. [DOI] [PubMed] [Google Scholar]
- 45. Gillies LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014; 115: 632–640. [DOI] [PubMed] [Google Scholar]
- 46. Duerksen-Hughes PJ, Hermiston TW, Wold WS, Gooding LR. The amino-terminal portion of CD1 of the adenovirus E1A proteins is required to induce susceptibility to tumor necrosis factor cytolysis in adenovirus-infected mouse cells. J Virol. 1991; 65: 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Westendorp MO, Frank R, Ochsenbauer C, et al. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995; 375: 497–500. [DOI] [PubMed] [Google Scholar]
- 48. Su F, Schneider RJ. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 1997; 94: 8744–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Norris KL, Youle RJ. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol. 2008; 82: 6232–6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jurak I, Schumacher U, Simic H, Voigt S, Brune W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J Virol. 2008; 82: 4812–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arnoult D, Skaletskaya A, Estaquier J, Dufour C, Goldmacher VS. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis. 2008; 13: 1100–1110. [DOI] [PubMed] [Google Scholar]
- 52. Manzur M, Fleming P, Huang DC, Degli-Esposti MA, Andoniou CE. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 2009; 16: 312–320. [DOI] [PubMed] [Google Scholar]
- 53. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013; 14: 727–736. [DOI] [PubMed] [Google Scholar]
- 54. Christofferson DE, Li Y, Hitomi J, et al. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012; 3: e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996; 15: 6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Y, Kim NS, Li X, et al. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J Neurochem. 2009; 110: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mandir AS, Poitras MF, Berliner AR, et al. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000; 20: 8005–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Andrabi SA, Kang HC, Haince JF, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011; 17: 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mandir AS, Przedborski S, Jackson-Lewis V, et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A. 1999; 96: 5774–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Osato K, Sato Y, Ochiishi T, et al. Apoptosis-inducing factor deficiency decreases the proliferation rate and protects the subventricular zone against ionizing radiation. Cell Death Dis. 2010; 1: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Piao CS, Loane DJ, Stoica BA, et al. Combined inhibition of cell death induced by apoptosis inducing factor and caspases provides additive neuroprotection in experimental traumatic brain injury. Neurobiol Dis. 2012; 46: 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sarnaik AA, Conley YP, Okonkwo DO, et al. Influence of PARP-1 polymorphisms in patients after traumatic brain injury. J Neurotrauma. 2010; 27: 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hisatomi T, Nakazawa T, Noda K, et al. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest. 2008; 118: 2025–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hagberg H, Wilson MA, Matsushita H, et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004; 90: 1068–1075. [DOI] [PubMed] [Google Scholar]
- 65. Zhu C, Qiu L, Wang X, et al. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003; 86: 306–317. [DOI] [PubMed] [Google Scholar]
- 66. Zhu C, Wang X, Huang Z, et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007; 14: 775–784. [DOI] [PubMed] [Google Scholar]
- 67. Culmsee C, Zhu C, Landshamer S, et al. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005; 25: 10262–10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Thal SE, Zhu C, Thal SC, Blomgren K, Plesnila N. Role of apoptosis inducing factor (AIF) for hippocampal neuronal cell death following global cerebral ischemia in mice. Neurosci Lett. 2011; 499: 1–3. [DOI] [PubMed] [Google Scholar]
- 69. Li X, Klaus JA, Zhang J, et al. Contributions of poly(ADP-ribose) polymerase-1 and -2 to nuclear translocation of apoptosis-inducing factor and injury from focal cerebral ischemia. J Neurochem. 2010; 113: 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]