

Abstract
Using a recently invented technique for gene expression mapping in the whole-anatomy context, termed transcriptome tomography, we have generated a dataset of 36,000 maps of overall gene expression in the adult-mouse brain. Here, using an informatics approach, we identified a broad co-expression network that follows an inverse power law and is rich in functional interaction and gene-ontology terms. Our framework for the integrated analysis of expression maps and graphs of co-expression networks revealed that groups of combinatorially expressed genes, which regulate cell differentiation during development, were present in the adult brain and each of these groups was associated with a discrete cell types. These groups included non-coding genes of unknown function. We found that these genes specifically linked developmentally conserved groups in the network. A previously unrecognized robust expression pattern covering the whole brain was related to the molecular anatomy of key biological processes occurring in particular areas.
An integrated understanding of biological structure and function based on knowledge of gene expression is critical in systemically approaching complex structures, such as the mammalian brain, where an estimated 25,000 genes are expressed1,2,3. The combinatorial number of randomly selected genes is greater than the number of cells in the brain. Indeed, a non-random combination of expressed genes is required for the formation of functional regions. The activation or suppression of specific subsets of genes, referred to as co-expressed or co-suppressed genes, is thought to regulate cell type- or region-specific brain functions. Thus, well-designed experimental systems for profiling combinatorial gene expression in relation to anatomical structures are required to reveal novel aspects that will allow an integrated understanding of brain structure and function.
Previous in situ hybridization (ISH)-based resources that were employed to comprehensively map co-expression networks in the mouse brain include the Anatomic Gene Expression Atlas (AGEA) of the Allen Mouse Brain Atlas (ABA), which is a database for searching expression similarity of approximately 4,500 genes that are selected by image processing of ABA expression maps as regionally expressed genes in the adult mouse brain, and Eurexpress, which offers co-expression clusters as well as annotated images of more than 18,000 gene expression patterns in E14.5 mouse embryos4,5. The systematic investigation of expression data within these databases provides detailed information in two dimensions at the cellular level for individual genes. However, information in the third dimension is difficult to obtain at the same resolution. Additionally, quantifying expression densities in a three-dimensional (3D) anatomical context and comparing these data across genes is difficult, despite time-consuming and labor-intensive experimental efforts.
An alternative approach is high-throughput analysis of gene expression data obtained through microarray or RNA sequencing methods. Weighted correlation network analysis (WGCNA) is a software package that performs computational analyses to reveal topological modules of co-expression that are based on microarray expression data6. Using archived microarray data, statistically significant co-expression patterns with biological relevance are revealed; and anatomical areas of co-expression are informed by anatomical descriptions associated with the data. High-throughput transcriptome analyses have been performed in a large number of selected anatomical areas in the brains of humans and mice7,8,9. However, due to the nature of these studies, the examined anatomical areas are restricted to key components, and an alternative strategy is, therefore, required to uncover gene expression networks across the brain.
Recently, we developed a system for high-throughput gene expression mapping in a broad 3D-anatomical context, denoted as transcriptome tomography10. In the data acquisition process, a block-face image is obtained before each section is cut. A serial set of sections is collected in batches as a “fraction”, and a series of fractions is obtained using a whole brain. The materials of the fractions are subjected to probe-based gene expression density measurements by microarray. At least three whole brains sectioned in each of three orthogonal planes are required for 3D reconstruction of the gene expression profiles (Supplementary Fig. S1). The block-face images are used to indicate the 3D outlines of the whole brains and their fractions. The outlines of the whole brains are used for transformation to locate all three in the same x-y-z coordinates. Values of the measured expression densities are assigned to voxels of the transformed fractions. The values in each voxel obtained from at least three fractions are averaged and the average represents the expression density of the voxel. Using this system, we have generated a 3D dataset that delivers a comprehensive spatial overview of gene expression patterns of the mouse brain in the ViBrism database (ViBrism DB: http://vibrism.neuroinf.jp/, including more than 36,000 maps of 25,000 genes). The first dataset was created using 61 fractions of six series of sectioning in total (two replicates of three orthogonal sectioning series). Because of the limited number of data points, the anatomical resolution is only so high, and the maps roughly show expression patterns in sub-anatomical regions. However, the expression maps cover the whole brain and expression density values for each gene-probe in the fractions can be correlated with data for any other probes, so that they are directly usable for probe-by-probe correlation analysis of co-expression. Here, we introduce a framework for comprehensively assessing co-expression patterns that are latent within expression maps. We elucidated functions associated with the co-expression of gene networks, discovering that these functions followed a power law and were enriched in functional gene groups, as defined by gene ontology (GO) terms and biological interactions. We focused on genes encoding transcription factors and related molecules and found that the combinatorial expression of genes functioning in cell differentiation during development also occurs in the adult brain and that each combination, including previously uncharacterized genes, is associated with a discrete spatial location (Supplementary Video S1).
Results
Co-expression network graphs following an inverse power law
To measure expression similarities between genes and to characterize expression patterns in a gene probe-based analysis, which used 36,558 total probes, we calculated the Pearson correlation coefficient, r, of the expression density values in the fractions between the gene probes as a measure of correlation. We then calculated κ, which is the number of similarly expressed gene-probes to a target probe including itself (SET), using each probe as a target (the SET search results are provided in the “Similarly Expressed Gene Search” database at the ViBrism website). The distribution of the number of probes exhibiting κ compared with κ was plotted (Fig. 1A). This κ distribution followed an inverse power law, which indicated formation of non-random robust clusters of similarly expressed genes: that is, the clusters contained a significant number of genes associated with a large number of gene-probes in the SET analysis, corresponding to co-expressed hub genes11.
Figure 1. Integrated analysis of graphs of co-expression networks and anatomical maps.

(A) The distribution of probe number in SETs Using the total probes, κ, the probe number in a SET (similarly expressed probes to a target probe) was calculated for each probe with a threshold of r > 0.5, 0.7 and 0.9. The value of κ is shown on the x-axis compared with the number of probes exhibiting κ on the y-axis (Log10 scales). Triangles, squares and green circles represent the results at the three thresholds, respectively. Dotted lines with formulas indicate the power law fit. (B) The distribution of probe number in SETs (κ) calculated in the non-randomized and randomized datasets Using the total probes, κ was calculated for each probe with a threshold of r > 0.85 in the non-randomized dataset that was the fraction data used in Figure A, and in the computationally randomized dataset (see Methods). The distributions of κ were then plotted in the same way as in figure A. Closed circles and cross marks represent the results of non-randomized and randomized datasets, respectively, and dashed lines and dotted lines with formulas indicate their respective power law fits: 95% confidence intervals are shown with chain lines, indicating that the κ distributions in the two datasets are significantly distinguished throughout the range of Log10 κ > 0. Probes accompanied by κ > 100 are only present in the non-randomized dataset (κ = 100 indicated with an arrow). (C) Graphs of co-expression networks of genes that are expressed in regions of the hypothalamus and the midbrain Ten genes that are expressed in regions of the hypothalamus and the midbrain were selected through a literature search, and graphs of their co-expression networks (r > 0.7) were depicted using a stochastic algorithm16. Three-dimensional expression maps are shown at the node positions, and the gene symbols are given at the bottom left. The map orientation is the same within a graph: A anterior, P: posterior, L: left, R: right, D: dorsal and V: ventral. The node positions of the genes that are selectively expressed in a type of cells are close in the graph. Consequently, the graphs represent anatomical and functional regions, as indicated by dotted lines and names12,13,14,15. Thick and thin lines: r > 0.85 and > 0.7, respectively, show that r > 0.85 is sufficiently high to reveal groups of genes that are present in particular regions or cell types. Abbreviations: LH: lateral hypothalamus, SO: supraoptic nucleus, Pa: paraventricular hypothalamic nucleus, Arc: arcuate nucleus. The scale for pseudo-colored expression densities with the cut-off filter is shown.
To evaluate the κ distribution, we computationally randomized the fraction data to produce a dataset representing 36,558 patterns of random expression for the total probes; then, using this dataset, we calculated κ values of the probes with a variety of r thresholds and compared the κ distributions of the randomized dataset to the κ distributions of the fraction data (randomized and non-randomized datasets, respectively, in Supplementary Fig. S2: r > 0.85, 0.75, and 0.65). With a threshold of r > 0.85, the κ distribution of the non-randomized dataset was statistically distinguishable from the distribution of a randomized dataset with a larger number of probes exhibiting κ throughout the range of κ values except for κ = 1 (p < 0.05). We concluded that the SETs with r > 0.85 were non-randomly formed with statistical significances in the fraction data. Thus, we decided to perform SET analysis with a threshold of r > 0.85 unless otherwise indicated. Indeed, at this r threshold, in the randomized dataset, no gene-probes were associated with κ larger than 100. Therefore, we decided to set a threshold of κ > 100 for co-expression hub genes in this study (Fig. 1B). Moreover, using an informatics approach, we inspected the r values between genes that are known to co-localize within cells or in closely located cells. As shown in sample analyses of genes in the hypothalamus and the midbrain12,13,14,15, genes that are co-expressed in serotonergic neurons of the raphe nuclei, in other monoamine neurons of the midbrain and in magnocellular neurons or other functional neurons of the hypothalamic nuclei displayed r values higher than 0.85 (Fig. 1C).
To investigate the underlying biological features of the observed non-random co-expression patterns, we searched databases for biological pathway analysis and GO terms. We revealed that co-expressed hub genes were enriched in functions related to biological interactions (p < 0.001, Supplementary Table S1-sheet 1), and probes exhibiting larger numbers of κ were related to more GO terms of neuronal cell components (p < 0.01, Supplementary Table S1-sheet 2). Moreover, we found that GO terms accumulated in SETs of area marker genes that were implied in a previous study10 (Supplementary Table S2 as spreadsheets). These findings support the idea that the SETs formed exhibited biological relevance.
Framework for integrated analysis of graphs of co-expression networks and anatomical maps
To further investigate the biological significance of these SETs, we developed a framework for integrating co-expression network analysis through cross-validation with spatial maps of gene expression. In our maps, anatomical space is defined by gene expression. For example, brain areas that are rich in cholinergic neurons are defined by the marker gene choline acetyltransferase, Chat10. A significant result of the integrated analysis framework was that a broad range of gene expression patterns can be quantified in a whole-brain context and that the graphs of the co-expression networks that were generated with a stochastic algorithm16 represented functional brain regions, without adding any anatomical information, as shown in the graphs of genes that are expressed in distinct cell types in the hypothalamus and the midbrain (Fig. 1C).
In the following studies, the framework proceeded with statistical significance, as follows: genes with an expression variance were chosen based on a variable V that is a false discovery rate (FDR) of ANOVA analysis, representing the variance of expression densities in the orthogonal series of fractions compared to the variance in the biological replicate fractions (see the Methods). We set the FDR value at < 0.05 to choose genes with variable expression10. These genes were then employed in searches for the associated SETs. Genes that are selectively expressed within the same cells should exhibit high r-values, theoretically r = 1, and these genes therefore represented a SET. Then, the cell type-specific functions of the genes were revealed through accumulated GO terms or literature searches. Finally, the regions of gene expression were visualized in maps, along with the graphs of co-expression networks (r > 0.85). We focused on homeodomain transcription factors, many of which play important roles in embryonic patterning and cell differentiation17.
Lhx and co-expressed genes in distinct cell types
LIM-homeodomain transcription factors (Lhx) developmentally specify discrete types of neurons18. All 12 genes of the Lhx family were detected in the adult brain, and all except for Lmx1b were variably expressed (FDR < 0.05, Fig. 2A). Lhx1 and Lhx5 (Lhx1/5) and Lhx8/Isl1 were co-expressed, whereas the remainder of the genes were not co-expressed with each other and displayed non-redundant expression patterns. Four genes, Lhx1/2/5/6, were co-expression hubs; however, no SETs were found for five of the genes, Lhx3/4, Isl2 and Lmx1a/1b.
Figure 2. Integrated analysis of graphs of co-expression networks and anatomical maps of Lhx genes.

(A) Expression maps of Lhx genes in the adult mouse brain All 12 genes are shown and the gene symbols are indicated at the bottom left of the maps. The maps are all presented in the same direction as the Lhx6 gene maps, A: anterior, P: posterior, D: dorsal and V: ventral region of the brain. All of the genes, except for Lmx1b, are variably expressed (FDR < 0.05). The expression areas are diverse; however, Lhx1/Lhx5 and Lhx8/Isl1 are found in the same SETs, as reported in developmental stages19,28. (B) Graph of the co-expression network of the Lhx8SET The graph (r > 0.85), composed of 15 genes, is depicted using the stochastic algorithm, and the expression maps are attached. The Lhx8 map is indicated with an arrow and 5 genes known to be regionally expressed in the mature CPu are indicated with blue-framed maps. Maps of the transcription factor genes Nkx2-1 and Isl1, which participate with Lhx8 in the differentiation of the CPu, are indicated with green frames. Maps of previously uncharacterized genes (Gm136 and D830015G02) are indicated with yellow frames. The expression area information indicated under maps of two genes was provided from the ABA. Abbreviations: CPu: caudate putamen, TH: thalamus, HY: hypothalamus. (C) Fates of the Nkx2-1-expressing cell lineage Upper panel: Pseudo-colored expression maps without the cut-off filter are used to show the whole-scale expression densities, including relatively low levels. Maps of the genes encoding Nkx2-1, Lhx8 and Isl1 are highlighted in the areas that are defined by Chat expression, as indicated with arrows. In parts of these areas, Nkx2-1 and Lhx8/Isl1 exhibit moderate (in yellow) and high (in red) expression densities, respectively. The high expression of Nkx2-1 caudal to the olfactory bulb, outside the highlighted areas, is indicated with an asterisk. The map orientation is shown as in A. Lower panel: the Chat expression map used to define the highlighted areas is shown. The scale for pseudo-colored expression densities with the cut-off filter is shown for Figure A-C, as otherwise indicated.
To demonstrate one advantage of this integrated analysis, we first mined genes in the SET of Lhx8 (also called Lhx7), which is an area marker gene for the caudate putamen (CPu)10. In total, 15 genes were included in the graph of the co-expression network for the SET, and five genes are known to be expressed and function in the CPu (Fig. 2B, the list of genes is provided in Supplementary Table S2: a spreadsheet for Lhx8)19,20,21,22,23. A subpopulation of cells in the adult CPu is composed of cholinergic neurons that develop as follows: post-mitotic progenitor cells expressing Nkx2-1 (a transcription factor also known as Titf1) begin migrating from the ventricular zone in the ventral part of the forebrain, and, during migration, the cells differentiate into mature neurons under the combined activity of Lhx8 and Isl1, which are associated with the suppression of Lhx619. In this study, the genes encoding these three transcription factors, Nkx2-1, Isl1 and Lhx8, were all included in this network graph, whereas Lhx6 was not.
Moreover, the expression densities that were observed in the maps could be accounted for by the developmentally programmed fate of the Nkx2-1-expressing cell lineage derived from the embryonic ventricular zone (Fig 2C). The moderately high expression of Nkx2-1 accompanied by the high expression of Lhx8/Isl1 was detected in the ventral part of the forebrain, which is rich in cholinergic neurons, as defined by the Chat expression. And the highest Nkx2-1 expression was observed in the area caudal to the olfactory bulb, where cells with neurogenesis potential in the adult brain that were derived from the ventricular zone are present24,25. We found that figures in of the open resource, GENSAT26, elicited Nkx2-1 expressing cells in these areas (Supplementary Fig. S3).
In addition, novel genes, including a predicted gene (Gm136) and a non-coding gene of unknown function (D830015G02), and Spint1, which regulates the proliferation and cell fate of neural progenitor cells27, were first revealed, to our knowledge, as highly expressed in these areas. We predict these genes to specifically function in the CPu and nearby areas.
We next investigated genes in the SETs of other Lhx family members, focusing on genes encoding morphogenetic transcription factors, morphogens and functional markers that are characteristic of cell functions, to determine whether we could obtain additional graphs of co-expression networks with distinct functions (Supplementary Table S3).
The Lhx1/5 proteins control Purkinje cell differentiation in the developing cerebellum28; Lhx1/5 were detected in the adult cerebellum in our maps, as shown in Figure 2A. We generated a graph of the co-expression network, which was composed of genes in the Lhx1/5 SETs (Fig. 3A). The graph included genes encoding molecules that are highly expressed in adult Purkinje cells, such as a morphogen, Wnt329 and a glutamate transporter, Slc1a630, in addition to genes encoding the Wnt receptor Fzd1, the transcription factor En2, which regulates the morphological and molecular patterning of the embryonic cerebellum31, the sodium/glucose co-transporter Slc5a1, which is regulated by En232, and the transcription factor Pax3, which is expressed in cells surrounding Purkinje cells, which presumably correspond to Bergmann glia in the adult cerebellum33. Because the gene expression in these cells was confirmed in the ISH figures of ABA (Supplementary Fig. S4A), we infer, based on the integrated analysis of anatomical maps and network graphs, that Fzd1 transduces the Wnt3 signal that is produced by Purkinje cells to the surrounding Bergmann-glial cells, wherein Pax3 is expressed. This local Wnt3/Fzd1/Pax3 signaling could regulate the differentiation of the glial cells, maintaining stem cell characteristics in the mature stage34 in relation to Purkinje cells. The Lhx1/5 SET contained more than 100 genes. Among these genes, we nominate Dao1 as a functional marker gene of this SET, in terms of Purkinje and Bergmann-glial cell interactions. Dao1 encodes an enzyme35 that is exclusively active in the mature cerebellum for the catabolization of a neuromodulator molecule, D-serine, which is produced in the Bergmann glia and modifies the glutamatergic synaptic plasticity of the Purkinje cells.
Figure 3. Graphs of co-expression networks that are composed of genes in the Lhx1/5, Lhx2/6 and Lhx9 SETs.

(A) Graph of the Lhx1/5 SET The SETs of Lhx1 and Lhx5 (the maps are indicated with arrows) contained more than 100 genes; therefore, we selected genes of morphogenetic molecules and markers characterizing cell functions. The co-expression graph was generated using the stochastic algorithm, and the maps are attached. Genes that are shown in blue- and green-framed maps are regionally expressed in immature Purkinje cells and the surrounding cells, respectively. Genes with brown- and purple-framed maps are functionally related. (B) Graph of the Lhx2/6 SET Using genes in the SETs of Lhx2 and Lhx6, network graphs were depicted in the same way as for the Lhx1/5 SETs. Genes shown with blue- and purple-framed maps are known to co-function in areas of the developmental neural system. Genes with green- and yellow-framed maps function in GABAergic neuron specification and GABAergic/glutamatergic transition, respectively. Maps of two cholinergic receptor genes are indicated with brown frames. (C) Graph of the Lhx9 SET Lhx9 is co-expressed with a Wnt mediator gene, Tcf7l2, in the dorsal midbrain. Expression maps are depicted using a pseudo-colored density scale with a filter.
Lhx2 specifies neurons in the upper layer of the cerebral neocortex and suppresses the organization of the hippocampus36,37. The graph of the Lhx2 co-expression network included genes encoding a forkhead transcription factor, Foxg1, and a homeodomain transcription factor, Emx2 (Fig. 3B). These three genes are expressed across the dorsolateral wall of the embryonic forebrain, and each gene plays crucial roles in specifying the location of the progenitor cells of projection neurons in the cerebral neocortex38. In our maps, these three genes were localized to the dorsolateral areas of the adult cerebrum. The co-expression graph also contained a gene encoding a transcription factor, Nr2e1, which controls the division of progenitor cells to generate subsets of neurons in the upper layer neocortex of the cerebrum39: Lhx2 and Nr2e1 are known to cooperate in demarcating the eye field40. A form of muscarinic cholinergic receptor gene, Chrm1, was included in the network as a functional marker gene. Chrm1 is involved in a distinct aspect of brain function: object recognition memory with delay41.
Lhx6 is a determinant of GABAergic interneurons in the CPu and parts of the cerebral cortex and a determinant of GABAergic projection pathway in subcortical regions of the ventral forebrain19,42,43. The Lhx6 co-expression network contained genes encoding Foxo6 and Wnt2, which play roles in GABAergic neuron specification during forebrain development44 (Fig. 3B). Intriguingly, this network also included genes encoding a zinc finger transcription factor, Fezf2, which can be used to experimentally generate glutamatergic projection neurons directly from GABAergic precursors in the CPu45, as well as a T-box transcription factor, Tbr1, which restrains Fezf2 activity to the lower-layer neocortex, where glutamatergic projection neurons normally originate46,47. A nicotinic cholinergic receptor gene, Chrna5, which regulates GABAergic and glutamatergic transmission48, was also present in this network and was considered a functional-marker gene, similar to Chrm1 in the Lhx2 network. Moreover, the co-expression networks of Lhx2 and Lhx6 were linked through Tbr1, which is expressed in both the upper and lower layers of the neocortex because of its function in regulating the layer identity of postmitotic neurons49 (Supplementary Fig. S5).
Lhx9 was found to be co-expressed with a Wnt signaling mediator gene, Tcf7l27 (Fig. 3C); both were present in the caudal forebrain and in the dorsal midbrain in the expression maps. Lhx9 determines cell differentiation in the caudal forebrain by regulating Wnt signals in the zebrafish50. In the mouse brain, Tcf7l2 is expressed in subcortical regions, the thalamus and the dorsal midbrain, where neurons are specialized for the integration of diverse sources of sensory information51. Therefore, we hypothesize that Lhx9 could determine the fate of the neurons that participate in the sensory input integration via Wnt/Tcf7l2 signaling and that this gene network continues to function in the adult brain. Indeed, we found experimental evidence that supported this hypothesis in figures of GENSAT26 (Supplementary Fig. S6). Lhx9 expressing cells are present in selective neuron groups of visual and auditory input system and of the integration system52,53 in the adult mouse brain.
Pax and co-expressed genes in subsets of cell types
To test how broadly developmentally programmed co-expression networks appeared in the adult brain, we investigated the expression of genes encoding other homeodomain transcription factors: Pax/PRD/Arx and ANTP, including Hox, Nkx, Dlx, Emx, Irx and En (Supplementary Table S3).
The expression of genes in the Pax family is developmentally programmed54. All nine genes were detected using our platform, and their spatial expression patterns were comparable to the areas originating from the cells expressing these genes in embryonic stages33 (Fig. 4A). The informatics approach to the co-expression networks indicated potential roles for the genes in neurogenesis or plasticity in local environments, as Pax3/Wnt3/Fzd1 were described above.
Figure 4. Analysis of Pax genes.
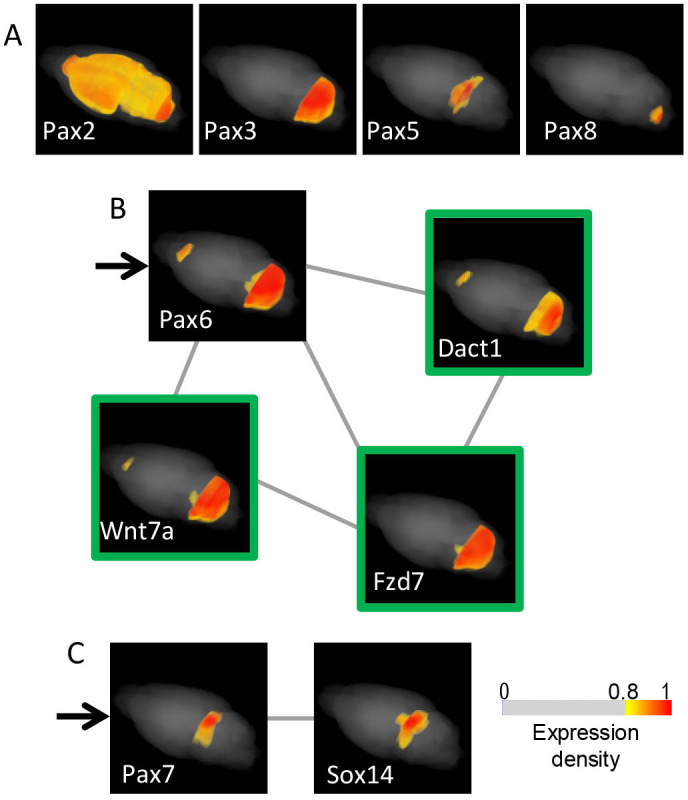
(A) Expression maps of Pax genes in the adult mouse brain Among the nine Pax family members, Pax1 and Pax9, which do not contain a homeodomain, and Pax4 were expressed with no statistically significant expression variance (FDR = 0.204). Maps of Pax6/7 are shown in Figure B and C, respectively; the other maps are shown in A. The areas of expression were comparable to the areas originating from the cells expressing the genes in embryonic stages: Pax3 was observed in the cerebellum, Pax5 in the midbrain, pax6 in the cerebellum and posterior to the olfactory bulb, Pax7 in the dorsal midbrain and Pax8 in the hindbrain; however, Pax2 was observed over a rather broad area54. No SET was found for Pax5. Graphs of the co-expression networks of Pax3/8 are provided in Figures 3A and 5B. (B and C) Graphs of co-expression networks that are composed of genes in the Pax6 and Pax7 SETs Pax6 (the map is indicated with an arrow) is a co-expression hub gene. The co-expression graph is depicted in the same manner as in Figure 3A. Genes encoding proteins that are involved in Wnt signaling are shown with green-framed maps. The Pax7 SET contains only Sox14. The scale for the expression densities with the cut-off filter is shown.
Pax6 was detected in an area caudal to the olfactory bulb and in the cerebellum. The co-expression network of Pax6 contained genes encoding Wnt signaling molecules, including Wnt7a, which controls central nervous system vasculature differentiation55, Fzd756 and Dact1 (also known as Frodo), which is downstream of Wnt and is essential for optic and neural development57 (Fig. 4B). Indeed, Fzd7 contributes to self-renewal signaling in embryonic stem cells, similar to the role of Wnt in neuronal stem cells56,58. In addition, the inhibition of Wnt signaling induces Pax6, consequently promoting the differentiation of retinal precursor cells from neuronal progenitor cells in vitro59. These experimental findings allow us to conclude that we can select a set of genes that are related to stem/progenitor cell regulation wherein local Wnt7a signaling via Fzd7/Dact1 restrains Pax6 from inducing excessive neurogenesis. This scenario appears to explain the regulatory mechanism of neurogenesis that takes place in the olfactory bulb areas60 and in the cerebellum61 (Supplementary Fig. S7). Because no genes were common to the Pax6 and Pax3 SETs, these two co-expression networks, though both were in the cerebellum, seem to participate in different local biological systems (Supplementary Fig. S4).
A gene for Pax7, which regionalizes the embryonic dorsal part of the midbrain62 and persists there in mature superior collicular visual-related neurons63, was detected in the superior colliculus of the dorsal midbrain in our map and was selected the area marker gene10. Pax7 was co-expressed with Sox14 (Fig. 4C). Sox14 organizes the development of the visual system, including the superior colliculus, and orchestrates the diurnal cycle in response to light and dark conditions64. We suggest that Pax7 may also participate in the diurnal cycle regulation. Pax8 was present in the hindbrain, as discussed below.
Genes encoding other homeodomain transcription factors expressed in subsets of cell types
Of the 18 Nkx family genes, 15 were expressed in the adult brain (Fig. 5A). The gene encoding Nkx6-1, which participates in motor neurogenesis65, was co-expressed with motor neuron-specific genes that play a role in synaptic plasticity, such as Grin3b, which encodes an NMDA-type ionotropic glutamate receptor66, and Calcb, which encodes a motor neuron-derived astrocyte activation protein67. Nkx6-1/6-3 were co-expressed. Both genes were co-expressed with Hox genes that display a co-linear expression density pattern of a posterior to anterior gradient and function in the segmental organization of the ventral hindbrain68 (Fig. 5B). Pax8 also participated in this network. Nkx2-2/6-2 and Nkx2-9 were hypothesized to be expressed in non-neuronal cells in the adult brain because these genes were co-expressed with genes encoding oligodendroglial cell markers, such as Mbp/Edg869, and astrocyte markers, such as Klk670, respectively (Fig. 5C). Nkx5-1/5-2 were co-expressed in the ventro-caudal forebrain and the ventral hindbrain. Nkx2-1/3-1 are mentioned elsewhere. No SETs were identified for the other Nkx genes.
Figure 5. Analysis of the Nkx and Arx genes.

(A) Expression maps of Nkx genes in the adult mouse brain Among the 18 Nkx family members, 15 genes are expressed: Nkx2-5/5-3 are expressed with no statistically significant expression variances (FDR = 0.928 and 0.864, respectively), and maps of four genes with no SETs are shown here. The maps of the rest are provided in figures B-D and Figure 3B, and are indicated with arrows when necessary. Nkx2-1, Nkx5-1, Nkx5-2 and Nkx5-3 are called Titf1, Hmx3, Hmx2 and Hmx1, respectively, in the ViBrism DB. (B and C) Graphs of co-expression networks that are composed of genes in the SET of Nkx6-1/6-3 and the SETs of Nkx2-2/2-9/5-1/5-2/6-3 The co-expression graph was generated in the same manner as in Figure 3A. Maps of genes expressed in motor neurons, Hox genes and an NMDA receptor gene are indicated with blue, green and red frames, respectively. (D) Graph of the co-expression network composed of genes in the Arx SET Arx, indicated with an arrowhead, is a co-expression hub gene. Maps of genes expressed in inter-neurons, Dlx genes and a NMDA receptor gene are indicated with blue, green and red frames, respectively. The color codes elicit analogous function of genes in the Arx SET to ones in the Nkx6-1 SET, modulating the plasticity of the inter- and motor-neurons via the NMDA receptors under the orchestration of the Dlx and Hox gradients, respectively. The scale for the pseudo-colored expression densities with the cut-off filter is shown.
Arx, which is a gene encoding a transcription repressor that is expressed in developing and mature interneurons under the influence of the transcription factors Dlx1/271,72, showed a co-expression network that was composed of genes with a similar function to genes that are present in the Nkx6-1 network (Fig. 5B and 5D). Arx was co-expressed with Nkx3-1, and its network included Dlx1/2, as expected. Among the 6 Dlx genes, Dlx1/2/5/6 displayed an expression density pattern of an anterior to posterior gradient, which was analogous to that found for Hox genes. The Arx network contained a gene encoding another form of NMDA-type receptor, Grin2b, which is localized to the forebrain and controls synaptic plasticity73, and a gene encoding a neural dendrite-specific adhesion molecule, Icam5, which regulates synapse formation74, analogous to Calcb in the Nkx6-1 network.
Emx1, which is an area marker gene for the dorsal forebrain during developmental stages, is inducible through Wnt signaling75. Emx1 was localized to the dorso-lateral forebrain and co-expressed with Wnt9a. Emx1 was also co-expressed with esPRC2p48, which encodes a polycomb complex protein (also called E130012A19Rik) that regulates stem cell differentiation to glutamatergic neurons in the cerebral cortex and the hippocampus76. Emx2 was described above, and Emx3 was not detected. En1, Irx3, Lrx4 and Irx6 were expressed in the hindbrain and displayed unique patterns. Irx1/2/5 formed a co-expression network in the cerebellum.
Combinatorial expression of transcription factor genes and previously uncharacterized coding and non-coding genes
Among 1332 probes for genes of transcription factors, 1251 probes (94%) were detected at least in one fraction of this study. The mean value of variable I10 that represented the intensity medians for the probes in the fractions of the 1251 probes (18.44 +/− 1.77) were 40% lower than the mean value of I for the total probes (30.97 +/− 0.62). Among the examined genes encoding homeodomain transcription factors17, all 12 Lhx, 9 Pax, 1 Arx, 2 Emx, 6 Irx and 2 En, 30 of 39 Hox, 15 of 18 Nkx and 5 of 6 Dlx genes, totaling 82 of 95 genes (86.3%), were detected in the adult brain, although the mean value of I (3.27 +/− 1.22) was approximately 10% of the value for the total probes. This result indicated that the sensitivity of our system was sufficient to detect low expression genes.
The examined genes formed co-expression gene groups (Supplementary Table S3). The combinatorial expression of distinct gene members of homeodomain transcription factor families, including Lhx, Pax/PRD/Arx and ANTP, as well as other transcription factor families was repeatedly observed as modules that constructed parts of the co-expression network, which followed an inverse power law. Approximately 40% of the genes encoding the homeodomain transcription factors covering the entire anatomical context of the adult brain have been described here. For further examination, the SETs of all genes expressed in the brain are searchable, and the 3D expression maps can be viewed and downloaded from the ViBrism DB website. Then, network graphs can be depicted as described in the methods.
Previously uncharacterized genes were involved in the SETs of transcription factor genes. In particular, six genes displayed extremely high r (> 0.95, Table 1) to the co-expression hub genes Lhx1/2/6 and Arx. To the best of our knowledge, our framework provided the first visualization of the location of action of the six genes and provided information about their function in relation to co-expressed genes (Fig. 6). Except for 2010300C02Rik, which encodes a putative protein with a rat ortholog, these gene sequences are not conserved in other species and are non-protein coding; thus, the functions of the genes are unique to the rodent or mouse brain. In the network graphs, five genes were in a position that linked the co-expression gene groups: BM939341 linked Fezf2/Tbr1 to Lhx6; Gm11549 and 2010300C02Rik linked Fezf2/Tbr1 to Lhx2/Emx2/Foxg1/Nr2e1; and AI835086 and 3110039M20Rik linked Lhx2/Emx2/Foxg1/Nr2e1 to Arx/Dlx1/2. This linkage suggests that these previously uncharacterized genes play roles in coordinating functions of the gene groups that are repeatedly observed and developmentally conserved and that consequently participate in creating mouse-specific neural designs.
Table 1. List of preciously uncharacterized genes in co-expression networks (r > 0.95).
| previously uncharacterized genes | |||||
|---|---|---|---|---|---|
| co-expression network of | r | gene symbol | gene ID | gene product | gene structure |
| Lhx1 | 0.983 | 1500016L03Rik | AK018772 | non protein coding transcript | 4 exons with no polyA in 3′ UTR |
| Lhx2 | 0.956 | 2010300C02Rik | BC029089 | putative protein similar to Rattus norvegicus putative protein (5S487) (RGD1310819), mRNA | 12 exons with polyA in 3′ UTR, including a protein coding region partially (3-1957 bp) similar to Mesocricetus auratus KIAA1211-like ortholog (Kiaa1211l), mRNA |
| 0.952 | Gm11549 | AK043581 | non protein coding transcript | 3 exons with no polyA in 3′ UTR | |
| Lhx6 | 0.964 | BM939341 | BM939341 | non protein coding transcript | 1 exon with no polyA in 3′ UTR |
| Arx | 0.984 | AI835086 | AK043585 | non protein coding transcript | 1 exon with no polyA in 3′ UTR |
| 0.951 | 3110039M20Rik | AK043991 | non protein coding transcript | 3 exons with no polyA in 3′ UTR | |
Figure 6. Graph of the co-expression network composed of previously uncharacterized genes and transcription factor genes in the Lhx2/6 and Arx SETs.

A network of similarly expressed genes (r > 0.95) is depicted with the stochastic algorithm and maps are attached. Maps of previously uncharacterized genes are indicated with yellow frames. A group of genes, Lhx2/Emx2/Foxg1/Nr2e1 (Supplementary Table S3), function in the neural pathway formation of upper-layer neurons in the cerebral cortex. Maps of the genes are shown with blue frames. Lhx6 SET (r > 0.85) is divided into two gene groups, Lhx6 and Fezf2/Tbr1, which function in the GABAergic and glutamatergic neural pathway formations, respectively. The gene maps are indicated with green and light-green frames. A group of genes, Arx/Dlx1/2, modulate inner neuron synaptic plasticity. The gene maps are indicated with magenta frames. An expression maps of 1500016L03Rik that is highly similarly expressed to Lhx1 is shown in the separate panel at the bottom left of the figure. The expression density scale is shown.
Discussion
We introduced a framework for the integrated analysis of 3D gene expression maps and graphs of co-expression networks and demonstrated how this framework can be used to gain insights into the underlying biology of co-expression networks, which were found to follow an inverse power law. The integration of expression maps and co-expression networks, which is not based on archived information of selected key areas in the brain but based on our whole brain analysis results were demonstrated in this study. Consequently, unexpectedly wide varieties of combinatorially expressed genes, which are known to be involved in the regulation of cell differentiation during development, were also observed in the adult brain. The gene combinations on which we focused included family members of homeodomain transcription factors, Wnt signaling molecules, functional markers and uncharacterized genes. Distinct members of the families were expressed in particular cell types. Consequently, analogous combinations appeared as modules in a variety of cells, for example: Lhx2/Foxg1/Nr2e1/Tbr1/Chrm1 in the upper-player neurons are analogous to Lhx6/Foxo6/Fezf2/Tbr1/Chrna5 in the lower-layer and subcortical neurons of the cerebrum; Pax3/Wnt3/Fzd1 in Purkinje/Bergmann cells are analogous to Pax6/Wnt7a/Fzd7 in stem/progenitor cells; and Nkx6-1/Nkx6-3/Hox/Calcb/Grin3b in the cranial motor neurons are analogous to Arx/Nkx3-1/Dlx/Icam5/Grin2b in the cerebral interneurons (Supplementary Table S3). Moreover, each module appeared to display a particular biological function: the first example is a module operating in neural pathway formation, the second functions in stem cell regulation, and the third functions in synaptic plasticity. Finally, we revealed that mouse-specific non-coding genes were in positions linking the conserved co-expression gene networks. This linkage suggests characteristics of these genes that coordinate multiple gene groups and create mouse-specific neurogenesis designs.
Genetic modules constituting the robust structures of co-expression networks are widely observed in various species as a mechanism underlying networks that are characterized by a power law1,6,11; however, such studies are limited to pre-determined anatomical areas or are based on retrieved datasets. Our framework enables the analysis of co-expression gene networks throughout the brain, along with an anatomical overview. Networks in complex areas that could not be described in anatomical terms, for instance, the Lhx2 network in Chrm1-expressing cells that are associated with delayed memory formation, are detectable, and the anatomical locations of the networks can be compared with areas that are defined by area marker genes. These advantages enable the visualization of previously unrecognized robust expression patterns.
An advantage of the data, which this study is built upon, is provided by the straightforward quantification of gene expression densities using our tomographic technique, compared to the AGEA data produced with elaborate image processing of ISH-based 2 dimensional datasets of ABA. The expression densities of a large number of genes are measured in a fraction. Consequently, all the 3D expression maps are reconstructed into the same 3D anatomical space, and the expression similarities of any measured genes can be detected using r as a similarity measure in the fractions. There is no need for spatial registration of expression maps in one-by-one manner and pre-selection of 4,500 regionally expressed genes out of approximately 25,000 gene maps by image processing, which is performed in the ABA dataset analysis. Therefore, novel genes that have not been mapped with other methods have been included in our network analysis and their functions were predicted. The pairwise comparison method using r as an expression similarity measure for network analysis can be implemented in any numerical datasets. It may be applied to the numerical data of ABA and may produce precise co-expression results.
The mapping resolution of the tomography technique depends on the number of data points. In this dataset, only 61 fractions are used for the mapping, and consequently the anatomical resolution of the maps is not high. In 40 anatomical sub-regions, as we have shown in the previous paper10, the 3D distributions of the expression maps are comparable to the maps in ABA. However, very selective expression cannot be mapped properly because of the limitation of the present fraction number. The number limitation is biologically due to the amount of RNA extracted from the fraction materials. A 50-μm-thick fraction is sufficient to obtain RNA materials for an analysis in the present molecular measurement technique. Therefore, 3D maps of higher resolution than existing ABA (200 μm at a distance in the sagittal plane) can be produced, and co-expression can be analyzed there, using the transcriptome tomography approach.
We believe that our framework will contribute to obtaining a gene expression-based understanding of complex brain structures and functions. Gene expression densities measured in the “fractions” are used for creating both 3D expression maps and co-expression networks, so that, the maps and the networks are directly integrated without any additional experimental procedures. Varieties of combinations of genes that are observed with the framework suggest a broader potential for neurogenesis and the plasticity of the adult brain. Whole gene expression maps and the SET search are provided in ViBrism DB. Transcriptome tomography, together with the framework for performing co-expression searches using the ViBrism DB platform, represents an important tool for the systematic investigation of the molecular anatomy of key biological processes that involve uncharacterized genes.
Methods
Ethics statement
All experiments on mice and their care were performed in accordance with the procedures that the RIKEN Regulations for Animal Experiments approved (approval ID: H19-1W009).
Transcriptome Tomography
The tomography methodology was described in a previous study10 and in the Introduction and Supplementary Fig. S1 in this paper. Briefly, two types of data were obtained from 1-mm-thick tissue fractions of 8-week-old male C57BL/6J mice brains via sequential cross-sectioning (5-μm-thick cross-sectioning slices × 200 times sectioning per fraction): 1) gene expression density data measured with microarray and 2) each block-face image of the cross-sectioning planes. This process was performed in six brains and resulted in six sectioning series (two biological replicates of three orthogonal series) of 61 fractions in total.
The gene expression densities in the fractions were measured as the intensities detected with microarray probes (Whole Mouse Genome 012694, Agilent, CA), as described in the previous paper10. They were per-chip normalized using 75 percentile normalization, which was robust enough for the batch effect in the study using a few batches, and subjected to probe-based analysis (36,558 probe data points from the 61 fractions, hereafter referred to as fraction data): Supplementary Table S4-sheet 1 showed probe information and sheet 2 shows quality control index values of extracted RNAs and microarray analyses. These probes are sufficient to detect most genes (approximately 25,000 genes) and splicing variants. The microarray data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus database77 and are accessible through GEO Series accession number GSE36408 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36408).
Fraction data analyses and statistics
Two sets of variables, I and V were calculated from the log-transformed fraction data10: briefly, I represented the intensity medians for the probes in the 61 fractions; mean +/−. sem of the indicated probes were shown; and the variable V, representing the variance of the probe intensity in the orthogonal series of fractions compared to the variance in the biological replicate fractions, was defined as FDR calculated through one-way ANOVA with multiple-testing Benjamini and Hochberg correction. FRD is a rate of false discovery of genes with expression variances of replicates as variably expressed genes (see Note in Supplementary Fig. S1). Another variable, κ, represents the number of similarly expressed probes to a target probe including itself (SET). The r of the intensity values in the 61 fractions between the probes was calculated and used as a correlation measure for the expression similarity. The applied threshold was r > 0.85, unless otherwise indicated. Power law fit lines and 95% confidence intervals of κ distributions are created with linear regression modeling of log scaled values. The hypergeometric p-values, with or without multiple-testing corrections, are shown for the GO analyses of the selected gene sets. The alpha level is 0.01 in all analyses.
Randomization of the fraction data to generate random expression patterns
To randomize the fraction data, the order of gene expression densities in fractions of a series were randomized for each probe using the Ruby programming language (http://www.ruby-doc.org/core-2.1.0/Random.html), on which a pseudo-random number generator is implemented using Mersenne Twister (http://www.math.sci.hiroshima-u.ac.jp/~m-mat/MT/mt.html). The density data in the paired fraction (see Note in Supplementary Fig. S1) were randomized in the same order in the series to be maintained as replicates in the randomized dataset. Consequently, in the statistical characteristics of the fraction data, variables I and V for the probes were not changed through the randomization process.
Network analyses
Graphs of co-expression networks at the indicated threshold of r were depicted with R version 3.0.078, using a stochastic, force-based graph layout algorithm described by Kamada-Kawai16. Biological pathways were analyzed using GeneSpring GX v12 software based on the open resource IntAct (http://www.ebi.ac.uk/intact/main.xhtml), together with Biopax-format data imported from Reactome (http://reactome.org/download/index.html) and the results of a PubMed search using a natural language processing algorithm. The biological meanings of the co-expression networks were manually curated. Probes for transcription factor genes were selected by association with GO:0003700, sequence specific transcription factor activity.
Anatomical map analysis
Gene expression maps were visualized using an 80% cut-off filter to reveal areas of high expression, unless otherwise indicated. The instructions for highlighting areas based on gene expression in 3D expression maps are provided in the Quick Manual for VCAT (downloadable from our website).
ISH of genes expressed in the mouse brain at postnatal stages
The spatial cellular expression patterns of the genes were analyzed by ISH histochemistry of male C57BL/6J mouse brains at postnatal stages of day 7 and 21. Sagittal sections of the brain were subjected to hybridization as previously described79. High resolution digital images of hybridized sections were used for this study.
Author Contributions
Y.O. designed the experiments and analyzed the data. K.S. created the database. M.N., S.T. and H.Y. created the 3D map datasets. A.S. and T.F. provided neonatal ISH images. Y.O. and H.Y. conceived the project plan. Y.O. wrote the manuscript.
Supplementary Material
Supplementary Video S1
Supplementary Fig. S1- S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Acknowledgments
We thank C. Armit (MRC Human Genetics Unit, University of Edinburgh, UK) for critical reading and help in manuscript editing, S. Onami (RIKEN Quantitative Biology Center, Japan) and S. Takagi (University of Tokyo, Japan) for suggestions regarding the network analyses and task force members of the International Neuroinformatics Coordinating Facility (INCF) Program on Digital Brain Atlasing in discussion of 2D-3D image analysis. This research was supported by funding from the RIKEN Strategic Programs for R&D (President's Discretionary Fund) to H.Y., Y.O., K.S. et, al.; JSPS KAKENHI Grant Number 25560428 and 26280110 to Y.O. and H.Y., and 23300137 and 258057 to T.F.; JST Acceleration Utilization of University IP Program to Y.O.; and RIKEN Neuroinformatics Japan Center to Y.O. and T.F.
Footnotes
H. Y., Y. O. and K. S. are co-inventors listed on patent applications associated with the method for constructing gene expression images and the system for constructing gene expression images (JP 2005248312, EP06796974.1 and US12/065014). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Stuart J. M., Segal E., Koller D. & Kim S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science (80-.). 302, 249–55 (2003). [DOI] [PubMed] [Google Scholar]
- Hobert O. Regulatory logic of neuronal diversity: terminal selector genes and selector motifs. Proc. Natl. Acad. Sci. U. S. A. 105, 20067–71 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–76 (2007). [DOI] [PubMed] [Google Scholar]
- Ng L. et al. An anatomic gene expression atlas of the adult mouse brain. Nat. Neurosci. 12, 356–62 (2009). [DOI] [PubMed] [Google Scholar]
- Diez-Roux G. et al. A High-Resolution Anatomical Atlas of the Transcriptome in the Mouse Embryo. PLoS Biol 9, e1000582– (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M. C. et al. Functional organization of the transcriptome in human brain. Nat. Neurosci. 11, 1271–82 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. B. et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasukawa T. et al. Quantitative expression profile of distinct functional regions in the adult mouse brain. PLoS One 6, e23228 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz M. J. et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura-Oho Y. et al. Transcriptome tomography for brain analysis in the web-accessible anatomical space. PLoS One 7, e45373 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A.-L. & Oltvai Z. N. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–13 (2004). [DOI] [PubMed] [Google Scholar]
- Liu Z. et al. Study of gene function based on spatial co-expression in a high-resolution mouse brain atlas. BMC Syst. Biol. 1, 19 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M. et al. Expression analysis of genes responsible for serotonin signaling in the brain. Neurobiol. Dis. 19, 378–85 (2005). [DOI] [PubMed] [Google Scholar]
- Silverman A. J. & Zimmerman E. A. Magnocellular neurosecretory system. Annu. Rev. Neurosci. 6, 357–80 (1983). [DOI] [PubMed] [Google Scholar]
- Kotz C. M. Integration of feeding and spontaneous physical activity: Role for orexin. Physiol. Behav. 88, 294–301 (2006). [DOI] [PubMed] [Google Scholar]
- Kamada T. & Kawai S. An Algorithm for Drawing General Undirected Graphs. Inf. Process. Lett. 31, 7–15 (1989). [Google Scholar]
- Holland P., Booth H. & Bruford E. Classification and nomenclature of all human homeobox genes. BMC Biol. 5, 47–75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. & Westphal H. Functions of LIM-homeobox genes. Trends Genet. 16, 75–83 (2000). [DOI] [PubMed] [Google Scholar]
- Fragkouli A., van Wijk N. V., Lopes R., Kessaris N. & Pachnis V. LIM homeodomain transcription factor-dependent specification of bipotential MGE progenitors into cholinergic and GABAergic striatal interneurons. Development 136, 3841–3851 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhais-Neto G. C. et al. Rere controls retinoic acid signalling and somite bilateral symmetry. Nature 463, 953–957 (2010). [DOI] [PubMed] [Google Scholar]
- Rahman Z. et al. Cloning and characterization of RGS9-2: a striatal-enriched alternatively spliced product of the RGS9 gene. J. Neurosci. 19, 2016–26 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasinska A. J. et al. Identification of brain transcriptional variation reproduced in peripheral blood: an approach for mapping brain expression traits. Hum. Mol. Genet. 18, 4415–4427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 7, e006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Tam M. & Anderson S. A. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol. 506, 16–29 (2008). [DOI] [PubMed] [Google Scholar]
- Lledo P.-M., Merkle F. T. & Alvarez-Buylla A. Origin and function of olfactory bulb interneuron diversity. Trends Neurosci. 31, 392–400 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S. et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925 (2003). [DOI] [PubMed] [Google Scholar]
- Koivuniemi R. et al. Hepatocyte growth factor activator inhibitor-1 is induced by bone morphogenetic proteins and regulates proliferation and cell fate of neural progenitor cells. PLoS One 8, e56117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. et al. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc. Natl. Acad. Sci. U.S.A. 104, 13182–6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas P. C., Fletcher C., Copeland N. G., Jenkins N. A. & Nusse R. Maintenance of Wnt-3 expression in Purkinje cells of the mouse cerebellum depends on interactions with granule cells. Development 120, 1277–86 (1994). [DOI] [PubMed] [Google Scholar]
- Kanai Y. & Hediger M. A. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflügers Arch. Eur. J. Physiol. 447, 469–79 (2004). [DOI] [PubMed] [Google Scholar]
- Sillitoe R. V., Stephen D., Lao Z. & Joyner A. L. Engrailed homeobox genes determine the organization of Purkinje cell sagittal stripe gene expression in the adult cerebellum. J. Neurosci. 28, 12150–62 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst M. I. et al. Engrailed-2 regulates genes related to vesicle formation and transport in cerebellar Purkinje cells. Mol. Cell. Neurosci. 38, 495–504 (2008). [DOI] [PubMed] [Google Scholar]
- Stoykova A. & Gruss P. Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J. Neurosci. 14, 1395–412 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile V., Li M. & Scotting P. J. Stem cell marker expression in the Bergmann glia population of the adult mouse brain. Brain Res. 1099, 8–17 (2006). [DOI] [PubMed] [Google Scholar]
- Kakegawa W. et al. D-serine regulates cerebellar LTD and motor coordination through the δ2 glutamate receptor. Nat. Neurosci. 14, 603–11 (2011). [DOI] [PubMed] [Google Scholar]
- Nakagawa Y., Johnson J. E. & O'Leary D. D. Graded and areal expression patterns of regulatory genes and cadherins in embryonic neocortex independent of thalamocortical input. J Neurosci. 19, 10877–85 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangale V. S. et al. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science (80-.). 319, 304–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux B. J., Arlotta P., Menezes J. R. L. & Macklis J. D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–37 (2007). [DOI] [PubMed] [Google Scholar]
- Land P. W. & Monaghan A. P. Expression of the transcription factor, tailless, is required for formation of superficial cortical layers. Cereb. cortex 13, 921–31 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber M. E. Eye-Field Transcription Factors. Encycl. Eye null, 121–127 (Elsevier, 2010). [Google Scholar]
- Bartko S. J. et al. Intact attentional processing but abnormal responding in M1 muscarinic receptor-deficient mice using an automated touchscreen method. Neuropharmacology 61, 1366–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski I., Mayer N. & Zecevic N. Multiple origins of human neocortical interneurons are supported by distinct expression of transcription factors. Cereb. cortex 21, 1771–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G. B. et al. Lhx6 delineates a pathway mediating innate reproductive behaviors from the amygdala to the hypothalamus. Neuron 46, 647–60 (2005). [DOI] [PubMed] [Google Scholar]
- Zhong J. et al. The Wnt receptor Ryk controls specification of GABAergic neurons versus oligodendrocytes during telencephalon development. Development 138, 409–19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouaux C. & Arlotta P. Fezf2 directs the differentiation of corticofugal neurons from striatal progenitors in vivo. Nat Neurosci 13, 1345–1347 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W. et al. TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc. Natl. Acad. Sci. U.S.A. 108, 3041–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna W. L. et al. Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J Neurosci 31, 549–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzer-Serhan U. H. & Leslie F. M. Expression of alpha5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J. Comp. Neurol. 481, 19–30 (2005). [DOI] [PubMed] [Google Scholar]
- Bedogni F. et al. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. U.S.A. 107, 13129–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert D., Weber S., Lumsden A. & Scholpp S. Lhx2 and Lhx9 determine neuronal differentiation and compartition in the caudal forebrain by regulating Wnt signaling. PLoS Biol. 9, e1001218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalski A. et al. Postnatal isoform switch and protein localization of LEF1 and TCF7L2 transcription factors in cortical, thalamic, and mesencephalic regions of the adult mouse brain. Brain Struct. Funct. 218, 1521–1549; 10.1007/s00429-012-0474-6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao A., Moser M.-B. & Moser E. I. Traces of experience in the lateral entorhinal cortex. Curr. Biol. 23, 399–405 (2013). [DOI] [PubMed] [Google Scholar]
- Nascimento E. S. et al. Retinal projections to the thalamic paraventricular nucleus in the rock cavy (Kerodon rupestris). Brain Res. 1241, 56–61 (2008). [DOI] [PubMed] [Google Scholar]
- Thompson J. A. & Ziman M. Pax genes during neural development and their potential role in neuroregeneration. Prog. Neurobiol. 95, 334–51 (2011). [DOI] [PubMed] [Google Scholar]
- Stenman J. M. et al. Canonical Wnt Signaling Regulates Organ-Specific Assembly and Differentiation of CNS Vasculature. Science (80-.). 322, 1247–1250 (2008). [DOI] [PubMed] [Google Scholar]
- Melchior K. et al. The WNT receptor FZD7 contributes to self-renewal signaling of human embryonic stem cells. Biol. Chem. 389, 897–903 (2008). [DOI] [PubMed] [Google Scholar]
- Gloy J., Hikasa H. & Sokol S. Y. Frodo interacts with Dishevelled to transduce Wnt signals. Nat. Cell Biol. 4, 351–357 (2002). [DOI] [PubMed] [Google Scholar]
- Kalani M. Y. et al. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 105, 16970–16975 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H. et al. Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 102, 11331–6 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C. & Alvarez-Buylla A. Long-distance neuronal migration in the adult mammalian brain. Science (80-.). 264, 1145–8 (1994). [DOI] [PubMed] [Google Scholar]
- Lee A. et al. Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci. 8, 723–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga E., Araki I. & Nakamura H. Role of Pax3/7 in the tectum regionalization. Development 128, 4069–77 (2001). [DOI] [PubMed] [Google Scholar]
- Thompson J. A., Zembrzycki A., Mansouri A. & Ziman M. Pax7 is requisite for maintenance of a subpopulation of superior collicular neurons and shows a diverging expression pattern to Pax3 during superior collicular development. BMC Dev. Biol. 8, 62 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delogu A. et al. Subcortical visual shell nuclei targeted by ipRGCs develop from a Sox14+-GABAergic progenitor and require Sox14 to regulate daily activity rhythms. Neuron 75, 648–62 (2012). [DOI] [PubMed] [Google Scholar]
- Prakash N. et al. Nkx6-1 controls the identity and fate of red nucleus and oculomotor neurons in the mouse midbrain. Development 136, 2545–55 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S. et al. Motoneuron-specific NR3B gene: no association with ALS and evidence for a common null allele. Neurology 70, 666–76 (2008). [DOI] [PubMed] [Google Scholar]
- Ringer C., Weihe E. & Schütz B. Pre-symptomatic alterations in subcellular betaCGRP distribution in motor neurons precede astrogliosis in ALS mice. Neurobiol. Dis. 35, 286–95 (2009). [DOI] [PubMed] [Google Scholar]
- Kiecker C. & Lumsden A. Compartments and their boundaries in vertebrate brain development. Nat. Rev. Neurosci. 6, 553–64 (2005). [DOI] [PubMed] [Google Scholar]
- Jaillard C. et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci 25, 1459–69 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarisbrick I. A. et al. Kallikrein 6 is a novel molecular trigger of reactive astrogliosis. Biol. Chem. 393, 355–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colasante G. et al. Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J Neurosci 28, 10674–86 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti P. R. & Noebels J. L. Interneuron, interrupted: molecular pathogenesis of ARX mutations and X-linked infantile spasms. Curr. Opin. Neurobiol. 22, 859–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Gaamouch F. et al. Interaction between αCaMKII and GluN2B controls ERK-dependent plasticity. J Neurosci 32, 10767–79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning L., Tian L. & Smirnov S. Interactions between Intercellular Adhesion Molecule-5 (ICAM-5) and b1 integrins regulate neuronal synapse formation. J. Cell Sci. 5, Advance Online Publication (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman M. et al. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev. Biol. 279, 155–68 (2005). [DOI] [PubMed] [Google Scholar]
- De Cegli R. et al. Reverse engineering a mouse embryonic stem cell-specific transcriptional network reveals a new modulator of neuronal differentiation. Nucleic Acids Res. 41, 711–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucl. Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. (2009).at <URL http://www.R-project.org>.
- Sato A. et al. Cerebellar development transcriptome database (CDT-DB): profiling of spatio-temporal gene expression during the postnatal development of mouse cerebellum. Neural Netw. 21, 1056–69 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video S1
Supplementary Fig. S1- S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4