

Abstract
Background
Nonsteroidal anti-inflammatory drugs (NSAIDs) include aspirin (acetylsalicylic acid, ASA). Long-term use of NSAIDs has been associated with lowered risk of colorectal cancer (CRC), but the use is hampered by adverse effects. Also, the anti-carcinogenic effects of NSAIDs are incompletely understood. Understanding biological effects of NSAIDs may help developing new preventive medical strategies.
Aim
To identify gene–environment interactions between genetic variation and NSAID use in relation to risk of CRC.
Methods
We performed a PubMed literature search and all studies reporting original data on interactions between NSAIDs and polymorphisms in relation to CRC were evaluated.
Results
We found indications that aspirin interacted with rs6983267 close to MYC (encoding a transcription factor involved in cell cycle progression, apoptosis and cellular transformation) and NSAIDs interacted with rs3024505 and rs1800872 in or close to IL10 (encoding IL-10) in preventing CRC. Homozygous carriers of the variant allele of rs6983267 (ca. 25% of the population) halved their risk for CRC by aspirin use compared to homozygous wildtype carriers who did not benefit from aspirin intake. No interaction between use of NSAIDs and PTGS-2 (encoding COX-2) in relation to CRC risk was detected. Other findings of interactions between genes in inflammatory and oncogenic pathways and NSAIDs were considered suggestive.
Conclusions
Knowledge of underlying biological effects of NSAIDs in relation to CRC is scarce and the basis for stratifying the patients for preventive treatment is not yet available. Further studies assessing interactions between long-term NSAID exposure and genetic variation in relation to CRC are warranted in large well-characterised prospective cohorts.
Introduction
Colorectal cancer (CRC) is a major health problem worldwide. CRC is the third most common cancer and the one with the highest mortality in Western populations.1 The incidence is increasing due to demographical changes and due to the implementation of Western lifestyle in the developing countries. Lifestyle factors, including diet, are considered to be the main cause of CRC. High intake of red meat, animal fat, alcohol and smoking has been associated with increased risk of CRC whereas high intake of dietary fibres, fruit and vegetables, and physical activity are considered protective in relation to CRC.2
Also medical preventive strategies may be considered.3 Nonsteroidal anti-inflammatory drugs (NSAIDs) include aspirin (acetylsalicylic acid, ASA). Long-term use of NSAIDs has been associated with lowered risk of CRC in prospective population-based studies.4–6 The primary anti-inflammatory effects of NSAIDs is competitive inhibition of cyclooxygenases (COX) 1 and 2 which catalyse the rate-limiting conversion of arachidonic acid (AA) to the pro- and anti-inflammatory prostaglandins and thromboxanes.7,8 The main cardiovascular effect of low-dose aspirin is acetylation of COX-1 leading to irreversible inactivation of COX-1 and thereby reducing COX-1-derived thromboxane A2 synthesis. However, the underlying biological mechanisms of action of the protective effect in relation to colorectal carcinogenesis are incompletely understood.5 It has been found that NSAID metabolites without ability to inhibit COX inhibited cell growth by stimulating apoptosis.9 Thus, NSAIDs may have COX-independent anti-carcinogenic effects which may involve peroxisome proliferator-activated receptor (PPAR) γ, nuclear factor κ-B (NFκB) and the transforming growth factor β (TGF-β) pathways10–12 (reviewed in 3,5,8,13). Some of these data come from animal and in vitro studies and may not be directly applicable in humans, e.g. due to the high doses of NSAIDs used. The use of NSAIDs in prevention of CRC has been hampered by the increased risk of cardiovascular and gastrointestinal side effects.5 Consequently, an important issue is to understand the biological mechanisms of action of NSAIDs in relation to colorectal carcinogenesis.
Identifying gene–environment interactions is a means to identify genes and biological pathways involved in the biological actions of aspirin and other NSAIDs. This strategy has proved valuable for the identifications of biological mechanisms underlying the effects of diet in CRC.14–20 We therefore reviewed the literature on interactions between NSAIDs and polymorphisms in relation to CRC to identify genes and pathways involved in the protective effects of NSAID use.
Materials and methods
A systematic review and meta-analysis were carried out according to the guidelines of PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement.21 PubMed was searched for various combinations of ‘NSAID’, ‘aspirin’, ‘colorectal cancer’, ‘snp(s)’, ‘gene variant’ and ‘polymorphisms’ resulting in 85 abstracts (December 2013). All studies suggesting that they presented original data on polymorphisms and NSAID interaction were retrieved (46 articles) and reviewed and all studies reporting original data on interaction between polymorphisms and NSAID use in relation to risk of CRC were included (seven prospective cohort studies14,15,17–20,22 and 21 case–control studies23–43). In addition, two studies were identified in the reference lists from found articles.44,45 Another study was identified by searching for related articles (January 2014).46 One study analysed two separate prospective cohorts46 and one case–control study included both a discovery and a validation cohort.27 Three studies were excluded due to small numbers of participants and missing data on NSAID use.25,44,45 Furthermore, previously unpublished data on interactions between NSAIDs and HMOX in relation to CRC from a published study was added.47 Figure 1 shows the search strategy. A new search (May 2014) identified no new studies in PubMed and one case–control study in Embase which was included in Table 2.48
Figure 1.
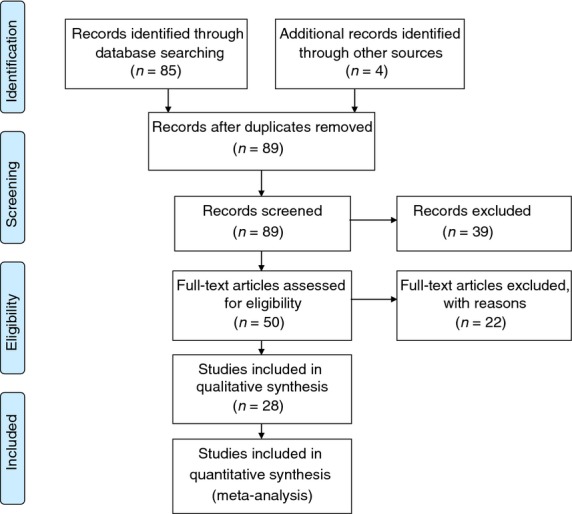
PRISMA flow diagram showing the search strategy.
Table 2.
Selected examples of study designs and NSAID definitions for case–control studies of interaction between NSAID and polymorphisms in relation to CRC
| Study design | Study population | NSAID use | ||
|---|---|---|---|---|
| KPMCP, Kaiser Permanente Medical Care Program of Northern California and Twin cities, Utah (DALS, Diet, Activity and Lifestyle Study) | Case–control study based on interview using standardised questionnaires within 2 years of diagnosis | NHW, non-Hispanic White | Use of aspirin or ibuprofen at least three times per week for 1 month or more27 | |
| CCFR, Colon Cancer Family Registry | Case–control study based on interview using standardised questionnaires within 5 years of diagnosis | NHW, non-Hispanic White | Use of aspirin or ibuprofen at least twice per week for 1 month or more27 | |
| NoSk, Northeast of Skotland | Case–control study using questionnaire, population-based controls matched on sex and age at diagnosis | Residents in Scotland | Use of aspirin every day for a month or more, presently or had ever taken43 | |
| SEER, Surveillance, Epidemiology and End Result Kentucky Cancer Registry | Case–control study using self-administered questionnaire with information on NSAID use. Controls were recruited via random digit dialling | No available information34 | ||
| NCCCS, North Carolina Colon Cancer Study | Case–control study, questionnaires administered by trained nurses. Controls from Division on Motor Vehicles or from Medicare with the same 5-year age, gender and ethnic group | African American | Any NSAID in the past 5 year for 3 days/week or more for 3 months or more40 | |
| UK | Multicentre case–control study, a questionnaires were administered by trained nurses, Age-, sex- and General Practitioner-matching controls. | Caucasian | Have you ever taken NSAID/aspirin on a regular basis for periods of 3 months or more (Y/N).42 | |
| CCFR2 (Colon Cancer Family Registry) | Multicentre case-unaffected sibling and populations-based controls study design, interviewed within 5 years of diagnosis | Non-Hispanic White | No available information23 | |
| GERM | Population-based case–control study with 22 hospitals in the area. Controls randomly selected from populations registries and frequency matched with cases by sex, county of residence and 5-year age group | Residents in Germany | Intake of NSAID two or more times per week for at least 1 year38 | |
| Diet, Health, and Cancer | Prospective cohort, nested case–subcohort study, questionnaires at entry | Danish | Intake of NSAID two or more times per month for at least 1 year at study entry19 | |
| The Rotterdam Study | Prospective populations-based cohort, nested case–control study, follow-up | Holland | Cumulative exposure to NSAIDs until the index date (date of cancer diagnosis)22 | |
| NHS & HPFS | Prospective cohort, nested case–control study within the Nurses' Health Study (NHS) and Health Professionals Follow-up Study (HPFS), follow-up | U.S. | Cumulative average intake of aspirin before diagnosis46 | |
P-value for interaction (Pint) between NSAID use and polymorphisms was retrieved and a P-value below 0.05 was considered statistically significant. P-values adjusted for confounders were chosen whenever possible as indicated in Tables 1 and S1. The retrieved P-values were not corrected for multiple testing.
Table 1.
Interactions between NSAID use (no, yes) and studied polymorphisms in relation to risk of colorectal cancer in prospective cohorts. Statistically significant interactions between NSAID use and a polymorphism in relation to CRC are shown in bold (Pint < 0.05)
| NSAID | IRR* 95% CI | IRR† 95% CI | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Yes | NSAID | NSAID | ||||||||||
| rsnumber | |||||||||||||
| Gene | NC | NS | NC | NS | No | Yes | No | Yes | Pint‡ | Ref | |||
| PTGS2 (COX-2) | rs689566 | A-1195G | AA-AG | 618 | 1169 | 16 | 19 | 1.00 | 1.01 (0.85–1.21) | 1.00 | 0.99 (0.83–1.19) | 19 | |
| GG | 31 | 43 | 274 | 529 | 1.23 (0.76–1.99) | 1.47 (0.63–3.44) | 1.27 (0.78–2.06) | 1.86 (0.89–3.88) | 0.39 | 19 | |||
| rs20417 | G-765C | GG | 477 | 874 | 218 | 391 | 1.00 | 1.09 (0.89–1.34) | 1.00 | 1.07 (0.87–1.32) | 19 | ||
| GC-CC | 165 | 330 | 68 | 152 | 0.96 (0.77–1.20) | 0.84 (0.61–1.15) | 0.93 (0.74–1.17) | 0.80 (0.58–1.10) | 0.18 | 19 | |||
| rs20417§ | G-765C | GG | 73 | 22 | |||||||||
| GC-CC | 27 | 0.67 | 22 | ||||||||||
| rs5275 | T8473C | TT | 292 | 514 | 134 | 215 | 1.00 (–) | 1.12 (0.86–1.46) | 1.00 (–) | 1.14 (0.87–1.49) | 19 | ||
| TC-CC | 346 | 695 | 151 | 326 | 0.88 (0.72–1.07) | 0.87 (0.68–1.11) | 0.87 (0.71–1.06) | 0.82 (0.64–1.06) | 0.21 | 19 | |||
| PPARG | rs3856806 | C1431T | CC | 191 | 376 | 85 | 173 | 1.00 (–) | 0.98 (0.71–1.35) | 1.00 (–) | 0.96 (0.70–1.33) | 20 | |
| CT-TT | 68 | 153 | 39 | 61 | 0.86 (0.61–1.21) | 1.26 (0.80–1.98) | 0.85 (0.60–1.21) | 1.22 (0.76–1.95) | 0.73 | 20 | |||
| rs1801282 | Pro12Ala | CC | 173 | 375 | 77 | 170 | 1.00 (–) | 1.02 (0.73–1.41) | 1.00 (–) | 1.01 (0.73–1.42) | 20 | ||
| CG and GG | 67 | 137 | 33 | 64 | 1.01 (0.71–1.43) | 1.30 (0.82–2.06) | 1.04 (0.72–1.48) | 1.29 (0.80–2.06) | 0.42 | 20 | |||
| IL10 | rs1800872 | C-592A | CC | 421 | 750 | 172 | 334 | 1.00 | 0.96 (0.76–1.20) | 1.00 | 0.97 (0.77–1.21) | 19 | |
| AC-AA | 230 | 460 | 118 | 217 | 0.89 (0.73–1.09) | 0.99 (0.76–1.29) | 0.91 (0.74–1.12) | 0.96 (0.73–1.25) | 0.58 | 19 | |||
| rs3024505 | CC | 432 | 840 | 210 | 366 | 1.00 | 1.14 (0.93–1.41) | 1.00 | 1.11 (0.90–1.38) | 19 | |||
| CT-TT | 214 | 384 | 81 | 188 | 1.07 (0.87–1.32) | 0.88 (0.66–1.18) | 1.08 (0.87–1.33) | 0.89 (0.66–1.19) | 0.05 | 19 | |||
| IL1B | rs4848306 | C-3737T | CC | 235 | 376 | 99 | 189 | 1.00 | 0.87 (0.65–1.18) | 1.00 | 0.82 (0.61–1.12) | 19 | |
| CT-TT | 409 | 834 | 190 | 361 | 0.76 (0.62–0.93) | 0.84 (0.66–1.08) | 0.74 (0.60–0.91) | 0.82 (0.64–1.06) | 0.04 | 19 | |||
| rs1143623 | G-1464C | GG | 322 | 652 | 129 | 279 | 1.00 | 0.99 (0.77–1.28) | 1.00 | 0.97 (0.75–1.25) | 19 | ||
| GC-CC | 326 | 561 | 161 | 270 | 1.19 (0.98–1.45) | 1.25 (0.98–1.59) | 1.20 (0.98–1.46) | 1.24 (0.97–1.58) | 0.65 | 19 | |||
| rs1143627 | T-31C | TT | 281 | 545 | 105 | 233 | 1.00 | 0.91 (0.69–1.20) | 1.00 | 0.90 (0.68–1.19) | 19 | ||
| TC-CC | 369 | 672 | 183 | 319 | 1.07 (0.88–1.30) | 1.16 (0.91–1.47) | 1.07 (0.88–1.31) | 1.13 (0.89–1.44) | 0.27 | 19 | |||
| HMOX | rs2071746 | A-413T | AA | 76 | 180 | 42 | 80 | 1.00 (–) | 1.20 (0.75–1.91) | 1.00 (–) | 1.14 (0.70–1.86) | 47 | |
| AT/TT | 183 | 349 | 82 | 154 | 1.22 (0.88–1.70) | 1.30 (0.88–1.92) | 1.20 (0.85–1.69) | 1.27 (0.85–1.88) | 0.73 | 47 | |||
| IL6 | rs1800795 | GG | 62 | 129 | 27 | 53 | 1.00 (–) | 1.27 (0.73–2.23) | 1.00 (–) | 1.23 (0.70–2.18) | 20 | ||
| G-174C | CG and CC | 178 | 383 | 83 | 181 | 1.04 (0.73–1.50) | 1.08 (0.72–1.62) | 1.04 (0.72–1.50) | 1.07 (0.70–1.62) | 0.48 | 20 | ||
| IL8 | rs4073 | T-251A | TT | 60 | 101 | 23 | 59 | 1.00 (–) | 0.77 (0.43–1.38) | 1.00 (–) | 0.74 (0.40–1.37) | 20 | |
| AT and AA | 180 | 411 | 87 | 175 | 0.80 (0.55–1.15) | 0.96 (0.63–1.45) | 0.81 (0.55–1.19) | 0.97 (0.63–1.49) | 0.08 | 20 | |||
| rs3856806 | C1431T | CC | 175 | 374 | 78 | 176 | 1.00 (–) | 0.98 (0.71–1.36) | 1.00 (–) | 0.99 (0.71–1.38) | 20 | ||
| CT and TT | 65 | 138 | 32 | 58 | 0.98 (0.69–1.39) | 1.37 (0.86–2.20) | 1.01 (0.71–1.45) | 1.35 (0.83–2.21) | 0.23 | 20 | |||
| ABCB1 (MDR1) | rs1045642 | 3435 | CC | 32 | 41 | 39 | 79 | 1.00 (–) | 2.21 (1.17–4.17) | 1.00 (–) | 2.34 (1.22–4.48) | 14 | |
| CT-TT | 82 | 204 | 198 | 449 | 0.92 (0.60–1.41) | 0.83 (0.52–1.33) | 0.99 (0.63–1.54) | 0.86 (0.53–1.39) | 0.001 | 14 | |||
| rs3789243 | in 3 | GG | 27 | 54 | 64 | 160 | 1.00 (–) | 1.35 (0.77–2.35) | 1.00 (–) | 1.31 (0.74–2.32) | 14 | ||
| GA-AA | 87 | 191 | 173 | 368 | 1.65 (1.15–2.38) | 1.65 (1.09–2.49) | 1.66 (1.15–2.41) | 1.61 (1.06–2.46) | 0.26 | 14 | |||
| ABCG2 (BCRP) | rs2231142 | 421 | CC | 97 | 199 | 178 | 414 | 1.00 (–) | 1.20 (0.88–1.63) | 1.00 (–) | 1.18 (0.85–1.62) | 14 | |
| CA-AA | 17 | 46 | 59 | 114 | 0.86 (0.58–1.27) | 0.62 (0.35–1.10) | 0.86 (0.57–1.29) | 0.60 (0.33–1.08) | 0.09 | 14 | |||
| ABCC2 (MRP2) | rs717620 | C-24T | CC | 167 | 350 | 93 | 158 | 1.00 | 1.26 (0.99–1.61) | 1.00 | 1.23 (0.96–1.58) | 18 | |
| CT and TT | 96 | 195 | 33 | 85 | 1.08 (0.85–1.39) | 0.85 (0.58–1.24) | 1.09 (0.85–1.41) | 0.85 (0.58–1.25) | 0.05 | 18 | |||
| rs2273697 | G1249A | GG | 162 | 331 | 76 | 149 | 1.00 | 1.10 (0.84–1.44) | 1.00 | 1.10 (0.84–1.44) | 18 | ||
| AG and AA | 101 | 214 | 50 | 94 | 0.98 (0.77–1.26) | 1.05 (0.77–1.44) | 0.99 (0.77–1.27) | 1.01 (0.74–1.38) | 0.75 | 18 | |||
| rs3740066 | C3972T | CC | 92 | 201 | 51 | 100 | 1.00 | 1.18 (0.84–1.64) | 1.00 | 1.13 (0.81–1.58) | 18 | ||
| CT and TT | 171 | 344 | 75 | 143 | 1.11 (0.87–1.43) | 1.15 (0.85–1.56) | 1.12 (0.88–1.44) | 1.16 (0.86–1.58) | 0.70 | 18 | |||
| SLC22A4 (OCTN) | C1672T | CC | 76 | 208 | 45 | 75 | 1.00 (–) | 1.62 (1.02–2.57) | 1.00 (–) | 1.60 (1.00–2.55) | 20 | ||
| CT and TT | 164 | 304 | 65 | 159 | 1.39 (1.00–1.93) | 1.20 (0.81–1.78) | 1.36 (0.97–1.91) | 1.16 (0.78–1.75) | 0.01 | 20 | |||
| SLC220A5 (OCTN) | G-209C | GG | 65 | 182 | 34 | 61 | 1.00 (–) | 1.49 (0.89–2.49) | 1.00 (–) | 1.44 (0.85–2.42) | 20 | ||
| GC and CC | 175 | 330 | 76 | 173 | 1.42 (1.01–2.00) | 1.34 (0.91–1.99) | 1.38 (0.97–1.95) | 1.30 (0.87–1.96) | 0.10 | 20 | |||
| (OCTN) | rs4705950 | CC | 81 | 210 | 42 | 74 | 1.00 (–) | 1.38 (0.86–2.19) | 1.00 (–) | 1.35 (0.84–2.16) | 20 | ||
| CT and TT | 159 | 302 | 68 | 160 | 1.25 (0.90–1.73) | 1.19 (0.81–1.74) | 1.22 (0.87–1.70) | 1.15 (0.77–1.71) | 0.13 | 20 | |||
| NOD2/CARD15 | rs20668441 | C2104T | CC | 226 | 479 | 101 | 221 | 1.00 (–) | 1.06 (0.80–1.41) | 1.00 (–) | 1.06 (0.79–1.42) | 20 | |
| CT | 14 | 33 | 9 | 13 | 0.93 (0.48–1.80) | 1.46 (0.60–3.55) | 1.01 (0.52–1.98) | 1.32 (0.53–3.25) | 0.55 | 20 | |||
| G2722C | GG | 236 | 496 | 109 | 233 | 1.00 (–) | 1.08 (0.82–1.42) | 1.00 (–) | 1.06 (0.80–1.41) | 20 | |||
| GC | 4 | 16 | 1 | 1 | 0.77 (0.29–2.05) | 1.15 (0.07–18.74) | 0.64 (0.23–1.76) | 1.49 (0.09–25.09) | 0.75 | 20 | |||
| 3020insC | 0 | 231 | 496 | 108 | 225 | 1.00 (–) | 1.11 (0.84–1.47) | 1.00 (–) | 1.10 (0.83–1.47) | 20 | |||
| 1 | 9 | 16 | 2 | 9 | 1.04 (0.45–2.42) | 0.68 (0.17–2.64) | 1.00 (0.41–2.48) | 0.52 (0.13–2.18) | 0.16 | 20 | |||
| DLG5 | G113A/R30Q | GG | 191 | 416 | 99 | 188 | 1.00 (–) | 1.22 (0.90–1.64) | 1.00 (–) | 1.21 (0.88–1.64) | 20 | ||
| AG/AA | 49 | 96 | 11 | 46 | 1.10 (0.74–1.62) | 0.62 (0.32–1.20) | 1.14 (0.76–1.71) | 0.63 (0.32–1.25) | 0.02 | 20 | |||
| NFKB1 | rs28362491 | -94ins/del | II | 84 | 211 | 38 | 96 | 1.00 (–) | 1.09 (0.69–1.73) | 1.00 (–) | 1.05 (0.66–1.67) | 15 | |
| ID-DD | 175 | 318 | 86 | 138 | 1.46 (1.06–2.02) | 1.60 (1.09–2.35) | 1.43 (1.02–2.00) | 1.56 (1.05–2.32) | 0.87 | 15 | |||
| NR1I2 (PXR) | rs1523127 | A-24381C | AA | 86 | 176 | 45 | 85 | 1.00 (–) | 1.10 (0.70–1.73) | 1.00 (–) | 1.11 (0.70–1.76) | 15 | |
| AC-CC | 173 | 353 | 79 | 149 | 0.98 (0.71–1.36) | 1.08 (0.74–1.59) | 1.00 (0.72–1.40) | 1.07 (0.72–1.58) | 0.85 | 15 | |||
| rs2276707 | C8055T | CC | 161 | 303 | 76 | 145 | 1.00 (–) | 1.04 (0.74–1.47) | 1.00 (–) | 1.00 (0.70–1.43) | 15 | ||
| CT-TT | 98 | 226 | 48 | 89 | 0.87 (0.64–1.19) | 1.04 (0.69–1.58) | 0.88 (0.64–1.20) | 1.06 (0.69–1.61) | 0.42 | 15 | |||
| rs6785049 | A7635G | AA | 91 | 184 | 46 | 80 | 1.00 (–) | 1.10 (0.70–1.72) | 1.00 (–) | 1.07 (0.67–1.70) | 15 | ||
| AG-GG | 168 | 345 | 78 | 154 | 0.97 (0.70–1.34) | 1.07 (0.73–1.57) | 0.95 (0.68–1.33) | 1.03 (0.69–1.54) | 0.96 | 15 | |||
| NR1H2 (LXR) | rs1405655 | CC | 121 | 239 | 53 | 109 | 1.00 (–) | 0.98 (0.65–1.46) | 1.00 (–) | 0.93 (0.61–1.41) | 15 | ||
| CT-TT | 138 | 290 | 71 | 125 | 0.90 (0.67–1.23) | 1.10 (0.75–1.60) | 0.89 (0.65–1.22) | 1.08 (0.74–1.58) | 0.24 | 15 | |||
| rs2695121 | TT | 85 | 166 | 32 | 61 | 1.00 (–) | 1.08 (0.65–1.80) | 1.00 (–) | 1.02 (0.59–1.75) | 15 | |||
| CT-CC | 174 | 363 | 92 | 173 | 0.91 (0.65–1.26) | 1.02 (0.70–1.47) | 0.89 (0.63–1.24) | 0.98 (0.67–1.44) | 0.72 | 15 | |||
| rs6983267¶ | rs6983267 | GG | 127 | 221 | 111 | 184 | 1.00 | 0.99 (0.71–1.38) | 1.00 | 0.99 (0.70–1.40) | 46 | ||
| GT | 250 | 339 | 155 | 411 | 1.00 | 0.60 (0.47–0.76) | 1.00 | 0.61 (0.47–0.79) | 46 | ||||
| TT | 104 | 201 | 60 | 207 | 1.00 | 0.55 (0.38–0.81) | 1.00 | 0.52 (0.35–0.78) | 0.01 | 46 | |||
Nc, number of cases; Ns, number of sub-cohort members.
Adjusted for sex and age.
In addition, adjusted for smoking status, alcohol, HRT status (women only), BMI, intake of red and processed meat, and dietary fibre.
P-value for interaction between the polymorphisms and NSAID use for the adjusted risk estimates (IRR†). IRR† indicates the slope of the line of the association between genotype and use of NSAID (“No” and “Yes”). The P-values in Table 1 indicate whether the slopes of the two lines are statistically significantly different.
This study was adjusted for age, gender, body mass index, C-reactive protein level, pack years of smoking, cholesterol, physical activity, total energy intake, rheumatoid arthritis, osteoarthritis and hormone replacement therapy.
This study was adjusted for age, race, sex, regular nonsterioidal anti-inflammatory drug use (yes or no), body mass index (in tertiles), physical activity (in tertiles), history of colorectal cancer in a parent or sibling (yes or no), smoking (never, former or current smoker), alcohol consumption (0–4.9 g, 5–9.9 g, 10–14.9 g or ≥15.0 g per day), post-menopausal hormone use (pre-menopausal, never, former or current user), consumption of beef, pork or lamb as a main dish (0–3 times per month, once a week, 2–4 times per week or ≥5 times per week), and energy-adjusted calcium and folate intake (in tertiles).
None of the studies was adjusted for multiple testing.
Results
In total, 28 studies were identified which reported original data on gene–environment interactions with NSAID use in relation to CRC risk. All retrieved studies were evaluated according to the validity of the data on NSAID use as prospective studies; i.e. where information on NSAID use was collected prior to the development of CRC or obtained from unbiased sources (prospective cohorts, nested case–control studies) and as case–control studies with possible recall bias; i.e. studies with retrospective collection of data on NSAID use (case–control studies). In total, eight prospective14,15,17–20,22,46 and 21 case–control studies23,24,26–43,48 were identified. Tables 1 and S1 show results on interactions between NSAID use and polymorphisms in relation to CRC from prospective and case–control studies respectively. Figure 2 shows examples of the effects of NSAID intake on the risk of CRC for carriers of the wildtype and the variant alleles respectively. The studies vary considerably in design and definition of NSAID use (Table 2) and, furthermore, in numbers of included cases and controls (Tables 1 and S1).
Figure 2.
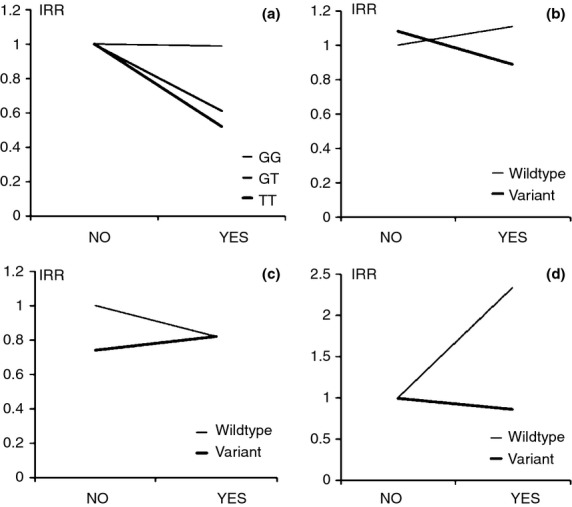
Effect of NSAID intake on risk of CRC for carriers of wildtype and variant alleles respectively. (a) rs6983267. (b) IL10 rs3024505, (c) IL1B rs4848306, (d) ABCB1 rs1945642. The P-values in Table 1 indicate whether the slopes of the two lines are different. An interaction effect between NSAID use and a polymorphism may theoretically result in considerable differential impact on the individual risk of CRC. Depending on the disease frequency in the population and the genotype distribution such interaction may also affect the disease frequency in the population. Variant allele frequencies are rs6983267: 0.50, IL10 rs3024505: 0.17, IL1B C-3737T: 0.43, ABCB1 C3435T: 0.59.14,19,46
Arachidonic acid pathway
PTGS1 (encoding COX-1) is constitutively expressed in many tissues, whereas PTGS2 (encoding COX-2) is induced by inflammatory stimuli such as cytokines. COX catalyses the rate-limiting conversion of AA to prostaglandins such as the pro-carcinogenic prostaglandin E2 (PGE2).7,8 Interactions between use of NSAIDs and PTGS1 and PTGS2, respectively, have been evaluated in prospective19,22 and case–control studies27,34,40 (Tables 1 and S1, respectively). No interaction between NSAID use and functional PTGS2 polymorphisms was found in two prospective studies (Table 1).19,22 A large case–control study including more than 3500 cases and 4900 controls evaluated six functional and marker polymorphisms in PTGS1 and one in PTGS2.27 They found interaction between NSAID use and PTGS1 rs3842787, rs10306135 and rs6478565 in relation to rectal cancer (P for interaction (Pint) = 0.05, 0.001 and 0.03 respectively) and PTGS2 rs20424 in relation to colon cancer (Pint = 0.001) in the discovery cohort, but no interactions were found in the validation cohort (Table S1).27 PTGS1 haplotype analysis was not performed.27 In addition, two other studies found no interaction between PTGS2 and NSAID use34,40 (Table S1).
ALOXs encodes for arachidonate lipoxygenases which convert AA into leukotrienes, including leukotriene B4 (LTB4), involved in colorectal cancer.7 Functional polymorphisms in ALOX5, AlOX12, ALOX15 and ALOX5AP (FLAP) encoding the arachidonate 5-lipoxygenase-activating protein were investigated (Table S1).28 Interactions with NSAID use were found for ALOX15 rs2619112, ALOX5AP rs9508832, rs9315053, rs4075692 and rs9551960 in relation to rectal cancer and ALOX5AP rs17239025 in relation to colon cancer (Table S1).28 The authors found that ALOX15 rs2619112 (G9562A) homozygous variant carriers benefitted more from NSAID use than wildtype allele carriers (Pint = 0.01).28 Variant allele carriers were at increased risk of rectal cancer compared to homozygous wildtype carriers (P = 0.05).28
Prostaglandins (PGE2, PGI2 and PGJ2) or their metabolites may be ligands for and activate PPARs encoded for by PPARD and PPARG.7 No interactions between NSAIDs and PPARD or PPARG were, however, found (Tables 1 and S1).15,41,42
The ultimate source of the essential fatty acid AA is diet. PLA2G1B encodes a calcium-dependent enzyme which catalyses the hydrolysis of fatty acids from dietary phospholipids in the intestine releasing AA for incorporation into the cellular membranes and eicosanoid synthesis.26 Interaction between NSAID use and PLA2G1B rs2070873 was found in relation to CRC (Table S1).26 The homozygous wildtype carriers benefitted more from NSAID use than variant allele carriers.26
ABCB1, ABCC2 and SLC22A4 encode transport proteins which may be involved in lipid transport such as phospholipids.49–51 Diet is a source of phospholipids which are converted to AA and may next lead to the formation of the pro-carcinogenic PGE2 and leukotriene B 4 (LTB4).7 Interactions between NSAID use and polymorphisms in ABCB1, ABCC2 and SLC22A4 in relation to risk of CRC were found in a prospective cohort (Table 1).14,18,20
These studies suggest interactions between NSAID use and PTGS1, PTGS2, ALOXs, PLA2G1B, ABCB1, ABCC2 and SLC22A4 polymorphisms in relation to risk of CRC, whereas no interaction was found for PTGS2 in other studies, including studies on prospective cohorts, and no interaction was found for PPAR.
Cytokines
The pro-inflammatory cytokines, interleukin (IL) IL-1B, IL-6, IL-8 and TNF-α, are early mediators of inflammatory response in the intestine. Leptin is involved in regulation of energy balance, immune and inflammatory responses. In case–control studies, interactions between NSAIDs and IL6 rs1800795 and LEP rs7799039 and rs2167270 were found (Table S1).36,37 Carriers of the variant IL6 rs1800795 G-174C C allele benefitted more from NSAID use than homozygous wildtype carriers.37 Likewise, carriers of the LEP rs7799039 wildtype allele and rs2167270 wildtype allele benefitted more from NSAID use than carriers of the other genotypes (Table S1).36 Interaction between NSAID and IL1B rs4848306 was found in a prospective study (Table 1).19 The IL1B homozygous wildtype genotype was associated with a higher risk of CRC and, furthermore, a reduced risk of CRC by intake of NSAIDs compared to the other genotypes.19 These findings suggest that NSAID use was more beneficial for those who had the highest risk of CRC, i.e. those with genetically determined high IL-1B activity.19 IL-1B induces COX-2 activity in response to inflammation.8,52
Furthermore, interactions between NSAIDs and the marker polymorphism near IL10 rs3024505 was found in a prospective cohort19 and between the functional IL10 polymorphism rs1800872 in a case–control study (Table 1 and Table S1, respectively).43 Thus, two different studies found interaction between NSAID use and genetic variation in or close to IL10.
Interactions between NSAID use and IL1B, LEP and IL6 in relation to CRC were suggested.
Wnt/β-catenin signalling
rs6983267 has been associated with risk of CRC in several studies.46 The most proximal gene in this region is MYC 335 kb downstream from rs6983267. MYC encodes a transcription factor involved in cell cycle progression, apoptosis and cellular transformation and the minor allele of rs6983267 has been found to up-regulate the MYC transcription leading to worsening of the prognosis of CRC.53 Interaction between aspirin use and rs6983267 was assessed in two prospective cohorts.46 Consistent results were found for the two cohorts.46 Compared with non-use, frequent aspirin use was associated with 48% risk reduction among carriers of the TT genotypes (95% confidence interval (95% CI) = 0.35–0.78) in contrast to carriers of the GG genotypes who had no effect of aspirin use (OR = 0.99, 95% CI = 0.70–1.40, Pint = 0.01).46 Thus, the protective effect of aspirin was confined to homozygous carriers of the protective T allele of rs6983267 who constitute ca. 25% of the population (variant allele frequency for rs6983267 is 0.5046). This study found interaction between aspirin use and rs6983267 near MYC in relation to CRC.
Other pathways
A case–control study found interaction between NSAIDs and transcription factor TCF7L2 in relation to risk of CRC. The TCF7L2 rs7903146 T-allele carriers were at reduced risk of CRC by NSAID use in contrast to the homozygous wildtype C-allele carriers (reference).35 A biological plausibility is suggested by another study which found association between TCF7L2 and risk of CRC.54
The SMAD7 gene encodes a nuclear protein which plays inhibitory roles in transforming growth factor-β pathway (TGF-β) (Table S1).23 No interaction between NSAID use and SMAD7 in relation to CRC was found.23
The mitogen-activated protein kinase (MAPK) pathways regulate cell proliferation and apoptosis.29 Interactions between NSAID use and MAP3K10 rs892117 in relation to risk of colon cancer, and between NSAID use and MAPK1 rs2548663, MAPK1 rs2298432, MAPK12 rs2272857 and MAPK14 rs851016 in relation to rectal cancer were found in a large case–control study (Table S1).29 However, the underlying biological effects were not readily interpretable as the functional effects of the polymorphisms are not known.
The NFκB pathway is involved in inflammatory response, apoptosis and cell proliferation.30 Interactions were found between NSAID use and NFKB1 (encoding NFκB) rs230490 in relation to risk of rectal cancer, and NSAID use and IKBKB (encoding IKKβ) rs6474387 and rs11986055 in relation to risk of colon cancer.30 No interaction was found between NSAID use and NFKB1 in a prospective cohort (Table 1).15
The proteins encoded by STAT6, TYK2 and JAK2 are members of the STAT family of transcription factors/tyrosine kinase pathway. In response to cytokines and growth factors, STAT family members are phosphorylated by the receptor-associated kinases, and then form homo- or heterodimers that translocate to the cell nucleus where they act as transcription activators. This pathway plays a central role in exerting IL-4-mediated biological responses. Interactions between NSAID and the marker polymorphisms TYK2 rs280521 in relation to colon cancer, and NSAID use and TYK2 rs280521, STAT6 rs324011 and JAK2 rs10815160 in relation to rectal cancer were found (Table S1).32 Carriers of the variant allele of TYK2 rs280521 benefitted most from NSAID use compared to wildtype carriers (Pint = 0.03 and Pint = 0.009 in relation to colon and rectal cancer respectively). The potential biological effect of TYK2 rs280521 is not known.
No interactions were found between NSAIDs and NR1I2 or NR1H2, encoding the xenobiotic receptors, members of the nuclear receptor superfamily, PXR and LXR, in relation to CRC in a prospective cohort (Table 1).15
The principal degradation pathway for aspirin is hydroxylation and glucuronidation by UGT1A6 whereas non-aspirin NSAIDs may be metabolised by cytochrome P450 enzymes followed by conjugation.42 Interaction was found between NSAID use and CYP2C9 polymorphisms in relation to risk of CRC in a large study.42 Enhanced protection by use of the NSAID ibuprofen among slower metabolising CYP2C9 variant carriers compared to fast metabolisers was found (Table S1).42 This result may suggest that slow metabolism of ibuprofen potentates the effects of the drug. Other studies found no interaction between NSAID use and polymorphisms in UGT1A6, CYP2C8 and CYP2C9 in relation to risk of CRC (Table S1).34,42
Toll like receptors (TLR) play a fundamental role in pathogen recognition and activation of innate immunity. They mediate host responses via NFκB activation and stimulation of the production of cytokines. One case–control study found that carriers of the variant alleles of TLR2 rs7656411 and TLR3 rs11721827 benefitted more from NSAID use than the homozygous wildtype carriers (Table S1).33
TERT encodes telomerase which is a ribonucleoprotein polymerase that maintains telomere ends by addition of the telomere repeat TTAGGG. Deregulation of telomerase expression in somatic cells may be involved in oncogenesis. Interaction between NSAID use and TERT in relation to risk of CRC was found in a large case–control study.31 TERT-CLPTM1L rs2853668 homozygous variant allele carriers benefitted more from NSAID use than wildtype carriers (Table S1).31
Interaction between NSAID use and TERT-CLPTM1L in relation to risk of CRC was suggested.
The DLG5 gene product localises to cell–cell adhesion sites and enhance degradation of TGF-β receptors.55 One study found that the DLG5 G113A A-allele carriers benefitted more from NSAID use than homozygous wildtype carriers (Table 1).20
Interaction between NSAID use and DLG5 in relation to risk of CRC was suggested.
The TP53 gene product regulates DNA repair, differentiation and apoptosis.38 Interaction between NSAID use and TP53 Arg72Pro and p53PIN3 intron 3 single repeat nucleotide polymorphisms was assessed in relation to risk of CRC (Table S1).38 For both polymorphisms, it was found that the homozygous wildtype carriers benefitted more from use of NSAIDs than variant allele carriers.38
Interactions between NSAID use and genes encoding transcription factors in oncogenic, inflammatory and drug metabolisms pathways in relation to risk of CRC were suggested.
Discussion
In this review, we evaluated interactions between NSAID use and polymorphisms in relation to CRC. Our aim was to identify biological pathways involved in the protective effects of NSAIDs and to identify polymorphisms that predict response to NSAIDs in relation to risk of CRC. Replication of previous findings is an essential step in genetic epidemiology. In this systematic review, one prospective and one case–control study identified the IL10 rs3024505 and rs1800872 respectively.19,43 In the study by NAN et al., two prospective cohorts were included and consistent results for the two cohorts were reported.46 We found indications that aspirin interacted with rs6983267 close to MYC and NSAIDs interacted with rs3024505 and rs1800872 in or close to IL10 in preventing CRC, whereas no interaction with PTGS2 was detected. Other findings of interaction between genes in inflammatory and oncogenic pathways with NSAIDs are considered suggestive.
One important finding was that there were no consistent significant interactions between PTGS (encoding COX) polymorphisms, and NSAID use in relation to risk of CRC. However, a large body of evidence implicates COX-2 in CRC. Animal studies indicate that COX-2 and the COX-2-derived PGE2 promote colorectal carcinogenesis.7,56 In humans, COX-2 was overexpressed in tumour tissue in a high percentage of CRC cases.57 Furthermore, increased COX-2 expression in tumour tissue was found to be associated with reduced survival among the CRC patients and aspirin use was found to selectively reduce risk and, furthermore, prolong survival of patients with COX-2 expressing CRC.58,59 Our genetic epidemiological studies showed that low-activity associated polymorphism PTGS2 A-1195G GG genotype was associated with increased risk of ulcerative colitis (a risk factor for CRC) and CRC indicating that genetically determined low COX-2 activity is a risk factor for CRC.19,60 Together, these studies indicate a role of COX-2 in colorectal carcinogenesis. Also, selective inhibition of COX-2 has been found to reduce risk of CRC in animal studies and observational studies.58 NSAIDs is a heterogeneous group of drugs.8 Perhaps, the most widely used daily NSAID is low-dose aspirin for cardiological prevention. The incidence and mortality rate due to colorectal cancer was significantly reduced when at least 75 mg of aspirin are taken daily for several years.59 Although the primary effects of non-aspirin NSAIDs are competitive inhibition of COX-1 and COX-2, aspirin leads to irreversible inactivation of COX-1. Low-dose aspirin leads to irreversible inactivation of COX-1 by acetylation of serine residues in contrast to non-aspirin NSAIDs. Recovery of COX activity will therefore depend on de novo synthesis. The platelets have no nucleus and aspirin treatment thus leads to permanent extinction of the COX activity. COX-1 is constitutively expressed in the intestine whereas COX-2 is induced during inflammation after stimulation by cytokines. However, colon cells are nucleated and therefore able to generate de novo COX and, furthermore, have a high cell turnover leading to renewal of the cell population every 5 days. The half-life of aspirin in the blood is 20 min.52 Luminal enteric-coated aspirin reaching the intestine may potentially contribute to the intestinal effects however, this topic is not illuminated. Moreover, aspirin has been found to be 170 times more potent in inhibiting COX-1 than COX-2.52 Thus, higher doses of aspirin and shorter dosing intervals are likely to be required to ensure inhibition of COX-2 than used for cardio-protective usage. Our results suggest that PTGS2 polymorphisms play a minor role in clinical effects of NSAIDs in relation to prevention of CRC. Our result is compatible with the finding that aspirin sufficient to inhibit COX-1 but not COX-2 was sufficient for inhibition of prostaglandin synthesis in the colon.5 Also, COX-2-independent NSAID mechanisms in relation to CRC prevention have been suggested, however, most of these have been based on animal or in vitro experiments which may not represent the clinical effects.5,61,62
The anti-inflammatory IL-10 is essential for maintaining intestinal homoeostasis by inducing the anti-inflammatory p50/p50 Nuclear Factor kappa Beta (NFκB) homodimer and thereby blocking induction of NFκB-derived pro-inflammatory cytokines such as IL-1B.63 Individuals carrying the IL10 rs1800872 (C-592T) variant allele had a significantly lower risk of CRC by the use of NSAIDs than homozygous carrier of the wildtype.43 The functional IL10 rs1800872 variant was associated with low IL-10 activity64–66 indicating that subjects with low genetically determined anti-inflammatory IL-10 response in the intestine may benefit from NSAID use whereas homozygous wildtype carriers were already protected.43
One of the most essential oncogenic pathways in CRC is WNT/cadherin-associated protein β1 (CTNNB1 or β-catenin) signalling. CTNNB1 is a part of the tight junctions which are required for the creation and maintenance of epithelial cell layers by regulating cell growth and adhesion between cells. CTNNB1 have been implicated in risk of CRC. Aspirin inhibits CTNNB1 signalling.46 rs6983267 on chromosome 8q24 is a CRC susceptibility locus (CRCS6, colorectal cancer susceptibility to, 6). Thus, the T allele is associated with 15% to 18% lower risk of CRC compared to the G allele.46 rs6983267 has been suggested to affect binding activity of transcription factor 7-like 2 (TCF7L2) to CTNNB1 thereby altering expression of target oncogenes, including MYC.46 Nan et al. found that the protective effect of aspirin was confined to carriers of the T allele of rs6983267 whereas carriers of the G allele had no effect of aspirin use.46 The authors furthermore performed in vitro functional analysis of the rs6983267 polymorphism on a LS174T cell line heterozygous for rs6983267. The analysis suggested that rs6983267 interacted with aspirin by affecting the binding of TCF7L2 in an allele-specific way.46 Higher doses of aspirin led to a stronger inhibition of the binding of the transcription factor to the T allele.46 Thus, rs6983267 seemed directly to modify the effect of aspirin. The finding of association in the two cohorts is thus supported by functional findings.
All others found interactions between NSAID use and polymorphisms must be regarded as suggestive.
Assessment of gene–environment interactions is dependent on objective and unbiased information on lifestyle factors such as NSAID use by, e.g. linkage to prescription registers. Prospective studies have the advantage that information from questionnaires is of the same quality. Furthermore, large studies are needed to have sufficient power to assess gene–environment interactions. On the other hand, pooling studies from different populations may reduce power for detecting gene–environment interactions because some differences in lifestyle factors cannot be accounted for in the studies. Another approach may be to search for gene–environment interactions in relation to adenomatous polyps. Thereby the gene effects may be evaluated in the colorectal adenoma–carcinoma sequence context. However, no studies were found on rs6983267 (close to MYC), rs3024505 or rs1800872 (in or close to IL10) and adenomatous polyps. The used definitions of NSAID intake and sample size varies considerable among the studies included in this review (Tables 2 and S1). Long-term daily use of NSAIDs has been found to have the most pronounced effect on CRC prevention4 whereas dosing every second day seems to be less effective.67,68 Adjusting for confounders is important, and all results on prospective cohorts reported in Table 1 were at least adjusted for age and sex (Table 1). Candidate gene studies have hypothesised interactions with NSAID in relation to CRC and, therefore, multiple testing was not applicable.69 Instead, replication of previous findings is considered an essential step in genetic epidemiology. Also, publication bias might have occurred; data on positive associations were reported in tables whereas data on negative results may be omitted. Furthermore, in this study, we were unable to separate aspirin and non-aspirin NSAIDs or distinguish between CRC in the proximal or distal colon.70,71
In conclusion, this study emphasised that identification of gene–environmental interactions is a valuable tool in the search for underlying pathways and genes involved in the actions of NSAIDs.
We found indications that aspirin interacts with rs6983267 close to MYC and NSAIDs interact with rs3024505 and rs1800872 in or close to IL10 in preventing CRC, whereas no interaction with PTGS-2 was detected. Other findings of interaction between genes in inflammatory and oncogenic pathways with NSAIDs were considered suggestive. An interaction between NSAID use and a polymorphism may theoretically result in considerably differential impact on the individual risk of CRC. Depending on the disease frequency and the genotype distribution in the population such interaction may potentially affect the disease frequency in the population. Although the basis for stratifying the patients for optimal treatment is not yet available, the results presented here illustrates the feasibility of the approach. Studies on interactions of long-term use of NSAIDs, polymorphisms and CRC in large well-characterised prospective cohorts are warranted. Also, the publishing of both positive and negative results from studies attempting to replicate previous findings is important to advance further.
Acknowledgments
Declaration of personal interests: Staff at the Libraries, Regional Hospital Viborg and Hospital of Southern Jutland, are thanked for help. VA has served as a consultant for MSD and Janssen.
Declaration of funding interests: None.
Authorship
Guarantor of the article: VA.
Author contributions: VA perceived the study concept and design, acquisition of data, analysis and drafting of the manuscript; both authors contributed to the interpretation and critical review of the manuscript for important intellectual content. All authors approved the final version of the article.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
Table S1. Case–control studies on interactions between NSAID and polymorphisms in relation to colorectal cancer (CRC), colon cancer only (CC) and rectal cancer only (RC).
References
- 1.World Cancer Research Fund International. http://www.wcrf.org/cancer_statistics/data_specific_cancers/index.php, 2014.
- 2.Huxley RR, Ansary-Moghaddam A, Clifton P, Czernichow S, Parr CL, Woodward M. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. Int J Cancer. 2009;125:171–80. doi: 10.1002/ijc.24343. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, DuBois RN. The role of anti-inflammatory drugs in colorectal cancer. Annu Rev Med. 2013;64:131–44. doi: 10.1146/annurev-med-112211-154330. [DOI] [PubMed] [Google Scholar]
- 4.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–13. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 5.Chan AT, Arber N, Burn J, et al. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev Res (Phila) 2012;5:164–78. doi: 10.1158/1940-6207.CAPR-11-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahin IH, Hassan MM, Garrett CR. Impact of non-steroidal anti-inflammatory drugs on gastrointestinal cancers: current state-of-the science. Cancer Lett. 2014;345:249–57. doi: 10.1016/j.canlet.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Wang D, DuBois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–93. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacchi S, Palumbo P, Sponta A, Coppolino MF. Clinical pharmacology of non-steroidal anti-inflammatory drugs: a review. Antiinflamm Antiallergy Agents Med Chem. 2012;11:52–64. doi: 10.2174/187152312803476255. [DOI] [PubMed] [Google Scholar]
- 9.Piazza GA, Rahm AL, Krutzsch M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995;55:3110–6. [PubMed] [Google Scholar]
- 10.Wang D, Xia D, DuBois RN. The crosstalk of PTGS2 and EGF signaling pathways in colorectal cancer. Cancers (Basel) 2011;3:3894–908. doi: 10.3390/cancers3043894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaish V, Piplani H, Rana C, Vaiphei K, Sanyal SN. NSAIDs may regulate EGR-1-mediated induction of reactive oxygen species and non-steroidal anti-inflammatory drug-induced gene (NAG)-1 to initiate intrinsic pathway of apoptosis for the chemoprevention of colorectal cancer. Mol Cell Biochem. 2013;378:47–64. doi: 10.1007/s11010-013-1593-y. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Baek SJ, Eling TE. The diverse roles of nonsteroidal anti-inflammatory drug activated gene (NAG-1/GDF15) in cancer. Biochem Pharmacol. 2013;85:597–606. doi: 10.1016/j.bcp.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tinsley HN, Grizzle WE, Abadi A, et al. New NSAID targets and derivatives for colorectal cancer chemoprevention. Recent Results Cancer Res. 2013;191:105–20. doi: 10.1007/978-3-642-30331-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersen V, Ostergaard M, Christensen J, Overvad K, Tjonneland A, Vogel U. Polymorphisms in the xenobiotic transporter Multidrug Resistance 1 (MDR1) gene and interaction with meat intake in relation to risk of colorectal cancer in a Danish prospective case-cohort study. BMC Cancer. 2009;9:407. doi: 10.1186/1471-2407-9-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen V, Christensen J, Overvad K, Tjonneland A, Vogel U. Polymorphisms in NFkB, PXR, LXR and risk of colorectal cancer in a prospective study of Danes. BMC Cancer. 2010;10:484. doi: 10.1186/1471-2407-10-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersen V, Holst R, Vogel U. Systematic review: diet-gene interactions and the risk of colorectal cancer. Aliment Pharmacol Ther. 2013;37:383–91. doi: 10.1111/apt.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen V, Egeberg R, Tjonneland A, Vogel U. Interaction between interleukin-10 (IL-10) polymorphisms and dietary fibre in relation to risk of colorectal cancer in a Danish case-cohort study. BMC Cancer. 2012;12:183. doi: 10.1186/1471-2407-12-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersen V, Egeberg R, Tjonneland A, Vogel U. ABCC2 transporter gene polymorphisms, diet and risk of colorectal cancer: a Danish prospective cohort study. Scand J Gastroenterol. 2012;47:572–4. doi: 10.3109/00365521.2012.668933. [DOI] [PubMed] [Google Scholar]
- 19.Andersen V, Holst R, Kopp TI, Tjonneland A, Vogel U. Interactions between Diet, Lifestyle and IL10, IL1B, and PTGS2/COX-2 Gene Polymorphisms in Relation to Risk of Colorectal Cancer in a Prospective Danish Case-Cohort Study. PLoS ONE. 2013;8:e78366. doi: 10.1371/journal.pone.0078366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogel U, Christensen J, Dybdahl M, et al. Prospective study of interaction between alcohol, NSAID use and polymorphisms in genes involved in the inflammatory response in relation to risk of colorectal cancer. Mutat Res. 2007;624:88–100. doi: 10.1016/j.mrfmmm.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siemes C, Visser LE, Coebergh JW, Hofman A, Uitterlinden AG, Stricker BH. Protective effect of NSAIDs on cancer and influence of COX-2 C(-765G) genotype. Curr Cancer Drug Targets. 2008;8:753–64. doi: 10.2174/156800908786733414. [DOI] [PubMed] [Google Scholar]
- 23.Jiang X, Castelao JE, Vandenberg D, et al. Genetic variations in SMAD7 are associated with colorectal cancer risk in the colon cancer family registry. PLoS ONE. 2013;8:e60464. doi: 10.1371/journal.pone.0060464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slattery ML, Lundgreen A, Welbourn B, Wolff RK, Corcoran C. Oxidative balance and colon and rectal cancer: interaction of lifestyle factors and genes. Mutat Res. 2012;734:30–40. doi: 10.1016/j.mrfmmm.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daraei A, Salehi R, Mohamadhashem F. PTGS2 (COX2) -765G>C gene polymorphism and risk of sporadic colorectal cancer in Iranian population. Mol Biol Rep. 2012;39:5219–24. doi: 10.1007/s11033-011-1319-8. [DOI] [PubMed] [Google Scholar]
- 26.Abbenhardt C, Poole EM, Kulmacz RJ, et al. Phospholipase A2G1B polymorphisms and risk of colorectal neoplasia. Int J Mol Epidemiol Genet. 2013;4:140–9. [PMC free article] [PubMed] [Google Scholar]
- 27.Makar KW, Poole EM, Resler AJ, et al. COX-1 (PTGS1) and COX-2 (PTGS2) polymorphisms, NSAID interactions, and risk of colon and rectal cancers in two independent populations. Cancer Causes Control. 2013;24:2059–75. doi: 10.1007/s10552-013-0282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinstein SE, Heath L, Makar KW, et al. Genetic variation in the lipoxygenase pathway and risk of colorectal neoplasia. Genes Chromosom Cancer. 2013;52:437–49. doi: 10.1002/gcc.22042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slattery ML, Lundgreen A, Wolff RK. MAP kinase genes and colon and rectal cancer. Carcinogenesis. 2012;33:2398–408. doi: 10.1093/carcin/bgs305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seufert BL, Poole EM, Whitton J, et al. IkappaBKbeta and NFkappaB1, NSAID use and risk of colorectal cancer in the Colon Cancer Family Registry. Carcinogenesis. 2013;34:79–85. doi: 10.1093/carcin/bgs296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellatt AJ, Wolff RK, Herrick J, Lundgreen A, Slattery ML. TERT's role in colorectal carcinogenesis. Mol Carcinog. 2013;52:507–13. doi: 10.1002/mc.21885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slattery ML, Lundgreen A, Kadlubar SA, Bondurant KL, Wolff RK. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol Carcinog. 2013;52:155–66. doi: 10.1002/mc.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slattery ML, Herrick JS, Bondurant KL, Wolff RK. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int J Cancer. 2012;130:2974–80. doi: 10.1002/ijc.26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson CL, Plummer SJ, Merkulova A, et al. No association between cyclooxygenase-2 and uridine diphosphate glucuronosyltransferase 1A6 genetic polymorphisms and colon cancer risk. World J Gastroenterol. 2009;15:2240–4. doi: 10.3748/wjg.15.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slattery ML, Folsom AR, Wolff R, Herrick J, Caan BJ, Potter JD. Transcription factor 7-like 2 polymorphism and colon cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:978–82. doi: 10.1158/1055-9965.EPI-07-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slattery ML, Wolff RK, Herrick J, Caan BJ, Potter JD. Leptin and leptin receptor genotypes and colon cancer: gene-gene and gene-lifestyle interactions. Int J Cancer. 2008;122:1611–7. doi: 10.1002/ijc.23135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slattery ML, Wolff RK, Herrick JS, Caan BJ, Potter JD. IL6 genotypes and colon and rectal cancer. Cancer Causes Control. 2007;18:1095–105. doi: 10.1007/s10552-007-9049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan XL, Nieters A, Hoffmeister M, Beckmann L, Brenner H, Chang-Claude J. Genetic polymorphisms in TP53, nonsteroidal anti-inflammatory drugs and the risk of colorectal cancer: evidence for gene-environment interaction? Pharmacogenet Genomics. 2007;17:639–45. doi: 10.1097/FPC.0b013e3280d5121c. [DOI] [PubMed] [Google Scholar]
- 39.Samowitz WS, Wolff RK, Curtin K, et al. Interactions between CYP2C9 and UGT1A6 polymorphisms and nonsteroidal anti-inflammatory drugs in colorectal cancer prevention. Clin Gastroenterol Hepatol. 2006;4:894–901. doi: 10.1016/j.cgh.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 40.Sansbury LB, Millikan RC, Schroeder JC, et al. COX-2 polymorphism, use of nonsteroidal anti-inflammatory drugs, and risk of colon cancer in African Americans (United States) Cancer Causes Control. 2006;17:257–66. doi: 10.1007/s10552-005-0417-0. [DOI] [PubMed] [Google Scholar]
- 41.Slattery ML, Curtin K, Wolff R, et al. PPARgamma and colon and rectal cancer: associations with specific tumor mutations, aspirin, ibuprofen and insulin-related genes (United States) Cancer Causes Control. 2006;17:239–49. doi: 10.1007/s10552-005-0411-6. [DOI] [PubMed] [Google Scholar]
- 42.McGreavey LE, Turner F, Smith G, et al. No evidence that polymorphisms in CYP2C8, CYP2C9, UGT1A6, PPARdelta and PPARgamma act as modifiers of the protective effect of regular NSAID use on the risk of colorectal carcinoma. Pharmacogenet Genomics. 2005;15:713–21. doi: 10.1097/01.fpc.0000174786.85238.63. [DOI] [PubMed] [Google Scholar]
- 43.Macarthur M, Sharp L, Hold GL, Little J, El-Omar EM. The role of cytokine gene polymorphisms in colorectal cancer and their interaction with aspirin use in the northeast of Scotland. Cancer Epidemiol Biomarkers Prev. 2005;14:1613–8. doi: 10.1158/1055-9965.EPI-04-0878. [DOI] [PubMed] [Google Scholar]
- 44.Lin HJ, Lakkides KM, Keku TO, et al. Prostaglandin H synthase 2 variant (Val511Ala) in African Americans may reduce the risk for colorectal neoplasia. Cancer Epidemiol Biomarkers Prev. 2002;11:1305–15. [PubMed] [Google Scholar]
- 45.Cox DG, Pontes C, Guino E, et al. Polymorphisms in prostaglandin synthase 2/cyclooxygenase 2 (PTGS2/COX2) and risk of colorectal cancer. Br J Cancer. 2004;91:339–43. doi: 10.1038/sj.bjc.6601906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nan H, Morikawa T, Suuriniemi M, et al. Aspirin Use, 8q24 Single Nucleotide Polymorphism rs6983267, and Colorectal Cancer According to CTNNB1 Alterations. J Natl Cancer Inst. 2013;105:1852–61. doi: 10.1093/jnci/djt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andersen V, Christensen J, Overvad K, Tjønneland A, Vogel U. Heme oxygenase-1 polymorphism is not associated with risk of colorectal cancer: a Danish prospective study. Eur J Gastroenterol Hepatol. 2011;23:282–5. doi: 10.1097/MEG.0b013e3283417f76. [DOI] [PubMed] [Google Scholar]
- 48.Angstadt AY, Hartman TJ, Lesko SM, et al. The effect ofUGT1AandUGT2Bpolymorphisms on colorectal cancer risk: Haplotype associations and gene-environment interactions. Genes Chromosom Cancer. 2014;53:454–66. doi: 10.1002/gcc.22157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coleman JA, Quazi F, Molday RS. Mammalian P4-ATPases and ABC transporters and their role in phospholipid transport. Biochim Biophys Acta. 2013;1831:555–74. doi: 10.1016/j.bbalip.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tarling EJ, de Aguiar Vallim TQ, Edwards PA. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol Metab. 2013;24:342–50. doi: 10.1016/j.tem.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quazi F, Molday RS. Lipid transport by mammalian ABC proteins. Essays Biochem. 2011;50:265–90. doi: 10.1042/bse0500265. [DOI] [PubMed] [Google Scholar]
- 52.Awtry EH, Loscalzo J. Aspirin Circulation. 2000;101:1206–18. doi: 10.1161/01.cir.101.10.1206. [DOI] [PubMed] [Google Scholar]
- 53.Takatsuno Y, Mimori K, Yamamoto K, et al. The rs6983267 SNP is associated with MYC transcription efficiency, which promotes progression and worsens prognosis of colorectal cancer. Ann Surg Oncol. 2013;20:1395–402. doi: 10.1245/s10434-012-2657-z. [DOI] [PubMed] [Google Scholar]
- 54.Hazra A, Fuchs CS, Chan AT, Giovannucci EL, Hunter DJ. Association of the TCF7L2 polymorphism with colorectal cancer and adenoma risk. Cancer Causes Control. 2008;19:975–80. doi: 10.1007/s10552-008-9164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sezaki T, Tomiyama L, Kimura Y, Ueda K, Kioka N. Dlg5 interacts with the TGF-beta receptor and promotes its degradation. FEBS Lett. 2013;587:1624–9. doi: 10.1016/j.febslet.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 56.Wang D, DuBois RN. An inflammatory mediator, prostaglandin E2, in colorectal cancer. Cancer J. 2013;19:502–10. doi: 10.1097/PPO.0000000000000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–8. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 58.Rostom A, Dube C, Lewin G, et al. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146:376–89. doi: 10.7326/0003-4819-146-5-200703060-00010. [DOI] [PubMed] [Google Scholar]
- 59.Fischer SM, Hawk ET, Lubet RA. Coxibs and other nonsteroidal anti-inflammatory drugs in animal models of cancer chemoprevention. Cancer Prev Res (Phila) 2011;4:1728–35. doi: 10.1158/1940-6207.CAPR-11-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andersen V, Nimmo E, Krarup HB, et al. Cyclooxygenase-2 (COX-2) polymorphisms and risk of inflammatory bowel disease in a Scottish and Danish case-control study. Inflamm Bowel Dis. 2011;17:937–46. doi: 10.1002/ibd.21440. [DOI] [PubMed] [Google Scholar]
- 61.Ferrandez A, Piazuelo E, Castells A. Aspirin and the prevention of colorectal cancer. Best Pract Res Clin Gastroenterol. 2012;26:185–95. doi: 10.1016/j.bpg.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Albeniz X, Chan AT. Aspirin for the prevention of colorectal cancer. Best Pract Res Clin Gastroenterol. 2011;25:461–72. doi: 10.1016/j.bpg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp Immunol. 2004;135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet. 1997;24:1–8. doi: 10.1111/j.1365-2370.1997.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 65.Yilmaz V, Yentur SP, Saruhan-Direskeneli G. IL-12 and IL-10 polymorphisms and their effects on cytokine production. Cytokine. 2005;30:188–94. doi: 10.1016/j.cyto.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 66.Crawley E, Kay R, Sillibourne J, Patel P, Hutchinson I, Woo P. Polymorphic haplotypes of the interleukin-10 5' flanking region determine variable interleukin-10 transcription and are associated with particular phenotypes of juvenile rheumatoid arthritis. Arthritis Rheum. 1999;42:1101–8. doi: 10.1002/1529-0131(199906)42:6<1101::AID-ANR6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 67.Cook NR, Lee IM, Gaziano JM, et al. Low-dose aspirin in the primary prevention of cancer: the Women's Health Study: a randomized controlled trial. JAMA. 2005;294:47–55. doi: 10.1001/jama.294.1.47. [DOI] [PubMed] [Google Scholar]
- 68.Gann PH, Manson JE, Glynn RJ, Buring JE, Hennekens CH. Low-dose aspirin and incidence of colorectal tumors in a randomized trial. J Natl Cancer Inst. 1993;85:1220–4. doi: 10.1093/jnci/85.15.1220. [DOI] [PubMed] [Google Scholar]
- 69.Perneger TV. What's wrong with Bonferroni adjustments. BMJ. 1998;316:1236–8. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruder EH, Laiyemo AO, Graubard BI, Hollenbeck AR, Schatzkin A, Cross AJ. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. Am J Gastroenterol. 2011;106:1340–50. doi: 10.1038/ajg.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rothwell PM, Wilson M, Elwin CE, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–50. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Case–control studies on interactions between NSAID and polymorphisms in relation to colorectal cancer (CRC), colon cancer only (CC) and rectal cancer only (RC).