

Abstract
Purpose
To determine the effect of PEG modification on pharmacologic and gene delivery properties of polymeric CXCR4 antagonist based on Plerixafor.
Methods
Polymeric Plerixafor (PAMD) was synthesized from Plerixafor (AMD3100) and grafted with different amounts of PEG (2 kDa). CXCR4 antagonism of the synthesized polymers was determined using receptor redistribution assay. Inhibition of cancer cell invasion by the polyplexes of the synthesized polymers was assessed using Boyden-chamber method. Transfection activity of DNA polyplexes formed with the synthesized polymers was evaluated in U2OS osteosarcoma and B16F10 melanoma cells.
Results
Our results demonstrate that modification of PAMD with PEG decreased toxicity of the polymers, while preserving their CXCR4 antagonism. Polyplexes prepared with PEG-PAMD inhibited invasion of cancer cells to an extent similar to the commercial CXCR4 antagonist Plerixafor. Negative effect of PEG on transfection activity of PEG-PAMD polyplexes could be overcome by using polyplexes formulated with a mixture of PAMD and PEG-PAMD.
Conclusion
Modification of PAMD with PEG is a viable strategy to preserve the desirable CXCR4 antagonism and ability to inhibit cancer cell invasion of PAMD, while improving safety and colloidal stability of the PAMD polyplexes.
Keywords: CXCR4 antagonists, Gene delivery, Polyplexes, Transfection
INTRODUCTION
Polyelectrolyte complexes of polycations with nucleic acids, commonly known as polyplexes, have received significant attention as promising gene delivery vectors. Polyplexes offer multiple potential advantages when compared with viral vectors, including lower toxicity, minimal immunogenicity, and easier manufacturing and functional modifications (1, 2). When prepared from simple polycations, polyplexes are positively charged nanoparticles that rely on a charge-mediated attachment to heparan sulfate proteoglycans on the cell surface to facilitate uptake into cells (3). However, high density of positive charges damages negatively charged cell membranes and contributes to cytotoxicity of polyplexes (4). Positively charged polyplexes are colloidally stabilized by electrostatic repulsion and frequently aggregate under physiological salt conditions. The positive surface charge and susceptibility to aggregation contribute to rapid elimination of polyplexes from blood circulation by the mononuclear phagocyte system (5-7). To improve in vivo applicability, polyplexes are often modified with nonionic polymers like poly(ethylene glycol) (PEG) to shield the surface charges and improve colloidal stability by steric stabilization (8-11). PEGylation typically minimizes the role of heparan sulfates in cellular uptake and interferes with endosomal escape of polyplexes, which decreases transfection activity. The PEG content has to be carefully balanced or a de-shielding strategy has to be employed in order to maintain sufficient transfection activity of polyplexes (12-14).
Chemokines and their receptors play a decisive part in the process of cancer metastasis (15). The role of chemokine networks is consistent with the seed-and-soil hypothesis of metastatic dissemination (16). Although malignant cells from different types of cancer have different expression profiles of chemokine receptors, CXC receptor 4 (CXCR4) is the most widely expressed chemokine receptor in human cancers, making it and its ligand SDF-1 the most-promising targets within the chemokine network for novel therapies (17). CXCR4 facilitates the metastatic spread of the disease to sites where SDF-1 is highly expressed (e.g., lung, liver, bone marrow, and brain). Furthermore, high expression of SDF-1 in primary tumors enhances growth and inflammation of the tumor by local autocrine and paracrine mechanisms (18-20). Binding of SDF-1 to CXCR4 activates several intracellular signaling transduction pathways that regulate proliferation, adhesion, and invasion of cancer cells (21, 22) (Scheme 1). There is growing clinical evidence that certain anticancer therapies increase CXCR4 expression and thus inadvertently enhance the metastatic potential of tumors (21). In particular, treatments that promote hypoxic environment are associated with an increase in CXCR4 expression, which is then correlated with a poorer overall prognosis (23, 24).
Scheme 1.
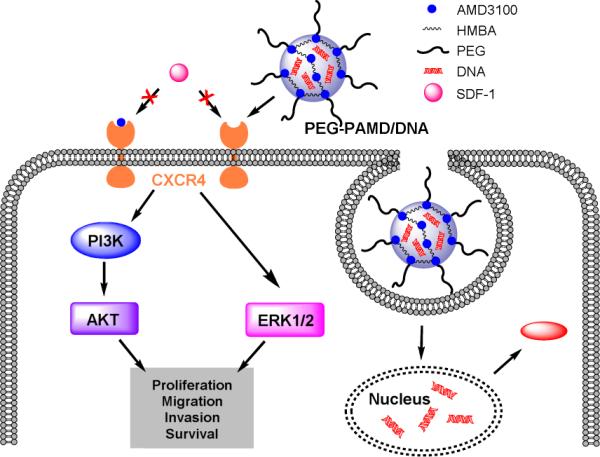
Mechanism of dual-function PEG-PAMD as gene delivery vector and CXCR4 antagonist inhibiting cancer cell invasion.
Inhibition of CXCR4 has the potential to prevent metastasis and limit tumor growth and vascularization, especially in combination with chemotherapy and radiotherapy. Chemokine networks are thus an important emerging target for development of novel drug delivery strategies (25). By devising systems capable of simultaneous CXCR4 inhibition and delivery of antitumor agents, it should be possible to improve the overall anticancer activity (26). As part of our long-term efforts to develop dually functioning polycations for combination drug/gene delivery (27, 28), we have recently reported synthesis of polycations based on a bicyclam CXCR4 antagonist Plerixafor (PAMD) (29, 30). The PAMD polymers showed dual functionality as efficient gene delivery vectors and CXCR4 antagonists that inhibited invasion of cancer cells. The goal of the present study was to improve physical properties and safety of PAMD by PEGylation. We set to evaluate how the presence of PEG affects CXCR4 antagonism, inhibition of cancer cell invasion, colloidal stability, safety, and transfection activity of the polymers and their polyplexes. The objective was to develop polyplex formulations that retain CXCR4 antagonism of PAMD, while exhibiting decreased cytotoxicity, improved transfection activity, and enhanced colloidal stability under physiologic conditions.
MATERIALS AND METHODS
Materials
N,N′-hexamethylenebisacrylamide (HMBA) was obtained from Polysciences, Inc. (Warrington, PA). AMD3100 was synthesized as published previously (31). mPEG-Acrylamide (2 kDa) was from Creative PEGworks (Winston-Salem, NC). Branched polyethylenimine (PEI, 25 kDa) was purchased from Sigma-Aldrich (St. Louis, MO). Plasmid DNA, gWiz high-expression luciferase (gWiz-Luc), containing luciferase reporter gene was from Aldevron (Fargo, ND). Dulbecco's Modified Eagle Medium (DMEM), Dulbecco's Phosphate Buffered Saline (PBS), and Fetal Bovine Serum (FBS) were from Thermo Scientific (Waltham, MA). Cell culture inserts for 24-well plates with 8.0 m pores (Translucent PET Membrane, cat# 353097) and BD Matrigel™ Basement Membrane Matrix (cat# 354234) were purchased from BD Biosciences (Billerica, MA). Human SDF-1 was from Shenandoah Biotechnology, Inc. (Warwick, PA). All other reagents were from Fisher Scientific and used as received unless otherwise noted.
Polymer Synthesis
Polymeric Plerixafor (PAMD) was synthesized by Michael polyaddition of equal molar ratio of AMD3100 and HMBA. Typically, 0.45 mmol of each reactant was dissolved in a glass vial containing 4 mL methanol/water (7/3 v/v) mixture. Polymerization was carried out under nitrogen atmosphere and in dark at 37 °C for 4 days. Then, additional 0.045 mmol of AMD3100 was added and the reaction mixture was stirred for further 12 h to consume all residual acrylamide groups. PAMD was isolated in overall 85% yield by double precipitation in diethyl ether, centrifugation, and drying in vacuum.
The synthesized PAMD was PEGylated by Michael addition between the secondary amines in the cyclam groups of PAMD and acrylamide group of mPEG-acrylamide. PAMD (72.3, 66.4 or 69.7 mg) and mPEG-acrylamide (8.0 mg, 35.8 mg or 69.7 mg) were dissolved in MeOH/water mixture (7/3 v/v) at a total concentration of 120 mg/mL and the solution was stirred for 2 days at 37 °C, followed by 1 day at 50 °C. The reaction was cooled to room temperature and the pH was adjusted to 4 with 1 M HCl. The resulting copolymers (PEG-PAMD) were isolated by precipitation in diethyl ether, centrifugation, and drying in vacuum, and finally dialyzed against deionized water (pH 4, membrane molecular weight cut-off 3.5 kDa). Typical yield was 70-80%.
Polymer characterization
The content of PEG in PEG-PAMD was determined using 1H-NMR on Varian INOVA (500 MHz). The molecular weight of the copolymers was analyzed by gel permeation chromatography (GPC) operated in 0.3 M sodium acetate buffer (pH 5) using Agilent 1260 Infinity LC system equipped with a miniDAWN TREOS multi-angle light scattering (MALS) detector and a Optilab T-rEX refractive index detector from Wyatt Technology (Santa Barbara, CA). Single-pore AquaGel™ columns (cat# PAA-202 and PAA-203, PolyAnalytik, London, ON, Canada) were used at a flow rate of 0.3 mL/min. GPC samples were prepared at 5 mg/mL. GPC data were analyzed using Astra 6.1 software from Wyatt Technology.
Ethidium bromide exclusion assay
The ability of the synthesized copolymers to condense DNA was determined by ethidium bromide (EtBr) exclusion assay by measuring the changes in EtBr/DNA fluorescence. 1 mL of DNA solution (20 μg/mL) was prepared in 10 mM HEPES buffer (pH 7.4) and mixed with EtBr (1 μg/mL). Fluorescence intensity was measured and set to 100% using an excitation wavelength of 540 nm and an emission wavelength of 590 nm. The fluorescence of ethidium bromide only in HEPES buffer was defined as background and set as 0%. Fluorescence readings were taken following a stepwise addition of polymer solution and condensation curves were constructed.
Preparation and characterization of DNA polyplexes
DNA solution at a concentration of 20 μg/mL was prepared in 10 mM HEPES buffer (pH 7.4). Polyplexes were formed by adding predetermined volume of polymer to the DNA solution to achieve a desired w/w ratio. The mixture was vigorously vortexed for 10 s and left standing at room temperature for 30 min before further analysis. To prepare mixed polyplexes, solutions of PAMD and PEG-PAMD were first mixed at desired ratios and then added to the DNA solution. Hydrodynamic diameter and zeta potential of the polyplexes were determined by dynamic light scattering (DLS) using a ZEN3600 Zetasizer Nano-ZS (Malvern Instruments Ltd., Worcestershire, UK). To evaluate colloidal stability of the polyplexes, 10× phosphate-buffered saline (PBS) was added to the polyplexes to obtain a final 1× PBS solution (pH 7.4) with the following composition: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4. The hydrodynamic diameter was then measured after 15 min, 1 h, and 12 h incubation at 25 °C. Results were expressed as mean ± standard deviation (SD) of three measurements.
Cell Culture
Human hepatocellular carcinoma HepG2 cells were purchased from ATCC (Manassas, VA). Mouse melanoma B16F10 cells were a kind gift from Dr. Rakesh Singh (UNMC). HepG2 cells were cultured at 37 °C and 5% CO2 in MEM supplemented with 10% FBS. B16F10 cells were maintained in DMEM supplemented with 10% FBS. Human epithelial osteosarcoma U2OS cells stably expressing functional EGFP-CXCR4 fusion protein were purchased from Fisher Scientific and cultured in DMEM supplemented with 2 mM L-glutamine, 1% Pen-Strep, 0.5 mg/mL G418 and 10% FBS.
Cytotoxicity
Cytotoxicity of the synthesized polycations was tested by MTS assay in HepG2 and U2OS cells. The cells were seeded in 96-well plates at a density of 10,000 cells/well (HepG2) and 8000 cells/well (U2OS) and incubated overnight. Culture medium was then replaced by 150 μL of serial dilutions of a polymer in serum-supplemented medium and the cells were incubated for 24 h. The medium was then aspirated and replaced by a mixture of 100 L serum-free media and 20 L of MTS reagent (CellTiter 96®AQueous Non-Radioactive Cell Proliferation Assay, Promega). After 1 h incubation, the absorbance [A] was measured using SpectraMax®M5e Multi-Mode Microplate Reader (Molecular Devices, CA) at a wavelength of 490 nm. The relative cell viability (%) was calculated as [A]sample/[A]untreated × 100%. The IC50 were calculated in GraphPad Prism using a built-in dose-response analysis as the polymer concentration that achieves 50% growth inhibition relative to untreated cells.
Transfection activity
All transfection experiments were conducted in 48-well plates with cells at logarithmic growth phase. B16F10 (40,000 cells/well) and U2OS (20,000 cells/well) cells were seeded 24 h prior to transfection. On the day of transfection, culture medium in each well was removed and replaced with 150 μL of antibiotic-free medium (+/− 10% FBS) before adding 20 μL of polyplexes prepared (DNA dose 0.4 μg/well). After 4 h incubation, polyplexes were completely removed and the cells were cultured in complete culture medium for 24 h prior to measuring luciferase expression. The medium was discarded and the cells were lysed in 100 μL of 0.5× cell culture lysis reagent buffer (Promega, Madison, WI) for 30 min. To measure the luciferase content, 100 μL of 0.5 mM luciferin solution was automatically injected into each well of 20 μL of cell lysate and the luminescence was integrated over 10 s using GloMax 96 Microplate Luminometer (Promega). Total cellular protein in the cell lysate was determined by the bicinchoninic acid protein assay using calibration curve constructed with standard bovine serum albumin solutions (Pierce, Rockford, IL). Transfection activity was expressed as relative light units (RLU)/mg cellular protein ± SD of triplicate samples.
CXCR4 antagonism
CXCR4 antagonism of the polycations and their DNA polyplexes was determined by CXCR4 redistribution assay using a high-content fluorescence microscopy analysis. U2OS cells stably expressing EGFP-CXCR4 receptor were seeded at a density of 8,000 cells/well in 96-well black plate with optical bottom 24 h before the experiment. On the day of the assay, cells were washed twice with 100 μL assay buffer (DMEM supplemented with 2 mM L-glutamine, 1% FBS, 1% Pen-Strep, and 10 mM HEPES) and incubated with different concentrations of the polycations, polyplexes, or AMD3100 in the assay buffer containing also 0.25% DMSO at 37 °C for 30 min. Then, SDF-1α solution was added to each well to make final concentration of 10 nM and the cells were incubated at 37 °C for 1 h. Cells were fixed with 4% formaldehyde at room temperature for 20 min, washed 4 times with PBS and stained in 1 μM Hoechst 33258 solution for 30 min before imaging by EVOS fl microscope (20×). Cellomics ArrayScan VT1 Reader and SpotDetectorV3 BioApplication software were used to quantify the internalization of the CXCR4 receptors. CXCR4 antagonistic activity of the polycations and polyplexes is expressed as mean % inhibition ± SD (n = 3). CXCR4 inhibitory activity of AMD3100 was considered as 100% and CXCR4 inhibitory activity in cells treated with SDF-1α only was set as 0%.
Cell invasion
Ice-cold Matrigel was diluted 1:3 (v:v) with serum-free medium (DMEM supplemented with 2 mM L-Glutamine) and 40 μL was added to each cell culture insert. The 24-well plates with Matrigel-coated inserts were then placed in 37 °C incubator for 2 h. 50,000 of U2OS cells were harvested and resuspended in serum-free medium containing AMD3100 (0.3 μM) or polyplexes (w/w 5, 2.5 μg/mL PAMD or its equivalent in PEG-PAMD). The treated cells were placed in the Matrigel-coated inserts and 20 nM SDF-1α in serum-free medium was added to the lower chamber of the wells. After 19 h incubation, the non-invaded cells on the top surface of the insert membrane were removed by cotton swabs and the invaded cells on the bottom surface of the insert membrane were fixed in 100% methanol and stained with 0.2% Crystal Violet solution for 10 min at room temperature. The number of invaded cells was counted under microscope set to 20x magnification. The results are expressed as average number of cells/imaging area ± SD (n = 5-10).
RESULTS AND DISCUSSION
We have recently reported synthesis of bioreducible poly(amido amine)s using commercial bicyclam CXCR4 antagonist Plerixafor as the main building block of the polymers (29). The synthesized polymers exhibited effective CXCR4 antagonism and demonstrated the ability to prevent invasion of cancer cells in vitro and metastasis in vivo (30). The synthesized polymers were positively charged because of the secondary amines in the cyclam ring of Plerixafor and were thus able to form polyplexes with plasmid DNA and facilitate efficient transfection. These initial studies suggested potential of the polymers as dual-function delivery systems suitable for combining antimetastatic effect of CXCR4 inhibition with antitumor effect of an appropriate therapeutic nucleic acid. As part of further development of this class of delivery vectors for in vivo use, this study investigates whether PEGylation can be used to enhance safety and colloidal stability of the polyplexes, while preserving pharmacologic activity and gene delivery capability (Scheme 1).
Synthesis and characterization of PEG-PAMD
We have synthesized polymeric Plerixafor (PAMD) in the form of a poly(amido amine) by Michael polyadditon of secondary amines present in Plerixafor to the CH2=CH– group of bisacrylamide HMBA (Scheme 2). Plerixafor functions as a hexafunctional monomer in the Michael polyaddition and its use leads to either insoluble crosslinked PAMD or soluble branched PAMD if the reaction is performed at low temperatures and low monomer concentrations (32, 33). Optimizing the reaction conditions allowed us to synthesize soluble PAMD with weight-average molecular weight of 10.6 kDa and unimodal distribution of molecular weights (Figure 1).
Scheme 2.
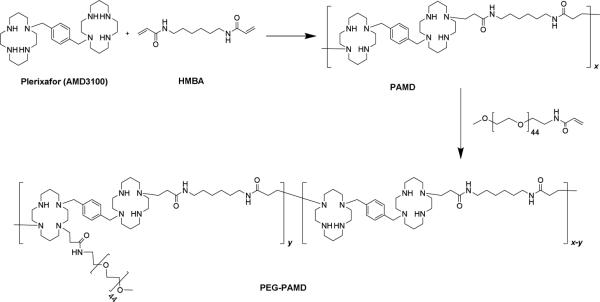
Synthesis of PAMD and PEG-PAMD.
Figure 1.
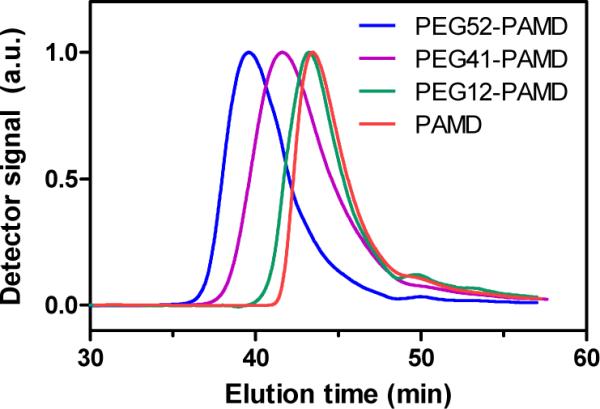
Gel permeation chromatograms of PAMD and PEG-PAMD.
PEG-PAMD copolymers were synthesized by the reaction of mPEG-acrylamide with the secondary amines of PAMD (Scheme 2). In contrast to common amide coupling, using Michael addition for the PEGylation allowed us to conserve the overall number of protonizable amines in PAMD. Three copolymers with increasing content of PEG were synthesized and named according to their PEG content (Table 1). The copolymers were isolated as hydrochloride salts after extensive dialysis. The content of PEG in the copolymers was determined from 1H-NMR integral intensity of the PEG methylene protons at 3.7 ppm and aromatic protons of Plerixafor at 7.4-7.8 ppm (Figure 2). Removal of the low-molecular-weight polymer fractions rich in PAMD during dialysis helps to explain why the PEG content in the final copolymers exceeded the feed content (Table 1). GPC analysis confirmed successful synthesis of PEG-PAMD as indicated by the shift to shorter elution times with increasing PEG content (Figure 1). The polydispersity index (PDI) of the polymers ranged from 1.1 to 1.4, indicating a good control of the polymerization.
Figure 2.
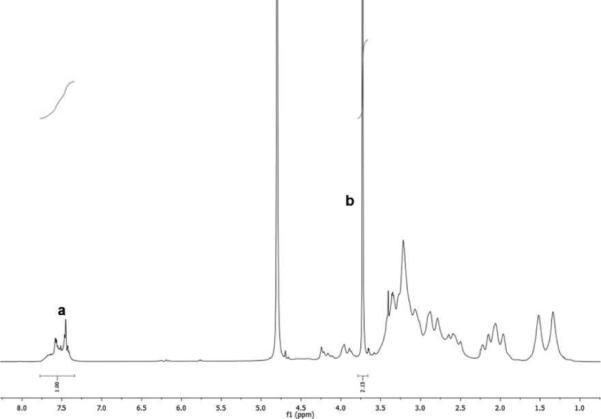
Typical 1H-NMR spectrum of PEG-PAMD (PEG41-PAMD in D2O) used in the determination of the PEG content (a – aromatic phenylene protons of AMD3100, b – methylene protons of PEG).
Preparation and characterization of PEG-PAMD polyplexes
The influence of PEGylation on the ability of PAMD to condense DNA into polyplexes was investigated by ethidium bromide exclusion assay (Figure 3). All the polymers displayed similar condensation curves with a typical sigmoidal shape. All PEG-PAMD were able to condense plasmid DNA to the same extent as PAMD as indicated by the same residual fluorescence at the highest polymer/DNA ratios. The DNA condensation curves shifted to higher w/w ratios with increasing PEG content because of the decreasing polycation content in the copolymers. However, when we considered only the polycation part of PEG-PAMD and re-plotted the data as relative fluorescence vs. N/P ratio, the shift was eliminated and all condensation curves were super-imposed (not shown). This suggests that PEG represented no significant steric hindrance for binding of the PAMD part of the copolymers to DNA, a finding similar to a recent report by Fitzsimmons and Uludağ (34).
Figure 3.
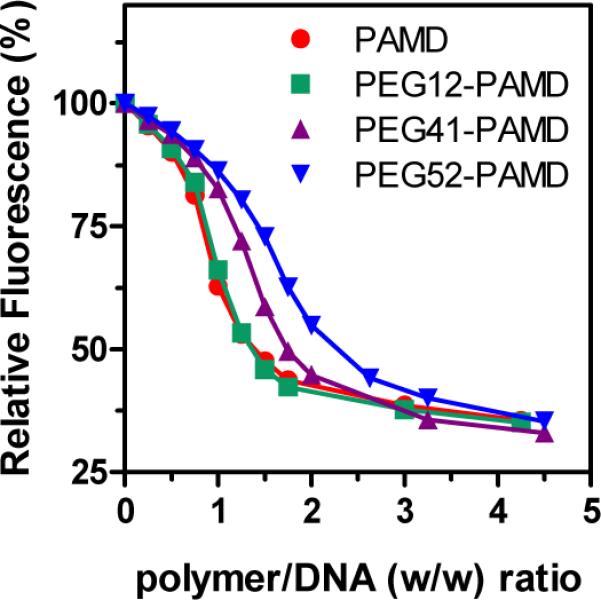
DNA condensation ability of PAMD and PEG-PAMD determined by ethidium bromide exclusion assay.
One of the key motivations for use of PEGylated polycations is to shield the positive surface charge of polyplexes. Our results confirmed that the use of PEG-PAMD significantly decreases surface charge of the polyplexes as documented by the decrease in the measured zeta potential (Figure 4). All PEG-PAMD polyplexes exhibited nearly neutral zeta potential in 10 mM HEPES buffer, pH 7.4. In contrast, the parent PAMD polyplexes showed a highly positive surface charge with zeta potential of +20 mV.
Figure 4.
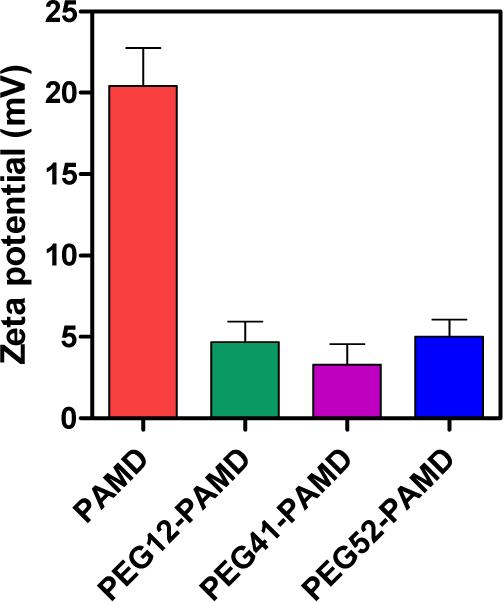
Zeta potential of DNA polyplexes prepared at polymer/DNA (w/w) ratio of 5 and measured in 10 mM HEPES (pH 7.4) (mean ± SD, n = 3).
The positive surface charge provides electrostatic stabilization to the polyplex nanoparticles at low concentration buffers but is ineffective at preventing aggregation at physiologic ionic strengths. Steric stabilization by PEG can typically overcome the problem of low colloidal stability of polyplexes (35). We have prepared PEG-PAMD and PAMD polyplexes in 10 mM HEPES buffer (pH 7.4) at polymer/DNA (w/w) ratio of 5 and measured their hydrodynamic diameter. Then, we added phosphate-buffered saline (PBS) (pH 7.4) to simulate physiologic ionic conditions and observed changes in polyplex size during the following period of 12 h (Figure 5). The results showed that PAMD polyplexes begin to aggregate immediately after PBS addition as documented by the increase of their size from ~60 nm to ~430 nm in the span of only 15 min. The size of PAMD polyplexes increased to nearly 1 μm within 1 h of PBS addition. In contrast, polyplexes prepared with PEG-PAMD exhibited markedly improved colloidal stability.
Figure 5.

Effect of PEG on colloidal stability of PAMD polyplexes (w/w 5) in phosphate-buffered saline (PBS). Results are shown as mean ± SD (n=3).
Our results show that although PEG-PAMD with the lowest PEG content improved short-term (1 h) colloidal stability, it was ineffective in long-term evaluation as documented by the increase in size from 58 nm to 690 nm within 12 h of PBS addition. Both PEG-PAMD with the higher PEG content were capable of effective stabilization of the polyplexes with nearly constant size displayed for the duration of the experiment.
Cytotoxicity of PEG-PAMD
Toxicity of polycations is strongly dependent on their molecular weight and charge density and for the most part follows a generally accepted two-phase model (36, 37). In the first acute phase, polycations disrupt cellular membranes and induce membrane leakage due to pore formation (36, 38, 39). The second phase originates from intracellular effects of polycations on mitochondrial membrane and is manifested by induction of apoptosis. The highly hydrophilic PEG forms a steric barrier in the form of a shell around a polycation. The shell creates a barrier that decreases the ability of the PEGylated polycations to perturb cellular membranes (34). As a result, PEGylation of polycations typically results in decreased cytotoxicity (40-45). We have evaluated cytotoxicity of PEG-PAMD by MTS assay in two cell lines, U2OS and HepG2 (Figure 6). In order to establish a safe, nontoxic working concentration range of PAMD, we have first tested U2OS cells which were then used throughout this study in evaluating CXCR4 antagonism, cell invasion inhibition, and transfection of PAMD. As expected, increasing the PEG content resulted in increased IC50 as a consequence of the relative decrease of the polycation content in the polymers. Importantly, similar trend was observed also when only the polycation part of the copolymers was considered in calculating IC50. In such case, the IC50 values for PAMD and PEG12-PAMD were indistinguishable at ~17 μg/mL but they increased to 25 μg/mL and 41 μg/mL in case of PEG41-PAMD and PEG52-PAMD, respectively. The cytotoxicity was then tested also in HepG cells, which are commonly used in drug development for prediction of liver toxicity (46). Although the toxicity profile of PEG-PAMD was similar in the two used cell lines, the HepG2 cells were overall less susceptible to the toxic effects of the polycations. The measured IC50 value for PAMD was 72 μg/mL. The IC50 values of PEG-PAMD were calculated considering only the PAMD polycation content. In such case, the cytotoxicity of PEG12-PAMD was similar to that of PAMD (77 μg/mL). The two copolymers with higher PEG content exhibited significantly decreased cytotoxicity with their IC50 values above the maximum tested polycation concentration of 100 μg/mL. For comparison, PEI was used as benchmark in this experiment and its IC50 was found to be 22 μg/mL. Unlike some existing reports, our results suggest that PEG decreases PAMD toxicity not only by simply decreasing the cationic content in the copolymers but also by directly affecting the toxicity of the PAMD part of the copolymers, most likely by reducing their interactions with cellular membranes and vital intracellular proteins (34, 43).
Figure 6.
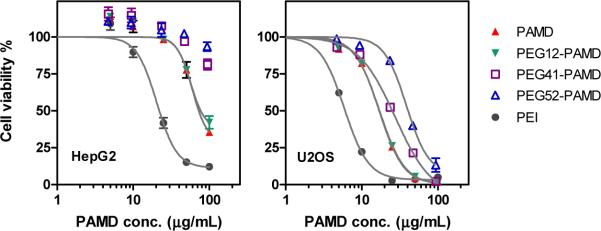
Effect of PEG on cytotoxicity of PAMD in HepG2 and U2OS cells. Cell viability was measured by MTS assay after 24 h incubation with increasing concentrations of polymers. Polymer concentration for PEG-PAMD copolymers is expressed as PAMD concentration only (i.e., excluding PEG). Results are expressed as mean ± SD (n=3).
CXCR4 antagonism and inhibition of cancer cell invasion by PEG-PAMD
Binding of polymers and other macromolecules such as proteins to receptors on the cell surface can be negatively affected by the steric barrier created by PEGylation. Binding of PAMD to CXCR4 receptor is required for the pharmacologic activity of the polymers. We evaluated the CXCR4 inhibition using CXCR4 receptor redistribution assay. The assay relies on high content screening (HCS) based on the inhibition of SDF1-triggered endocytosis of EGFP-tagged CXCR4 receptor. HCS is a phenotypic assay that in this case uses automatic image analysis to quantify the extent of EGFP-CXCR4 internalization into the cells. The difference in the fluorescence pattern of EGFP-CXCR4 between untreated cells and cells treated with CXCR4 inhibitor is illustrated in Figure 7a. The control small-molecule CXCR4 antagonist AMD3100 inhibits EGFP-CXCR4 internalization, as documented by the diffuse pattern of fluorescence. In contrast, untreated cells display punctate fluorescence indicative of EGFP-CXCR4 internalization into endosomes. Activity of PEG-PAMD was analyzed and expressed as % CXCR4 antagonism relative to the control AMD3100 (Figure 7b). In order to permit direct evaluation of the effect of PEGylation on CXCR4 antagonism of PAMD, we have tested activity of the synthesized polymers at equal concentrations of the polycationic (PAMD) content. At the lowest concentration tested (0.05 μg/mL), PEG-PAMD exhibited CXCR4 antagonism that ranged from 57 to 77% of AMD3100 activity. The differences between PEG-PAMD and PAMD were not statistically significant in a one-way ANOVA analysis. At the two higher tested concentrations, all PEG-PAMD and PAMD reached CXCR4 inhibitory activity that was equal to that of the control AMD3100, indicating that the polymers could fully inhibit CXCR4 at concentrations higher than 0.5 g/mL. We then tested if PEG-PAMD polyplexes prepared at two different w/w ratios retained the CXCR4 inhibitory functionality of the free polymers under practically relevant experimental conditions employed in transfection assays (Figure 7c). The polyplexes were prepared at w/w 1.5 and 5, which corresponded to the total PAMD concentration of 0.75 and 2.5 μg/mL. As suggested by the EtBr condensation assay, only a minimum amount of free polycation was present in polyplexes at w/w 1.5 (Figure 3) and thus this formulation is mostly reflective of the ability of the polyplexes to inhibit CXCR4. In contrast, polyplexes prepared at w/w 5 contained significant amount of free polycation, which was most likely the dominant contributor to the observed CXCR4 antagonism. Nevertheless, both polyplex formulations achieved nearly 100% CXCR4 inhibition suggesting that PEGylation does not negatively affect pharmacologic activity of PAMD even in polyplex formulations.
Figure 7.

CXCR4 antagonism of PAMD polycations and polyplexes. (a) Effect of AMD3100 on redistribution of EGFP-CXCR4 receptor in U2OS cells. CXCR4 antagonism of PAMD and PEG-PAMD (b) and their polyplexes (c). The results are shown as mean % CXCR4 inhibition relative to AMD3100 ± SD (n=3).
The CXCR4/SDF-1 axis is involved in migration of multiple types of cancer cells, and CXCR4 antagonists like AMD3100 are known to inhibit invasion of those cancer cells towards SDF-1 concentration gradient. We have previously shown that bioreducible PAMD and PAMD/DNA polyplexes effectively inhibit cancer cell invasion mediated by SDF-1 (29). Using a Boyden chamber method, here we investigated how PEGylation affects the ability of PAMD polyplexes to inhibit invasion of cancer cells (Figure 8). Control AMD3100 was able to prevent 81% cancer cells from invading and migrating through the layer of Matrigel. Both PAMD and PEG-PAMD polyplexes prepared at equivalent PAMD/DNA w/w ratio 5 achieved similar activity as they inhibited invasion of 77-80% cancer cells. Thus, PEGylation did not negatively impact the ability of PAMD/DNA polyplexes to inhibit cancer cell invasion, indicating that PEG-PAMD could be well-suited for applications in treatments that aim at preventing or delaying metastasis.
Figure 8.

Inhibition of cancer cell invasion by PAMD polyplexes. Polyplexes were prepared at w/w ratio of 5 (total PAMD concentration 2.5 μg/mL). Cells were allowed to invade through a layer of Matrigel toward SDF-1 concentration gradient for 19 h before fixation and imaging. Average numbers of invaded cells were counted in randomly selected 5-10 imaging areas at 20× magnification (Scale bar = 200 μm).
Transfection of PEG-PAMD polyplexes
The effect of PEGylation on transfection of PAMD polyplexes was studied using luciferase reporter plasmid in B16F10 and U2OS cell lines (Figure 9). Transfection activities of polyplexes formed with PAMD and PEG12-PAMD were fully comparable, but the transfection declined greatly for polyplexes prepared with PEG41-PAMD and PEG52-PAMD. For example, transfection of PEG52-PAMD polyplexes decreased more than 6,000-times in B16F10 and ~500-times in U2OS cells when compared with non-PEGylated PAMD polyplexes. These findings are fully consistent with previous reports on the effect of PEGylation on transfection activity of polyplexes and are due to decreased cellular uptake and compromised endosomal escape of polyplexes due to restricted interaction with cellular membranes (35, 41).
Figure 9.
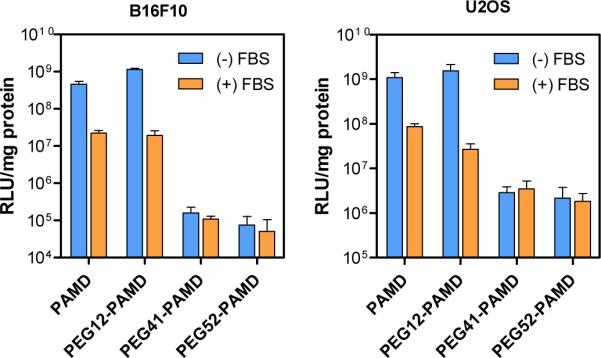
Transfection activity of PAMD polyplexes in B16F10 and U2OS cells. Polyplexes were prepared at PAMD/DNA (w/w) ratio 10 in B16F10 transfections and 5 in U2OS transfections. Results are expressed as luciferase expression in RLU/mg protein ± SD (n=3).
As discussed above, several strategies have been developed to overcome the negative effect of PEGylation on transfection activity. Here, we have utilized a mixed polyplex strategy to prepare polyplexes with acceptable colloidal stability, near-neutral zeta potential, and high transfection activity. The mixed polyplex strategy relies on using a mixture of non-PEGylated and PEGylated polycations. The non-PEGylated polycation provides effective DNA condensation and facilitates endosomal escape of the polyplexes, while the PEGylated polycation equips the polyplexes with favorable surface properties and colloidal stability. This strategy has been successfully used with several different types of polycations (14, 47, 48). We used a mixture of PAMD with PEG52-PAMD to optimize the polyplex formulation (Figure 10). We found that the DNA condensation curves of the mixed formulations shifted to higher w/w ratios with increasing PEG content (Figure 10a), however, the condensation efficiency of PAMD part in the formulation was not affected because the curves overlaid when re-plotted as relative fluorescence vs. N/P ratio (not shown). Using a mixture consisting of 80% PAMD and 20% PEG52-PAMD resulted in positively charged polyplexes with poor colloidal stability as indicated by rapid increase in size and aggregation in PBS (Figure 10b). Increasing the content of PEG52-PAMD in the mixture to 40 and 70% resulted in decreased zeta potential and formulation of colloidally stable polyplexes. The transfection results revealed that even polyplexes prepared with 70% PEG52-PAMD retained transfection activity that was similar to the transfection activity of non-PEGylated PAMD polyplexes (Figure 10c). This effect was particularly evident in experiments conducted in the presence of 10% serum. These results confirm that the mixed polyplex strategy is a suitable approach to prepare polyplexes that exhibit high transfection activity, while showing good colloidal stability and low surface charge.
Figure 10.

Properties of mixed PAMD/PEG-PAMD polyplexes. (a) DNA condensation ability of the PAMD/PEG-PAMD mixture determined by ethidium bromide exclusion assay. (b) Colloidal stability (left) and zeta potential (right) of polyplexes (w/w 5) prepared with increasing content of PEG52-PAMD in a mixture with PAMD. (c) Transfection activity of the mixed polyplexes.
CONCLUSIONS
In summary, this study demonstrates the potential of polymeric Plerixafor as a dual-function vector capable of preventing cancer cell invasion and facilitating efficient transfection. Our results suggest that combination of PEGylation of PAMD with using a mixed polyplex approach is a viable strategy to prepare formulations that retain CXCR4 antagonism and cancer cell inhibitory activity of PAMD, while at the same time, exhibit favorable colloidal stability and high transfection activity.
1.
Table PEG content in PEG-PAMD determined by 'H-NMR.
| Polymer | PEG content (wt %) | |
|---|---|---|
| in feed | in copolymer | |
| PAMD | 0 | 0 |
| PEG12-PAMD | 10 | 12 |
| PEG41-PAMD | 35 | 41 |
| PEG52-PAMD | 50 | 52 |
Acknowledgment
This work was supported in part by NIH grant EB014570. We thank Dr. Hazeldine for synthesizing Plerixafor.
REFERENCES
- 1.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 2.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4(7):581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 3.Payne CK, Jones SA, Chen C, Zhuang X. Internalization and Trafficking of Cell Surface Proteoglycans and Proteoglycan-Binding Ligands. Traffic. 2007;8(4):389–401. doi: 10.1111/j.1600-0854.2007.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter AC. Molecular hurdles in polyfectin design and mechanistic background to polycation induced cytotoxicity. Adv Drug Deliv Rev. 2006;58(14):1523–1531. doi: 10.1016/j.addr.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Q, Wu C, Manickam DS, Oupicky D. Evaluation of pharmacokinetics of bioreducible gene delivery vectors by real-time PCR. Pharm Res. 2009;26(7):1581–1589. doi: 10.1007/s11095-009-9847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke RS, Pun SH. Extracellular barriers to in vivo PEI and PEGylated PEI polyplex-mediated gene delivery to the liver. Bioconjugate Chem. 2008;19(3):693–704. doi: 10.1021/bc700388u. [DOI] [PubMed] [Google Scholar]
- 7.Smith AE, Sizovs A, Grandinetti G, Xue L, Reineke TM. Diblock glycopolymers promote colloidal stability of polyplexes and effective pDNA and siRNA delivery under physiological salt and serum conditions. Biomacromolecules. 2011;12(8):3015–3022. doi: 10.1021/bm200643c. [DOI] [PubMed] [Google Scholar]
- 8.Merdan T, Kunath K, Petersen H, Bakowsky U, Voigt KH, Kopecek J, Kissel T. PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice. Bioconjugate Chem. 2005;16(4):785–792. doi: 10.1021/bc049743q. [DOI] [PubMed] [Google Scholar]
- 9.Merkel OM, Librizzi D, Pfestroff A, Schurrat T, Buyens K, Sanders NN, De Smedt SC, Béhé M, Kissel T. Stability of siRNA polyplexes from poly(ethylenimine) and poly(ethylenimine)-g-poly(ethylene glycol) under in vivo conditions: Effects on pharmacokinetics and biodistribution measured by Fluorescence Fluctuation Spectroscopy and Single Photon Emission Computed Tomography (SPECT) imaging. J Controlled Rel. 2009;138(2):148–159. doi: 10.1016/j.jconrel.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Oupicky D, Ogris M, Howard KA, Dash PR, Ulbrich K, Seymour LW. Importance of lateral and steric stabilization of polyelectrolyte gene delivery vectors for extended systemic circulation. Mol Ther. 2002;5(4):463–472. doi: 10.1006/mthe.2002.0568. [DOI] [PubMed] [Google Scholar]
- 11.Oupicky D, Carlisle RC, Seymour LW. Triggered intracellular activation of disulfide crosslinked polyelectrolyte gene delivery complexes with extended systemic circulation in vivo. Gene Ther. 2001;8(9):713–724. doi: 10.1038/sj.gt.3301446. [DOI] [PubMed] [Google Scholar]
- 12.Walker GF, Fella C, Pelisek J, Fahrmeir J, Boeckle S, Ogris M, Wagner E. Toward Synthetic Viruses: Endosomal pH-Triggered Deshielding of Targeted Polyplexes Greatly Enhances Gene Transfer in vitro and in vivo. Mol Ther. 2005;11(3):418–425. doi: 10.1016/j.ymthe.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Fella C, Walker GF, Ogris M, Wagner E. Amine-reactive pyridylhydrazone-based PEG reagents for pH-reversible PEI polyplex shielding. Eur J Pharm Sci. 2008;34(4-5):309–320. doi: 10.1016/j.ejps.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Brumbach JH, Lin C, Yockman J, Kim WJ, Blevins KS, Engbersen JFJ, Feijen J, Kim SW. Mixtures of Poly(triethylenetetramine/cystamine bisacrylamide) and Poly(triethylenetetramine/cystamine bisacrylamide)-g-poly(ethylene glycol) for Improved Gene Delivery. Bioconjugate Chem. 2010;21(10):1753–1761. doi: 10.1021/bc900522x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 17.Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226(2):148–157. doi: 10.1002/path.3029. [DOI] [PubMed] [Google Scholar]
- 18.Gangadhar T, Nandi S, Salgia R. The role of chemokine receptor CXCR4 in lung cancer. Cancer Biol Ther. 2010;9(6):409–416. doi: 10.4161/cbt.9.6.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wald O, Izhar U, Amir G, Avniel S, Bar-Shavit Y, Wald H, Weiss ID, Galun E, Peled A. CD4+CXCR4highCD69+ T Cells Accumulate in Lung Adenocarcinoma. J of Immunol. 2006;177(10):6983–6990. doi: 10.4049/jimmunol.177.10.6983. [DOI] [PubMed] [Google Scholar]
- 20.Wald O, Izhar U, Amir G, Kirshberg S, Shlomai Z, Zamir G, Peled A, Shapira OM. Interaction between neoplastic cells and cancer-associated fibroblasts through the CXCL12/CXCR4 axis: Role in non–small cell lung cancer tumor proliferation. J Thorac Cardiovasc Surg. 2011;141(6):1503–1512. doi: 10.1016/j.jtcvs.2010.11.056. [DOI] [PubMed] [Google Scholar]
- 21.Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11(9):597–606. doi: 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- 22.Burger M, Glodek A, Hartmann T, Schmitt-Graff A, Silberstein LE, Fujii N, Kipps TJ, Burger JA. Functional expression of CXCR4 (CD184) on small-cell lung cancer cells mediates migration, integrin activation, and adhesion to stromal cells. Oncogene. 2003;22(50):8093–8101. doi: 10.1038/sj.onc.1207097. [DOI] [PubMed] [Google Scholar]
- 23.Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1α)-CXCR4/CXCR7 Pathway Inhibition: An Emerging Sensitizer for Anticancer Therapies? Clin Cancer Res. 2011;17(8):2074–2080. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips RJ, Mestas J, Gharaee-Kermani M, Burdick MD, Sica A, Belperio JA, Keane MP, Strieter RM. Epidermal growth factor and hypoxia-induced expression of CXC chemokine receptor 4 on non-small cell lung cancer cells is regulated by the phosphatidylinositol 3-kinase/PTEN/AKT/mammalian target of rapamycin signaling pathway and activation of hypoxia inducible factor-1α. J Biol Chem. 2005;280(23):22473–22481. doi: 10.1074/jbc.M500963200. [DOI] [PubMed] [Google Scholar]
- 25.Guo P, You J-O, Yang J, Jia D, Moses MA, Auguste DT. Inhibiting Metastatic Breast Cancer Cell Migration via the Synergy of Targeted, pH-triggered siRNA Delivery and Chemokine Axis Blockade. Mol Pharm. 2014 doi: 10.1021/mp4004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egorova A, Kiselev A, Hakli M, Ruponen M, Baranov V, Urtti A. Chemokine-derived peptides as carriers for gene delivery to CXCR4 expressing cells. J Gene Med. 2009;11(9):772–781. doi: 10.1002/jgm.1366. [DOI] [PubMed] [Google Scholar]
- 27.Dong Y, Zhu Y, Li J, Zhou Q-H, Wu C, Oupický D. Synthesis of Bisethylnorspermine Lipid Prodrug as Gene Delivery Vector Targeting Polyamine Metabolism in Breast Cancer. Mol Pharm. 2012;9(6):1654–1664. doi: 10.1021/mp300001m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong YM, Li J, Wu C, Oupicky D. Bisethylnorspermine Lipopolyamine as Potential Delivery Vector for Combination Drug/Gene Anticancer Therapies. Pharm Res. 2010;27(9):1927–1938. doi: 10.1007/s11095-010-0197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Zhu Y, Hazeldine ST, Li C, Oupicky D. Dual-function CXCR4 antagonist polyplexes to deliver gene therapy and inhibit cancer cell invasion. Angew Chem Int Ed Engl. 2012;51(35):8740–8743. doi: 10.1002/anie.201203463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Oupicky D. Effect of biodegradability on on CXCR4 antagonism, transfection efficacy and antimetastatic activity of polymeric Plerixafor. Biomaterials. 2014;35(21):5572–5579. doi: 10.1016/j.biomaterials.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu KK, Sakya SM, O'Donnell CJ, Flick AC, Li J. Synthetic approaches to the 2009 new drugs. Bioorg Med Chem. 2011;19(3):1136–1154. doi: 10.1016/j.bmc.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Zhu Y, Hazeldine ST, Firestine SM, Oupicky D. Cyclam-based polymeric copper chelators for gene delivery and potential PET imaging. Biomacromolecules. 2012;13(10):3220–3227. doi: 10.1021/bm3009999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong CY, You YZ, Wu DC, Liu Y, Pan CY. Thermal control over the topology of cleavable polymers: From linear to hyperbranched structures. J Am Chem Soc. 2007;129(17):5354–5355. doi: 10.1021/ja070871+. [DOI] [PubMed] [Google Scholar]
- 34.Fitzsimmons RE, Uludağ H. Specific effects of PEGylation on gene delivery efficacy of polyethylenimine: interplay between PEG substitution and N/P ratio. Acta Biomater. 2012;8(11):3941–3955. doi: 10.1016/j.actbio.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 35.Mishra S, Webster P, Davis ME. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur J Cell Biol. 2004;83(3):97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
- 36.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11(6):990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 37.Parhamifar L, Larsen AK, Hunter AC, Andresen TL, Moghimi SM. Polycation cytotoxicity: a delicate matter for nucleic acid therapy-focus on polyethylenimine. Soft Matter. 2010;6(17):4001–4009. [Google Scholar]
- 38.Hong S, Leroueil PR, Janus EK, Peters JL, Kober M-M, Islam MT, Orr BG, Baker JR, Banaszak Holl MM. Interaction of Polycationic Polymers with Supported Lipid Bilayers and Cells: Nanoscale Hole Formation and Enhanced Membrane Permeability. Bioconjugate Chem. 2006;17(3):728–734. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Hessler JA, Putchakayala K, Panama BK, Khan DP, Hong S, Mullen DG, DiMaggio SC, Som A, Tew GN, Lopatin AN, Baker JR, Holl MMB, Orr BG. Cationic Nanoparticles Induce Nanoscale Disruption in Living Cell Plasma Membranes. J Phys Chem B. 2009;113(32):11179–11185. doi: 10.1021/jp9033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong Z, Feijen J, Lok MC, Hennink WE, Christensen LV, Yockman JW, Kim YH, Kim SW. Low molecular weight linear polyethylenimine-b-poly(ethylene glycol)-b-polyethylenimine triblock copolymers: synthesis, characterization, and in vitro gene transfer properties. Biomacromolecules. 2005;6(6):3440–3448. doi: 10.1021/bm050505n. [DOI] [PubMed] [Google Scholar]
- 41.Sung SJ, Min SH, Cho KY, Lee S, Min YJ, Yeom YI, Park JK. Effect of polyethylene glycol on gene delivery of polyethylenimine. Biol Pharm bull. 2003;26(4):492–500. doi: 10.1248/bpb.26.492. [DOI] [PubMed] [Google Scholar]
- 42.Hong JW, Park JH, Huh KM, Chung H, Kwon IC, Jeong SY. PEGylated polyethylenimine for in vivo local gene delivery based on lipiodolized emulsion system. J Control Release. 2004;99(1):167–176. doi: 10.1016/j.jconrel.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 43.Casettari L, Vllasaliu D, Mantovani G, Howdle SM, Stolnik S, Illum L. Effect of PEGylation on the Toxicity and Permeability Enhancement of Chitosan. Biomacromolecules. 2010 doi: 10.1021/bm100522c. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Pan SR, Hu HM, Wu GF, Feng M, Zhang W, Luo X. Poly(ethylene glycol)-block-polyethylenimine copolymers as carriers for gene delivery: effects of PEG molecular weight and PEGylation degree. J Biomed Mater Res A. 2008;84(3):795–804. doi: 10.1002/jbm.a.31343. [DOI] [PubMed] [Google Scholar]
- 45.Tang GP, Zeng JM, Gao SJ, Ma YX, Shi L, Li Y, Too HP, Wang S. Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent. Biomaterials. 2003;24(13):2351–2362. doi: 10.1016/s0142-9612(03)00029-2. [DOI] [PubMed] [Google Scholar]
- 46.Mersch-Sundermann V, Knasmuller S, Wu XJ, Darroudi F, Kassie F. Use of a human-derived liver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology. 2004;198(1-3):329–340. doi: 10.1016/j.tox.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 47.Uchida S, Itaka K, Chen Q, Osada K, Ishii T, Shibata M-A, Harada-Shiba M, Kataoka K. PEGylated polyplex with optimized PEG shielding enhances gene introduction in lungs by minimizing inflammatory responses. Mol Ther. 2012;20(6):1196–1203. doi: 10.1038/mt.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of PEGylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjugate Chem. 2001;12(4):529–537. doi: 10.1021/bc0001488. [DOI] [PubMed] [Google Scholar]