

Abstract
Objectives
Gout is one of the most common forms of arthritis. It is well established that urate lowering therapy that aims for a serum urate less than at least 0.36mmol/l (6mg/dL) is required for successful management of gout. Allopurinol, a xanthine oxidase (XO) inhibitor is the most commonly used urate lowering therapy. However, many patients fail to achieve the target serum urate on allopurinol, these patients can be considered to have “inadequate response” to allopurinol. Herein we examine the potential mechanisms and implications of inadequate response to allopurinol.
Methods
The literature was reviewed for potential causes for failure to reach target serum urate in patients receiving allopurinol.
Results
The two most common causes of inadequate response to allopurinol are poor adherence and under-dosing of allopurinol. Adherent patients who fail to achieve target serum urate on standard doses of allopurinol form a group that could be considered to be “partially resistant” to allopurinol. There are four potential mechanisms for partial allopurinol resistance: decreased conversion of allopurinol to oxypurinol; increased renal excretion of oxypurinol; abnormality in XO structure and or function such that oxypurinol is rendered less effective, and/or drug interactions.
Conclusions
It is important to determine the reasons for failure to achieve treatment targets with allopurinol, particularly as newer agents become available. The knowledge of the mechanisms for inadequate response may help guide the clinician toward making a therapeutic choice that is more likely to result in achieving the serum urate target.
Introduction
Gout is a common and challenging problem. Based on the National Health and Nutrition Examination Survey 2007–2008 the prevalence of gout in US adults was estimated to be ~3.9% (~8.3 million people). In New Zealand, gout is particularly common affecting 3.2%, 6.1% and 7.6% of Europeans, Māori, and Pacific adult New Zealanders respectively (1). Elevation in serum urate (SU) (hyperuricaemia) is the biochemical cause of gout. Inadequately treated gout leads to recurrent acute attacks, formation of tophi and joint damage. Significant time off work, poor health related quality of life and disability are common (2, 3).
The aim of gout treatment is sustained reduction in SU to 6mg/dl or lower (≤0.36mmol/L) (4). There are three potential mechanisms for urate lowering: 1) inhibition of urate production through the use of xanthine oxidase inhibitors (XOI); 2) increasing renal uric acid excretion through the use of uricosuric agents; and 3) metabolism of urate to the more water soluble and readily excretable allantoin through use of recombinant uricases. Xanthine oxidase (XO) inhibition is the first line recommendation for urate lowering in patients with gout (5).
Until the recent development and approval of febuxostat, allopurinol was the only available XOI for urate lowering therapy. Allopurinol continues to be the most commonly used urate lowering therapy. However, a large number of patients do not reach the target serum urate despite therapy with allopurinol. For example, in the Febuxostat vs. Allopurinol Controlled Trial (FACT) study only 21% of patients receiving allopurinol 300mg/d achieved the primary endpoint of the last three serum urates being ≤6mg/dl (6). While the cost of newer agents remains high, allopurinol is likely to remain the most commonly used urate lowering therapy. Thus it is important that we determine the reasons for failure to achieve treatment targets with allopurinol. This is especially important as newer agents become available, because the knowledge of the mechanism for sub-optimal response may help guide the clinician toward making a therapeutic choice that is more likely to result in achieving the serum urate target.
Terminology – inadequate response and allopurinol resistance
Patients who fail to reach the target SU on allopurinol can be considered to have an “inadequate response” to allopurinol. There are several potential causes for inadequate response to allopurinol including poor-adherence, under-dosing, or “partial resistance”. The definition of resistance is “the capacity to withstand something”. Thus adherent patients who fail to achieve target SU form a group that could be considered to be “partially resistant” to allopurinol. Complete resistance, that is absolutely no reduction in serum urate with allopurinol, appears to occur very rarely, if at all (7).
How allopurinol under-dosing fits within these definitions needs to be considered. For example, if patients have not undergone allopurinol dose escalation in a treat-to-target manner can they truly be considered to be partially resistant to allopurinol? Many patients who fail to achieve target SU on creatinine clearance (CrCL)-based allopurinol doses will respond to higher doses (7). Therefore, the clinical management of those patients who fail to achieve target SU on a “standard” dose of allopurinol i.e. ≤300mg daily, and those that fail to achieve target on higher doses e.g. 600mg daily may be different (Table 1).
Table 1.
Definitions of treatment failure
| Definition | Patient group | Possible mechanisms |
|---|---|---|
| Inadequate response | Patients who fail to reach target SU on allopurinol |
Non-adherence Under dosing Partial resistance |
| Partial resistance | Adherent patients on “standard” allopurinol doses (≤300mg daily) who fail to achieve target SU |
Impaired absorption Decreased conversion of allopurinol to oxypurinol Efficient renal excretion of oxypurinol Abnormality in XO structure and or function such that oxypurinol is rendered less effective Drug interactions |
Herein we examine the potential mechanisms of “inadequate response” and “partial resistance” to allopurinol.
Inadequate response to allopurinol
The two most common causes of inadequate response to allopurinol are poor adherence and under-dosing of allopurinol. Partial resistance is another cause.
Poor adherence
Poor adherence with urate lowering therapy is recognised as a significant problem in patients with gout. In a recent study of 7664 patients receiving allopurinol only 17% (1331) were adherent as defined by proportion of days covered (PDC) >80%. 2745 patients (36%) were partially adherent (20%<PDC<80%) and 3568 (47%) were poorly adherent (PDC<20%) (8). Plasma oxypurinol measurement, where available, may be helpful in determining adherence with allopurinol. A low steady-state plasma oxypurinol (<20µmol/l) in patients receiving allopurinol regularly over a long period (i.e. months), suggests that adherence may be poor or absorption is inadequate. Further efforts at engaging with and educating the patient as to the importance of adherence should be undertaken in the first instance.
Under-dosing of allopurinol
Allopurinol dosing remains controversial, especially in patients with renal impairment. The association between allopurinol dose and renal impairment in the development of the rare but potentially fatal allopurinol hypersensitivity syndrome (AHS) was highlighted by Hande et al (9). This led to the development of creatinine clearance (CrCL) based allopurinol dosing (9). This CrCL-based dosing has been accepted and broadly followed by many medical practitioners. However, there is no direct evidence that using lower allopurinol doses in patients with renal impairment reduces the risk of AHS. Furthermore, the relationship between elevated oxypurinol concentration and the development of AHS remains unproven. The most important consequence of CrCL-based dosing is that many patients with gout, particularly those with renal impairment, fail to achieve the target urate concentration. For example, in a study from New Zealand only 19.1% of patients achieved the target urate on the CrCL-based dose of allopurinol (10, 11). This lack of willingness to treat to target is further highlighted in recent data from Europe where, aside from the UK, very few patients received allopurinol at a dose of >300mg/day with a consequent failure to achieve target SU (Table 2).
Table 2.
Allopurinol dose and target SU in five European countries (31)
| France (n=193) |
Germany (n=208) |
Italy (n=194) |
Spain (n=197) |
UK (n=173) |
|
|---|---|---|---|---|---|
| Allopurinol dose >300mg/d |
5% | 5% | 10% | 6% | 34% |
| Percentage patients not achieving SU target (<6mg/dl) |
54% | 62% | 53% | 72% | 31% |
We have previously shown that systematic allopurinol dose escalation above the CrCL-based dose can result in the vast majority (88.8%) of patients achieving the treatment target (7). Perez-Ruiz et al have also shown that higher allopurinol doses are more effective in lowering SU with the target of <0.36mmol/L being achieved in 55% of patients when allopurinol dose was increased from 300mg/day to 450mg/day (12). Larger clinical trials with a particular focus on the safety of such an approach are underway. In the mean-time, for those patients who fail to achieve target serum urate on the CrCL-based dose the physician can opt to either gradually increase the dose of allopurinol with close monitoring, switch to febuxostat where it is available, or add a uricosuric agent such as probenecid.
Partial Resistance to Allopurinol
We have previously shown considerable inter-individual variability in plasma oxypurinol concentrations for an individual allopurinol dose (Figure 1) (13, 14).
Figure 1.

Allopurinol dose and oxypurinol concentrations in patients with gout from (13)
There is also a subgroup of patients who require doses of allopurinol >400mg/d to achieve target SU (Figure 1). These patients have a significantly smaller increase in plasma oxypurinol per mg increase in allopurinol dose compared to the majority of patients who require lower doses (≤400mg/day) to achieve target SU (13). This suggests that these patients have reduced absorption of allopurinol, can not convert allopurinol to oxypurinol as efficiently and/or excrete oxypurinol via the kidneys more effectively. There is also a subgroup of patients who fail to achieve the target SU despite high plasma oxypurinol concentrations (>100μmol/L), suggesting that oxypurinol is less effective in inhibiting XO in this subgroup. Finally there are some drug interactions that may contribute to partial allopurinol resistance. Thus there are four potential mechanisms for partial allopurinol resistance:
decreased conversion of allopurinol to oxypurinol,
increased renal excretion of oxypurinol,
abnormality in XO structure and or function such that oxypurinol is rendered less effective, and/or
drug interactions
Decreased conversion of allopurinol to oxypurinol
It has been widely assumed that XO is responsible for the conversion of allopurinol to oxypurinol. The metabolism of allopurinol to oxypurinol should be saturable if XO is primarily responsible for the metabolism of allopurinol. However, steady-state plasma oxypurinol concentrations increase in a linear fashion allopurinol dose which suggests that metabolism of allopurinol to oxypurinol is not saturable and another enzyme must be involved (15). The involvement of the closely related enzyme aldehyde oxidase (AOX) is supported by the ability of patients who lack XO but who do have AOX to metabolise allopurinol to oxypurinol, while those patients who lack both XO and AOX cannot convert allopurinol to oxypurinol (16, 17).
To function, both XO and AOX require the conversion of the oxo-form of molybdenum cofactor (MOCO) to the sulfido form by MOCO sulfurase (MOCOS) (18). A recent study identified two single nucleotide polymorphisms (SNPs), AOX1 3404A>G and MOCOS 2107A>C, which were associated with altered azathioprine treatment outcome in inflammatory bowel disease (19). SNPs in AOX1 have been reported to have effects on conversion of some aldehyde substrates (20). For example one common SNP AOX1 rs55754655 encodes an Asn to Ser change that results in enzyme that has 2–4-fold greater efficacy than wildtype AOX1, conferring a fast metaboliser phenotype (20). However, this and other SNPs in AOX1 have not previously been examined in relation to allopurinol response or conversion of allopurinol to oxypurinol. Based on the functional data, AOX1 rs55754655 is worthy of further investigation and may be a robust marker of allopurinol response. The minor allele frequency of this SNP is 0.1 in Caucasians included in the 1000 Genomes Project (21).
From a clinical perspective patients with poor conversion of allopurinol to oxypurinol may be best served with an alternate urate lowering drug as first line therapy. Febuxostat does not require conversion by AOX and therefore may be an appropriate alternative. Likewise, uricosuric agents would be appropriate.
Increased renal oxypurinol excretion
The primary route of elimination of oxypurinol is via the kidneys. It is known that URAT1, which is involved in renal urate reabsorption, is also involved in reabsorption of oxypurinol (22). OAT4, another important renal urate transporter may also have a role. The genes encoding these transporters are therefore candidates for variation that may influence oxypurinol clearance. Supporting this hypothesis, common variation in these genes influences serum urate concentrations (23).
How allopurinol response is affected by renal urate transporter genotype remains unknown. However, baseline serum urate appears to be an important determinant of final allopurinol dose (24) so that genetic determinants of urate may also be involved in allopurinol response. From a clinical perspective patients with increased clearance of oxypurinol may be best served with an alternative urate-lowering drug as first line therapy, such as febuxostat or an uricosuric agent. Currently quantification of oxypurinol clearance is not routine therefore this decision would need to be based on a clinical decision to try an alternative agent. However, future models that can calculate or predict clearance may guide the clinician.
Abnormality in XO structure and/or function
In vitro studies have shown that SNPs in XO may affect enzyme activity. For example SNPs Arg149Cys and Thr910Lys are associated with deficiency in XO activity compared to wild type (25). There are currently no frequency data on these SNPs nor is it known whether these SNPs or others are associated with allopurinol response. From a clinical perspective, first-line urate-lowering therapy for patients with abnormalities in XO structure and/or function may be a uricosuric drug, rather than a XOI.
Drug interactions
Co-morbidities such as hypertension and heart failure are common in patients with gout. As such many patients are receiving concomitant medications that might alter uric acid excretion or interact with allopurinol. Furosemide, a loop diuretic, is one such medication. Its effects on decreasing urinary uric acid excretion and thereby contributing to hyperuricaemia are well recognised. However, furosemide also interacts with allopurinol. Concomitant use of furosemide results in a significantly higher plasma oxypurinol concentration for any given allopurinol dose (13) (Figure 2). Thus the net effect of the interaction is retention of both urate and oxypurinol. Patients receiving furosemide also require relatively higher doses of allopurinol to achieve the target serum urate of <6mg/dl (7). This suggests that the effects of furosemide on increasing urate are greater than the urate lowering effects of allopurinol.
Figure 2.

Effect of furosemide on oxypurinol concentrations in patients with gout. From (13)
The mechanism of this interaction is unclear. There are complex interactions between urate and oxypurinol transport mechanisms within the kidney, particularly URAT1, which may be altered by co-administration of diuretics such as furosemide. Polymorphisms within genes coding for renal transporters (SCL2A9, NPT1, NPT3, URAT1, OAT4) have been associated with gout and hyperuricaemia (26, 27). There is biochemical evidence for interaction of diuretics with uric acid transporters (28) and evidence that SLC2A9 and OAT4 interact with diuretics in gout risk (29).
From a clinical perspective understanding such drug interactions may help guide the choice of urate lowering therapy. For example, a uricosuric agent may be more appropriate in patients receiving diuretics where renal urate excretion is impaired.
Can we predict partial resistance to allopurinol?
It would be useful to be able to predict inadequate response to allopurinol prior to commencing urate lowering therapy. Whilst it may not be possible to predict non-adherence, it may be possible to predict the dose of allopurinol most likely to result in the patient achieving target serum urate. Graham et al have developed a pharmacokinetic model that suggests that baseline urate prior to treatment is the single most important factor in final allopurinol dose (24). A similar association has been observed between baseline urate and achieving target with probenecid (30). If the predicted dose is higher than the clinician feels comfortable using in an individual then an alternate first line urate lowering therapy may need to be considered. However, as clinicians gain more experience with use of the older and cheaper agent allopurinol at higher doses the threshold for changing to an alternate agent based solely on dose may rise. Whether genetic markers influence this model remains to be determined. Ultimately a model that incorporates baseline urate, other indicators of likely partial resistance such as diuretic use or renal function, and genes that influence serum urate and allopurinol metabolism, excretion or mechanism of action may guide the clinician in choosing the most appropriate first line urate lowering therapy (Figure 3).
Figure 3.
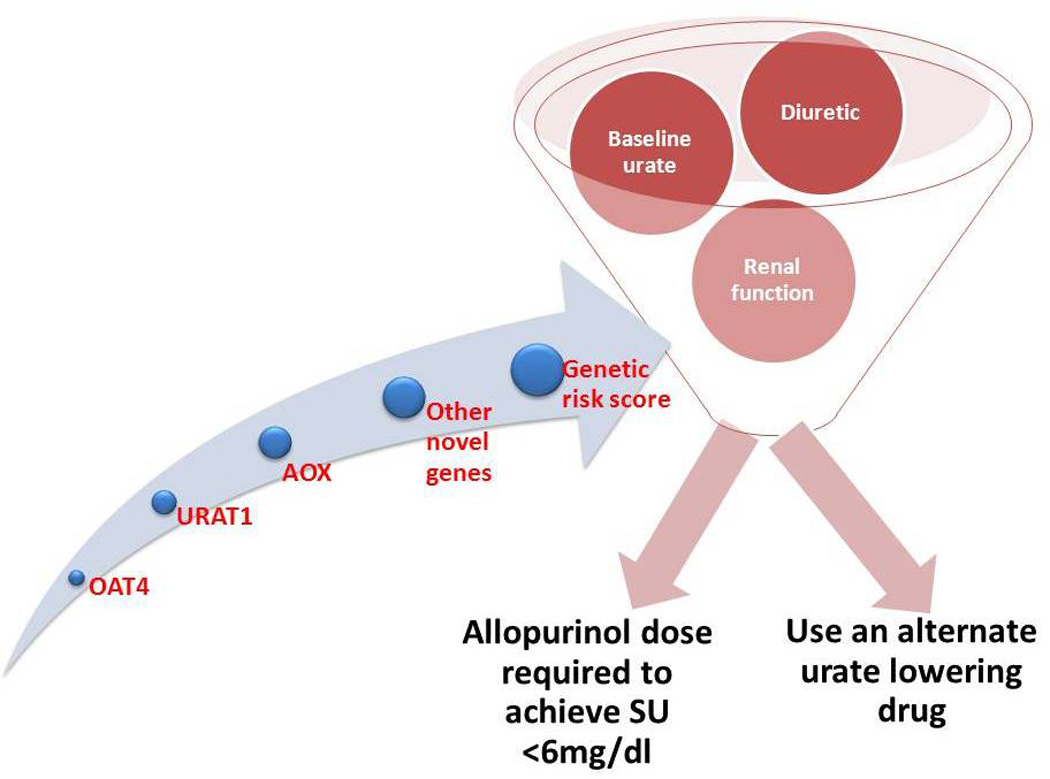
Factors that may be involved in predicting response to urate lowering therapy
Conclusion
Inadequate response to allopurinol is common when treating gout. There are a number of potential mechanisms for this occurrence. Understanding these mechanisms should provide clinicians with a basis for making rational therapeutic decisions (Figure 4). Further research is required to determine whether the mechanisms proposed herein are clinically relevant and whether inadequate response or partial resistance to allopurinol can be predicted.
Figure 4.
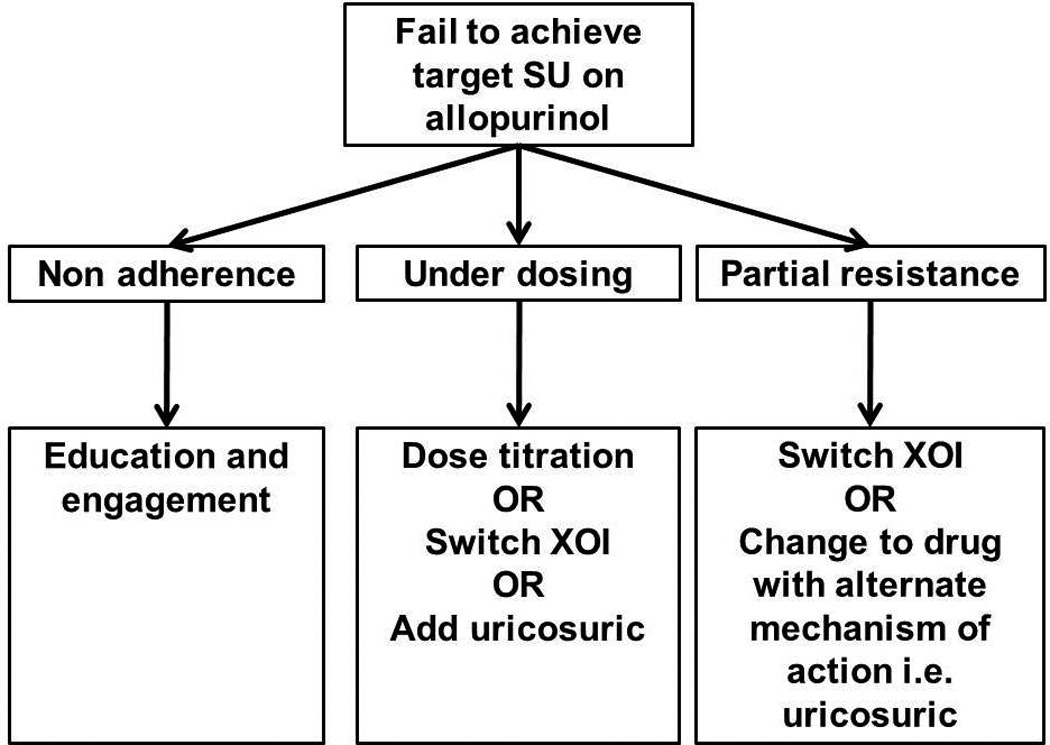
Mechanisms for failure to reach target serum urate in patients receiving allopurinol (middle boxes), and potential clinical responses (lower boxes). SU: serum urate, XOI: xanthine oxidase inhibitor.
Acknowledgments
Grant support:
This material is the result of work supported by a grant from the Division of Rheumatology at the University of Alabama at Birmingham. J.A.S. is supported by the resources and use of facilities at the Birmingham VA Medical Center, Alabama, USA. J.A.S. is also supported by grants from the Agency for Health Quality and Research Center for Education and Research on Therapeutics (CERTs), the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS), the National Institute of Aging (NIA) and the National Cancer Institute (NCI).
ND, LS and TM are supported by the Health Research Council of New Zealand.
Footnotes
Financial Conflict:
L.K.S has received consultant fees from Astra Zeneca
ND has received consultant or speaker fees from Takeda, Savient, Menorini, Astra Zeneca, Ardea, Novartis, Metabolex and Fonterra.
J.A.S. has received research and travel grants from Takeda and Savient; and consultant fees from Savient, Takeda, Regeneron and Allergan.
References
- 1.Winnard D, Wright C, Taylor W, Jackson G, Te Karu L, Gow P, et al. National prevalence of gout derived from administrative health data in Aotearoa New Zealand. Rheumatology. 2012;51(5):901–909. doi: 10.1093/rheumatology/ker361. [DOI] [PubMed] [Google Scholar]
- 2.Becker M, Schumacher HR, Benjamin K, Gorevic P, Greenwald M, Fessel J, et al. Quality of life and disability in patients with treatment-failure gout. J Rheumatol. 2009;36(5):1041–1048. doi: 10.3899/jrheum.071229. [DOI] [PubMed] [Google Scholar]
- 3.Dalbeth N, Collis J, Gregory K, Clark B, Robinson E, McQueen F. Tophaceous joint disease strongly predicts hand function in patients with gout. Rheumatology. 2007;46:1804–1807. doi: 10.1093/rheumatology/kem246. [DOI] [PubMed] [Google Scholar]
- 4.Li-Yu J, Clayburne G, Sieck M, Beutler A, Rull M, Eisner E, et al. Treatment of chronic gout. Can we determine when urate stores are depleted enough to prevent attacks of gout? J Rheumatol. 2001;28:577–580. [PubMed] [Google Scholar]
- 5.Khanna D, Fitzgerald J, Khanna P, Sangmee B, Singh M, Neogi T, et al. 2012 American College of Rheumatology Guidelines for the Management of Gout. Part 1: Systematic Nonpharmacologic and Pharmacologic Therapeutic Approaches to Hyperuricaemia. Arthritis Care Res. 2012;64(10):1431–1446. doi: 10.1002/acr.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker M, Schumacher HR, Wortmann R, MacDonald P, Eustace D, Palo W, et al. Febuxostat compared with allopurinol in patients with hyperuricaemia and gout. N Engl J Med. 2005;353:2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 7.Stamp L, O’Donnell J, Zhang M, James J, Frampton C, Barclay M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rheum. 2011;63(2):412–421. doi: 10.1002/art.30119. [DOI] [PubMed] [Google Scholar]
- 8.Zandman-Goddard G, Amital H, Shamrayevsky N, Raz R, Shalev V, Chodick G. Rates of adherence and persistence with allopurinol therapy among gout patients in Israel. Rheumatology. 2013;52:1126–1131. doi: 10.1093/rheumatology/kes431. [DOI] [PubMed] [Google Scholar]
- 9.Hande K, Noone R, Stone W. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76:47–56. doi: 10.1016/0002-9343(84)90743-5. [DOI] [PubMed] [Google Scholar]
- 10.Dalbeth N, Kumar S, Stamp LK, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricaemia in patients with gout. J Rheumatol. 2006;33(8):1646–1650. [PubMed] [Google Scholar]
- 11.Dalbeth N, House M, Horne A, Petrie K, McQueen F, Taylor W. Prescription and dosing of urate-lowering therapy, rather than patient behaviours, are the key modifiable factors associated with targeting serum urate in gout. BMC Musculoskelet Disord. 2012;13:174. doi: 10.1186/1471-2474-13-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57:545–549. doi: 10.1136/ard.57.9.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamp L, Barclay M, O’Donnell J, Zhang M, Drake J, Frampton C, et al. Relationship between serum urate and plasma oxypurinol in the management of gout: determination of minimum plasma oxypurinol concentration to achieve a target serum urate level. Clin Pharm Ther. 2011;90(3):392–398. doi: 10.1038/clpt.2011.113. [DOI] [PubMed] [Google Scholar]
- 14.Wright D, Stamp L, Merriman T, Barclay M, Duffull S, Holford N. A population pharmacokinetic model for allopurinol and oxypurinol in patients with gout. Eur J Clin Pharmacol. 2013;69:1411–1421. doi: 10.1007/s00228-013-1478-8. [DOI] [PubMed] [Google Scholar]
- 15.Graham S, Day R, Wong H, McLachlan A, Bergendal L, Miners J, et al. Pharmacodynamics of oxypurinol after administration of allopurinol to healthy subjects. Br J Clin Pharmacol. 1996;41:299–304. doi: 10.1046/j.1365-2125.1996.03116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiter S, Simmonds HA, Zollner N, Braun SL, Knedel M. Demonstration of a combined deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxipurinol. Clin Chim Acta. 1990 Mar 15;187(3):221–234. doi: 10.1016/0009-8981(90)90107-4. [DOI] [PubMed] [Google Scholar]
- 17.Shibutani Y, Ueo T, Yamamoto T, Takahashi S, Moriwaki Y, Higashino K. A case of classical xanthinuria (type 1) with diabetes mellitus and Hashimoto's thyroiditis. Clin Chim Acta. 1999 Jul;285(1–2):183–189. doi: 10.1016/s0009-8981(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 18.Schwarz G. Molybdenum cofactor biosynthesis and deficiency. Cell Mol Life Sci. 2005;63:2792–2810. doi: 10.1007/s00018-005-5269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith M, Marinaki A, Arenas M, Shobowale-Bakre M, Lewis C, Ansari A, et al. Novel pharmacogenetic markers for treatment outcome in azathioprine-treated inflammatory bowel disease. Aliment Pharm Therapeut. 2009;30:375–384. doi: 10.1111/j.1365-2036.2009.04057.x. [DOI] [PubMed] [Google Scholar]
- 20.Hartmann T, Terao M, Garattini E, Teutloff C, Alfaro J, Jones J, et al. The impact of single nucleotide polymorphisms on human aldehyde oxidase. Drug Metab Dispos. 2012;40(5):856–864. doi: 10.1124/dmd.111.043828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature Genetics. 2010;467 doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwanaga T, Kobayashi D, Hirayama M, Maeda T, Tamai I. Involvement of uric acid transporter in increased renal clearance of the xanthine oxidase inhibitor oxypurinol induced by a uricosuric agent, benzbromarone. Drug Metab Dispos. 2005;33:1791–1795. doi: 10.1124/dmd.105.006056. [DOI] [PubMed] [Google Scholar]
- 23.Kottgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nature Genetics. 2013;45:145–154. doi: 10.1038/ng.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham G, Kannangara D, Stocker S, Portek I, Pile K, Indraratna P, et al. Understanding the dose-response relationship of allopurinol: predicting the optimal dosage. Br J Clin Pharmacol. 2013;76(6):932–938. doi: 10.1111/bcp.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kudo M, Moteki T, Sasaki T, Konno Y, Ujiie S, Onose A, et al. Functional characterization of human xanthine oxidase allelic variants. Pharmacogenet Genomics. 2008;18:243–251. doi: 10.1097/FPC.0b013e3282f55e2e. [DOI] [PubMed] [Google Scholar]
- 26.So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120(6):1791–1799. doi: 10.1172/JCI42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merriman T, Dalbeth N. The genetic basis of gout. Joint Bone Spine. 2011;78:35–40. doi: 10.1016/j.jbspin.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Hagos Y, Stein D, Ugele B, Burckhardt G, Bahn A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J Am Soc Nephrol. 2007;18:430–439. doi: 10.1681/ASN.2006040415. [DOI] [PubMed] [Google Scholar]
- 29.McAdams-Demarco M, Maynard J, Baer A, Kao L, Kottgen A, Coresh J. A urate gene-by-diuretic interaction and gout risk in participants with hypertension: results from the ARIC study. Ann Rheum Dis. 2013;72:701–706. doi: 10.1136/annrheumdis-2011-201186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pui K, Gow P, Dalbeth N. Efficacy and tolerability of probenecid as urate-lowering therapy in gout; clinical experience in high-prevalence population. J Rheumatol. 2013;40(6):872–876. doi: 10.3899/jrheum.121301. [DOI] [PubMed] [Google Scholar]
- 31.Khanna P, Hagerty D, Mischler R, Morlock R. Adherence to EULAR recommendations for the treatment of gout. Ann Rheum Dis. 2012;71(Suppl 3):448. [Google Scholar]