

Abstract
Background
The tumor suppressor p53 is a sequence-specific transcription factor that regulates an extensive network of coding genes, long non-coding RNAs and microRNAs, that establish intricate gene regulatory circuits influencing many cellular responses beyond the prototypical control of cell cycle, apoptosis and DNA repair.
Methods
Using bioinformatic approaches, we identified an additional group of candidate microRNAs (miRs) under direct p53 transcriptional control. To validate p53 family-mediated responsiveness of the newly predicted target miRs we first evaluated the potential for wild type p53, p63β and p73β to transactivate from p53 response elements (REs) mapped in the miR promoters, using an established yeast-based assay.
Results
The REs found in miR-10b, -23b, -106a, -151a, -191, -198, -202, -221, -320, -1204, -1206 promoters were responsive to p53 and 8 of them were also responsive to p63β or p73β. The potential for germline p53 mutations to drive transactivation at selected miR-associated REs was also examined. Chromatin Immuno-Precipitation (ChIP) assays conducted in doxorubicin-treated MCF7 cells and HCT116 p53+/+ revealed moderate induction of p53 occupancy at the miR-202, -1204, -1206, -10b RE-containing sites, while weak occupancy was observed for the miR-23b-associated RE only in MCF7 cells. RT-qPCR analyses cells showed modest doxorubicin- and/or Nutlin-dependent induction of the levels of mature miR-10b, -23b, -151a in HCT116 p53+/+ and MCF7 cells. The long noncoding RNA PVT1 comprising miR-1204 and −1206 was weakly induced only in HCT116 p53+/+ cells, but the mature miRs were not detected. miR-202 expression was not influenced by p53-activating stimuli in our cell systems.
Conclusions
Our study reveals additional miRs, particularly miR-10b and miR-151a, that could be directly regulated by the p53-family of transcription factors and contribute to the tuning of p53-induced responses.
Keywords: p53 family, miRNA, Promoter occupancy, Transcription regulation
Background
The master regulator p53 is a prominent tumor suppressor gene, functioning in the cell as a tetrameric (dimer of dimers) sequence-specific transcription factor, able to bind to two copies of a decameric sequence with the RRRCWWGYYY consensus (where R stands for a purine, W for A/T and Y for a pyrimidine) representing the so called p53-response element (p53-RE) [1]. p53 is known to be inducible in response to a large number of cellular stress signals that, besides genotoxic stress, include carbon and oxygen deficiencies, perturbations of the translation apparatus, excessive proliferation signals, alteration in microtubule dynamics [2,3]. There are >100 established p53 targets genes that link p53 to cell cycle arrest, apoptosis, DNA repair and inhibition of angiogenesis [3-6]. More recently, p53 was demonstrated to modulate the expression of genes able to modify glucose as well as lipid metabolism, induction of autophagy, immune responses and cell motility [7-11].
A direct role of p53 on the activation of microRNA (miRs) expression as well as a role on selective maturation of microRNA precursors has been recently established [12,13]. miRs are small non coding RNAs typically of 21–25 nucleotides in length that regulate gene expression by inhibiting translation or repressing stability of target messenger RNAs including those coding for oncogenes and tumor suppressor proteins [14]. Dysregulation in miR expression has been reported in various cancers and can contribute to tumorigenesis [15]. The first evidence of a p53-dependent regulation of miR genes was provided by He et al., who identified a family of miRs, namely miR-34a-c, whose expression reflected the p53 status [16]. The authors demonstrated that genes encoding miR-34 family cluster were direct transcriptional targets of p53 and that their induced expression levels upon genotoxic or oncogenic stress was dependent on p53 expression, both in vitro and in vivo. Moreover, He et al. identified the DNA sequences responsible for the p53 responsiveness of those miRs. A year later another group of miRs, (miR-192, -194 and −215) was identified as targets of p53 and their ability to increase the level of CDKN1A (p21CIP1) and to function as drivers of cell cycle arrest was established [17]. Examples of feedback loops or regulatory circuits comprising p53, a target miR and target mRNAs were discovered. For example, p53-directed repression of c-Myc has also been linked to p53-dependent induction of miR-145 [18]. miR-107 was demonstrated to be activated by p53 and to cooperate in its cancer suppressive function through the inhibition of HIF-1β and, consequently, tumor angiogenesis [19]. The p53 targeted miR-34a was shown to modulate SIRT1 [20]. More recently, Jin et al. surprisingly found that p53 directly induced the transcription of miR-149*, which in turn can target the glycogen synthase kinase-3α mRNA, resulting in elevated expression of Mcl-1 and resistance to apoptosis in melanoma cells, thus providing a rational explanation for the poor ability of p53 to suppress melanoma progression [21].
Furthermore, it has been demonstrated that p53 itself can be indirectly activated by the miR-29 family members (miR-29a, -29b and -29c), which inhibit the expression of p85 alpha (the regulatory subunit of the phosphatidylinositol-3 kinase, PI3K) and CDC42 (Cell division cycle 42, a Rho family GTPase), thereby decreasing their inhibitory effect on p53 [22]. Alternatively, miRs can also negatively regulate p53 expression as observed for miR-1285, miR-504, miR-33, miR-380, miR-30d, miR-25 and miR-125b [23-28].
The mechanisms regulating in vivo p53 transactivation specificity still need to be fully understood, but require in most cases the interaction of p53 with its response element sequences (REs) at target promoters [4]. Recent evidences, including our studies using functional as well as DNA binding assays in yeast or mammalian cells or with cell extracts, demonstrated that maximal transactivation potential requires adjacent dimer binding sites [29-34]. A spacer between dimer sites even of 1 or 2 nucleotides conferred a negative impact, particularly for the p53-related protein p73 [31,35,36]. We also established that p53 can stimulate transcription, albeit at a reduced levels, from noncanonical response elements (half-sites and ¾ sites) [31], that do not provide for a p53 tetramer binding site. The same sequence-specific requirements that were shown to maximize the transactivation potential from full-site REs, appeared to be valid for the half-site REs [31]. This information is relevant to optimize pattern-based motif searches aiming at identifying functional p53 response elements within genomes [37-39].
In this study we used a regression-based predictor for p53 transactivation [39], to identify additional p53-target miRs through the presence of functional p53-REs in their promoter regions or in promoter regions of long noncoding RNA that are precursors of those miRs. We then used a yeast-based functional assay to determine the relative transactivation capacity of p53 family proteins towards the identified REs and Chromatin Immuno-Precipitation (ChIP) assays in human cells to investigate genotoxic-stress dependent p53 occupancy at the chromosomal sites containing those REs. Changes in the expression levels for mature miRs or precursors were measured by real-time qPCR using cell lines and treatments probing the direct involvement of p53. We propose miR-10b, -23b and -151a to be included in the list of direct p53 target miRs contributing to the fine-tuning of p53-induced responses.
Methods
Yeast reporter strains and media
We constructed a panel of 16 reporter strains in the budding yeast Saccharomyces cerevisiae containing the Firefly luciferase gene under the control of putative p53-REs (see Table 1 for the RE sequence and location with respect to miR genomic coordinates) predicted to control the expression of miR -10b, -23b, -34a, -106a, -145, -151a, -191, -198, -202, -221, -320, -328, -455, -671, -1204, -1206. To this aim we took advantage of the methodology of the well-established delitto perfetto approach for in vivo mutagenesis using oligonucleotides [40] starting with the master reporter strain yLFM-ICORE [41]. The strain contains the luciferase cDNA integrated at the chromosome XV downstream a minimal promoter derived from the CYC1 gene. The ICORE cassette is located 5′ to the minimal promoter and enables high efficiency targeting of the locus by oligonucleotides that contain desired RE sequences. The targeting events were followed by phenotypic selection and clones examined by colony PCR and direct DNA sequencing (BMR Genomics, Padua, Italy).
Table 1.
List of predicted p53-REs mapped nearby miR genes
| miR name | p53-RE Genomic location | p53-RE distance miRNA | Overlapping transcript | Distance from sense transcript TSS |
p53- RE organization |
||
|---|---|---|---|---|---|---|---|
| DIMER 1 | Spacer | DIMER 2 | |||||
|
Consensus |
- |
- |
- |
- |
RRRCATGYYY |
N = 0 - 13 |
RRRCATGYYY |
| miR-34a |
Chr 1 - 9242203 |
- 30,476 bp |
Intergenic |
+ 123 bp |
GGGCTTGCCT |
- |
GGGCTTGTTC |
| miR-10b |
Chr 2 - 177013750 |
- 1,281 bp |
HOXD3 |
+ 12,065 bp |
TAACTCGTTG |
GCTTTGACCTGTCT |
GAACAAGTCG |
| miR-23b |
Chr 9 - 97818687 |
- 28,803 bp |
C9orf3 |
+ 135 bp |
AGGTCAGTCA |
TG |
GGACATGTCC |
| miR-106a |
Chr X - 133309906 |
- 5,678 bp |
Intergenic |
- |
GTTATGTTC |
ATGTGCTCAT |
GTGCATGCCC |
| miR-145 |
Chr 5 - 148786372 |
- 23,837 bp |
Intergenic |
+ 68 bp |
GCACCCGCCT |
CGCCCCAATACG |
GGGCTTGCCT |
| miR-151a |
Chr 8 - 141734774 |
- 2,349 bp |
PTK2 |
- 75,874 bp |
TGGCTTGTTT |
- |
TGGCAAGTTC |
| miR-191 |
Chr 3 - 49063594 |
- 5,543 bp |
DARLD3 |
+ 5,103 bp |
GACCTTGTCT |
TGCTTCC |
AGCCATGTCA |
| miR-198 |
Chr 3 - 120112741 |
+ 1,774 bp |
FSTL1 |
- 57,359 bp |
AGGCAAGCTT |
- |
CAACAAGCCG |
| miR-202 |
Chr 10 - 135058647 |
+ 2,368 bp |
RP13-49115.5 |
- 2,748 bp |
GGGCATGTCT |
- |
TGGCAAGCCT |
| miR-221 |
Chr X - 45599946 |
+ 5,639 bp |
Intergenic |
- |
GAACATGCAT |
- |
GCACATGTTT |
| miR-455 |
Chr 9 - 116880600 |
- 91,114 bp |
COL27A1 |
- 49,395 bp |
CTTCCTGCAT |
AAGGCTTGGCGG |
GCGCAAGCCC |
| miR-320a |
Chr 8 - 22095461 |
+ 7,014 bp |
Intergenic |
- |
AGGCATGGTG |
- |
CGGCATGCCT |
| miR-328 |
Chr 16 - 67331546 |
- 95,322 bp |
ELMO3 |
+ 98,475 bp |
CGGCAAGTCC |
C |
CAGCCAGTTC |
| miR-671 |
Chr 7 - 150896632 |
- 38,875 bp |
CHPF2 |
- 32,953 bp |
GGTCCAGCCC |
TCTGGCCCCC |
CAACAAGTCT |
| miR-1204 |
Chr 8 - 128808017 |
- 191 bp |
PVT1 |
+ 1,238 bp |
CGACAAGTTG |
- |
AGACTTGTTC |
| miR-1206 | Chr 8 - 129002467 | - 18,677 bp | PVT1 | + 1,037 bp | GGGCTAGTGA | - | AGGCATGTCT |
miR name, genomic location of the predicted p53-RE (including chromosome number and genomic coordinates according to BLAT tool in UCSC Genome Browser, http://genome.ucsc.edu/), presence and distance from an overlapping primary transcript, p53 RE sequence and position relative to the miR sequence (obtained through the miRBase tool, http://www.mirbase.org/) are presented. p53-RE organization is shown grouping nucleotides in Dimer 1, Spacer (if any) and Dimer 2, according to the p53 consensus (in bold).
Yeast cells were grown in 1% yeast extract, 2% peptone, 2% dextrose with the addition of 200 mg/L adenine (YPDA medium). Selective minimal plates lacking tryptophan or leucine but containing adenine (200 mg/L) and dextrose as carbon sources were used (transformation in yeast and luciferase-based assay).
Yeast expression vectors
For the expression of p53 family protein in yeast we used available CEN/ARS expression vectors (pTSG- or pLSG-based) harbouring alternatively the cDNA wild-type of p53, p63 or p73 under the control of the GAL1,10 inducible promoter [31]. Among the various isoforms of p53 family members, we selected the full-length wild type p53 and the TA-p63β and TA-p73β isoforms as they showed the maximal transactivation potential in our experimental settings. The expression levels were modulated by the concentration of galactose in the culture medium (0.008%, 0.128% or 1%). The whole panel of 104 p53 germline missense mutants from the IARC R11 database (http://p53.iarc.fr) cloned in the pLS-Ad vector (providing for a constitutive expression of p53 mutants) [42] were used to test transactivation capability towards the miR-34a p53 RE. The pRS-314 or pRS-315 empty vectors were included as controls; these vectors contain respectively the TRP1 (as pTSG-) or LEU2 (as pLSG-) yeast selectable markers.
Luciferase assays in yeast
The p53 family responsiveness of miRNA-associated REs was examined by transforming the panel of yLFM-RE strains with the p53 expression vectors. Transformants were obtained using the LiAc method and selected on minimal plates lacking tryptophan or leucine but containing adenine (200 mg/L) and dextrose as carbon source to allow respectively the growth of white colonies of normal shape and to keep the expression of p53 family members inhibited. After 3 days of growth at 30°C, transformants were streaked onto the same plates and allowed to grow for an additional day. For each reporter strain, the basal luciferase activity was measured from pRS314- or pRS315-transformants and used to calculate the fold of induction due to the expression of each p53 family member. For the luciferase assay we exploited the recently developed miniaturized yeast assay [43]. Transformant colonies were grown in 100 μl of the selective medium with 2% raffinose as carbon source (raffinose result in basal level of expression from the GAL1,10 promoter), supplemented with different concentrations of galactose (0.008% and 0.128% for p53; 0.008% and 1% for the other members of the family, to obtain a low, moderate or high expression of the proteins) in a transparent 96-well plate and incubated under 150 rpm shaking for 16–24 hours at 30°C. OD600 was measured directly in the multi-well plate to normalize for cell density using a multilabel plate reader (Infinite M200-Pro, Tecan, Milan, Italy). 10 μl of cells suspensions were transferred to a white 384-well plate (Brand, Milan Italy) and mixed with an equal volume of PLB buffer 2X (Passive Lysis Buffer, Promega, Milan, Italy). After 15 minutes of shaking at room temperature, 10 μl of Firefly luciferase substrate (Bright Glo Luciferase Reporter Assay, Promega) were added. Luciferase activity was measured at the plate reader, using a luminescence program with 1” integration time.
Cell lines and treatments
The human breast adenocarcinoma-derived MCF7 cell line was obtained from the InterLab Cell Line Collection bank, ICLC (Genoa, Italy). The colon adenocarcinoma HCT116 (p53+/+) cell line and its p53−/− derivative were obtained from B. Vogelstein’s group (The Johns Hopkins Kimmel Cancer Center, Baltimore, Maryland, USA). Cells were normally maintained in DMEM or RPMI supplemented with 10% FCS and antibiotics (100 units/ml penicillin plus 100 mg/ml streptomycin).
Cells were treated for 24 hours with 1.5 μM doxorubicin (DXR) or 10 μM Nutlin-3A for p53 stabilization/activation. Stock solutions of Nutlin-3A were dissolved in 100% DMSO (stock solution 10 mM); DXR was dissolved in H2O (stock 10 mM). DXR was bought from Sigma-Aldrich® (Milan, Italy) whereas Nutlin-3A from Alexis® Biochemicals (Enzo Life Sciences, Exeter, UK). All the treatments were performed when cells reached 70-80% of confluence.
RNA extraction and RT-qPCR
RNA extraction from human cell lines was done with Trizol reagent (InVitrogen, Life Technologies, Milan, Italy) following the manufacturer’s protocol. Cells were treated with 1.5 μM DXR or 10 μM Nutlin-3A for 24 hours prior to Trizol extraction. Quantification of mature microRNA expression was carried out using TaqMan MicroRNA Assay kits according to the manufacturer’s protocol (Applied Biosystems, Life Technologies), as described in Barbareschi et al. [44]. Specifically, ready-made TaqMan MicroRNA Assays (containing microRNA-specific forward and reverse PCR primers and microRNA-specific Taqman MGB probe) were used for the investigation of miR-34a (ABI P/N 000426), miR-10b (ABI P/N 000388), miR-23b (ABI P/N 000400), miR-151a (ABI P/N 002254), and miR-202 (ABI P/N 002363). We also quantified the U6 small nuclear RNA (RNU6B) (ABI P/N 4373381) as an endogenous control to normalize the levels of target microRNA. Complementary DNA was generated using the Taqman MicroRNA Reverse Transcription (RT) Kit (ABI P/N 4366596) according to the manufacturer’s instructions. Reverse transcriptase reactions contained 10 ng of total RNA as the template, 3 μl of gene specific stem-loop RT primer, 1.5 μl of 10X RT buffer, 0.15 μl of 100 mM dNTPs, 1 μl of MultiScribe reverse transcriptase, and nuclease-free water. The 15 μl reactions were incubated on a GeneAmp PCR System (Bio-Rad, Hercules, CA) for 30 minutes at 16°C, 30 minutes at 42°C, 5 minutes at 85°C, and then kept at 4°C. Quantitative RT-PCR was carried out using the Rotorgene 6000 (Corbett Life Science, Ancona, Italy) in 20 μl PCR reactions containing 1.33 μl of RT product, 10 μl of FastStart TaqManProbe Master (Roche, Milan, Italy, P/N04673417001), 7.67 μl of nuclease–free water, and 1 μl of MicroRNA Assay (Applied Biosystems, Life Technologies) buffer. Reactions were incubated at 95°C for 10 minutes, followed by 40 cycles of incubation at 95°C for 15 seconds and at 60°C for 1 minute. The quantification of protein coding mRNAs was performed using a Sybr green RT-qPCR approach. Total RNAs extracted with Trizol were converted using the RevertAid™ First Strand cDNA Synthesis Kit containing the M-MuLV Reverse Transcriptase following the manufacture’s recommendation (Thermo Scientific Inc., M-Medical, Milan, Italy). qPCR were carried out using the KAPA Sybr Green PCR mix (Kapa Biosystems, Resnova, Rome, Italy) with 12.5 ng of cDNA on the CFX384 real-time PCR detection system (BioRad, Milan, Italy). Primers were picked using the Primer-BLAST online tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/); sequences are available upon request. The Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β2-microgobulin (B2M) were used as reference genes for normalization.
In all the qPCR assays, the threshold cycle data (Ct) and baselines were determined using auto settings. The Ct value was defined as the fractional cycle number at which the fluorescence passed a fixed threshold. Fold changes were calculated using the comparative Ct method (ΔΔCt).
Western blot analysis
To evaluate p53, p63 or p73 protein levels in yeast we cultured transformant colonies for 24 hours using selective medium containing 0.128% (p53) or 1% (p63β and p73β) galactose to induce the expression. Yeast cells were harvested, washed in ddH2O and lysed mechanically with glass beads as previously described [45]. 15 μg (p53) and 75 μg (p63β and p73β) were loaded on a 7.5% Acrylamide gel and separated by SDS-PAGE. DO-1, 4A4 (Santa Cruz Biotechnology, Milan, Italy) and ER-15 (Calbiochem, Merck, Millipore, Milan, Italy) antibodies were used for p53, p63 and p73 immunodetection, respectively. PhosphoGlycerate Kinase 1 (PGK1, detected by Ab clone number 22C5D8, InVitrogen, Life Technologies) was used as loading control.
To demonstrate p53 stabilization and activation upon treatment with doxorubicin or Nutlin-3A, MCF7, HCT p53+/+ and HCT p53−/− cells were harvested 16–18 hours after the treatments and lysed using RIPA (Radio Immuno Precipitation Assay) buffer (150 mM sodium chloride; 1.0% NP-40; 0.5% sodium deoxycholate; 0.1% SDS, sodium dodecyl sulphate; 50 mM Tris, pH 8.0) supplemented with Protease Inhibitors cocktail (Roche, Milan, Italy) [46]. 50 μg of the soluble extracts were loaded on a 12% Acrylamide gel and separated by SDS-PAGE. p53 and p21 endogenous protein levels were detected with incubation with monoclonal antibodies (DO-1 and C-19 clones against p53 and p21 respectively, Santa Cruz Biotechnology). Glyceraldehyde 3-phosphate dehydrogenase protein (GAPDH, Ab clone 6C5, Santa Cruz Biotechnology) served as loading control. All antibodies were diluted in 1% non-fat skim milk dissolved in PBS-0.1% Tween20.
Chromatin immunoprecipitation (ChIP) analysis
HCT116 p53+/+ and HCT116 p53−/− or MCF7 cells were grown on 150-mm dishes and treated with 1.5 μM doxorubicin for 24 hours. Proteins were cross-linked with DNA by addition of 1% formaldehyde. After 10 minutes incubation at room temperature the reaction was stopped by addition of glycine at a final concentration of 0.125 M followed by additional incubation for 5 minutes. Cells were washed twice with 10 ml cold PBS, harvested in 1 ml PBS plus protease inhibitors (PI) (Complete EDTA-free, Roche), and lysed using an SDS lysis buffer (1% SDS, 50 mM Tris–HCl pH 8, 20 mM EDTA, 0.1 mg/mL Sonicated Salmon sperm DNA, 1X PI). In order to eliminate soluble p53 protein, lysates were incubated with gently shaking for 10 min and insoluble material was collected by centrifugation at 800 g at 4°C for 5 min. Pellets were resuspended in 0.5 ml of sonication buffer containing 0.25% SDS, 200 mM NaCl, 100 mg/ml of sonicated salmon sperm DNA and protease inhibitors and were sonicated to shear DNA to lengths between 150 and 500 base pairs (bp) using a Misonix S-4000 sonicator with a plate horn (Misonix, Newtown, Connecticut, USA). After 10-fold dilution in ChIP dilution buffer (16.7 mM Tris, 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl), IPs were carried out overnight at 4°C with 2 μg of anti-p53 (DO-1, Santa Cruz Biotechnology) or 2 μg of normal IgG as a negative control. Fifty microliters of Dynabeads protein G magnetic beads (Invitrogen, Life Technologies) were added to each sample for 2–3 h, and the beads were then washed as indicated in the Upstate Biotechnology ChIP protocol. DNA was eluted firstly with 100 μL of TE with 1% SDS for 10 min at 65°C and a second time with 150 μL of TE with 0.67% SDS for an additional 10 min at 65°C. The cross-links were reversed overnight at 65°C. RNase A was added and incubated at 37°C for 30 min and then Proteinase K for 2 h at 56°C. DNA was then purified by QIAquick PCR purification KIT columns (Qiagen, Milan, Italy). Immunoprecipitated DNA was analyzed for p53 occupancy on selected chromosomal sites surrounding the predicted miR-associated p53 REs by RealTime-qPCR and fold enrichment of p53 binding to the respective DNA sequences was calculated by the comparative Ct method as described previously [47]. RealTime-qPCR was carried out with the KAPA SYBR Green PCR mix (Kapa Biosystems, Resnova) and all primers were checked for equal amplification efficiency. All PCR results were normalized to input controls. Three different DNA loci were used as ChIP negative controls (the CCNB1 exon 9, the Actin and GAPDH promoter). Sequences of all ChIP primers are available upon request.
Results and discussion
Identification of functional p53 response elements in miR genes
We applied a predictor tool for p53 RE-transactivation potential [39] to identify candidate p53 REs within regulatory regions of miR genes or promoter regions of long noncoding RNAs containing pri-miR clusters. Based on this analysis several novel p53 target miRs can be predicted (Table 1). To confirm p53 responsiveness of the identified p53 REs we first applied a well-established quantitative reporter assay in the budding yeast Saccharomyces cerevisiae[37,43]. This assay was chosen as it provides a defined experimental system to measure transactivation potential of a panel of REs each cloned at the same chromosomal location in isogenic derivative reporter strains where wild type or mutant p53, as well as p53-related proteins p63 and p73, can be expressed, one at a time, from a inducible promoter [43]. 15 candidate miR-associated p53 REs were studied. The validated miR-34a p53 RE was included as a positive control. The results indicated that most of the identified p53-REs could be transactivated at low to moderate levels by wild type p53 (Figure 1A). As expected the responsiveness was proportional to p53 expression levels (Figure 1A and B). Based on results obtained both with low and high p53 expression, REs from miR-34a, -10b, -202, -1204 were highly responsive (HR), from miR-23b, -151a, -221, -320, -1206 were moderately-responsive (MR), from -106a, -191, -198 were weakly responsive. Putative REs from miR-145, -328, -455, -671 were not responsive to p53 in the yeast-based assay.
Figure 1.
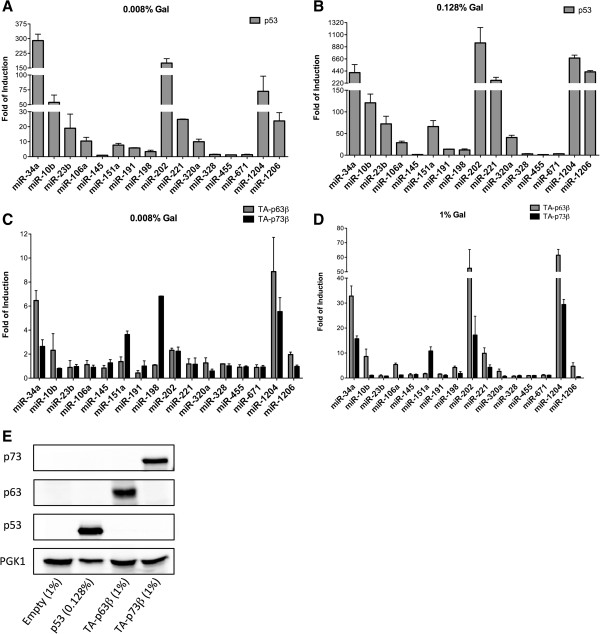
p53 family members can transactivate p53-REs found in miR-associated promoter regions. A-B) Transactivation potential of p53 protein tested on a panel of 15 putative p53-dependent miR-REs and miR-34a-RE (a positive control) using the yeast functional assay. The expression of p53 was modulated by increasing concentrations of galactose in the culture medium (A, 0.008%: a moderate p53 expression; B, 0.128%: high p53 expression). C-D) The same panel of p53 miR-REs was tested using the other members of the p53 family p63 and p73 (gray and black bars respectively). The expression of p63β and p73β isoforms was induced using two different concentrations of galactose (C, 0.008%: moderate expression; D, 1%: maximal induction). Results are presented as fold of induction calculated over an empty expression vector. Bars plot the averages and standard deviations of at least three independent biological repeats. (E) Western Blot establishing galactose-dependent expression of p53, p63 or p73 in yeast. PGK1 was used as reference.
Next we tested p63 and p73 responsiveness of the same panel of REs, using the transactivation competent, TA-p63β and TA-p73β isoforms for these proof-of-principle experiments, as they exhibit higher relative transactivation potential compared to N-terminal truncated ΔN- and also compared to TA-p63α and TA-p73α isoforms [37]. As expected the transactivation potential of p63 β and p73β were much lower compared to p53 (Figure 1C and D). Only a subset of p53-responsive REs was active with p63 and p73 and included miR-34a, -202 and −1204 REs. Furthermore, differences in relative transactivation potential were observed in the comparison of the three family members. For example, miR-34a and −1204 were more responsive to p63 than to p73. Furthermore, we observed examples of selective lack of responsiveness: p73 towards miR-10b and −221 REs; p63 towards mir-151a. To verify the protein expression of the three p53 family members in yeast after galactose induction (0.128% and 1% galactose for p53 or p63 and p73, respectively, to match the experimental conditions used for the luciferase assays) we performed a western blot using antibodies specific for each transcription factor (Figure 1E).
For 5 REs, representative of strong, moderate, weak, and nearly-absent responsiveness to wild type p53, the functional assay was extended to five p53 missense germline mutations, of which three retain partial function (A138S, C141Y, R337C) and two are loss of function (A138P, R175H) [42]. Similar results were obtained with the responsive REs, confirming the functional classification of the p53 mutants examined (Figure 2); for example, A138S was near-wild type, while R337C was weakly active (<10% of wt activity) and 141Y almost inactive (<5% of wt activity). Given the important role of miR-34a as a positive modulator of p53-mediated responses [48,49] and our recent studies indicating that p53 mutant transactivation capacity can correlate with clinical variables in Li-Fraumeni patients [42,50], we decided to use the miR-34a reporter strain to examine the entire panel of 104 germline p53 alleles described in the R11 release of the p53-mutant IARC database [42]. The vast majority, 83 out of the total 104 alleles (79.8%) were considered loss of function (LOF, defined for the cases where a residual transactivation was lower than 25% of the wild type p53 activity). Eight (7.7%) retained a partial function phenotype (activity between 75% to 25% of wt p53), while 9 (8.7%) had the same transactivation potential as the wild-type. Interestingly, 4 alleles (R337H, H365Y, S366A, P82L) showed a transcriptional activity higher than wild-type p53 (>125%) and can be considered as super-transactivating alleles (ST). All the results are summarized in the Additional file 1: Table S1. As expected, while the vast majority of the LOF-p53 alleles hit the central portion of the p53 protein, associated with DNA binding capacity, the WT-, PT-, and ST-alleles were preferentially confined to the extremities (both N- or C-terminal domains). Compared to our previous classification based on four p53 REs derived from P21, MDM2, BAX and PUMA, 8 additional alleles are classified as LOF with miR-34a, while one (I305M) would change from LOF to partial function.
Figure 2.
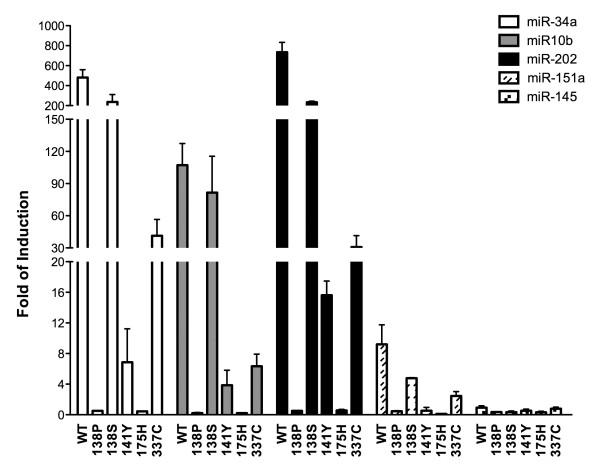
p53-REs found in miR promoters can be used to classify p53 germline alleles associated with Li-Fraumeni Syndrome. Five p53 missense mutants representative of partial function (A138S, C141Y, R337C) or loss-of-function (A138P, R175H) germline p53 alleles were tested for transactivation from 5 p53 miR-REs using the yeast functional assay. Yeast transformants were cultured over-night in selective medium and the luciferase activity was measured using the miniaturized assay format [43]. Bars represent the averages and standard deviations of at least three independent biological repeats.
Overall these results identify a panel of p53 REs that based on the comparison with well-established REs from coding genes [31,37] suggest the potential for p53-mediated control of miR gene expression in vivo. Further, results suggested that miRs could be selectively or more specifically (e.g. -10b, -151a; -198) targeted by individual p53 family proteins, possibly contributing to the distinctiveness of the regulated networks and biological outcomes. Finally, the identified p53 miR-REs can be used to refine the functional classification of cancer associated p53 mutant alleles.
p53-occupancy at endogenous miR-associated promoters in human cells
As the yeast functional data provided us information predominantly on the nature of the mapped p53-REs, specifically on the transactivation potential in an isogenic, ectopic context, we analyzed the correspondence of our results with the ability of p53 to physically interact with those sequences in their natural context in mammalian cells treated with a genotoxic agent (doxorubicin, DXR) known to result in p53 stabilization and activation [51,52]. To this aim we treated HCT116 p53+/+, HCT116 p53−/− and MCF7 (p53 wt) cells with 1.5 μM DXR for 24 hours and performed ChIP assays. We selected all the miR promoters with REs classified as HR and some of the MR according to the p53 responsiveness in the yeast assay. In HCT116 p53+/+ cells, DXR-induced p53 occupancy was observed for all chromosomal coordinates surrounding miR-associated REs with the only exception of miR-23b. The relative increase in occupancy was comparable for miR-202 and miR-1204 sites to the well-established p53 target P21-5′ RE region and the p53-miR-34a target (Figure 3A and B). As expected in HCT116 p53−/− cells we did not find any occupancy, confirming the specificity of the assay. The experiment was repeated in another p53 wild-type cell line, MCF7, using IgG as a control of IP specificity. Doxorubicin-induced occupancy was observed for all sites examined, including miR-23b. In particular, miR-202 and miR-10b promoters showed the highest relative induction of p53 occupancy (Figure 3C and D).
Figure 3.
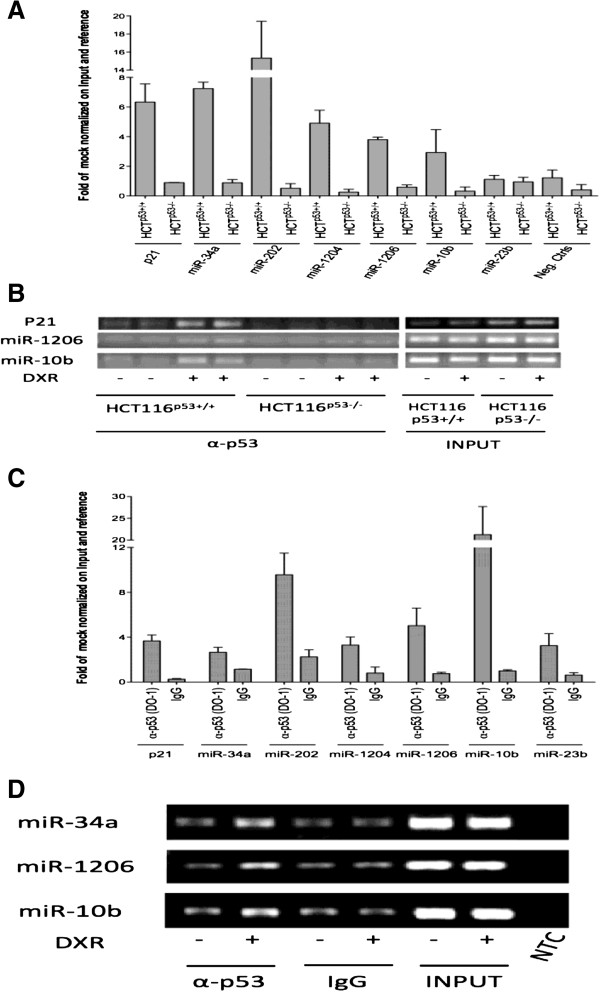
p53 can bind chromatin region surrounding the identified p53 REs in miR genes. A) ChIP assays were performed in HCT116 p53+/+(gray bars) and HCT116 p53−/− (black bars) upon doxorubicin treatment for 24 hours. The results of Real-Time qPCR are presented as fold of mock treatment, normalized with respect to the signal obtained with Input DNA. The results from three control locations corresponding to promoter regions of Actin, GAPDH and exon 9 of CCNB1 genes were averaged and are also presented in panel A (Neg. Ctrls). P21 and miR-34a occupancy were measured as positive controls. Bars represent average and standard deviations of three independent experiments. C) ChIP assays of MCF7 cells treated with doxorubicin for 24 hours. Results obtained after ChIP with an antibody against IgG were included as a negative control for the p53 miR-REs. Examples of agarose gel analysis of standard ChIP-PCR are given in panels B and D. Specifically, panel B shows experiments performed in HCT116 p53+/+ and p53−/− cells, while panel D presents results from MCF7 cells. The DO-1 p53 antibody was used for immunoprecipitation; NTC, no template control. Regions surrounding the established P21 and miR-34a p53 REs were examined as positive controls.
Downstream of and consistent with the yeast-based results, ChIP assays further supported the putative function of the identified p53 REs in modulating p53-mediated responsiveness of miR genes. However, the correlation between occupancy and transactivation is not direct, nor linear [4]. p63 and p73 occupancy was not investigated and awaits further studies to clarify the contribution of p53 family proteins on miR gene expression.
Doxorubicin responsiveness of identified p53 target miRs in p53 wild type human cells
With the yeast-based assays we established the potential for p53-mediated transactivation of p53 REs associated with miR sites, while ChIP experiments established accessibility and potential recruitment of p53 at those sites. Next we examined if the expression levels of mature or precursor miR transcripts could be modulated by treatments resulting in p53 activation using again the HCT116 p53+/+, HCT116 p53−/− and MCF7 cell line systems. The results indicated that of miR-10b, -151a and -23b are p53-responsive (Figure 4A). Consistent with ChIP analysis higher induction levels of mature miR-10b and -23b in response to DXR were observed in MCF7 than in HCT116 p53+/+ cells. The treatment did not result in miR induction in HCT116 p53−/− cells, in fact some repression was apparent, particularly for miR-23b (Figure 4A). In contrast to RE transactivation potential and p53 occupancy studies, miR-202 expression did not change after the genotoxic treatment (Figure 4). Unfortunately, we were not able to measure miR-1204 or miR-1206 as the expression in these cells appeared to be below the detection limit of the qPCR in these cell lines (data not shown).
Figure 4.
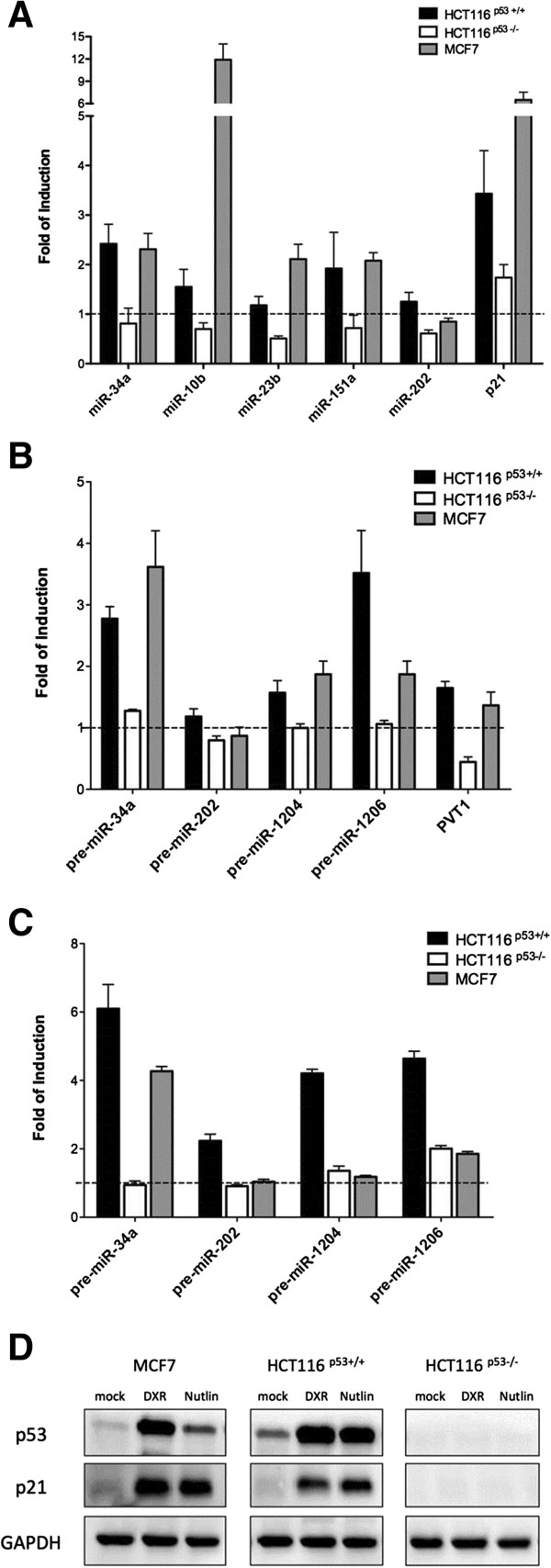
p53-induced expression of mature and pre-miR genes. RT-qPCR were performed in HCT116 p53+/+(black bars), HCT116 p53−/− (white bars) and MCF7 (gray bars) cells upon doxorubicin (A, B) or Nutlin (C) for 24 hours. The expression of the processed mature miR (A) or of the pre-miR RNA (B, C) was tested. p21 mRNA expression was measured as p53-dependent positive control. PVT1 non-coding RNA expression levels were measured as additional evidence of p53-dependent expression of miR-1204 and miR-1206. Results are presented as fold of induction with respect to the mock condition. Bars plot average and standard deviations of three independent experiments. (D) Western Blot establishing stabilization of p53 protein in doxorubicin and Nutlin treated cells and the induction of the p53 target gene p21. GAPDH was used [6] as reference.
To exclude any impact of the miR maturation processes or low sensitivity of the mature miR assay systems, we also selected primers that can amplify the pre-miR RNA and performed RT-qPCR for miR-1204, miR-1206, miR-202 and miR-34a (Figure 4B). We also analyzed the expression of PVT1, the long non-coding RNA transcript comprising the miR-1204 cluster [53]. Weak, DXR-dependent induction was observed for PVT1, pre-miR-1204 and pre-miR-1206 in HCT116 p53+/+ and MCF7 cells. No changes were observed in HCT116 p53−/− or repression of PVT1 (Figure 4B). To further confirm the direct involvement of p53 in the transcriptional regulation of those miRs we also treated the cells with the MDM2 specific inhibitor Nutlin-3A (Figure 4C). Except for pre-miR-34a, pre-miR-1204, -1206 and even −202 were responsive to Nutlin treatment only in the HCT116 p53+/+ cell line, highlighting cell type and treatment dependencies in the expression regulation. The effect of the treatments on p53 stabilization and activation was examined using western blot (Figure 4D).
miR expression analysis in doxorubicin treated cells differing for p53 status supported p53-mediated responsiveness for miR-10b, -151a and, limited to MCF7 cells, also -23b. The levels of induction were in general comparable to those of miR-34a. Despite the high transactivation potential of the associated p53 REs and the p53 occupancy analysis, the mature miR-202 was not responsive to p53-inducing treatment. This discrepant finding could be related to the relatively large distance between the mapped p53 REs and the pri-miR-202 transcript start site and/or to the inaccessibility of the site due chromatin structure. The p53 RE sequence does not fall within DNAse sensitive sites based on ENCODE data. We were not able to confirm the p53-dependent induction of mature miR-1204 and −1206 in our cell lines, although we detected weak induction of the long noncoding RNA containing the miR-1204 cluster and possibly evidence for an internal transcript comprising pre-miR-1206. A recent study established p53-dependent induction of Plasmacytoma Variant Translocation 1 gene PVT1 and miR-1204 [53] in HCT116 p53 wild type cells treated with doxorubicin. Our results confirm those findings and also suggest p53 recruitment internally to the PVT1 gene locus to possibly further modulate miR-1206 independently or in addition to the activation of the entire miR-1204-1208 cluster. Further studies are needed, including the use of cell lines expressing higher basal levels of PVT1 to examine whether miR-1206, and possibly −1207 and −1208 downstream, can be modulated by p53 family proteins also independently from PVT1 gene transcription. A link between p53 and modulation of miR-23b was also recently described and indirectly related to human papillomavirus (HPV)-mediated responses through inhibition of p53 function [54]. Our results further confirm miR-23b as a p53 target miR in other cancer–derived cell lines. A previous link between p53 and miR-151a, as well as FAK pre-mRNA that contains miR-151a, was proposed based on transient silencing of p53 in the hepatocellular carcinoma-derived HepG2 cells resulting in FAK and miR-151a up-regulation (Figure S4 in [55]). Our results in different cell models indicate instead the potential for positive modulation of this miR by doxorubicin treatment in p53 wild type cells. Bioinformatics-based predictions, transactivation potential of RE, occupancy and mature miR expression changes in doxorubicin treated cells, consistently indicate, to our knowledge for the first time, miR-10b as a p53 target gene.
An expanded role of p53 in the modulation of microRNA expression
The study of the p53 gene transcriptional networks continues to raise particular interest in the field due to the increasing complexity of regulatory circuits and the functions of the extensive list of target genes spanning a myriad of different biological pathways. The discovery of p53-target miRs has led to the identification of several feedback and feed-forward loops that can lead to fine-tuning of p53-mediated responses. A few p53 target miRs, more prominently miR-34a, have been shown to act as bona-fide tumor suppressor genes [56,57]. Multiple evidence, comprising gene expression, ChIP-seq and phenotypic studies upon gene silencing or targeting in cell and animal models indicate a complex crosstalk between p53 and the related p63 and p73 proteins at the level of common and exclusive coding gene targets [58-61]. An integrated view of common and p53-family protein specific regulation of miR genes is however largely missing.
This work led to the identification of new p53 target miRs (miR-10b; -151a) and also confirmed or extended recent evidence from the literature (miR-1204, -1206; -23b). Proof-of-principle experiments also suggested miR genes worth of further analysis to ascertain a specific or selective role for p63 or p73 transcription in their expression. The weak p53-responsiveness towards p53 REs associated with miR-106a, -191, -198, -221 and −320 was not pursued in this study and awaits further investigation.
Perhaps surprising is the fact that the miR genes we propose (miR-10b; -151a) or confirm more in detail (miR-23b; -1204; -1206) as direct p53 targets do not fit intuitively with the expected p53-mediated functions. In fact all these miRs have been proposed to exhibit oncogenic activities or at least their over-expression has been correlated to aggressive cancer phenotypes in some tissues [55,62-66]. For example, the established potential for miR-10b to target both CDKN1A and CDKN2A mRNAs could in principle result in a p53-directed attenuation circuit of cell cycle arrest and senescence [62]. However, KLF4 mRNA has been described as a miR-10b target and KLF4 down-regulation in breast cancer cells has been reported to restore p53 functions leading to apoptosis [67]. Hence, in specific cellular contexts, it is possible that the p53-dependent regulation of miR-10b we discovered could result in a positive feedback loop stimulating p53 activity. Further, CpG islands upstream from the miR10b/10b* locus were found to be hyper-methylated in breast cancers and through ectopic expression an important role for miR-10b* in cell cycle inhibition was established [68].
It is known that miR functions can be highly context and tissue-dependent and their p53-mediated control in normal cells could potentially affect biological responses also not directly related to cell cycle control or apoptosis. For example, low levels of miR-23b resulting in higher levels of its target urokinase-type plasminogen activator could promote cervical cancer cell migration [54]. Finally, increasing evidence link p53 functions to innate and adaptive immunity and it could be speculated that miR-23b as well as PVT1 and the miR-1204 cluster regulation could be relevant in this context [53,69]. Interestingly, functional enrichment analyses of predicted targets of both miR-10b and -151a showed enrichment for neuron generation/development and brain-related phenotypes (uncorrected p-value < 0.05; data not shown).
Conclusions
In our study, bioinformatics-based predictions, transactivation potential of putative p53 REs, p53 occupancy at the endogenous RE positions, and mature miR expression changes in cell lines differing for p53 status, were combined to identify miRs that are direct transcriptional targets of wild type p53. We established that miR-10b and miR-151a are new p53 target genes and also confirmed cis-mediated regulation by p53 of miR-1204, -1206 and -23b. Further studies are warranted to establish the biological implications of the newly identified p53 target miRs.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AB, AGJ, AI and YC designed research; AB, VDS, VDV, MAD, AGJ, AI and YC performed research; AB, VDS, MAD, AGJ, AI and YC analyzed data; AB, MAD, AGJ, AI and YC wrote the paper. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Transactivation potential of 104 Li-Fraumeni associated germline p53 missense mutations towards the miR-34a p53-RE.
Contributor Information
Alessandra Bisio, Email: bisio@science.unitn.it.
Veronica De Sanctis, Email: desanctis@science.unitn.it.
Valerio Del Vescovo, Email: valerio@science.unitn.it.
Michela A Denti, Email: denti@science.unitn.it.
Anil G Jegga, Email: anil.jegga@cchmc.org.
Alberto Inga, Email: inga@science.unitn.it.
Yari Ciribilli, Email: ciribilli@science.unitn.it.
Acknowledgement
We thank Gilberto Fronza and Paola Monti for sharing the panel of germline Li-Fraumeni derived p53 mutant expression plasmids. This work was partially supported by the AIRC, Italian Association for Cancer Research (Associazione Italiana per la Ricerca sul Cancro, #IG9086 to AI) and by CIBIO start-up funds. YC is supported by a Marie-Curie/Autonomous-Province-of-Trento (PAT) cofund grant (#40101712). VDV is recipient of an Outgoing Marie-Curie/Autonomous-Province-of-Trento (PAT) cofund grant. VDS was supported by a Pezcoller Foundation fellowship.
References
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1(1):45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Appella E, Anderson CW. Dynamic interactions of proteins in complex networks. FEBS J. 2009;276(19):5380. doi: 10.1111/j.1742-4658.2009.07252.x. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13(6):1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- Espinosa JM. Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene. 2008;27(29):4013–4023. doi: 10.1038/onc.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Vousden KH. Outcomes of p53 activation–spoilt for choice. J Cell Sci. 2006;119(Pt 24):5015–5020. doi: 10.1242/jcs.03293. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Rotter V. Regulation of lipid metabolism by p53 - fighting two villains with one sword. Trends Endocrinol Metab. 2012;23(11):567–575. doi: 10.1016/j.tem.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, Komune T, Sato T, Saya H, Li JD. et al. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol. 2008;28(21):6557–6567. doi: 10.1128/MCB.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, Bergmann A, Johnson RL, Barton MC. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009;106(28):11612–11616. doi: 10.1073/pnas.0813177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Miyazono K. Dynamics of microRNA biogenesis: crosstalk between p53 network and microRNA processing pathway. J Mol Med (Berl) 2010;88(11):1085–1094. doi: 10.1007/s00109-010-0650-1. [DOI] [PubMed] [Google Scholar]
- Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012;12(9):613–626. doi: 10.1038/nrc3318. [DOI] [PubMed] [Google Scholar]
- Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107(7):823–826. doi: 10.1016/S0092-8674(01)00616-X. [DOI] [PubMed] [Google Scholar]
- Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302(1):1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D. et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun CJ, Zhang X, Savelyeva I, Wolff S, Moll UM, Schepeler T, Orntoft TF, Andersen CL, Dobbelstein M. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008;68(24):10094–10104. doi: 10.1158/0008-5472.CAN-08-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106(9):3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, Huso D, Lowenstein CJ. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci U S A. 2010;107(14):6334–6339. doi: 10.1073/pnas.0911082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M, Ferlito M, Lowenstein C. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Hu WL, Jiang CC, Wang JX, Han CC, Chu P, Zhang LJ, Thorne RF, Wilmott J, Scolyer RA. et al. MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc Natl Acad Sci U S A. 2011;108(38):15840–15845. doi: 10.1073/pnas.1019312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16(1):23–29. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- Tian S, Huang S, Wu S, Guo W, Li J, He X. MicroRNA-1285 inhibits the expression of p53 by directly targeting its 3′ untranslated region. Biochem Biophys Res Commun. 2010;396(2):435–439. doi: 10.1016/j.bbrc.2010.04.112. [DOI] [PubMed] [Google Scholar]
- Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, Lodish HF, Lim B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23(7):862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Chan CS, Wu R, Zhang C, Sun Y, Song JS, Tang LH, Levine AJ, Feng Z. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell. 2010;38(5):689–699. doi: 10.1016/j.molcel.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera-Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, Gonzalez S. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle. 2010;9(16):3277–3285. doi: 10.4161/cc.9.16.12598. [DOI] [PubMed] [Google Scholar]
- Swarbrick A, Woods SL, Shaw A, Balakrishnan A, Phua Y, Nguyen A, Chanthery Y, Lim L, Ashton LJ, Judson RL. et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med. 2010;16(10):1134–1140. doi: 10.1038/nm.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Lu Z, Takwi AA, Chen W, Callander NS, Ramos KS, Young KH, Li Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene. 2011;30(7):843–853. doi: 10.1038/onc.2010.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLure KG, Lee PW. How p53 binds DNA as a tetramer. EMBO J. 1998;17(12):3342–3350. doi: 10.1093/emboj/17.12.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Levine AJ. Probing potential binding modes of the p53 tetramer to DNA based on the symmetries encoded in p53 response elements. Nucleic Acids Res. 2007;35(22):7733–7747. doi: 10.1093/nar/gkm890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan JJ, Menendez D, Inga A, Noureddine M, Bell DA, Resnick MA. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008;4(6):e1000104. doi: 10.1371/journal.pgen.1000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokino T, Thiagalingam S, el-Deiry WS, Waldman T, Kinzler KW, Vogelstein B. p53 tagged sites from human genomic DNA. Hum Mol Genet. 1994;3(9):1537–1542. doi: 10.1093/hmg/3.9.1537. [DOI] [PubMed] [Google Scholar]
- Veprintsev DB, Fersht AR. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008;36(5):1589–1598. doi: 10.1093/nar/gkm1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z. et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124(1):207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Ethayathulla AS, Tse PW, Monti P, Nguyen S, Inga A, Fronza G, Viadiu H. Structure of p73 DNA-binding domain tetramer modulates p73 transactivation. Proc Natl Acad Sci U S A. 2012;109(16):6066–6071. doi: 10.1073/pnas.1115463109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9(10):724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- Jegga AG, Inga A, Menendez D, Aronow BJ, Resnick MA. Functional evolution of the p53 regulatory network through its target response elements. Proc Natl Acad Sci U S A. 2008;105(3):944–949. doi: 10.1073/pnas.0704694105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha AU, Kaimal V, Chen J, Jegga AG. Dissecting microregulation of a master regulatory network. BMC Genomics. 2008;9:88. doi: 10.1186/1471-2164-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrisankar S, Jegga AG. Regression based predictor for p53 transactivation. BMC Bioinforma. 2009;10:215. doi: 10.1186/1471-2105-10-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Resnick MA. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006;409:329–345. doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
- Tomso DJ, Inga A, Menendez D, Pittman GS, Campbell MR, Storici F, Bell DA, Resnick MA. Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proc Natl Acad Sci USA. 2005;102(18):6431–6436. doi: 10.1073/pnas.0501721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti P, Perfumo C, Bisio A, Ciribilli Y, Menichini P, Russo D, Umbach DM, Resnick MA, Inga A, Fronza G. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol Cancer Res. 2011;9(3):271–279. doi: 10.1158/1541-7786.MCR-10-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti V, Ciribilli Y, Monti P, Bisio A, Lion M, Jordan J, Fronza G, Menichini P, Resnick MA, Inga A. p53 transactivation and the impact of mutations, cofactors and small molecules using a simplified yeast-based screening system. PLoS One. 2011;6(6):e20643. doi: 10.1371/journal.pone.0020643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbareschi M, Cantaloni C, Del Vescovo V, Cavazza A, Monica V, Carella R, Rossi G, Morelli L, Cucino A, Silvestri M. et al. Heterogeneity of large cell carcinoma of the lung: an immunophenotypic and miRNA-based analysis. Am J Clin Pathol. 2011;136(5):773–782. doi: 10.1309/AJCPYY79XAGRAYCJ. [DOI] [PubMed] [Google Scholar]
- Ciribilli Y, Monti P, Bisio A, Nguyen HT, Ethayathulla AS, Ramos A, Foggetti G, Menichini P, Menendez D, Resnick MA. et al. Transactivation specificity is conserved among p53 family proteins and depends on a response element sequence code. Nucleic Acids Res. 2013;41(18):8637–8653. doi: 10.1093/nar/gkt657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viiri J, Amadio M, Marchesi N, Hyttinen JM, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D’Agostino VG. et al. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One. 2013;8(7):e69563. doi: 10.1371/journal.pone.0069563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciribilli Y, Andreotti V, Menendez D, Langen JS, Schoenfelder G, Resnick MA, Inga A. The coordinated p53 and estrogen receptor cis-regulation at an FLT1 promoter SNP is specific to genotoxic stress and estrogenic compound. PLoS One. 2010;5(4):e10236. doi: 10.1371/journal.pone.0010236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ. et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26(5):745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26(5):731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Monti P, Ciribilli Y, Jordan J, Menichini P, Umbach DM, Resnick MA, Luzzatto L, Inga A, Fronza G. Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin Cancer Res. 2007;13(13):3789–3795. doi: 10.1158/1078-0432.CCR-06-2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froelich-Ammon SJ, Osheroff N. Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J Biol Chem. 1995;270(37):21429–21432. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74(6):957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Barsotti AM, Beckerman R, Laptenko O, Huppi K, Caplen NJ, Prives C. p53-Dependent induction of PVT1 and miR-1204. J Biol Chem. 2012;287(4):2509–2519. doi: 10.1074/jbc.M111.322875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au Yeung CL, Tsang TY, Yau PL, Kwok TT. Human papillomavirus type 16 E6 induces cervical cancer cell migration through the p53/microRNA-23b/urokinase-type plasminogen activator pathway. Oncogene. 2011;30(21):2401–2410. doi: 10.1038/onc.2010.613. [DOI] [PubMed] [Google Scholar]
- Ding J, Huang S, Wu S, Zhao Y, Liang L, Yan M, Ge C, Yao J, Chen T, Wan D. et al. Gain of miR-151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol. 2010;12(4):390–399. doi: 10.1038/ncb2039. [DOI] [PubMed] [Google Scholar]
- He X, He L, Hannon GJ. The guardian’s little helper: microRNAs in the p53 tumor suppressor network. Cancer Res. 2007;67(23):11099–11101. doi: 10.1158/0008-5472.CAN-07-2672. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Perez CA, Ott J, Mays DJ, Pietenpol JA. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene. 2007;26(52):7363–7370. doi: 10.1038/sj.onc.1210561. [DOI] [PubMed] [Google Scholar]
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416(6880):560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17(6):901–911. doi: 10.1038/cdd.2010.35. [DOI] [PubMed] [Google Scholar]
- Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, Lane DP, Callen DF. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget. 2011;2(12):1203–1217. doi: 10.18632/oncotarget.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriely G, Yi M, Narayan RS, Niers JM, Wurdinger T, Imitola J, Ligon KL, Kesari S, Esau C, Stephens RM. et al. Human glioma growth is controlled by microRNA-10b. Cancer Res. 2011;71(10):3563–3572. doi: 10.1158/0008-5472.CAN-10-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Luo A, Cai Y, Su Q, Ding F, Chen H, Liu Z. MicroRNA-10b promotes migration and invasion through KLF4 in human esophageal cancer cell lines. J Biol Chem. 2010;285(11):7986–7994. doi: 10.1074/jbc.M109.062877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Wessely O, Marcusson E, Ivan C, Calin G, Alahari SK. Pro-oncogenic factors miR-23b- and miR-27b are regulated by Her2/Neu, EGF, and TNFalpha in breast cancer. Cancer Res. 2013;280(16):16030–16037. doi: 10.1158/0008-5472.CAN-12-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu F, Chang C, Byon J, Papayannopoulou T, Deeg HJ, Marcondes AM. Transcriptional regulation of miR-10a/b by TWIST-1 in myelodysplastic syndromes. Haematologica. 2013;98(3):414–419. doi: 10.3324/haematol.2012.071753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7(11):1074–1082. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- Biagioni F, Bossel Ben-Moshe N, Fontemaggi G, Canu V, Mori F, Antoniani B, Di Benedetto A, Santoro R, Germoni S, De Angelis F. et al. miR-10b*, a master inhibitor of the cell cycle, is down-regulated in human breast tumours. EMBO Mol Med. 2012;4(11):1214–1229. doi: 10.1002/emmm.201201483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, O’Connell RM. MiR-23b is a safeguard against autoimmunity. Nat Med. 2012;18(7):1009–1010. doi: 10.1038/nm.2849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transactivation potential of 104 Li-Fraumeni associated germline p53 missense mutations towards the miR-34a p53-RE.