

Abstract
5q31.3 microdeletion syndrome is characterized by neonatal hypotonia, encephalopathy with or without epilepsy, and severe developmental delay, and the minimal critical deletion interval harbors three genes. We describe 11 individuals with clinical features of 5q31.3 microdeletion syndrome and de novo mutations in PURA, encoding transcriptional activator protein Pur-α, within the critical region. These data implicate causative PURA mutations responsible for the severe neurological phenotypes observed in this syndrome.
Main Text
To date, several individuals have been described as having 5q31.3 microdeletion syndrome.1–4 Shimojima et al.1 reported two individuals with profound neonatal hypotonia, feeding difficulties, an abnormal electroencephalogram (EEG), hypomyelination, and severe psychomotor delay.1 Lennox-Gastaut syndrome was diagnosed in one of these individuals, for whom seizures started at 12 months of age. Additional affected individuals were subsequently reported as presenting with marked hypotonia, respiratory insufficiency with central or obstructive sleep apnea, swallowing dysfunction, seizure-like episodes, epilepsy, and neuroimaging abnormalities.2,3 Although the deletion size varied from 2.6 to 5.0 Mb in the original report,1 subsequent individuals with smaller deletions narrowed the critical region; the shortest overlapping segment was approximately 101 kb and encompassed purine-rich element binding protein A (PURA [MIM 600473; RefSeq accession number NM_005859.4]), IgA-inducing protein (IGIP), and cysteine-rich transmembrane module containing 1 (CYSTM1).3 Although PURA has earlier been proposed as a candidate gene with mutations responsible for the neurological phenotypes in this syndrome,3 no individuals with point mutations in this gene have been described.
We identified 11 individuals with de novo heterozygous mutations in the single-exon gene PURA among 2,908 consecutive subjects referred to the Whole Genome Laboratory at Baylor College of Medicine from October 2011 to April 2014 for clinical exome sequencing.5 Approximately 70% of the individuals were pediatric subjects with a variable spectrum of neurodevelopmental disorders. Sequencing and data analysis were conducted as previously described (Yang et al.5) and targeted ∼20,000 genes, including the coding and UTR exons. Average coverage for targeted regions was greater than 130×, and more than 95% of the target bases were covered by at least 20 reads. To clarify the role of PURA in 5q31.3 microdeletion syndrome, we analyzed previously unreported PURA variants found by clinical exome sequencing. Subjects were enrolled in research studies approved by the institutional review board of Baylor College of Medicine or the University of Rochester, and informed consent was obtained. Sanger sequencing confirmed the PURA mutations in all individuals and validated the absence of the relevant mutations in both parents for each of the 11 probands. For each proband, samples were obtained from the biological parents, and the transmission of multiple rare variants was verified by Sanger sequencing confirming the stated parental relationship. Of the identified PURA mutations, four were truncating (three nonsense and one frameshift), five were missense, and two were in-frame deletions (Table 1; Figure 1A). None of these mutations were detected in 1000 Genomes (release 20110521), dbSNP134, the NHLBI Exome Sequencing Project Exome Variant Server, or the database of Atherosclerosis Risk in Communities (exome data of ∼6,000 subjects). No additional contributing mutations in other genes were identified in the 11 individuals with de novo PURA variants.
Table 1.
Clinical Features of Individuals with PURA Mutations
|
Subject |
Summary | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| Gender | male | male | male | female | female | female | female | female | male | female | female | four males, seven females |
| Age at evaluation | 6 months | 7 months | 10 months | 21 months | 23 months | 2 years | 2 years | 5 years | 12 years | 12 years | 15 years | NA |
| Mutation | c.812_814delTCT (p.Phe271 del) | c.307_308delTC (p.Ser103Hisfs∗97) | c.556C>T (p.Gln186∗) | c.289A>G (p.Lys97Glu) | c.299T>C (p.Leu100Pro) | c.363C>G (p.Tyr121∗) | c.783C>G (p.Tyr261∗) | c.470T>A (p.Met157Lys) | c.265G>C (p.Ala89Pro) | c.263_265delTCG (p.Ile88_Ala89delinsThr) | c.596G>C (p.Arg199Pro) | four truncating, seven nontruncating |
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | 11/11 |
| Hypotonia | + | + | + | + | + | + | + | + | + | + | + | 11/11 |
| Feeding difficulties | + | + | + | + | + | + | + | + | + | + | + | 11/11 |
| Respiratory difficulties | + | + | + | + | + | + | + | + | + | + | ND | 10/10 |
| Seizures | + | − | + | + | + | + | + | − | + | + | + | 9/11 |
| Seizure onset | 3 weeks | − | neonatal | ND | 5 months | 3 months | neonatal | − | 3 years | 4 years | ND | NA |
| Seizure type | myoclonic, secondary generalized tonic | − | myoclonic jerks | myoclonic jerks, exaggerated startle | generalized tonic clonic | myoclonic jerks | myoclonic jerks | − | myoclonic, generalized tonic and atonic | Lennox-Gastault syndrome | Lennox-Gastault syndrome | NA |
| Abnormal EEG | + | − | + | ND | ND | + | + | ND | + | + | + | NA |
| Psychomotor delay | + | + | + | + | + | + | + | + | + | + | + | 11/11 |
| Nonverbal | ND | ND | ND | + | + | + | + | + | + | + | + | 8/8 |
| Ambulatory | ND | ND | ND | − | − | − | − | − | − | walked at 3.5 years, ataxia | − | 1/8 |
| Craniofacial features | nondysmorphic | high arched palate, upturned nose, simple lobulation of ears, intermittent exotrophia | nondysmorphic | myopathic facies, strabismus | myopathic facies, tented upper lip, nystagmus | myopathic facies | myopathic facies, left-eye esotropia | nondysmorphic, nystagmus, strabismus | myopathic facies, high arched palate, nystagmus | myopathic facies, high arched palate, wide nasal bridge and tip, hypoplastic alae nasi, strabismus, myopia | nondysmorphic, palpebral fissures long, nystagmus | NA |
See Table S1 for additional details of clinical presentations. Abbreviations are as follows: EEG, electroencephalogram; NA, not applicable; and ND, not determined because of availability or applicability.
Figure 1.
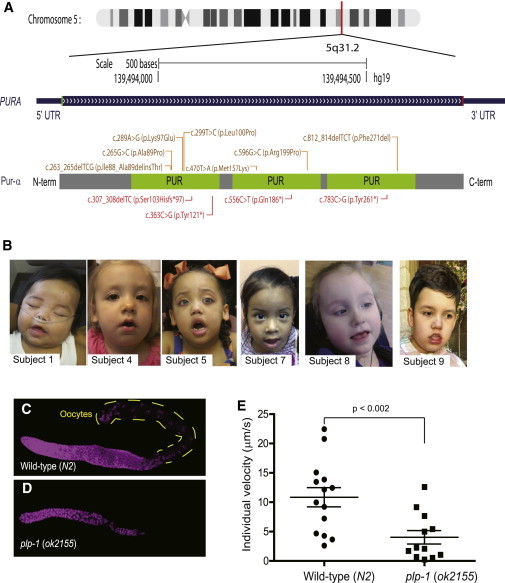
PURA Mutations in the Affected Human Subjects and Functions of the PURA Ortholog in Model System C. elegans
(A) PURA is located in chromosomal region 5q31.2 (according to the UCSC Genome Browser). A single exon (blue) encodes a protein with three PUR motifs, as shown in green. On the protein diagram, missense and in-frame-deletion mutations are brown, and the truncating mutations are shown in red.
(B) Six subjects with de novo PURA mutations are shown. The craniofacial features are mainly characterized by myopathic facies. For whom clinical photographs are shown, informed consent specifically agreeing to publish these photographs in medical publications was obtained.
(C) DNA staining in the germline of a wild-type C. elegans animal. Yellow dashed lines indicate differentiated oocytes.
(D) Oocytes were absent in the plp-1-mutant animal with smaller gonads. Wild-type (N2) and RB1711 (plp-1(ok2155)IV) animals were obtained from the Caenorhabditis Genetics Center. C. elegans were grown on standard nematode growth media plates (containing 5 mg/l cholesterol) with E. coli OP50 at 25°C according to standard protocols. DAPI staining was performed at room temperature. The germline was dissected out from young adult worms.
(E) Average speed of individual worm movement (wild-type n = 14, plp-1 n = 12). The p value was obtained by a one-tailed Student’s t test. Error bars in the scattered dot plot represent the mean and the SEM. Spontaneous movement of age-synchronized worms on standard worm plates was recorded for 15 s with an SMZ1500 stereo microscope (Nikon) connected to a C11440 camera (Hamamatsu). Individual worms were tracked with NIS Elements AR 2D tracking imaging software (Nikon), and the average velocity (μm/s) was calculated. Representative recording of wild-type worms and plp-1 mutants are shown in Movies S1 and S2, respectively.
The phenotype of these 11 probands (Table 1; Table S1, available online) is distinctly similar to that of 5q31 microdeletions encompassing this gene.1–3 All 11 individuals presented at birth with significant hypotonia. Respiratory insufficiency, including central and obstructive sleep apnea and recurrent pulmonary aspiration, were frequently observed. Early-onset feeding difficulties with moderate dysphagia and evidence of tracheal aspiration often necessitated nasogastric or gastric-tube feeding. Myopathic facies with an open mouth and high arched palate were common features (Figure 1B), as observed in the subjects with 5q31.3 microdeletion syndrome.1–3 Hypomyelination or myelin-maturation delay was noted on brain-imaging studies in 4/11 individuals. Myoclonic jerks in infancy were frequent, leading 6/11 children to progress to develop epilepsy. An abnormal EEG was documented in most individuals. Lennox-Gastaut syndrome was diagnosed in two individuals. Almost all of the affected children were nonverbal and nonambulatory at the time of evaluation. Prior to exome sequencing, many of these individuals had undergone extensive diagnostic evaluations, including DNA sequencing and biochemical studies for genetic conditions, such as Prader-Willi and Angelman syndromes, spinal muscular atrophy, congenital disorders of glycosylation, peroxisomal disorders, Rett syndrome, and mitochondrial disease. Our results suggest that individuals with PURA mutations share a recurring pattern of clinical features composing a recognizable phenotype and potentially establish a link between the severe neurodevelopmental profile of 5q31.3 microdeletion syndrome and transcriptional activator protein Pur-α.6
PURA encodes a highly conserved 322 amino acid multifunctional protein, Pur-α, that has important roles in DNA replication, DNA transcription, and mRNA trafficking.7 Pur-α is required for postnatal development of the murine brain and is involved in both neuronal proliferation and maturation of dendrites.8,9 It has also been shown to be important for the transport of specific mRNA molecules to sites of translation in hippocampal neurons.10 Homozygous Pura−/− mice appear normal at birth, but they develop tremors and seizures at 2 weeks and die by 4 weeks of age.8 The number of neurons is severely lower in the cerebellum, cortex, and hippocampus of these mice than in those of age-matched Pura+/+ controls.8 The development of dendrites is also abnormal in the cerebellum and hippocampus of Pura−/− mice.10 Pur-α has been found to colocalize with fragile X mental retardation protein 1 homolog (FMRP) at several sites throughout dendrites in mice.10 Pur-α, the Drosophila ortholog of PURA, has been shown to interact with the RNA transcript of FMR1 at the rCGG repeats and has been implicated in neurodegeneration in the model of fragile-X-associated tremor/ataxia syndrome.11 It is postulated that this interaction might be necessary for FMR1 mRNA transport into dendrites. Similarly, Pur-α has been shown to be involved in the transport of mRNAs into the growing oocytes in Drosophila, given its expression in both follicle cells and germline cells.12 The crystal structure of the protein reveals that the functionality of Pur-α relies on the three conserved PUR motifs.13 The first and second PUR motifs function in binding of single-stranded DNA or RNA, whereas the third PUR motif is involved in the dimerization of Pur-α. In previous studies, targeted mutations in critical codons within the PUR motifs abolished the ability of Pur-α to bind DNA or RNA and impaired the translocation of Pur-α from follicle or nurse cells into oocytes.12,13 The mutation in codon 97 of subject 4 overlaps the Drosophila critical codon 80 mutation (causing p.Arg80Ala), previously12,13 shown to result in reduced nucleic acid binding without affecting correct folding. The missense and in-frame-deletion mutations in our study are all located at conserved sites within PUR motifs (Figure S1). Two in silico prediction tools, PolyPhen-2 and SIFT, predict that the five missense mutations have damaging effects (Table S2). Interestingly, given that this is a single-exon gene and thus not likely subject to nonsense-mediated decay, all truncating mutations found in our series will result in loss or partial loss of PUR motif(s).
To further elucidate the biological roles of PURA, we employed the model organism Caenorhabditis elegans, whose PURA ortholog is plp-1. The plp-1-null allele, ok2155, has a 1,107 bp deletion encompassing part of the promoter region and the first 255 bp of cDNA sequence involving two exons (Figure S2). We found that homozygous plp-1 (ok2155) mutants were sterile. DAPI staining showed that oocytes were absent in the plp-1 mutant (Figures 1C and 1D), suggesting a requirement of plp-1 for germline differentiation. The plp-1 mutants had defective locomotion, marked by a 3-fold reduction in speed in comparison to wild-type animals (Figure 1E; Movies S1 and S2). Compared to age-matched wild-type worms, which showed normal movement, the mutants exhibited minimal forward movement across the culture medium. The observed defects might be related to the impaired mRNA trafficking during oogenesis and neuronal activities. Together, these results suggest the essential functions of plp-1 in both somatic and germline tissues. Human PURA is expressed in the brain and ovarian tissues (Figure S3) and might have roles similar to those of plp-1.
In conclusion, we characterize the clinical features and report de novo heterozygous PURA mutations in multiple individuals with a significant neurodevelopmental phenotype recapitulating 5q31.3 microdeletion syndrome. Considering all 2,117 pediatric subjects who had a neurodevelopmental disorder and were studied by clinical exome sequencing in our laboratory, the 11 heterozygous mutations in PURA account for ∼0.52% and are exceeded only by those in well-established genes AT-rich interactive domain 1B (SWI1-like) (ARID1B [MIM 614556]; 21 subjects [0.99%]) and ankyrin repeat domain 11 (ANKRD11 [MIM 611192]; 13 subjects [0.61%]). Notably, our observed frequency of pathogenic events in ARID1B is consistent with the 0.9% rate previously reported by Hoyer et al.14 PURA mutations should be considered in the evaluation of infants with profound neonatal hypotonia, myoclonic jerks, respiratory insufficiency, and an abnormal EEG. Although further investigations are essential for understanding the effects of PURA haploinsufficiency on encephalopathy with or without epilepsy, this study is an important step toward the clinical and molecular characterization of this disease, which is typically associated with a significant neurological outcome.
Acknowledgments
We are grateful to the families for participating in the study. This study was supported in part by the Doris Duke Foundation and Gillson-Longenbaugh Foundation to S.R.L., by the Joan and Stanford Alexander Family Foundation to C.P.S., and by NIH grants to R.A.G. (U54HG003273), J.R.L. (U54HG006542), M.C.W. (R01AG045183), and L.C.B. and M.J. (T32GM07526). The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from genetic testing offered in the Whole Genome Laboratory and Medical Genetics Laboratories. J.R.L. is a paid consultant for Athena Diagnostics, holds stock ownership in 23andMe and Ion Torrent Systems, and is a coinventor on United States and European patents related to molecular diagnostics. M.N.B. is a founder of Codified Genomics.
Contributor Information
Seema R. Lalani, Email: seemal@bcm.edu.
Fan Xia, Email: fxia@bcm.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Atherosclerosis Risk in Communities Study, http://www2.cscc.unc.edu/aric/
Baylor College of Medicine Whole Genome Laboratory, https://www.bcm.edu/research/medical-genetics-labs/wholegenomelab
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
Accession Numbers
The ClinVar accession numbers for the DNA variant data reported in this paper are SCV000189113, SCV000189114, SCV000189115, SCV000189116, SCV000189117, SCV000189118, SCV000189119, SCV000189120, SCV000189121, SCV000189122, and SCV000189123.
References
- 1.Shimojima K., Isidor B., Le Caignec C., Kondo A., Sakata S., Ohno K., Yamamoto T. A new microdeletion syndrome of 5q31.3 characterized by severe developmental delays, distinctive facial features, and delayed myelination. Am. J. Med. Genet. A. 2011;155A:732–736. doi: 10.1002/ajmg.a.33891. [DOI] [PubMed] [Google Scholar]
- 2.Hosoki K., Ohta T., Natsume J., Imai S., Okumura A., Matsui T., Harada N., Bacino C.A., Scaglia F., Jones J.Y. Clinical phenotype and candidate genes for the 5q31.3 microdeletion syndrome. Am. J. Med. Genet. A. 2012;158A:1891–1896. doi: 10.1002/ajmg.a.35439. [DOI] [PubMed] [Google Scholar]
- 3.Brown N., Burgess T., Forbes R., McGillivray G., Kornberg A., Mandelstam S., Stark Z. 5q31.3 Microdeletion syndrome: clinical and molecular characterization of two further cases. Am. J. Med. Genet. A. 2013;161A:2604–2608. doi: 10.1002/ajmg.a.36108. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld J.A., Drautz J.M., Clericuzio C.L., Cushing T., Raskin S., Martin J., Tervo R.C., Pitarque J.A., Nowak D.M., Karolak J.A. Deletions and duplications of developmental pathway genes in 5q31 contribute to abnormal phenotypes. Am. J. Med. Genet. A. 2011;155A:1906–1916. doi: 10.1002/ajmg.a.34100. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergemann A.D., Johnson E.M. The HeLa Pur factor binds single-stranded DNA at a specific element conserved in gene flanking regions and origins of DNA replication. Mol. Cell. Biol. 1992;12:1257–1265. doi: 10.1128/mcb.12.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White M.K., Johnson E.M., Khalili K. Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle. 2009;8:1–7. doi: 10.4161/cc.8.3.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khalili K., Del Valle L., Muralidharan V., Gault W.J., Darbinian N., Otte J., Meier E., Johnson E.M., Daniel D.C., Kinoshita Y. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell. Biol. 2003;23:6857–6875. doi: 10.1128/MCB.23.19.6857-6875.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hokkanen S., Feldmann H.M., Ding H., Jung C.K., Bojarski L., Renner-Müller I., Schüller U., Kretzschmar H., Wolf E., Herms J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum. Mol. Genet. 2012;21:473–484. doi: 10.1093/hmg/ddr476. [DOI] [PubMed] [Google Scholar]
- 10.Johnson E.M., Kinoshita Y., Weinreb D.B., Wortman M.J., Simon R., Khalili K., Winckler B., Gordon J. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J. Neurosci. Res. 2006;83:929–943. doi: 10.1002/jnr.20806. [DOI] [PubMed] [Google Scholar]
- 11.Jin P., Duan R., Qurashi A., Qin Y., Tian D., Rosser T.C., Liu H., Feng Y., Warren S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aumiller V., Graebsch A., Kremmer E., Niessing D., Förstemann K. Drosophila Pur-α binds to trinucleotide-repeat containing cellular RNAs and translocates to the early oocyte. RNA Biol. 2012;9:633–643. doi: 10.4161/rna.19760. [DOI] [PubMed] [Google Scholar]
- 13.Graebsch A., Roche S., Niessing D. X-ray structure of Pur-alpha reveals a Whirly-like fold and an unusual nucleic-acid binding surface. Proc. Natl. Acad. Sci. USA. 2009;106:18521–18526. doi: 10.1073/pnas.0907990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoyer J., Ekici A.B., Endele S., Popp B., Zweier C., Wiesener A., Wohlleber E., Dufke A., Rossier E., Petsch C. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.