

Abstract
Background
Primary distal renal tubular acidosis (dRTA) caused by mutations in the genes that codify for the H + −ATPase pump subunits is a heterogeneous disease with a poor phenotype-genotype correlation. Up to now, large cohorts of dRTA Tunisian patients have not been analyzed, and molecular defects may differ from those described in other ethnicities. We aim to identify molecular defects present in the ATP6V1B1, ATP6V0A4 and SLC4A1 genes in a Tunisian cohort, according to the following algorithm: first, ATP6V1B1 gene analysis in dRTA patients with sensorineural hearing loss (SNHL) or unknown hearing status. Afterwards, ATP6V0A4 gene study in dRTA patients with normal hearing, and in those without any structural mutation in the ATP6V1B1 gene despite presenting SNHL. Finally, analysis of the SLC4A1 gene in those patients with a negative result for the previous studies.
Methods
25 children (19 boys) with dRTA from 20 families of Tunisian origin were studied. DNAs were extracted by the standard phenol/chloroform method. Molecular analysis was performed by PCR amplification and direct sequencing.
Results
In the index cases, ATP6V1B1 gene screening resulted in a mutation detection rate of 81.25%, which increased up to 95% after ATP6V0A4 gene analysis. Three ATP6V1B1 mutations were observed: one frameshift mutation (c.1155dupC; p.Ile386fs), in exon 12; a G to C single nucleotide substitution, on the acceptor splicing site (c.175-1G > C; p.?) in intron 2, and one novel missense mutation (c.1102G > A; p.Glu368Lys), in exon 11. We also report four mutations in the ATP6V0A4 gene: one single nucleotide deletion in exon 13 (c.1221delG; p.Met408Cysfs*10); the nonsense c.16C > T; p.Arg6*, in exon 3; and the missense changes c.1739 T > C; p.Met580Thr, in exon 17 and c.2035G > T; p.Asp679Tyr, in exon 19.
Conclusion
Molecular diagnosis of ATP6V1B1 and ATP6V0A4 genes was performed in a large Tunisian cohort with dRTA. We identified three different ATP6V1B1 and four different ATP6V0A4 mutations in 25 Tunisian children. One of them, c.1102G > A; p.Glu368Lys in the ATP6V1B1 gene, had not previously been described. Among deaf since childhood patients, 75% had the ATP6V1B1 gene c.1155dupC mutation in homozygosis. Based on the results, we propose a new diagnostic strategy to facilitate the genetic testing in North Africans with dRTA and SNHL.
Keywords: Distal renal tubular acidosis, ATP6V1B1, ATP6V0A4, Tunisian population
Background
Distal renal tubular acidosis (dRTA) is defined by the inability of the kidney to acidify urine, in presence of metabolic acidosis [1,2]. Patients present with hyperchloraemic metabolic acidosis, hypokalaemia [3], bone disease and, commonly, nephrocalcinosis. Some affected individuals also exhibit hearing impairment [3,4]. Malfunction of the H + −adenosine triphosphatase (ATPase) pumps, or Cl/HCO3- (anion exchanger AE1) at the α-intercalated cells of the distal nephron causes failure to excrete acid or reabsorb bicarbonate, respectively, and leads to primary dRTA [1,5].
Primary dRTA caused by mutations of H + −ATPase pump subunits is wide genetic and clinically heterogeneous [6]. In 1999, Karet et al demonstrated that, in families with classic dRTA and early sensorineural hearing loss (SNHL), which became clinically evident from birth to late childhood, the disease was caused by mutations in the ATP6V1B1 gene, encoding the B subunit of the H + ATPase pump [7]. Later, in 2000, Smith et al found that some families with dRTA and normal audiometry presented with mutations in the ATP6V0A4 gene, which encodes for the A4 subunit of the H + ATPase pump [8].
Despite those original molecular descriptions distinguished between ATP6V1B1 and ATP6V0A4 gene mutations based on the presence or absence of early SNHL, phenotype overlapping between both subgroups of patients makes that classification inaccurate. Indeed, Stover et al demonstrated that young adult patients with ATP6V0A4 mutations could develop mild SNHL in the long term [9]. Further, some dRTA patients without early SNHL were found to present mutations in the ATP6V1B1 gene as well [10,11].
Further, autosomal dominant (AD) and autosomal recessive (AR) dRTA have also been associated with mutations in the SLC4A1 gene encoding the human AE1 [12-15], which also plays a central role in the maintenance of acid–base balance [16,17]. Overall, lack of phenotype-genotype correlation supports the need of genetic analysis in dRTA patients.
Up to now, small series of patients with dRTA from different ethnic backgrounds had been genetically studied [7-9,11,18]. Despite a few data on Tunisians have been reported [8,9,11], to our knowledge large cohorts of Tunisian patients with dRTA have not yet been analyzed. In order to establish the genetic diagnosis of primary dRTA in Tunisia, this study aimed to identify the molecular defects that occur in ATP6V1B1, ATP6V0A4 and SLC4A1 genes using a proposed algorithm.
Methods
Patients
We studied 25 children (19 boys and 6 girls) from 20 unrelated families of Tunisian origin, who had been clinically diagnosed of primary dRTA [19]. The study group was recruited after deep review of every medical record with the diagnosis of dRTA, and recall of those affected subjects in four Tunisian hospitals: Hospital Fattouma Bourguiba of Monastir (13 patients from 10 families), Hospital Ibn al-Jazzar of Kairouan (9 patients from 7 families), Hospital Mohamed Ben Sassi of Gabes (2 patients from 2 families), and Hospital Tahar Sfar of Mahdia (one patient). After the four Medical Ethical Committees belonging to the hospitals from which patients were recruited approved the project, and after informed consent was given, we recorded patients’ data regarding family history, symptoms, acid–base and biochemistry at presentation, as well as kidney ultrasound. Clinical diagnosis of dRTA was based on the simultaneous presence of non-gap metabolic acidosis (plasma bicarbonate <20 mEq/L) and inability to reduce urinary pH below 5.5 [20]. Additional features were dehydration, failure to thrive, polyuria, and hypokalemia as described [19]. Hearing was assessed by pure-tone audiometry and/or auditory evoked responses, and the presence of SNHL was graded according to the “Bureau International d’Audiophonologie” recommendations (http://www.biap.org). Moreover, 17 asymptomatic patients’ parents without metabolic acidosis were also studied.
DNA analysis
Peripheral blood samples from the patients were collected on EDTA tubes. DNAs were extracted by the standard method of phenol/chloroform [21]. We planned to optimize the study of the three genes described as being responsible for primary dRTA (ATP6V1B1, OMIM *192132; ATP6V0A4, OMIM *605239 and SLC4A1, OMIM +109270) using the following algorithm: first, we analyzed the ATP6V1B1 gene for those patients with dRTA and SNHL or unknown hearing status. Afterwards, we studied the ATP6V0A4 gene in those patients with dRTA but normal hearing, and in those who were negative for ATP6V1B1 gene mutation screening despite presence of SNHL. Finally, we planned to investigate the SLC4A1 gene in those remaining patients with dRTA but no identified mutations in either ATP6V1B1 or ATP6V0A4 genes (Figure 1).
Figure 1.
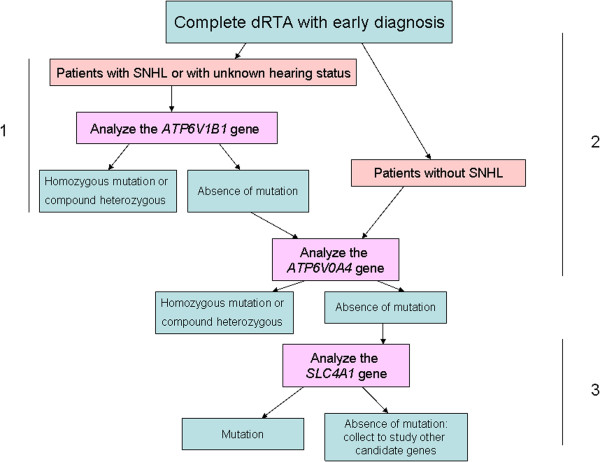
Algorithm followed for the genetic analysis. 1, 2 and 3 (the different steps of the analysis).
We used previously reported primers [7] for the polymerase chain reaction (PCR) amplification of the exonic and flanking intronic regions of the ATP6V1B1 gene. Amplification was conducted for the coding regions and flanking intronic sequences of ATP6V0A4 using a set of newly designed primers pairs. Structural analysis of the SLC4A1 gene was performed using both previously described [22] and newly designed primers (for the amplification of the three first exons). Two overlapping regions in SLC4A1 gene intron 3 (one for the potential kidney promoter and another for the possible 5’ sequence of the transcript expressed in kidney) were also analyzed [22]. In all cases, primer sequences and PCR conditions are available on request. Amplified products were purified using ExoSap (USB, Cleveland, OH, USA) and sequenced in both strands on an ABI3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences were analyzed with Sequencing Analysis software v.5.2 (Applied Biosystems) and compared with the references sequences of each gene (Ensembl identifiers: ATP6V1B1: ENSG00000116039, ATP6V0A4: ENSG00000105929 and SLC4A1: ENSG00000004939), using SeqScape software v.2.5 (Applied Biosystems).
Mutation prediction
The potential pathogenic effect of novel missense mutations was analyzed in silico using SIFT (http://sift.jcvi.org/) and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) software packages, which are able to predict whether an amino acid substitution may affect protein function, based on sequence homology and physical properties of substituted amino acids.
Results
dRTA kindreds
Twenty five Tunisian children (19 males) of 8 (0.6-20) (median, range) years of age were diagnosed with primary dRTA at 6.27 (0.8-36) months of life. At diagnosis, most relevant symptoms were feeding difficulties, dehydration, and failure to thrive (Table 1). One of the patients (case 7.2) was asymptomatic at recruitment; he was diagnosed by molecular familiar screening, as he had an affected brother (case 7.1) and has developed the symptoms later. All patients presented with a non-gap metabolic acidosis (plasma pH 7.23 ± 0.15; bicarbonate 9.67 ± 0.62 mEq/L), and inability to renal acidification, as shown by alkaline urine (urine pH 7 ± 0.67). Further, common findings were hypokalemia (15 cases had K < 3.5 mEq/L), diffuse nephrocalcinosis which was demonstrated in all but three children (nephrocalcinosis was not observed in subjects 7.1 and 19.1 and ultrasound data was not available in case 11.1), rickets (5 cases), and total hearing loss/cophosis (16 cases). Unexpected hyperkalemia was observed in a 1 month old patient (case 13.1), possibly due to extreme acidosis and potential inadequate blood draw (Table 1). The disease was inherited following an AR pattern, and remarkably consanguinity rate was very high in the study group (80%) (4 families in first degree of consanguinity, 5 families in second degree, 3 families in third degree and 4 families reported consanguinity in an unknown status).
Table 1.
Clinical presentation of the 25 Tunisian children with dRTA
| Case | Symptoms | Age at diagnosis (months) | P pH | P Bicarbonate mEq/L | P K mEq/L | U pH | Nephro calcinosis |
|---|---|---|---|---|---|---|---|
| 1.1 |
Vomits, growth failure |
6 |
7.3 |
10.4 |
2.26 |
7 |
Yes |
| 2.1 |
Vomits, growth failure |
7 |
7.22 |
9.7 |
3 |
6.5 |
Yes |
| 3.1 |
Growth failure |
1.3 |
7.14 |
7 |
2.3 |
6 |
Yes |
| 3.2 |
Speech delay, growth failure, rickets |
36 |
7.21 |
12.3 |
2.7 |
7 |
Yes |
| 4.1 |
Dehydration |
1.3 |
7.21 |
10.4 |
4.1 |
6.5 |
Yes |
| 5.1 |
Vomits, diarrhea, growth failure |
3 |
7.3 |
6.9 |
4.06 |
7.5 |
Yes |
| 5.2 |
Growth failure, dehydration |
3.5 |
7.25 |
8.3 |
4.65 |
6 |
Yes |
| 6.1 |
Growth failure, fever |
3 |
7.34 |
12.4 |
3.82 |
7 |
Yes |
| 7.1 |
Loss of weight, dehydration, fever, vomits, diarrhea |
4 |
7.29 |
12.1 |
2.43 |
6 |
No |
| 7.2 |
Growth failure, dehydration, polyuria, urine infection |
11 |
7.17 |
8.5 |
2.9 |
7 |
Yes |
| 8.1 |
Fever, vomits, growth failure |
3.3 |
7.3 |
8.6 |
2.68 |
7 |
Yes |
| 9.1 |
Vomits, dehydration, vitamin D-resistant rickets |
1 |
7 |
NA |
4.22 |
8 |
Yes |
| 10.1 |
Vomits, dehydration, leukocyturia |
3 |
7.3 |
11.4 |
2 |
7 |
Yes |
| 10.2 |
Dehydration, vomits |
6 |
7.4 |
14.5 |
3.2 |
6.5 |
Yes |
| 11.1 |
Diarrhea, vomits, polyuria, rickets |
16 |
7.32 |
11.6 |
3.12 |
NA |
NA |
| 12.1 |
Vomits, dehydration, growth failure |
5 |
7.28 |
NA |
2.4 |
7.5 |
Yes |
| 13.1 |
Vomits after each feeding |
1 |
7.23 |
11.3 |
7.2 |
7 |
Yes |
| 14.1 |
Dehydration, respiratory distress |
0.8 |
7.1 |
3.4 |
2 |
8 |
Yes |
| 15.1 |
Dehydration, vomits, feeding difficulties |
1.5 |
7.19 |
8.4 |
3.5 |
7 |
Yes |
| 16.1 |
Vomits, feeding difficulties, severe dehydration, diarrhea, vitamin D-resistant rickets, severe osteopenia |
18 |
7.18 |
9.6 |
NA |
7.5 |
Yes |
| 16.2 |
Hypotonia, failure to thrive, severe vitamin D-resistant rickets, inability to walk, dumb |
17 |
7.15 |
9.4 |
4.1 |
NA |
Yes |
| 17.1 |
Vomits, dehydration, respiratory infection, breathing difficulties |
0.9 |
7.29 |
12.5 |
3.11 |
8 |
Yes |
| 18.1 |
Dehydration, vomits, polyuria |
2 |
7.15 |
6.1 |
3.9 |
7 |
Yes |
| 19.1 |
Bronchiolitis and cough |
3 |
7.13 |
8.8 |
3.8 |
8 |
No |
| 20.1 |
Dehydration, axial hypotonia, hypotrophy |
2.3 |
7.39 |
9 |
2.2 |
8 |
Yes |
| |
|
6.27 (0.8–36)* |
7.23 ± 0.15** |
9.67 ± 0.62** |
3.3 ± 0.83** |
7 ± 0.67** |
|
| *** (mEq/L) | 7.35-7.45 | 22-28 | 3.5-5 | 5.5-6.5 |
Abbreviations: P (plasma), U (urine), NA (not available). *:median (range); **:mean value ± SD; ***:normal ranges.
Mutation detection
All molecular results are indicated in Table 2. Following the strategy described above (Figure 1), we analyzed the ATP6V1B1 gene in 16 index cases who presented dRTA and SNHL (12 cases) or an unknown hearing status (4 cases). Mutations were detected in homozygosis in 13 probands. Afterwards, we investigated the ATP6V0A4 gene in the remaining 3 patients without ATP6V1B1 mutations, and in those 4 index cases with no hearing loss. Mutations were detected in all 7 probands, 5 of them in homozygosis (4 with normal hearing and 1 with unknown hearing status) and two in heterozygosis, in which loss of hearing was present in one case and the other had an unknown hearing status. Further, in those two patients with isolated heterozygous ATP6V0A4 gene mutations, we screened for the second mutations in the SLC4A1 gene, in order to complete the genetic diagnosis of all known causative genes of primary dRTA. We did not find any SLC4A1 gene mutation, and therefore, the second mutation in those two siblings, if present, remained unidentified.
Table 2.
Genetic results of the 25 Tunisian patients with dRTA, according to family history and presence of sensorineural hearing loss (SNHL)
| Case | Origin | Consanguinity | SNHL | Gene | Mutation | Protein |
|---|---|---|---|---|---|---|
| 1.1 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.175-1G > C |
p.? |
| 2.1 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 3.1 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 3.2 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 4.1 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 5.1 |
Monastir |
Yes |
NA |
ATP6V1B1 |
c.1102G > A |
p.Glu368Lys (p.E368K) |
| 5.2 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1102G > A |
p.Glu368Lys (p.E368K) |
| 6.1 |
Monastir |
No |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 7.1 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 7.2 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 8.1 |
Monastir |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 9.1 |
Kairouan |
No |
NA |
ATP6V1B1 |
c.175-1G > C |
p.? |
| 10.1 |
Kairouan |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 10.2 |
Kairouan |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 11.1 |
Kairouan |
Yes |
NA |
ATP6V1B1 |
c.175-1G > C |
p.? |
| 12.1 |
Gabes |
No |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 13.1 |
Gabes |
Yes |
Yes |
ATP6V1B1 |
c.1155dupC |
p.Ile386Hisfs*56 (p.I386Hfs*56) |
| 14.1 |
Mahdia |
Yes |
No |
ATP6V0A4 |
c.1221delG |
p.Met408Cysfs*10 (p.M408Cfs*10) |
| 15.1 |
Kairouan |
Yes |
NA |
ATP6V0A4 |
c.16C > T |
p.Arg6* (p.R6*) |
| 16.1 |
Kairouan |
Yes |
Yes |
ATP6V0A4 |
c.2035G > T |
p.Asp679Tyr (p.D679Y) heterozygous |
| 16.2 |
Kairouan |
Yes |
Yes |
ATP6V0A4 |
c.2035G > T |
p.Asp679Tyr (p.D679Y) heterozygous |
| 17.1 |
Kairouan |
Yes |
No |
ATP6V0A4 |
c.16C > T |
p.Arg6* (p.R6*) |
| 18.1 |
Monastir |
Yes |
No |
ATP6V0A4 |
c.16C > T |
p.Arg6* (p.R6*) |
| 19.1 |
Monastir |
No |
NA |
ATP6V0A4 |
c.1739 T > C |
p.Met580Thr (p.M580T) heterozygous |
| 20.1 | Kairouan | Yes | No | ATP6V0A4 | c.16C > T | p.Arg6* (p.R6*) |
Otherwise stated, mutations are present in homozygosis.
cDNA and protein numbering according to Ensembl identifiers: ATP6V1B1: ENSG00000116039, and ATP6V0A4: ENSG00000105929.
Abbreviations: SNHL (sensorineural hearing loss), NA (Not available).
On the basis of this genetic diagnosis strategy in patients with dRTA, which is based on the presence of SNHL, the initial ATP6V1B1 gene screening resulted in a mutation detection rate of 81.25% (26/32 alleles). After ATP6V0A4 gene analysis, the mutation detection rate increased up to 95% of the probands (38/40 alleles). That diagnostic approach was reinforced by the fact that no phenotypic difference, apart from the presence or absence of hearing impairment, was observed between patients with ATP6V1B1 vs. ATP6V0A4 mutations in the studied cohort. All the mutations identified in this study were distributed throughout both ATP6V1B1 and ATP6V0A4 genes (Figure 2), so no hot-spot was identified in any of the genes.
Figure 2.
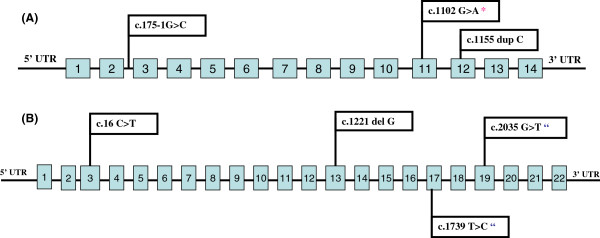
Schematic representation of the ATP6V1B1 (A) and the ATP6V0A4 (B) genes with the localization of the mutations found in this study. *: Novel mutation. “Heterozygous.
Mutations in the ATP6V1B1 gene
Analysis of the coding regions and flanking intronic sequences of the ATP6V1B1 gene showed the presence of three different mutations in 13 families (Table 2). One of them was a missense mutation (c.1102G > A; p.Glu368Lys), another was a frameshift mutation (c.1155dupC; p.Ile386fs), and the third was a splice-site mutation (c.175-1G > C; p.?). The first one had not yet been described.
The novel missense mutation in exon 11 (c.1102G > A; p.Glu368Lys) was identified in two siblings (cases 5.1, 5.2) in the current study. That guanine to adenine substitution leads to the change of the amino acid glutamine to lysine. We did not perform a functional analysis of the mutated protein, but a molecular difference between these two residues (glutamic acid is an acidic amino acid, whereas lysine is a basic one) supports the hypothesis that this replacement may affect the structure of the protein subunit, thus the function, folding, or trafficking of that protein. Indeed, glutamic acid in position 368 remains highly conserved among species in proton ATPases. Data obtained using SIFT and Polyphen-2 bioinformatics software provided additional support for the pathogenic effect of this mutation. Furthermore, we did not find this mutation in 110 Tunisian healthy subjects.
Remarkably, the c.1155dupC mutation was observed in homozygosis in 12 patients from 9 families (cases 2.1, 3.1, 3.2, 4.1, 6.1, 7.1, 7.2, 8.1, 10.1, 10.2, 12.1 and 13.1). In 11 of these 12 patients, SNHL was evident since childhood, but in one of them with normal hearing capability at diagnosis (case 7.2, diagnosed by molecular familiar screening), hypoacusia appeared later. This frameshift mutation results in the generation of a premature stop codon after a short segment from the generated novel triplet. Parents of the siblings 10.1, 10.2, 12.1 and 13.1 were heterozygous, thus carriers for that mutation. The other parents’ samples were not available for genetic testing.
Another mutation in intron 2, affecting the splice acceptor site (c.175-1G > C; p.?), was identified in three probands (1.1, 9.1 and 11.1) from three different families. Mother of 9.1 (father’s sample was not available) and both parents of 11.1 were found to be heterozygous for this mutation. This mutation predicts the loss of the acceptor splice site [11].
In addition, we found a novel heterozygous polymorphic change in intron 5 (c.368-27 C > T) in four patients from four unrelated families (cases 1.1, 8.1, 11.1 and 14.1).
Mutations in the ATP6V0A4 gene
Screening for ATP6V0A4 mutations in 7 probands (patients 14.1, 15.1, 16.1, 17.1, 18.1, 19.1 and 20.1) showed the presence of four different mutations in these 7 families (Table 2): two missense (c.1739 T > C; p.Met580Thr in exon 17 and c.2035G > T; p.Asp679Tyr in exon 19), one nonsense (c.16C > T; p.Arg6* in exon 3) and one frameshift (c.1221delG; p.Met408Cysfs*10 in exon 13). The latter is a single-base deletion leading to a premature stop codon, predicting the consequent synthesis of a truncated protein, lacking all the trans-membrane domains. This mutation found in our series (case 14.1) has previously been published as a case report [23]. Patient’s mother was found to be heterozygous, thus carrier for the mutation (father’s sample was not available).
The missense c.2035G > T mutation was found in two siblings (cases 16.1 and 16.2) in heterozygosis (their father was also heterozygous for this mutation). It leads to the change of aspartic acid in position 679 by tyrosine [11]. In this family, we were not able to find the second mutation responsible for recessive dRTA.
The second missense c.1739 T > C mutation was found in one proband (case 15.1) in heterozygosis. This mutation was described to be located in a predicted transmembrane segment [8]. The substitution of methionine in position 580 by threonine is likely to disrupt secondary structure, function or both.
The nonsense c.16C > T mutation was identified in four families (cases 15.1, 17.1, 18.1 and 20.1). It causes the substitution of Arg6 by a premature stop codon. Prediction analysis showed that this nonsense mutation should lead to an abnormally short protein of only 6 amino acids (p.Arg6*). Further, mother of 17.1, father of 18.1 and both parents of 20.1 were found to be heterozygous for the mutation.
In addition, we found three polymorphic changes in both heterozygosis or homozygosis: (c.5 T > C; p.Val2Ala) in exon 3 in 7 patients (14.1, 15.1, 16.2, 17.1, 18.1, 19.1 and 20.1), c.1662C > T; p.Phe554Phe in exon 16 in three patients (16.1, 16.2 and 19.1) and c.1812 T > C; p.His604His in exon 17 in 8 patients (14.1, 15.1, 16.1, 16.2, 17.1, 18.1, 19.1 and 20.1).
Discussion
This current study is, to our knowledge, the first large Tunisian dRTA cohort that has been genetically studied so far. As a whole, we identified three different ATP6V1B1 gene mutations (in 13 families), and four ATP6V0A4 mutations (in 7 families) in 25 Tunisian children. Remarkably, one novel mutations was observed, c.1102G > A; p.Glu368Lys in the ATP6V1B1 gene. On the other hand, most studied patients were deaf since early childhood and, remarkably, 75% of them (12/16) had the ATP6V1B1 gene c.1155dupC mutation [7] in homozygosis, a finding that may improve genetic diagnosis in Tunisia and close populations.
Despite primary dRTA is a rare disease of unknown prevalence, we found a surprising large number of affected Tunisian patients, a country with an estimated population of 10,4 million inhabitants and a mean endogamy rate of 26% [24]. This can be explained, almost in part, by the high rate of consanguinity found in our study cohort, up to 80%. Patients’ clinical presentation was consistent with the prototypic picture of primary dRTA, and every patient showed inability to acidify urine in presence of non-gap metabolic acidosis [1,3,19]. In our series, ATP6V1B1 gene causative mutations were 2 time-fold more frequent than ATP6V0A4 gene mutations: 13 vs. 7. This finding may be related not only to ethnicity or the studied population’s characteristics, but also to an easier recognizable pattern of severe acidosis associated with early onset SNHL, leading straightforward to the disease clinical diagnosis [1,7].
Similar presentation with early onset of non-gap metabolic acidosis, hypokalemia and nephrocalcinosis was observed in almost all patients despite carrying different mutations. No phenotype-genotype correlation in dRTA with SNHL was detected in this cohort, as previously described [10]. On the other hand, this study confirms the association between ATP6V1B1 mutations and dRTA with early onset SNHL since childhood, and ATP6V0A4 mutations with dRTA but normal hearing (at least until young adulthood), as reported in the original disease genetic descriptions [7,8,18,25,26], but conflicts with some other reports [9,11], which described ATP6V0A4 gene mutations to be more frequent than ATP6V1B1 ones.
However, with more recent clinical follow up data, we have become aware that hearing impairment may represent a spectrum in dRTA, and hearing loss may also develop at an older age in a number of those affected patients with ATP6V0A4 mutations, but likely less severe than in those with ATP6V1B1 mutations [9]. Indeed, two siblings (cases 16.1 and 16.2) carrying ATP6V0A4 mutations had dRTA with SNHL since childhood, as described [11]. Reversely, patients with ATP6V1B1 mutations may not present hearing loss [11]. One of our patients with the ATP6V1B1 gene c.1155dupC mutation (case 7.2) developed hypoacusia after diagnosis. In general, these findings raise the question regarding potential mechanisms involved in the effect of different subunit mutations of the same proton pump over hearing ability [9].
Importantly, one of the most interesting findings of this study was the detection of ATP6V1B1 c.1155dupC mutation in homozygosis in 12 patients from 9 families. In all probands, SNHL was evident since childhood, as classically described [7]. This mutation is present in 12 out of 16 (75%) patients with SNHL. This c.1155dupC mutation had previously been described in 12 families, of which 7 are from North Africa [7,9,19]. Therefore, if we consider these studies, performed in North African patients with dRTA , it turns out that approximately 55% of patients suffering from dRTA with early onset SNHL (before 4 years of age) carry this mutation.
On the basis of similar patients’ geographic origin, it could be hypothesized that there might be a founder effect for the frameshift c.1155dupC mutation (Table 3). Nevertheless, a previous study [11] performed in six families with the recurrent p.Ile386fs mutation found they presented with different haplotypes at the ATP6V1B1 locus, concluding that a founder effect was not the cause of the high prevalence of this mutation in their population. Unfortunately, we could not study ATP6V1B1 haplotypes in our cohort due to the lack of available relatives, but we plan to test this issue in the future. It is also possible that Ile386 appears as a potential “mutation hotspot” due to a string of seven cytosine residues at the surrounding positions, suggesting a DNA polymerase slippage during replication.
Table 3.
Geographic distribution of previously reported dRTA populations carrying the ATP6V1B1 and ATP6V0A4 gene mutations described in this study
| Gene | Mutation | Origin | Number of families | Reference |
|---|---|---|---|---|
|
ATP6V1B1
|
c.1155dupC |
Sweden |
1 |
[7] |
| Spain |
2 |
|||
| Saudi Arabia |
1 |
[9] |
||
| Sicily |
1 |
|||
| Morocco |
1 |
|||
| Tunisia |
1 |
[11] |
||
| Algeria |
5 |
|||
| Tunisia |
9 |
Current study |
||
| c.175-1G > C |
Tunisia |
1 |
[11] |
|
| Algeria |
2 |
|||
| Tunisia |
3 |
Current study |
||
| c.1102G > A |
Tunisia |
1 |
Current study |
|
| ATP6V0A4 | c.16C > T |
Pakistan |
1 |
[11] |
| Tunisia |
4 |
Current study |
||
| c.1739 T > C |
Turkey |
1 |
[8] |
|
| Tunisia |
1 |
Current study |
||
| c.2035G > T |
Pakistan |
1 |
[11] |
|
| Tunisia |
1 |
Current study |
||
| c.1221delG | Tunisia | 1 | Current study and [23] |
Anyhow, this finding may facilitate and help for rapid genetic diagnosis of dRTA in North African patients; and therefore, we propose that in patients with dRTA and SNHL from Northern Africa countries, c.1155dupC mutation in the ATP6V1B1 gene should be screened first and, then, analysis should continue with additional mutation screening only in those affected subjects in whom c.1155dupC is not found.
Regarding the origin of the other mutations, the splice-site (c.175-1G > C; p.?) defect was described for the first time in three families from Algeria and Tunisia [11]. The nonsense c.16C > T mutation was previously described in a Pakistani family [11]. The two missense c.2035G > T and c.1739 T > C mutations were previously described in one Pakistani [11] and one Turkish family [8], respectively (Table 3).
On the other hand, the newly identified mutation (c.1102G > A; p.Glu368Lys) may provide in the future some insights into A1 subunit structure and function, which are presently unknown in mammals.
The diagnostic strategy based on the presence of SNHL was successfully used in the current study to determine which gene to test first. Vargas-Poussou et al performed linkage studies to identify the disease locus in consanguineous families, and then used the age of onset of SNHL to determine which gene to test first in the non-consanguineous families [11]. Comparing with the algorithm followed by these authors, our strategy resulted in a higher mutation detection global rate: 95% versus 79.5%.
Nevertheless, our mutation screening partially failed in three patients (two of them are siblings). We only identified two already described heterozygous mutations (c.2035G > T and c.1739 T > C) in the ATP6V0A4 gene. We were unable to find the second mutation responsible for the recessive inheritance of the disease, as described by other authors [11], even after screening the SLC4A1 gene [12,27,28]. We admit that SLC4A1 gene was a poor candidate for screening as our patients presented severe metabolic acidosis early in life, but we wanted to exclude the possibility of presenting any mutation in the SLC4A1 gene, as compound heterozygosity has been described [14,29]. Anyway, we cannot rule out the possibility of a potential undetected deletion involving one or more exons of the analyzed genes [30]. Other authors have hypothesized about the possibility of mutations occurring in an unknown regulatory element of either gene, like promoters [31] or that some intronic variants, especially those affecting the normal splicing sites, may also be involved in the pathogenesis of dRTA [11]. More plausible, there might be other genes involved in tubular acidification ability not yet discovered. Haplotype linkage studies should be performed in those affected patients in whom the second mutation is missed and we plan to continue with that project in the future.
Conclusion
In this report, the genetic analysis of a Tunisian cohort of dRTA patients as well as a new diagnostic strategy to facilitate the genetic testing in North Africans with dRTA and SNHL is presented. The putative dRTA mutation was found in 90% of the index cases (18/20, the other two being heterozygous), thus genetically confirming the clinical diagnosis.
Our findings confirm the association of ATP6V1B1 mutations and early onset severe SNHL since childhood, and identify delayed hearing loss as a feature associated with ATP6V0A4 mutations in some patients as well. Based on our results, we propose that in patients of Northern Africa origin with dRTA and SNHL, c.1155dupC mutation in the ATP6V1B1 gene should be screened first. Further studies with more patients would be needed to confirm this result.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DEH made acquisition of the data (the samples), coordinated the data collection, participated in the conception and design of the study, carried out of the genetic studies, interpreted data and wrote the manuscript. GPN participated in the conception and design of the study, carried out of the genetic studies, analysis data, revised critically and made major contribution to the manuscript. SC, SH, AM, MT, NL, WBR, NG, FEA, SM carried out substantial acquisition of the clinical data. JBC, LC, AHK, GA conceived the study, analyzed and interpreted the data and introduce major corrections to the manuscript. All authors revised the manuscript and gave their final approval.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Donia Elhayek, Email: hayek.donia@yahoo.fr.
Gustavo Perez de Nanclares, Email: gustavo.perezdenanclaresleal@osakidetza.net.
Slaheddine Chouchane, Email: chouchane_slaheddine@yahoo.fr.
Saber Hamami, Email: hamami_sabeur@yahoo.fr.
Adnène Mlika, Email: adnene_mlika@yahoo.fr.
Monia Troudi, Email: monia_troudi@yahoo.fr.
Nadia Leban, Email: lebannadia@yahoo.fr.
Wafa Ben Romdane, Email: wafadoc_fr@yahoo.fr.
Mohamed Neji Gueddiche, Email: neji.guediche@rns.tn.
Féthi El Amri, Email: fethiamri@yahoo.fr.
Samir Mrabet, Email: samirmrabet@yahoo.fr.
Jemni Ben Chibani, Email: jemnichibani2@yahoo.fr.
Luis Castaño, Email: LUISANTONIO.CASTANOGONZALEZ@osakidetza.net.
Amel Haj Khelil, Email: hajkhelil@yahoo.fr.
Gema Ariceta, Email: gariceta@vhebron.net.
Acknowledgements
We thank patients and families, and collaborating physicians for participating in this study.
This research study was supported by PI09/90888 and PI11/01412 grants, from the Instituto de Salud Carlos III (Spain), by BIO08/ER/020 grant, from the EITB Maratoia-Bioef (Basque Foundation for Health Innovation and Research) and by the Tunisian Ministry of Scientific Research (Research Unit code 05/UR-09-04, University of Monastir) for DEH mobility.
References
- Rodríguez-Soriano J. New insights into the pathogenesis of renal tubular acidosis – from functional to molecular studies. Pediatr Nephrol. 2000;14:1121–1136. doi: 10.1007/s004670000407. [DOI] [PubMed] [Google Scholar]
- Bastani B, Gluck SL. New insights into the pathogenesis of distal renal tubular acidosis. Miner Electrolyte Metab. 1996;22:396–409. [PubMed] [Google Scholar]
- Fry AC, Karet FE. Inherited renal acidosis. Physiology. 2007;22:202–211. doi: 10.1152/physiol.00044.2006. [DOI] [PubMed] [Google Scholar]
- Tasic V, Korneti P, Gucev Z, Hoppe B, Blau N, Cheong HI. Atypical presentation of distal renal tubular acidosis in two siblings. Pediatr Nephrol. 2008;23:1177–1181. doi: 10.1007/s00467-008-0796-z. [DOI] [PubMed] [Google Scholar]
- Wagner S, Vogel R, Lietzke R, Koob R, Drenckhahn D. Immunochemical characterization of a band 3-like anion exchanger in collecting duct of human kidney. Am J Physiol. 1987;253(2 Pt 2):F213–21. doi: 10.1152/ajprenal.1987.253.2.F213. [DOI] [PubMed] [Google Scholar]
- Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012;27:3691–3704. doi: 10.1093/ndt/gfs442. [DOI] [PubMed] [Google Scholar]
- Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H + −ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A, Voskarides K, Demosthenous P, Michalopoulou A, Malliarou MA, Georgaki E, Athanasiou Y, Patsias C, Pierides A, Deltas C. Founder mutations in the ATP6V1B1 gene explain most Cypriot cases of distal renal tubular acidosis: first prenatal diagnosis. Nephron Clin Pract. 2011;117:c206–12. doi: 10.1159/000320192. [DOI] [PubMed] [Google Scholar]
- Vargas-Poussou R, Houillier P, Le Pottier N, Strompf L, Loirat C, Baudouin V, Macher MA, Déchaux M, Ulinski T, Nobili F, Eckart P, Novo R, Cailliez M, Salomon R, Nivet H, Cochat P, Tack I, Fargeot A, Bouissou F, Kesler GR, Lorotte S, Godefroid N, Layet V, Morin G, Jeunemaître X, Blanchard A. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol. 2006;17:1437–1443. doi: 10.1681/ASN.2005121305. [DOI] [PubMed] [Google Scholar]
- Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJA. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest. 1997;100:1693–1707. doi: 10.1172/JCI119694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce LJ, Wrong O, Toye AM, Young MT, Ogle G, Ismail Z, Sinha AK, McMaster P, Hwaihwanje I, Nash GB, Hart S, Lavu E, Palmer R, Othman A, Unwin RJ, Tanner MJ. Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. Biochem J. 2000;350:41–51. doi: 10.1042/0264-6021:3500041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrong O, Bruce LJ, Unwin RJ, Toye AM, Tanner MJ. Band 3 mutations, distal renal tubular acidosis, and Southeast Asian ovalocytosis. Kidney Int. 2002;62:10–19. doi: 10.1046/j.1523-1755.2002.00417.x. [DOI] [PubMed] [Google Scholar]
- Fry AC, Su Y, Yiu V, Cuthbert AW, Trachtman H, Karet FE. Mutation conferring apical-targeting motif on AE1 exchanger causes autosomal dominant distal RTA. J Am Soc Nephrol. 2012;23:1238–1249. doi: 10.1681/ASN.2012020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkin A, Kurtz I. SLC4 base (HCO3−, CO32−) transporters: classification, function, structure, genetic diseases, and knockout models. Am J Physiol Renal Physiol. 2006;290:580–599. doi: 10.1152/ajprenal.00252.2005. [DOI] [PubMed] [Google Scholar]
- Kollert-Jöns A, Wagner S, Hübner S, Appelhans H, Drenckhahn D. Anion exchanger 1 in human kidney and oncocytoma differs from erythroid AE1 in its NH2 terminus. Am J Physiol. 1993;265:813–821. doi: 10.1152/ajprenal.1993.265.6.F813. [DOI] [PubMed] [Google Scholar]
- Borthwick KJ, Kandemir N, Topaloglu R, Kornak U, Bakkaloglu A, Yordam N, Ozen S, Mocan H, Shah GN, Sly WS, Karet FE. A phenocopy of CAII deficiency: a novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J Med Genet. 2003;40:115–121. doi: 10.1136/jmg.40.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13:2160–2170. doi: 10.1097/01.ASN.0000023430.92674.E5. [DOI] [PubMed] [Google Scholar]
- Batlle DC, Hizon M, Cohen E, Gutterman C, Gupta R. The use of the urinary anion gap in the diagnosis of hyperchloremic metabolic acidosis. N Engl J Med. 1988;318:594–599. doi: 10.1056/NEJM198803103181002. [DOI] [PubMed] [Google Scholar]
- Grunebaum L, Cazenave JP, Camerino G, Kloepfer C, Mandel JL, Tolstoshev P, Jaye M, De la Salle H, Lecocq JP. Carrier detection of Hemophilia B by using restriction site polymorphism associated with the coagulation Factor IX gene. J Clin Invest. 1984;73:1491–1495. doi: 10.1172/JCI111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasuvattakul S, Yenchitsomanus PT, Vachuanichsanong P, Thuwajit P, Kaitwatcharachai C, Laosombat V, Malasit P, Wilairat P, Nimmannit S. Autosomal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosis. Kidney Int. 1999;56:1674–82. doi: 10.1046/j.1523-1755.1999.00756.x. [DOI] [PubMed] [Google Scholar]
- Hayek DE, Bouzidi H, Perez De Nanclares G, Soua H, Chibani JB, Ariceta G, Castaño L, Haj Khelil A. Novel ATP6V0A4 mutation described in a Tunisian patient with distal renal tubular acidosis. Clin Nephrol. 2012. Aug 1, [Epub ahead of print] [DOI] [PubMed]
- El Moncer W, Esteban E, Bahri R, Gayà-Vidal M, Carreras-Torres R, Athanasiadis G, Moral P, Chaabani H. Mixed origin of the current Tunisian population from the analysis of Alu and Alu/STR compound systems. J Hum Genet. 2010;55:827–833. doi: 10.1038/jhg.2010.120. [DOI] [PubMed] [Google Scholar]
- Ruf R, Rensing C, Topaloglu R, Guay-Woodford L, Klein C, Vollmer M, Otto E, Beekmann F, Haller M, Wiedensohler A, Leumann E, Antignac C, Rizzoni G, Filler G, Brandis M, Weber JL, Hildebrandt F. Confirmation of the ATP6B1 gene as responsible for distal renal tubular acidosis. Pediatr Nephrol. 2003;18:105–109. doi: 10.1007/s00467-002-1018-8. [DOI] [PubMed] [Google Scholar]
- Hahn H, Kang HG, Ha IS, Cheong HI, Choi Y. ATP6B1 gene mutations associated with distal renal tubular acidosis and deafness in a child. Am J Kidney Dis. 2003;41:238–243. doi: 10.1053/ajkd.2003.50014. [DOI] [PubMed] [Google Scholar]
- Jarolim P, Shayakul C, Prabakaran D, Jiang L, Stuart-Tilley A, Rubin HL, Simova S, Zavadil J, Herrin JT, Brouillette J, Somers MJ, Seemanova E, Brugnara C, Guay-Woodford LM, Alper SL. Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band 3) Cl–/HCO3– exchanger. J Biol Chem. 1998;273:6380–6388. doi: 10.1074/jbc.273.11.6380. [DOI] [PubMed] [Google Scholar]
- Yenchitsomanus PT, Kittanakom S, Rungroj N, Cordat E, Reithmeier RA. Molecular mechanisms of autosomal dominant and recessive distal renal tubular acidosis caused by SLC4A1 (AE1) mutations. J Mol Genet Med. 2005;1(2):49–62. doi: 10.4172/1747-0862.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenchitsomanus PT. Human anion exchanger 1 mutations and distal renal tubular acidosis. Southeast Asian J Trop Med Public Health. 2003;34:651–658. [PubMed] [Google Scholar]
- Miura K, Sekine T, Takahashi K, Takita J, Harita Y, Ohki K, Park MJ, Hayashi Y, Tajima A, Ishihara M, Hisano M, Murai M, Igarashi T. Mutational analyses of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosis. Nephrol Dial Transplant. 2013;28(8):2123–30. doi: 10.1093/ndt/gft216. [DOI] [PubMed] [Google Scholar]
- Myers RM, Tilly K, Maniatis T. Fine structure genetic analysis of a beta-globin promoter. Science. 1986;232(4750):613–618. doi: 10.1126/science.3457470. [DOI] [PubMed] [Google Scholar]