

Abstract
Multiple sclerosis (MS) is a neuroinflammatory disease characterized by demyelination and axonal damage of the central nervous system. The pathogenesis of MS has also been linked to vascular inflammation and local activation of the coagulation system, resulting in perivascular fibrin deposition. Treatment of experimental autoimmune encephalomyelitis (EAE), a model of human MS, with antithrombotic and antiinflammatory activated protein C (APC) reduces disease severity. Since recombinant APC (Drotecogin alfa), originally approved for the treatment of severe sepsis, is not available for human MS studies, we tested the hypothesis that pharmacologic activation of endogenous protein C could likewise improve the outcome of EAE. Mice were immunized with murine myelin oligodendrocyte glycoprotein (MOG) peptides and at the onset of EAE symptoms, were treated every other day with either WE thrombin (25μg/kg; i.v.), a selective recombinant protein C activator thrombin analog, or saline control. Mice were monitored for changes in disease score until euthanized for ex vivo analysis of inflammation. Administration of WE thrombin significantly ameliorated clinical severity of EAE, reduced inflammatory cell infiltration and demyelination, suppressed the activation of macrophages comprising the CD11b+ population and reduced accumulation of fibrin(ogen) in the spinal cord. These data suggest that symptomatic MS may respond to a treatment strategy that involves temporal pharmacological enhancement of endogenous APC generation.
Keywords: multiple sclerosis, thrombin, antithrombotics, activated protein C
Introduction
Multiple sclerosis (MS) is a neuroinflammatory disease of unknown etiology affecting approximately 0.1% of the population (Olek 2014). MS is characterized by oligodendrocyte apoptosis, progressive destruction of myelin sheaths and axonal damage (Kutzelnigg and Lassmann 2014). While all symptomatic MS patients exhibit some form of neurological deficit, there is considerable variation in clinical presentation, disease course and pathological features. MS has been classified into four disease courses depending on whether symptoms are acute, remittent, relapsing or chronic progressive. Temporal worsening of the symptoms in acute or remittent MS can be particularly devastating. Current treatment of such exacerbations with plasmapheresis, interferon or antiinflammatory drugs is reasonably effective; however, patients with progressive forms of MS may show little or no benefit (La Mantia et al. 2012; Ciccone et al. 2008).
Abnormalities of the cerebral blood perfusion have been associated, though without evidence of causality, to MS pathogenesis. Disruption of the blood-brain barrier (BBB) is a pathogenic feature of MS linked to clinical relapse (Kermode et al. 1990). Histopathological analyses of pre-demyelinating MS lesions indicate that one of the earliest events associated with BBB disruption is activation of microglia, the resident immune cells of the central nervous system (CNS) and the leakage of blood proteins into CNS (Vos et al. 2005; Marik et al. 2007; Nimmerjahn et al. 2005). Among the proteins that traverse into the CNS are members of the coagulation cascade, including procoagulant modulators fibrinogen, tissue factor and protein C inhibitor (Gay et al. 1997; Han et al. 2008; Kwon and Prineas 1994).
Blood vessel injuries are strong promoters of blood coagulation activation and thrombogenesis. Analysis of brain tissue from MS patients has revealed the presence of fibrinogen and fibrin within MS lesions and localized around blood vessels (Claudio et al. 1995; Gay et al. 1997; Kwon and Prineas 1994). Fibrin is the product of fibrinogen cleavage by the serine protease thrombin. Early studies indicate that limiting thrombin via administration of heparin, an indirect inhibitor of thrombin, may reduce the number of MS exacerbations (Courville 1959a, b; Maschmeyer et al. 1961), suggesting that anticoagulant treatment may be beneficial for MS therapy. Although animal models of MS do not replicate the human disease, they have been used in the evaluation of antithrombotic approaches to the treatment of MS. Such animal studies also suggest that anticoagulation with heparin (Chelmicka-Szorc and Arnason 1972; Lider et al. 1989) or the direct thrombin inhibitor, hirudin (Han et al. 2008) improve the outcome of experimental autoimmune/allergic encephalomyelitis (EAE). Likewise, complementary studies performed in fibrinogen-deficient mice (Akassoglou et al. 2004) or fibrin depletion by prophylactic administration of anticoagulants (Paterson 1976) or the defibrinogenating agent batroxobin (Yang et al. 2011) have suggested that fibrinogen is at least one of the thrombin substrates that drives EAE progression and severity. To date, however, randomized controlled clinical studies have not been conducted to explore the utility of anticoagulants in the treatment of MS.
A more recent study evaluated the utility of recombinant activated protein C (APC), a serine protease with potent anticoagulant and antiinflammatory properties, in EAE (Han et al. 2008). Endogenous APC is generated from the receptor-bound zymogen protein C on the endothelial cell surface by the molecular complex of thrombomodulin and thrombin. In addition to reducing intravascular thrombin generation, APC functions as a signaling molecule, exhibiting cytoprotective, anti-inflammatory and anti-apoptotic effects via activation of the protease-activated receptor, PAR-1 and endothelial protein C receptor, EPCR (Mosnier et al. 2007). In animal models, APC administration exerts beneficial effects in the CNS and periphery, suppressing pro-inflammatory NF-κB signaling and apoptosis in endothelial, neuronal and immune cell populations (Gorbacheva et al. 2010; Joyce and Grinnell 2002; Seol et al. 2011). Mutant APC analogs with reduced anticoagulant activity also exhibit neuroprotective properties in animal models of CNS injury (Wang et al. 2013a; Wang et al. 2013b). The ability of exogenous APC administration to reduce neurological deficits and suppress inflammation in the CNS and periphery in EAE (Han et al. 2008) indicates that augmenting this pathway may have therapeutic potential for MS. However, exogenous APC (Drotecogin alfa, Lilly) is no longer marketed and available for clinical evaluation in human patients. We therefore contemplate that pharmacologic activation of endogenous protein C with an enzyme, WE thrombin, currently in preclinical development for the antithrombotic treatment of cardiovascular emergencies, may represent a safe alternative to exploring the use of exogenous APC administration or systemic anticoagulants in MS.
WE (W215A/E217A) thrombin is a recombinant thrombin analog that contains two amino acid mutations, generating an enzyme with significantly reduced procoagulant activity. WE thrombin activity toward fibrinogen and the thrombin receptor, protease activated receptor-1 (PAR-1) in vitro is reduced 19,000- and 1,200-fold, respectively (Cantwell and Di Cera 2000). Importantly, WE thrombin retains 10% of the thrombomodulin-dependent anticoagulant function of thrombin. Due to this re-design of the molecule, WE thrombin selectively activates protein C in the presence of its endothelial receptor, thrombomodulin, to form APC. Administration of low dose WE thrombin to non-human primates has been shown to cause activation of endogenous protein C and inhibit acute vascular graft thrombosis without enhancement of wound bleeding or other adverse events (Gruber et al. 2002; Gruber et al. 2007). Interestingly, the concentration of APC detected in the plasma after administration of WE thrombin is significantly lower than the concentration of circulating exogenous APC required to generate a comparable antithrombotic effect.
Activation of endogenous protein C by WE thrombin administration improves disease outcomes in animal models of various diseases that affect tissue perfusion, including vascular graft thrombosis, ischemic stroke, carotid artery occlusion and collagen-induced arthritis (Berny-Lang et al. 2011; Flick et al. 2011; Vicente et al. 2012). These observations, coupled with studies demonstrating that the thrombin product APC has potent cytoprotective and antiinflammatory effects, lend support to the hypothesis that treatment of MS with a thrombin analog with drastically reduced activity for fibrinogen and PAR-1 but retained activity for protein C could attenuate disease progression.
Experimental Procedures
Expression and purification of WE thrombin
WE (W215A/E217A) prethrombin-2 was expressed in E. coli as inclusion bodies. Following lysis of inclusion bodies, the prethrombin-2 precursor was refolded, purified by affinity chromatography on heparin-sepharose, activated by proteolytic cleavage using ecarin and further purified by heparin affinity and ion exchange chromatography. The protein solution was treated with Detoxi-gel Endotoxin Removing Gel (Pierce) to remove potential endotoxin contamination. The Detoxi-gel treated pool was subsequently concentrated and diafiltered into 20mM Tris, 300mM NaCl, pH 8 for storage. Enzymatic activity of the resulting protein was assessed by chromogenic assay as described (Cantwell and Di Cera 2000).
Animals
Wild-type male mice were housed in the Animal Resource Facility at the Portland Veterans Affairs Medical Center in accordance with institutional guidelines. The study was conducted in accordance with National Institutes of Health guidelines for the use of experimental animals, and the protocols were approved by the Institutional Animal Care and Use Committee.
Induction of active EAE and treatment with WE thrombin
EAE was induced using mouse myelin oligodendrocyte glycoprotein, peptides 35-55 (MOG35-55). MOG35-55 was combined with complete Freund’s adjuvant containing heat-inactivated M. tuberculosis as described (Sinha et al. 2010). All mice were injected with 75ng and 200ng pertussis toxin (Ptx) intraperitoneally on days 0 and 2 relative to immunization. The mice were assessed for signs of EAE according to the following scale: 0, normal; 1, limp tail or mild hind limb weakness; 2, moderate hind limb weakness or mild ataxia; 3, moderately severe hind limb weakness; 4, severe hind limb weakness or mild forelimb weakness or moderate ataxia; 5, paraplegia with no more than moderate forelimb weakness; and 6, paraplegia with severe forelimb weakness or severe ataxia or moribund condition. At the onset of clinical signs of EAE (between days 10–13 when the clinical scores were ≥ 2.5), mice were divided into two groups and treated intravenously every other day with 50ng of WE thrombin in 100μl volume (25μg/kg) or saline vehicle at the same volume. The dose and dosing regimen selected were based on previous studies with WE thrombin (Berny-Lang et al. 2011; Flick et al. 2011). Mice were monitored for changes in disease score until they were euthanized for ex vivo analyses.
Fluorescence-activated cell-sorting (FACS)
Four-color fluorescence flow cytometry analyses were performed to determine the phenotypes of cells following general antibody staining procedures as described (Dziennis et al. 2011). For splenocytes, single cell suspensions of spleens were prepared by homogenizing the tissue through a fine mesh screen. Mononuclear cells from the CNS were isolated by Percoll gradient centrifugation as described (Bebo et al. 1996). Due to low numbers, CNS cells from 3 animals per treatment group were pooled for analysis. 1×106 cells were washed with staining medium (PBS containing 0.1% NaN3 and 1% bovine serum albumin (Sigma)) and incubated with the combinations of the following monoclonal antibodies: CD4, CD45, ICAM-1 and CD11b for phenotype analysis. All antibodies were purchased from previously described sources (Dziennis et al. 2011).
Histopathology
Mice were anesthetized with isoflurane, heparinized and perfused with 100ml of 4% paraformaldehyde in 0.1M sodium phosphate buffer (pH 7.4), and fixed at 4°C for 24 hr. The spinal cords were dissected from the spinal columns and sections 1–2 mm in length sampled from the thoracic and lumbar region. Tissues were fixed in 5% glutaraldehyde in 0.1M sodium phosphate buffer (pH 7.4) for three days at 4°C, post-fixed with 1% osmium tetroxide for 3.5hr, rehydrated in ethanol, and embedded in plastic as described (Wang et al. 2009). Semithin sections (0.5μm) were stained with toluidine blue and photographed under 20x magnification using a light microscope. Images were compiled to create a complete construction of the entire spinal cord section. Stained tissue sections were analyzed by an investigator blinded to treatment status, and axonal damage and demyelination identified and quantified in one representative section for each treatment. As described (Wang et al. 2006), regions in the white matter area containing tissue damage, including disrupted compact myelin, demyelinated axons and degenerating axons, were circumscribed on photomontages of the entire spinal cord. The total white matter area and the total damaged area within the white matter were measured to determine the % area of damage.
Immunohistochemistry
Fibrin and fibrinogen (fibrin(ogen)) deposition in one representative lumbar section for each group was detected using a rabbit polyclonal antibody against human fibrin(ogen). Sections were fixed, embedded in paraffin, sectioned, and dewaxed. Slides were blocked in 10% NGS, 1% BSA, 0.025% triton-X for 45 minutes at room temperature, followed by incubation with rabbit polyclonal antibody to fibrin(ogen) (MP Biomedicals, Santa Ana, CA) overnight at 4°C diluted 1:50 in goat serum, and then incubated with goat anti-rabbit IgG (Molecular Probes; Alexa Fluor 488). Slides were washed and mounted under aqueous media. Negative control slides were treated as above without primary antibody. Sections were photographed with a Zeiss Axiovert fluorescent microscope under 20x and images processed under identical conditions using Slidebook (version 5.5 for Windows, Intelligent Imaging Innovations, Inc., Denver, CO).
Analysis of fibrin(ogen) accumulation was performed using the following method. Fluorescence intensity in a 10-μm2, non-cellular region was measured, averaged to yield a mean background fluorescence and subtracted from every image. Fibrin(ogen) signal intensity was thresholded by measuring nonspecific staining in a negative control (absence of primary antibody). This value was used to set the lower limit of the calibration scale for all subsequent measurements. Thus, only fluorescence above background and separate from nonspecific staining was visualized. Images were compiled in Photoshop to create a complete construction of the entire spinal cord section. Fibrin(ogen) staining in one representative lumbar section from each treatment group was evaluated by measuring the total area of the spinal cord section in μm2. The area of positive signal above threshold within this area was subsequently measured to determine the percent area positive for signal and the total signal per μm2. Image analyses were performed using a custom program in MATLAB (The Mathworks, Inc., Natick, MA).
Statistical Analysis
Data are expressed as means ± SEM, with the exception of pooled analysis in brain samples, assessment of spinal cord damage and fibrin(ogen) staining quantification. Statistical difference between vehicle and treatment group for disease score was evaluated by Mann-Whitney U test. Cumulative disease index and cell frequency were analyzed using students t test. A p value of less than 0.05 was considered significant. Statistical analyses were made using GraphPad Prism (version 5.01 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com).
Results
WE thrombin reverses symptoms of EAE
Compared with vehicle-treated control mice, performance scores for WE thrombin-treated mice were significantly improved (Fig. 1a). The peak disease score for WE thrombin-treated mice was also significantly reduced as compared to vehicle-treated mice (3.7±0.2 vs 2.5±0.2; p <0.05). Average EAE scores were calculated for each day and summed for the entire experiment to yield a Cumulative Disease Index (CDI), representing total disease load. Control mice had a CDI of 27.4±2.2, whereas WE thrombin-treated mice demonstrated a significantly reduced CDI of 14.3±2.2 (Fig. 1b). Thus, WE thrombin reduced the disease load by nearly 50% over the evaluation period.
Fig. 1. WE thrombin attenuates the symptoms of EAE.
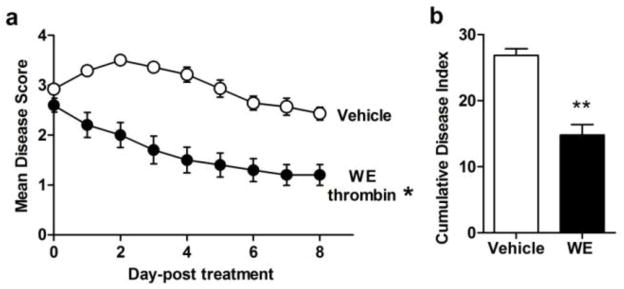
Male mice were immunized with MOG/CFA/Ptx and assessed daily and scored as outlined in the Materials and Methods. At peak disease onset, when disease scores reached > 2.5 (range from 0 = asymptomatic to 6 = paraplegia with severe forelimb weakness or severe ataxia or moribund condition), mice were treated every other day with vehicle or WE thrombin (25μg/kg; i.v.). Compared to vehicle, mean disease scores for WE thrombin-treated mice were significantly lower (a). The cumulative disease index for WE thrombin-treated mice was significantly lower than vehicle treated mice (b). Data presented are the means ± SEM of two independent experiments, with n = 7–10, from which three mice per group were sampled for ex vivo analysis. Statistical difference between vehicle and treatment group for disease score was evaluated by Mann-Whitney U test, p<0.05. Cumulative disease index and was analyzed using students t test, p<0.01.
WE thrombin reduces inflammatory markers in EAE
We next designed experiments to evaluate the anti-inflammatory function of WE thrombin. As accumulation of macrophages is a hallmark of CNS inflammation, we quantified the population of mononuclear cells in pooled brain of vehicle- and WE thrombin-treated mice. Our results indicate that WE thrombin significantly reduced the absolute numbers of activated macrophages/microglia (CD11b/CD45high cells) from 18880 to 9912 cells in vehicle vs. WE thrombin-treated mice, respectively. The frequency of CD11b/CD45high cells decreased from 5.9% to 3.5% in vehicle vs. WE thrombin-treated mice, respectively. The frequency of resting microglia (CD11b/CD45low cells) were increased from 61.1% in vehicle treated to 91.4% in WE thrombin-treated mice. Furthermore, treatment with WE thrombin significantly reduced both the absolute number (from 11552 to 3192) and percentage (from 3.6% to 1.1%) of CD4+ T cells in the brain. We next determined whether WE thrombin attenuated activation of the macrophage subpopulation of splenocytes during EAE. Our data indicate that treatment with WE thrombin reduced both TNFα, and ICAM-1 expression on CD11b+ macrophages in EAE (Figs. 2a,b). Collectively, these results demonstrate that WE thrombin reduces the inflammatory markers in the brain and spleen of mice during EAE.
Fig. 2. Inflammatory response in the spleen is attenuated by WE thrombin treatment of EAE.

Mice were sacrificed on day 30 and splenocytes subjected to FACS analysis as described (Dziennis et al. 2011). To evaluate the macrophage subpopulation, cells were gated for CD11b+. Compared to vehicle treated mice, the frequency of TNFα (a) and ICAM-1 (b) labeled macrophages was significantly lower in WE thrombin-treated mice. Data are the means ± SEM of n = 3 mice. Significant difference between vehicle and WE thrombin-treated groups were determined using unpaired t-test, p<0.01.
WE thrombin reduces CNS pathology in mice immunized with MOG35-55
We examined the efficacy of WE thrombin to reduce axonal damage and demyelination in spinal cord tissue. Consistent with the pathology associated with EAE, the spinal cord from vehicle-treatment showed axonal injury and demyelination (Fig. 3). The spinal cord from WE thrombin treatment showed a dramatic decrease in demyelination and injury. The damaged area within the white matter regions of a representative section from each treatment group was quantified, revealing that the percent of white matter area damaged was reduced from 15.7% in vehicle to 2.43% in WE thrombin.
Fig. 3. WE thrombin treatment reduces axonal damage and demyelination in the spinal cord.

Thoracic spinal cords sampled from one representative animal per treatment group were fixed, embedded in plastic, stained with toluidine blue and assessed for axonal damage as described (Wang et al. 2006). Semithin sections were photographed with a light microscope under 20x and images were compiled to create a complete construction of the entire spinal cord section. Shown are representative photographs of thoracic spinal cords of MOG immunized mice treated with either vehicle (a) or WE thrombin (b). In whole sections, areas of tissue damage are circled in red. The white rectangles demarcate area magnified and presented below whole sections. In whole sections, scale bar is 100μm and in high power (63x) view, scale bar is 10μm.
Fibrin(ogen) deposition
Given the importance of BBB disruption in disease activity, we sought to examine whether or not WE thrombin affected fibrin(ogen) accumulation in the CNS. Immunofluorescence microscopy revealed positive staining for fibrin(ogen) in lumbar spinal cord sections. Distribution of fibrin(ogen) in spinal cord sections was consistent with previous data demonstrating fibrin in white matter of EAE mice (Yang et al. 2011). Compared to vehicle control, there appeared to be reduced signal for fibrin(ogen) in WE thrombin-treatment (Fig. 4). To quantify fibrin(ogen), the percent of area positive was measured in a representative section from each treatment group. Our data indicate that fibrin(ogen) staining was reduced from 14.3% in vehicle to 10.5% in WE thrombin. Similarly, the total signal area per μm2 was reduced to nearly half from 74.7 to 40.5 in vehicle and WE thrombin groups, respectively.
Fig. 4. WE thrombin treatment reduces fibrin accumulation in the spinal cord.

Spinal cords sampled from the lumbar region of one representative animal per treatment group were fixed, paraffin embedded, sectioned and rehydrated. Fibrin(ogen) deposition was detected using a rabbit polyclonal antibody against fibrin(ogen) as described in the Materials and Methods. Sections were photographed under a fluorescent microscope under 20x and images processed under identical conditions. Images were compiled in Photoshop to create complete construction of the entire spinal cord section. Shown are representative photographs of lumbar spinal cords labeled with an antibody to fibrin(ogen) treated with either vehicle (left) or WE thrombin (right). Fibrin(ogen) accumulation was quantified by measuring the percent of positively stained area (white). Scale bar is 100μm.
Discussion
In the present study, we examined the effect of endogenous protein C activation by WE thrombin administration in a mouse model of multiple sclerosis. Compared with vehicle control, treatment with WE thrombin attenuated neurological impairment associated with EAE. These findings are consistent with previous work demonstrating that administration of recombinant APC suppresses disease progression in PLP-induced EAE (Han et al. 2008). The two-fold reduction in CDI (Fig. 1a) that we observed with WE thrombin treatment is comparable with other treatments evaluated in EAE, including a small molecule inhibitor of macrophage migration inhibitory factor (Kithcart et al. 2010) and recombinant T-cell receptor ligands (RTL) (Vandenbark et al. 2003; Benedek et al. 2013; Wang et al. 2006; Burrows et al. 2012), the latter having advanced as a drug candidate for multiple sclerosis and tested in a human phase 1 setting (Yadav et al. 2012). Thus, WE thrombin is as effective in reducing disease severity in EAE as other potential drug candidates for MS.
Inflammation of the central nervous system contributes to disease severity in MS, therefore we examined the extent to which WE thrombin reduced inflammation in the brain. Both the innate and the adaptive immune responses contribute to disease severity in MS and EAE. As part of the innate immune response in MS and EAE, macrophages are a major source of pro-inflammatory cytokines, such as TNFα, and play a central role in local tissue damage. Likewise, T cells participate in the adaptive immune response in MS and EAE, augmenting the immune response by stimulating antibody production, producing cytokines that activate other T cells and destroying cells that present antigenic material. We observed that the number and frequency of activated macrophages/microglia (CD11b+/CD45high) and T cells (CD4) within the brain was significantly reduced in WE thrombin treated mice. The CD45low population comprising resting microglia was significantly increased in WE thrombin-treated mice, suggesting an overall dampening of inflammation.
We next examined whether immune cell reductions in the brain were associated with reduced macrophage activation in the spleen. In the spleen, WE thrombin reduced expression of TNFα and intercellular adhesion molecule-1 (ICAM-1) on CD11b+ macrophages (Figs. 2a,b). During inflammation, both TNFα and ICAM-1 are upregulated on splenic macrophages, resulting in an activated phenotype. Adhesion molecule ICAM-1 contributes to immune cell adhesion and infiltration of macrophages. Therefore, reductions in brain macrophages in WE thrombin-treated mice may be consequent of reduced ICAM-1 expression. Based on these data, we conclude that treatment with WE thrombin suppresses inflammation in the brain, possibly due to reduced recruitment of activated macrophages.
The beneficial effect on WE thrombin treatment on EAE-induced inflammation is most likely the result of endogenous APC generation. It is known that activation of protein C by WE thrombin improves disease outcomes in other disease models (Berny-Lang et al. 2011; Flick et al. 2011; Vicente et al. 2012). Likewise, exogenous administration of APC has beneficial effects in the CNS and periphery, suppressing pro-inflammatory and apoptosis in endothelial, neuronal and immune cell populations (Gorbacheva et al. 2010; Joyce and Grinnell 2002; Seol et al. 2011). APC has well known anti-inflammatory effects on endothelial cells and leukocytes via inhibiting inflammatory mediator release and down-regulating adhesion molecules, thereby reducing immune cell infiltration into tissues and reducing damage.
The spinal cords of WE thrombin treated mice had less damage and had fewer demyelinated axons (Fig. 3). Notably, we observed fewer inflammatory cells within the CNS, consistent with reduced activation and recruitment. Thus, reductions in axonal injury and demyelination may be partly due to reduced inflammation. Given the cytoprotective and antiapoptotic effects of APC, WE thrombin-mediated generation of endogenous APC may have had a mitigating effect on axonal injury within the spinal cord.
We also observed that WE thrombin reduced the accumulation of fibrin(ogen) in the spinal cord. In addition to its role in coagulation, fibrinogen contains binding sites for cellular receptors regulating inflammatory processes. In particular, ICAM-1 has also been identified as a receptor for fibrinogen (Languino et al. 1993). By binding ICAM-1, fibrinogen acts as a bridging molecule, thereby enhancing leukocyte-endothelium interactions. Our data indicated that ICAM-1 expression was reduced on splenic macrophages, concomitant with reduced immune cell infiltration in the brain and fibrinogen in the spinal cord. Early pathological changes occurring in normal-appearing white matter in MS patients point to the activation of microglial cells as an early pathogenic event, occurring before demyelinating lesions (van der Valk and Amor 2009). In vitro, fibrinogen activates microglia via integrin receptor Mac-1 (CD11b/CD18), inducing differentiation and leading to increased phagocytosis (Adams et al. 2007). Leakage of fibrinogen into the spinal cords of mice undergoing EAE induces release of reactive oxygen species in microglia and promotes the formation of perivascular microglial clusters and axon damage (Davalos et al. 2012). Inhibiting this interaction in EAE via pharmacologic or genetic approaches attenuates microglia activation and suppresses disease course and relapse (Adams et al. 2007). Thus it may be that reductions in spinal cord fibrin(ogen) and activated macrophages/microglia in the brain account partly for the beneficial effects of WE thrombin on disease score.
In MS patients, perivascular deposition of fibrin is coupled to BBB disruption (Claudio et al. 1995; Gay et al. 1997; Kwon and Prineas 1994), with colocalization of lesions near areas of BBB disruption (Minagar and Alexander 2003). APC is known to have barrier stabilizing effects on injured endothelium (Feistritzer and Riewald 2005; Finigan et al. 2005). Thus it is conceivable that endogenous protein C activation near BBB disruption may account for the reduced fibrin(ogen) observed in the WE thrombin group. This is consistent with our data showing reductions in inflammatory cells and frequency of activated macrophages in the brain. Taken together, these results suggest that WE thrombin may limit the progression of MS via local generation of APC, leading to reduced vascular leak/BBB disruption and/or inflammatory cell activation.
Our results indicating that WE thrombin suppresses inflammation and improves disease outcome in EAE are consistent with the observation that WE thrombin attenuates the development of inflammatory joint disease in mice (Flick, Chauhan et al. 2011). In this model of collagen-induced arthritis, inflammatory cell infiltration and synovial hyperplasia were reduced in the knee joints of transgenic mice engineered to express WE thrombin, compared to wildtype littermates. This study also found that daily intravenous administration of WE thrombin reduced arthritic symptoms in wildtype mice. Coupled with the findings herein, these studies indicate that WE thrombin may confer therapeutic opportunities for limiting inflammatory disease processes.
In conclusion, we have shown that administration of a protein C activator, WE thrombin reduces disease severity of EAE, dampens the inflammatory response in the brain and spleen, and reduces axonal injury and deposition of fibrinogen in the spinal cord. Taken together, our data suggest that temporal enhancement of endogenous APC activity in the neurovascular unit may represent a novel treatment strategy for MS and other neuroinflammatory disease patients.
Acknowledgments
We thank Gil Benedek and Arthur Vandenbark for insightful comments and technical assistance. We acknowledge Enrico Di Cera, Leslie Pelc, and David Wood for contributions to the development of WE thrombin.
Funding
This work was supported by grants from the National Multiple Sclerosis Society (PP1900), the National Institutes of Health (R44HL117589 and R01HL101972) and the Oregon Clinical and Translational Research Institute (OCTRI), grant number (UL1TR000128) from the National Center for Advancing Translational Sciences. O.J.T. McCarty is an American Heart Association Established Investigator (13EIA12630000).
Footnotes
Competing Interests
A.G., N.G.V., E.I.T., and Oregon Health & Science University may have financial interest in the results of this research. This potential conflict of interest has been reviewed and managed by the Oregon Health & Science University Conflict of Interest in Research Committee. The remaining authors declare no competing financial interests.
References
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. The fibrin-derived gamma377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. The Journal of experimental medicine. 2007;204 (3):571–582. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, Probert L, Strickland S. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101 (17):6698–6703. doi: 10.1073/pnas.0303859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebo BF, Jr, Vandenbark AA, Offner H. Male SJL mice do not relapse after induction of EAE with PLP 139–151. Journal of neuroscience research. 1996;45 (6):680–689. doi: 10.1002/(SICI)1097-4547(19960915)45:6<680::AID-JNR4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Benedek G, Zhu W, Libal N, Casper A, Yu X, Meza-Romero R, Vandenbark AA, Alkayed NJ, Offner H. A novel HLA-DRalpha1-MOG-35-55 construct treats experimental stroke. Metabolic brain disease. 2013 doi: 10.1007/s11011-013-9440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berny-Lang MA, Hurst S, Tucker EI, Pelc LA, Wang RK, Hurn PD, Di Cera E, McCarty OJ, Gruber A. Thrombin mutant W215A/E217A treatment improves neurological outcome and reduces cerebral infarct size in a mouse model of ischemic stroke. Stroke; a journal of cerebral circulation. 2011;42 (6):1736–1741. doi: 10.1161/STROKEAHA.110.603811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows GG, Meza-Romero R, Huan J, Sinha S, Mooney JL, Vandenbark AA, Offner H. Gilt required for RTL550-CYS-MOG to treat experimental autoimmune encephalomyelitis. Metabolic brain disease. 2012;27 (2):143–149. doi: 10.1007/s11011-012-9289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantwell AM, Di Cera E. Rational design of a potent anticoagulant thrombin. The Journal of biological chemistry. 2000;275 (51):39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- Chelmicka-Szorc E, Arnason BG. Partial suppression of experimental allergic encephalomyelitis with heparin. Archives of neurology. 1972;27 (2):153–158. doi: 10.1001/archneur.1972.00490140057009. [DOI] [PubMed] [Google Scholar]
- Ciccone A, Beretta S, Brusaferri F, Galea I, Protti A, Spreafico C. Corticosteroids for the long-term treatment in multiple sclerosis. The Cochrane database of systematic reviews. 2008;(1):CD006264. doi: 10.1002/14651858.CD006264.pub2. [DOI] [PubMed] [Google Scholar]
- Claudio L, Raine CS, Brosnan CF. Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta neuropathologica. 1995;90 (3):228–238. doi: 10.1007/BF00296505. [DOI] [PubMed] [Google Scholar]
- Courville CB. The effects of heparin in acute exacerbations of multiple sclerosis. Observations and deductions. Bulletin of the Los Angeles Neurological Society. 1959a;24:187–196. [PubMed] [Google Scholar]
- Courville CB. Multiple sclerosis as an incidental complication of a disorder of lipid metabolism. III. Treatment with heparin of acute exacerbations of the disease. Bulletin of the Los Angeles Neurological Society. 1959b;24 (2):89–105. [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nature communications. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziennis S, Akiyoshi K, Subramanian S, Offner H, Hurn PD. Role of dihydrotestosterone in post-stroke peripheral immunosuppression after cerebral ischemia. Brain, behavior, and immunity. 2011;25 (4):685–695. doi: 10.1016/j.bbi.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105 (8):3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. The Journal of biological chemistry. 2005;280 (17):17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Flick MJ, Chauhan AK, Frederick M, Talmage KE, Kombrinck KW, Miller W, Mullins ES, Palumbo JS, Zheng X, Esmon NL, Esmon CT, Thornton S, Becker A, Pelc LA, Di Cera E, Wagner DD, Degen JL. The development of inflammatory joint disease is attenuated in mice expressing the anticoagulant prothrombin mutant W215A/E217A. Blood. 2011;117 (23):6326–6337. doi: 10.1182/blood-2010-08-304915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay FW, Drye TJ, Dick GW, Esiri MM. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain : a journal of neurology. 1997;120 (Pt 8):1461–1483. doi: 10.1093/brain/120.8.1461. [DOI] [PubMed] [Google Scholar]
- Gorbacheva L, Pinelis V, Ishiwata S, Strukova S, Reiser G. Activated protein C prevents glutamate- and thrombin-induced activation of nuclear factor-kappaB in cultured hippocampal neurons. Neuroscience. 2010;165 (4):1138–1146. doi: 10.1016/j.neuroscience.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Gruber A, Cantwell AM, Di Cera E, Hanson SR. The thrombin mutant W215A/E217A shows safe and potent anticoagulant and antithrombotic effects in vivo. The Journal of biological chemistry. 2002;277 (31):27581–27584. doi: 10.1074/jbc.C200237200. [DOI] [PubMed] [Google Scholar]
- Gruber A, Marzec UM, Bush L, Di Cera E, Fernandez JA, Berny MA, Tucker EI, McCarty OJ, Griffin JH, Hanson SR. Relative antithrombotic and antihemostatic effects of protein C activator versus low-molecular-weight heparin in primates. Blood. 2007;109 (9):3733–3740. doi: 10.1182/blood-2006-07-035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451 (7182):1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Joyce DE, Grinnell BW. Recombinant human activated protein C attenuates the inflammatory response in endothelium and monocytes by modulating nuclear factorkappaB. Critical care medicine. 2002;30 (5 Suppl):S288–293. doi: 10.1097/00003246-200205001-00019. [DOI] [PubMed] [Google Scholar]
- Kermode AG, Thompson AJ, Tofts P, MacManus DG, Kendall BE, Kingsley DP, Moseley IF, Rudge P, McDonald WI. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenetic and clinical implications. Brain : a journal of neurology. 1990;113 (Pt 5):1477–1489. doi: 10.1093/brain/113.5.1477. [DOI] [PubMed] [Google Scholar]
- Kithcart AP, Cox GM, Sielecki T, Short A, Pruitt J, Papenfuss T, Shawler T, Gienapp I, Satoskar AR, Whitacre CC. A small-molecule inhibitor of macrophage migration inhibitory factor for the treatment of inflammatory disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24 (11):4459–4466. doi: 10.1096/fj.10-162347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzelnigg A, Lassmann H. Pathology of multiple sclerosis and related inflammatory demyelinating diseases. Handbook of clinical neurology. 2014;122:15–58. doi: 10.1016/B978-0-444-52001-2.00002-9. [DOI] [PubMed] [Google Scholar]
- Kwon EE, Prineas JW. Blood-brain barrier abnormalities in longstanding multiple sclerosis lesions. An immunohistochemical study. Journal of neuropathology and experimental neurology. 1994;53 (6):625–636. doi: 10.1097/00005072-199411000-00010. [DOI] [PubMed] [Google Scholar]
- La Mantia L, Vacchi L, Di Pietrantonj C, Ebers G, Rovaris M, Fredrikson S, Filippini G. Interferon beta for secondary progressive multiple sclerosis. The Cochrane database of systematic reviews. 2012;1:CD005181. doi: 10.1002/14651858.CD005181.pub3. [DOI] [PubMed] [Google Scholar]
- Languino LR, Plescia J, Duperray A, Brian AA, Plow EF, Geltosky JE, Altieri DC. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell. 1993;73 (7):1423–1434. doi: 10.1016/0092-8674(93)90367-y. [DOI] [PubMed] [Google Scholar]
- Lider O, Baharav E, Mekori YA, Miller T, Naparstek Y, Vlodavsky I, Cohen IR. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. The Journal of clinical investigation. 1989;83 (3):752–756. doi: 10.1172/JCI113953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain : a journal of neurology. 2007;130 (Pt 11):2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschmeyer J, Shearer R, Lonser E, Spindle DK. Heparin potassium in the treatment of chronic multiple sclerosis. Bulletin of the Los Angeles Neurological Society. 1961;26:165–171. [PubMed] [Google Scholar]
- Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Multiple sclerosis. 2003;9 (6):540–549. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109 (8):3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308 (5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Olek MJ. Epidemiology and clinical features of multiple sclerosis in adults. Wolters Kluwer Health; 2014. [Accessed February 24 2014]. http://www.uptodate.com/contents/epidemiology-and-clinical-features-of-multiple-sclerosis-in-adults#H1. [Google Scholar]
- Paterson PY. Experimental allergic encephalomyelitis: role of fibrin deposition in immunopathogenesis of inflammation in rats. Federation proceedings. 1976;35 (13):2428–2434. [PubMed] [Google Scholar]
- Seol JW, Lee YJ, Jackson CJ, Sambrook PN, Park SY. Activated protein C inhibits bisphosphonate-induced endothelial cell death via the endothelial protein C receptor and nuclear factor-kappaB pathways. International journal of molecular medicine. 2011;27 (6):835–840. doi: 10.3892/ijmm.2011.649. [DOI] [PubMed] [Google Scholar]
- Sinha S, Subramanian S, Emerson-Webber A, Lindner M, Burrows GG, Grafe M, Linington C, Vandenbark AA, Bernard CC, Offner H. Recombinant TCR ligand reverses clinical signs and CNS damage of EAE induced by recombinant human MOG. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2010;5 (2):231–239. doi: 10.1007/s11481-009-9175-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Valk P, Amor S. Preactive lesions in multiple sclerosis. Current opinion in neurology. 2009;22 (3):207–213. doi: 10.1097/WCO.0b013e32832b4c76. [DOI] [PubMed] [Google Scholar]
- Vandenbark AA, Rich C, Mooney J, Zamora A, Wang C, Huan J, Fugger L, Offner H, Jones R, Burrows GG. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35-55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. Journal of immunology. 2003;171 (1):127–133. doi: 10.4049/jimmunol.171.1.127. [DOI] [PubMed] [Google Scholar]
- Vicente CP, Weiler H, Di Cera E, Tollefsen DM. Thrombomodulin is required for the antithrombotic activity of thrombin mutant W215A/E217A in a mouse model of arterial thrombosis. Thrombosis research. 2012;130 (4):646–648. doi: 10.1016/j.thromres.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos CM, Geurts JJ, Montagne L, van Haastert ES, Bo L, van der Valk P, Barkhof F, de Vries HE. Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiology of disease. 2005;20 (3):953–960. doi: 10.1016/j.nbd.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. Journal of immunology. 2009;182 (5):3294–3303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Gold BG, Kaler LJ, Yu X, Afentoulis ME, Burrows GG, Vandenbark AA, Bourdette DN, Offner H. Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE. Journal of neurochemistry. 2006;98 (6):1817–1827. doi: 10.1111/j.1471-4159.2006.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sinha RK, Mosnier LO, Griffin JH, Zlokovic BV. Neurotoxicity of the anticoagulant-selective E149A-activated protein C variant after focal ischemic stroke in mice. Blood cells, molecules & diseases. 2013a;51 (2):104–108. doi: 10.1016/j.bcmd.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao Z, Chow N, Rajput PS, Griffin JH, Lyden PD, Zlokovic BV. Activated protein C analog protects from ischemic stroke and extends the therapeutic window of tissue-type plasminogen activator in aged female mice and hypertensive rats. Stroke; a journal of cerebral circulation. 2013b;44 (12):3529–3536. doi: 10.1161/STROKEAHA.113.003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V, Bourdette DN, Bowen JD, Lynch SG, Mattson D, Preiningerova J, Bever CT, Jr, Simon J, Goldstein A, Burrows GG, Offner H, Ferro AJ, Vandenbark AA. Recombinant T-Cell Receptor Ligand (RTL) for Treatment of Multiple Sclerosis: A Double-Blind, Placebo-Controlled, Phase 1, Dose-Escalation Study. Autoimmune diseases. 2012;2012:954739. doi: 10.1155/2012/954739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Tian SJ, Wu L, Huang DH, Wu WP. Fibrinogen depleting agent batroxobin has a beneficial effect on experimental autoimmune encephalomyelitis. Cellular and molecular neurobiology. 2011;31 (3):437–448. doi: 10.1007/s10571-010-9637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]