

Abstract
Drug-loaded nanoparticles (NPs) are of particular interest for efficient cancer therapy due to their improved drug delivery and therapeutic index in various types of cancer. However, the encapsulation of many chemotherapeutics into delivery NPs is often hampered by their unfavorable physicochemical properties. Here, we employed a drug reform strategy to construct a small library of SN-38 (7-ethyl-10-hydroxycamptothecin)-derived prodrugs, in which the phenolate group was modified with a variety of hydrophobic moieties. This esterification fine-tuned the polarity of the SN-38 molecule and enhanced the lipophilicity of the formed prodrugs, thereby inducing their self-assembly into biodegradable poly(ethylene glycol)-block-poly(d,l-lactic acid) (PEG-PLA) nanoparticulate structures. Our strategy combining the rational engineering of prodrugs with the pre-eminent features of conventionally used polymeric materials should open new avenues for designing more potent drug delivery systems as a therapeutic modality.
Keywords: antitumor activity, drug delivery, nanoparticles, prodrugs, SN-38
In the continuing search for effective cancer treatments, nanoparticle (NP)-mediated drug targeting offers great promise for improving therapeutic efficacy.[1] Compared to free drugs, drug-loaded nanocarriers can preferentially accumulate within solid tumors due to the enhanced permeability and retention (EPR) effect, thereby improving drug efficacy and minimizing the associated off-target drug effects.[2] Toward the development of nanomedicines, amphiphilic copolymer materials such as poly(ethylene glycol)-block-poly(d,l-lactic acid) (PEG-PLA) and poly(ethylene glycol)-block-poly(lactic-co-glycolic acid) (PEG-PLGA) have been widely explored and have now been approved for clinical use by the US Food and Drug Administration (FDA).[3] For example, several PLA- or PLGA-based nanoparticles carrying drug payloads (e.g., docetaxel) are under development in clinical trials, including Nanoxel-PM and BIND-014.[4] Unfortunately, many chemotherapeutic agents are often incompatible with these delivery materials, resulting in limited entrapment efficiency and suboptimal release kinetics. While most efforts have focused on developing novel multifunctional delivery materials,[5] little attention has been paid to fine-tuning the chemical structures of these therapeutics to reformulate them into ideal polymeric nanocarriers.[6] A pioneering example has been demonstrated for a platinum(IV)-prodrug in which the structure was tailored to facilitate encapsulation in a targetable nanoparticulate system.6a,b
SN-38 (7-ethyl-10-hydroxycamptothecin) is an inhibitor of DNA topoisomerase I with low nanomolar potency, but its clinical application has been hampered by its extremely low water solubility and dose-limiting side effects.[1] Irinotecan hydrochloride (CPT-11), a water-soluble prodrug of SN-38, has been clinically approved for treating a variety of solid tumors. After administration, CPT-11 is converted to the active metabolite SN-38 by carboxylesterases, but the conversion rate is inefficient and usually yields less than 8 %.[8] SN-38 exhibits 100- to 1000-fold more potent cytotoxic activity against various cancer cells in vitro compared to CPT-11. Directly harnessing SN-38 would therefore bypass the inefficient enzymatic activation and thereby improve the therapeutic index for cancer treatment. However, physical encapsulation of this molecule into amphiphilic polymeric NPs is a challenge due to the unfavorable physicochemical properties of the drug, which are attributed to the intrinsically planar structure and moderate polarity of this molecule.[9] Thus, a sophisticated strategy is needed to adapt this potent agent for incorporation into a favorable polymeric delivery platform. We noticed that the phenolate moiety (10-OH) on SN-38 may account for its poor compatibility with polymeric NPs. Given this possibility, we hypothesized that it might be possible to improve the drug’s stability and thereby its retention in the particles by appropriate drug engineering.
Motivated by this rationale, we set out to design and synthesize a small library of novel SN-38 derivatives with the phenolate group on SN-38 shielded by a variety of hydrophobic moieties aimed at enhancing the drug lipophilicity. Sufficient lipophilicity imparted by these moieties would be expected to drive these prodrugs into assembling into the hydrophobic core of NPs in an aqueous environment (Figure 1 a and b). Specifically, we systematically studied these prodrugs for their loading capacity, stability, and release kinetics using a model PEG-PLA material system. The prodrug-loaded NPs exhibited a sustained release profile and exerted cytotoxic activity against each tested cell line upon hydrolysis of the phenyl ester. Furthermore, we showed that the optimized prodrug nanoformulations can be exploited to achieve a high therapeutic index in a human colorectal tumor xenograft model.
Figure 1.

a) Prodrugs were engineered for combination with amphiphilic copolymer-based nanoparticulate drug delivery platforms. b) Structures of a small library of SN-38 prodrugs. A variety of hydrophobic moieties were conjugated to the 10-hydroxy group through the formation of phenyl ether (1) or ester (2–15) bonds. c) Assessment of the stability of the prodrug-encapsulated, self-assembled PEG-PLA nanoparticles (PEG-PLA/prodrug weight ratios fixed at 20:1).
We primarily used condensation reactions to functionalize the phenolate moiety (10-OH) on SN-38 (Figure 1 b). The phenyl ester bond is liable to hydrolyze under physiological conditions and thus liberate free SN-38. By comparison, prodrug 1 was generated with a non-hydrolyzable ether bond. Based on these two chemistries, we selected a variety of hydrophobic moieties, including fatty acids, vitamin E, Boc-protected amino acids, artesunate, cholesterol, and deoxycholic acid, to shield the 10-hydroxy group of SN-38. These modifications are expected to alleviate the polarity of the SN-38 molecules and enhance the lipophilicity of prodrugs, which could facilitate their self-assembly into amphiphilic copolymer-based NPs within inner hydrophobic core structures. The corresponding prodrugs 1–15 were obtained from commercially available materials by a single- or two-step reaction in moderate to high yields of 41–77 %.
To assess the ability of these prodrugs to incorporate into polymeric NPs, we chose to use an amphiphilic PEG-PLA copolymer that is both biodegradable and approved by the US FDA as a building block for clinical use. Nanoparticles derived from PEG-PLA exhibit several characteristics such as low critical micelle concentration (CMC), long circulating times following systemic administration and improved accumulation at targeted tumor sites, that make them a promising class of potential drug delivery vehicles.[3] The hydrophobic moieties incorporated into the SN-38 structure are expected to enhance the noncovalent interaction between prodrugs and the PLA core, which could improve solubility and prodrug retention in the particles. To test this hypothesis, we prepared the prodrug-encapsulated NPs by using a nanoprecipitation method[10] to promote the self-assembly of PEG-PLA with varying molecular weights and prodrugs 1–15. During this procedure, the nonpolar, hydrophobic prodrugs are expected to incorporate into the hydrophobic core by interacting with the PLA segment, whereas the hydrophilic PEG spontaneously forms the corona in aqueous environments. When the PEG-PLA/prodrug weight ratios were fixed at 20:1, the stabilities of the prodrug-loaded NPs with a concentration of 0.5 mg mL−1 (SN-38 equivalent) were monitored during and after preparation. As a result of the first round of screening, we successfully obtained 28 prodrug-formulated NPs from a library of total 75 types of NPs that are stable at this concentration (Figure 1 c). Because higher prodrug concentration was required for in vivo experiments, a second round of selection was performed to further optimize these NPs. This effort resulted in five formulations of prodrugs 6, 7, 8, 12, and 13-NPs with SN-38 equivalent concentrations above 1.6 mg mL−1 (Table S1). In contrast to the poor water solubility of SN-38 (≈2 μg mL−1 at pH 7), we were able to dissolve 50 mg mL−1 of prodrug-loaded NPs (5 % prodrugs by weight), representing at least an 800-fold increase in the solubility of SN-38. In sharp contrast, when attempting to encapsulate free SN-38, it was found to precipitate, resulting in less than 10 μg mL−1 solubility with the PEG-PLA copolymers. We could attribute this incompatibility to the highly planar overall structure of SN-38 and the high polarity of its phenolate moiety. Furthermore, to demonstrate the shielding role of 10-OH during the assembly into NPs, we switched the Boc-glycine group from 10-OH to 20-OH (compound 16, Figure S1 in the Supporting Information, SI) and assessed its NP loading capacity. Prodrug 16 also lost its retention ability in these NPs, strongly suggesting that the stability of prodrug-formulated NPs is closely associated with the reduced polarity derived from the phenolate moiety.
Given the success of nanoprecipitation for SN-38 prodrugs (6, 7, 8, 12, and 13), we next characterized their morphology by transmission electron microscopy (TEM) and dynamic light scattering (DLS) analysis. In TEM images, these NP libraries exhibited homogenous populations of similarly spherical shapes with average diameters of 16–43 nm (Figure 2 a–e). DLS analysis showed the monomodality and narrow size distribution (polydispersity index<0.2) of the prodrug-loaded NPs. Compared to the TEM results, the hydrodynamic diameters (dh) measured by DLS were slightly larger, but all were smaller than 100 nm (Figure 2 f). In particular, prodrug 12 encapsulated NPs had a very small diameter (dh≈20 nm, n=4) compared to the other prodrug-NPs, indicating the formation of more compact core–shell structures. Previous studies suggested that the tumor penetration and accumulation of nanoparticles is highly dependent on the overall size and that nanomedicines in the sub-100 nm range have superior antitumor efficacy in various solid tumors.[11] Therefore, we envisioned that this fabrication technology for SN-38 prodrugs could potentially increase intratumoral delivery.
Figure 2.

In vitro characterization of SN-38 prodrug-loaded polymeric nanoparticles. a–e) Transmission electron microscopic (TEM) images of SN-38 prodrugs 6-, 7-, 8-, 12-, and 13-loaded NPs (scale bars=100 nm). f) Size distribution of the prodrug-loaded NPs measured by dynamic light scattering (DLS). Except for 7- and 12-loaded NPs (with average dh≈82 and 20 nm, respectively), the formulations of 6, 8, and 13 produce a superimposable histogram with average dh of ca. 43 nm.
The liberation of free SN-38 from the prodrug-loaded NPs requires two steps, in which the release of prodrugs from the NPs is concurrent with the pH-dependent hydrolytic cleavage of the phenyl ester bond. We first verified the kinetics of active SN-38 release at pH 7.4 using 6-, 7-, 8-, 12-, and 13-loaded NPs. The controlled release of free SN-38 molecules was monitored by high-performance liquid chromatography (HPLC). As shown in Figure S2 in SI, an initial burst release was observed within 2 h that yielded less than 20 % of the total SN-38 amounts at pH 7.4. Thereafter, a period of sustained release occurs for up to two days, during which up to 80 % of the free drug is released. Interestingly, distinct release profiles were observed among these NPs. NPs encapsulating prodrugs 8, 12, and 13 exhibited slower release kinetics than 6- and 7-loaded NPs. To further elucidate the correlation between the release and the hydrolysis kinetics of prodrugs, we studied the hydrolysis of 7 and 8 as model compounds at neutral pH. Prodrug 8 was found to be more kinetically inert to hydrolysis than 7 (see Figure S3 in SI). This result is consistent with the release profile for free SN-38 and indicates that this engineered prodrug platform could deliver a sufficient quantity of active agents to the target tumor sites by a simple hydrolysis process that avoids the inefficient enzymatic activation required for CPT-11. The prodrugs with enhanced lipophilicity might readily bind to endogenous albumin, which would lead to the destruction of the NPs. We thus verified the stability of 7- and 12-NPs in the presence of serum by DLS analysis. 7-loaded NPs exhibited a partial peak shift in size distributions but still remained stable within several hours. In contrast, 12-loaded NPs were quite stable during the longtime incubation with 50 % serum (see Figure S4 in SI), indicating that the drugs can arrive at the tumor sites together with nanoparticles through the EPR effect.
We next evaluated the cytotoxic effects of prodrug-loaded NP candidates by measuring the half maximal inhibitory concentration (IC50) of cell proliferation. Cell viability was quantified using the standard MTT assay after 48 h of treatment with prodrug-NPs. As shown in Figure 3 a,b and Table 1, these SN-38 prodrug-NPs exhibited high cytotoxicity to all tested cancer cells and were approximately two orders of magnitude more effective than CPT-11. Unexpectedly, the antiproliferative activities induced by incubation with 8-, 12-, and 13-NPs occurred at much lower concentrations than for free SN-38 administered in DMSO in HCT-116, SW480, and A549 cells (e.g., in the best case showing 26-fold potency). The superior activity observed in vitro is impressive given that the prodrug-NPs must undergo two steps to release active SN-38 molecules as discussed above. This requirement in general will result in reduced in vitro cytotoxicity in comparison to the free drug. By contrast, the nonhydrolyzable ether-linked SN-38 prodrug 1-NP exerted less cytotoxicity to all tested cancer cells, with IC50 values greater than 3 μM.
Figure 3.
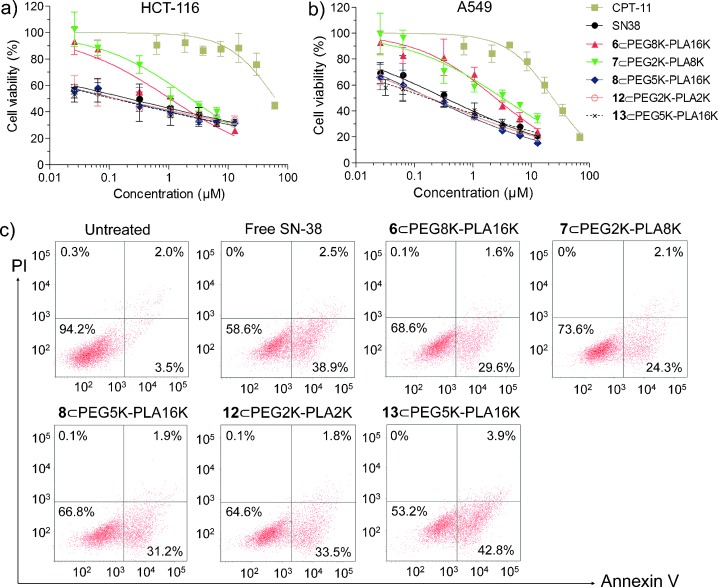
Cell viability for CPT-11, free SN-38, and prodrug-NPs in HCT-116 (a) and A549 (b) cells measured by the MTT assay (Mean±SD). c) Apoptotic analysis of HCT-116 cells determined by FACS using Alexa Fluor 488 Annexin V/PI staining kit after 24 h drug treatments. Four distinct phenotypes: viable cells (lower left quadrant); early apoptotic cells (lower right quadrant); late apoptotic cells (upper right quadrant); necrotic or dead cells (upper left quadrant).
Table 1.
Antitumor potential of SN-38 prodrug-loaded PEG-PLA NPs after 48 h of incubation (expressed as IC50±standard deviation in μM).a
| Cell line | HCT-116 | SW480 | A549 | MCF-7 |
|---|---|---|---|---|
| CPT-11 | 21.6±1.8 | 28.6±2.5 | 20.4±2.0 | 88.5±20.8 |
| SN-38 | 0.22±0.05 | 0.26±0.06 | 0.31±0.02 | 1.43±0.22 |
| 6⊂PEG8K-PLA16K | 1.05±0.17 | 0.98±0.25 | 2.11±0.06 | 2.69±0.49 |
| 7⊂PEG2K-PLA8K | 2.19±0.34 | 1.63±0.20 | 4.53±0.08 | 2.27±0.61 |
| 8⊂PEG5K-PLA16K | 0.13±0.02 | 0.02±0.01 | 0.18±0.03 | 1.52±0.46 |
| 12⊂PEG2K-PLA2K | 0.13±0.05 | 0.04±0.02 | 0.19±0.02 | 1.21±0.27 |
| 13⊂PEG5K-PLA16K | 0.11±0.06 | 0.01±0.004 | 0.17±0.03 | 1.36±0.31 |
Determined by MTT assay.
To determine whether the inhibition of cancer cell proliferation by these prodrug-NPs was a consequence of SN-38-induced apoptosis, we conducted an Alexa Fluor 488 Annexin V/propidium iodide (PI) double-staining assay in HCT-116 cells. The exposure of phosphatidylserine on the outer leaflet of the cell membrane is an essential event in apoptosis that can be specifically detected by the binding of fluorescently labeled annexin V.[12] After exposure to CPT-11 (3 μM), free SN-38 (3 μM), and prodrug-NPs (3 μM, SN-38 equivalent doses) for 12 or 24 h, cells were analyzed by fluorescence-activated cell sorting (FACS). Indeed, a high level of apoptosis after 12 or 24 h of treatment was induced by these prodrug-loaded NPs, which was comparable to the level induced by free SN-38 (Figure 3 c and Figure S5 in SI). Together, these cell-based experiments clearly suggest that these prodrug-loaded NPs can perform similarly to free SN-38 agents in vitro in effectively inducing apoptosis in cancer cells.
To establish the clinical translation potential of prodrug-encapsulated NPs, we performed therapeutic studies in vivo using a HCT-116 colorectal xenograft model. To decrease the number of required animals, only prodrug 6-, 7-, 8-, 12-, and 13-loaded NPs were tested and compared due to their high in vitro activities and stability of formulation. Unfortunately, the 13-formulated NP caused immediate death of mice during intravenous injection; we thus terminated the in vivo therapeutic procedure. The antitumor efficacy of 6-, 7-, 8-, and 12-loaded NPs is illustrated in Figure 4 a, c and d. The tumor growth was remarkably inhibited after the successive intravenous injection of all prodrug-loaded NPs (at 10 mg kg−1 SN-38 equivalent dose) as compared to saline and CPT-11 (12 mg kg−1) controls, thus demonstrating the superiority of combining the drug reform strategy with nanoparticle-based delivery platforms. In particular, the group treated with prodrug 12-loaded NPs produced a more drastic decrease in the tumor progression, resulting in a mean tumor volume of 215 mm3 versus 708 mm3 for CPT-11-treated control (n=7, p<0.01). By comparison, untreated mice from the saline group showed rapid tumor growth, with tumor volume reaching approximately 1293 mm3 by day 20. The in vivo distribution of drugs and consequently of their antitumor efficacies rely heavily on factors such as the nanoparticle size, surface characteristics, and shape.[13] Considering the similar surface properties of the prodrug-encapsulated NPs (e.g., shapes and zeta potentials), we might partially attribute the superior outcome of 12-loaded NPs to their higher cytotoxicity in vitro and relatively small particle size (approximately 20 nm), which exceeds the 5 nm cutoff for clearance by the kidney but may exhibit preferential accumulation in the tumor site.[14] It was also notable that the in vivo efficacy of the prodrug-formulated NPs was closely correlated with their in vitro cytotoxicity, highlighting the value of using this parameter in designing more efficient chemotherapeutics in future work.
Figure 4.
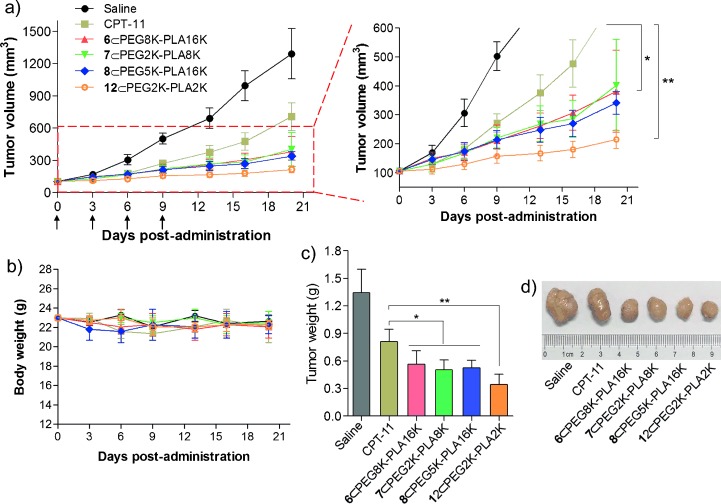
In vivo antitumor efficacy of prodrug-loaded NPs against HCT-116 colorectal xenograft model. a) Tumor growth curve of different groups. Mice received four i.v. injections of 10 mg kg−1 (SN-38 equivalent dose) on days 0, 3, 6, and 9. Each point represents the mean of tumor size±standard deviation (n=7). *P<0.05; **P<0.01. b) Body weights (mean±standard deviation). c) Tumor masses of drug-treated groups 20 days after treatment. d) Representative images of HCT-116 tumors of the mice after treatment with saline, CPT-11, or prodrug-loaded NPs at day 20.
The body weights of the mice receiving treatment with NPs all remained stable, suggesting a low systemic toxicity of these prodrugs and related delivery materials (Figure 4 b). Although animal experiments with higher doses of prodrug-loaded NPs were not conducted, elevated doses should be expected to improve cancer therapy.[14]
In summary, as a proof of principle, we presented a drug reform strategy for constructing a small library of lipophilic SN-38 derivatives and screened their ability to be incorporated into amphiphilic NP formulations. Compared to the incompatibility of the SN-38 molecule with NPs, the esterification markedly reinforced the stability of prodrugs inside the hydrophobic PLA core. Encapsulating therapeutics in NPs is a promising way of protecting them from rapid clearance, degradation in blood, and more efficiently promoting their accumulation in the target tumor site. Using the optimized prodrugs 6-, 7-, 8-, and 12-loaded NPs, we also demonstrated their potential as novel antitumor chemotherapeutic modalities in a colorectal xenograft model. The coupling of the EPR effect derived from NP formulations with direct liberation of biologically active SN-38 allowed the treatments to bypass enzymatic activation, contributing to their considerable inhibition of tumor growth in comparison to CPT-11. As many promising compounds have been abandoned in the preclinical stages due to pharmacologic challenges,[16] it should be interesting to employ this approach to rescue them. Overall, our results are encouraging and the combination of structure-oriented rational prodrug engineering and nanoparticle-based technology may facilitate the further development of effective drug delivery systems.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- 1.pp. 2971–3010.; Kamaly N, Xiao Z, Valencia PM, Fadovic-Moreno AF, Farokhzad OC. Chem. Soc. Rev. 41 doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]; Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat. Nanotechnol. 2012;2 doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Angew. Chem. Int. Ed. 2007;48 doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]; Petros RA, DeSimone JM. Nat. Rev. Drug Discovery. 2009;9 doi: 10.1038/nrd2591. 2010. [DOI] [PubMed] [Google Scholar]
- 2.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J. Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 3.pp. 315–319.; Kang N, Perron M-È, Prud’homme RE, Zhang Y, Gaucher G, Leroux J-C. Nano Lett. 5 doi: 10.1021/nl048037v. [DOI] [PubMed] [Google Scholar]; Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Chem. Soc. Rev. 2005;42 doi: 10.1039/c2cs35265f. 20133a 3b. [DOI] [PubMed] [Google Scholar]
- 4.pp. 262–271.; Lee S-W, Yun M-H, Jeong SW, In C-H, Kim J-Y, Seo M-H, Pai C-M, Kim S-O. J. Controlled Release. 155 doi: 10.1016/j.jconrel.2011.06.012. [DOI] [PubMed] [Google Scholar]; Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, et al. Sci. Transl. Med. 2011;4:39. doi: 10.1126/scitranslmed.3003651. 4a 4b 2012. [DOI] [PubMed] [Google Scholar]
- 5.pp. 991–1003.; Mura S, Nicolas J, Couvreur P. Nat. Mater. 12 doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]; Cohen Stuart MA, Huck WTS, Genzer J, Müller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Nat. Mater. 2013;9 doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]; Bae Y, Fukushima S, Harada A, Kataoka K. Angew. Chem. Int. Ed. 2010;42 doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003;115 5a 5b 5c 2003. [Google Scholar]
- 6.pp. 17356–17361.; Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc. Natl. Acad. Sci. USA. 105 doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Proc. Natl. Acad. Sci. USA. 2008;108 doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]; Harrisson S, Nicolas J, Maksimenko A, Bui DT, Mougin J, Couvreur P. Angew. Chem. Int. Ed. 2011;52 doi: 10.1002/anie.201207297. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]; Sengupta P, Basu S, Soni S, Pandey A, Roy B, Oh MS, Chin KT, Paraskar AS, Sarangi S, Connor Y, Sabbisetti VS, Kopparam J, Kulkarni A, Muto K, Amarasiriwardena C, Jayawardene I, Lupoli N, Dinulescu DM, Bonventre JV, Mashelkar RA, Sengupta S. Proc. Natl. Acad. Sci. USA. 2013;109 doi: 10.1073/pnas.1203129109. 6a 6b 6c 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pommier Y. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 8.pp. 245–259.; Chabot GG. Clin. Pharmacokinet. 33 doi: 10.2165/00003088-199733040-00001. [DOI] [PubMed] [Google Scholar]; Slatter JG, Schaaf LJ, Sams JP, et al. Drug. Metab. Dispos. 1997;28 8a 8b 2000. [PubMed] [Google Scholar]
- 9.Kasai H, Murakami T, Ikuta Y, Koseki Y, Baba K, Oikawa H, Nakanishi H, Okada M, Shoji M, Ueda M, Imahori H, Hashida M. Angew. Chem. Int. Ed. 51:10315–10318. doi: 10.1002/anie.201204596. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 2012. [Google Scholar]
- 10.pp. 389–400.; Chorny M, Fishbein I, Danenberg HD, Golomb G. J. Controlled Release. 83 doi: 10.1016/s0168-3659(02)00211-0. [DOI] [PubMed] [Google Scholar]; Landfester K. Angew. Chem. Int. Ed. 2002;48 [Google Scholar]; Angew. Chem. 2009;121 10a 10b 2009. [Google Scholar]
- 11.pp. 815–823.; Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Nat. Nanotechnol. 6 doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]; Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Nano Lett. 2011;9 doi: 10.1021/nl900031y. 11a 11b 2009. [DOI] [PubMed] [Google Scholar]
- 12.pp. 1415–1420.; Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Blood. 84 [PubMed] [Google Scholar]; Logue SE, Elgendy M, Martin SJ. Nat. Protoc. 1994;4 doi: 10.1038/nprot.2009.143. 12a 12b 2009. [DOI] [PubMed] [Google Scholar]
- 13.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nat. Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.pp. 761–769.; Wang B, He X, Zhang Z, Zhao Y, Feng W. Acc. Chem. Res. 46 doi: 10.1021/ar2003336. [DOI] [PubMed] [Google Scholar]; Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol. Pharm. 2013;5 doi: 10.1021/mp800051m. 14a 14b 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S, Hamaguchi T, Matsumura Y. Cancer Res. 2006;66:10048–10056. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 16.pp. 8230–8235.; Karve S, Werner ME, Sukumar R, Cummings ND, Copp JA, Wang EC, Li C, Sethi M, Chen RC, Pacold ME, Wang AZ. Proc. Natl. Acad. Sci. USA. 109 doi: 10.1073/pnas.1120508109. [DOI] [PMC free article] [PubMed] [Google Scholar]; Turk B. Nat. Rev. Drug Discovery. 2012;5 doi: 10.1038/nrd2092. 11a 11b 2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.