

Abstract
Rhizomelic chondrodysplasia punctata (RCDP) is a group of disorders with overlapping clinical features including rhizomelia, chondrodysplasia punctata, coronal clefts, cervical dysplasia, congenital cataracts, profound postnatal growth retardation, severe intellectual disability, and seizures. Mutations in PEX7, GNPAT, and AGPS, all involved in the plasmalogen-biosynthesis pathway, have been described in individuals with RCDP. Here, we report the identification of mutations in another gene in plasmalogen biosynthesis, fatty acyl-CoA reductase 1 (FAR1), in two families affected by severe intellectual disability, early-onset epilepsy, microcephaly, congenital cataracts, growth retardation, and spasticity. Exome analyses revealed a homozygous in-frame indel mutation (c.495_507delinsT [p.Glu165_Pro169delinsAsp]) in two siblings from a consanguineous family and compound-heterozygous mutations (c.[787C>T];[1094A>G], p.[Arg263∗];[Asp365Gly]) in a third unrelated individual. FAR1 reduces fatty acids to their respective fatty alcohols for the plasmalogen-biosynthesis pathway. To assess the pathogenicity of the identified mutations, we transfected human embryonic kidney 293 cells with plasmids encoding FAR1 with either wild-type or mutated constructs and extracted the lipids from the cells. We screened the lipids with gas chromatography and mass spectrometry and found that all three mutations abolished the reductase activity of FAR1, given that no fatty alcohols could be detected. We also observed reduced plasmalogens in red blood cells in one individual to a range similar to that seen in individuals with RCDP, further supporting abolished FAR1 activity. We thus expand the spectrum of clinical features associated with defects in plasmalogen biosynthesis to include FAR1 deficiency as a cause of syndromic severe intellectual disability with cataracts, epilepsy, and growth retardation but without rhizomelia.
Main Text
Peroxisomes are eukaryotic membrane-bound subcellular organelles that catalyze a large number of critical metabolic reactions, including peroxisomal fatty-acid β-oxidation, plasmalogen (ether phospholipid) biosynthesis, fatty-acid α-oxidation, and glyoxylate detoxification.1 Their crucial role in human health is exemplified by the increasing number of genetic disorders resulting from defects in peroxisomal metabolism, including peroxisomal biogenesis (e.g., Zellweger syndrome [MIM 214100]) and peroxisome function (e.g., rhizomelic chondrodysplasia punctata [RCDP1 (MIM 215100), RCDP2 (MIM 222765), and RCDP3 (MIM 600121)]).1 In our study, we focused on the plasmalogen-biosynthesis pathway in peroxisomes. Defects in plasmalogen synthesis have been described and lead to low levels of plasmalogens and to symptoms of RCDP.2–4 Low levels of plasmalogens were described in other peroxisomal disorders, such as severe forms of Zellweger syndrome, and also in more common neurodegenerative disorders, such as Alzheimer disease (MIM 104300) and trisomy 21 (MIM 190685). Nevertheless, plasmalogen deficiencies in these disorders have been implicated as a secondary effect.2,5–10
Plasmalogens are essential membrane components that have many diverse roles, including in protection against reactive oxygen species,6 in membrane-fusion-mediated events,2,11,12 and as precursors for the platelet-activating factor and cannabinoid receptor ligands.13,14 Plasmalogen synthesis is a multistep process that involves several enzymes. The first few steps take place in the peroxisome, and final processing occurs in the endoplasmic reticulum (ER).2,12,15 Mutations in three genes, PEX7 (peroxisomal biogenesis factor 7 [MIM 601757]), GNPAT (glyceronephosphate O-acyltransferase [MIM 602744]), and AGPS (alkylglycerone phosphate synthase [MIM 603051]), all involved in plasmalogen synthesis in the peroxisome, lead to RCDP1, RCDP2, and RCDP3, respectively.16–20 RCDP is characterized by decreased levels of plasmalogens in red blood cells, rhizomelia, chondrodysplasia punctata, coronal clefts, cervical dysplasia, congenital cataracts, profound postnatal growth retardation, severe intellectual disability, cerebellar atrophy, and seizures. Although most affected children do not survive the first decade of life, a milder phenotype with variable rhizomelia, less growth retardation and developmental delays, congenital cataracts, and sometimes cerebellar hypoplasia has been reported.21–23
Here, we describe two families affected by mutations in FAR1 (fatty acyl-CoA reductase 1). FAR1 reduces fatty acyl-CoAs to their respective fatty alcohols and is therefore a critical enzyme in plasmalogen biosynthesis.24,25 We show that mutations in FAR1 result in an autosomal-recessive peroxisomal disorder of severe, syndromic intellectual disability with congenital cataracts, growth retardation, and epilepsy but without rhizomelia or the skeletal changes that are seen in individuals with RCDP.
This study was approved by the ethics committees of the Universities of Bonn and Erlangen-Nuremberg in Germany and the University of Calgary in Canada. Informed written consent was obtained from all examined individuals or their guardians.
Family A is of Syrian descent and has two affected children of consanguineous parents (Figure 1). The affected female (individual V-1; Figure 1) was born in the seventh month of pregnancy, and the placenta had calcifications. She was hypotonic in the neonatal period, and at the age of 13 months she developed epileptic seizures that were controlled with medication. She was last assessed at 5 years and 3 months of age, at which time she had profound intellectual disability and only rudimentary interaction with her family. She had significant microcephaly with a head circumference of 44 cm (−6.6 SDs), weighed 11 kg (−4.8 SDs), and appeared small for her age. The examining clinical geneticist did not see obvious cataracts, but there was no examination by an ophthalmologist, so smaller cataracts have not been excluded. The affected male (individual V-2; Figure 1) was born at term without any complications. Shortly thereafter, a bilateral congenital cataract was diagnosed and required surgical removal. In the neonatal period, he was hypotonic, which later progressed to spasticity of the upper and lower extremities. At the age of 13 months, he developed epileptic seizures that were also controlled with medication. At the time of assessment, he was 3 years and 7 months old and had profound intellectual disability and only rudimentary interaction with his family. He had significant microcephaly with a head circumference of 40.5 cm (−8.7 SDs) and was small for his age. Investigations of individual V-2 from family A included a normal karyotype, no evidence of congenital infections, normal amino acid concentrations, normal blood gases and electrolytes, and no hints of a mitochondrial or metabolic disorder. He had a normal ultrasound examination of the heart and abdomen and a normal computed-tomography scan of the brain at the age of 5 months. Nevertheless, brain MRI at the age of 3 years and 10 months showed a Dandy-Walker variant.
Figure 1.
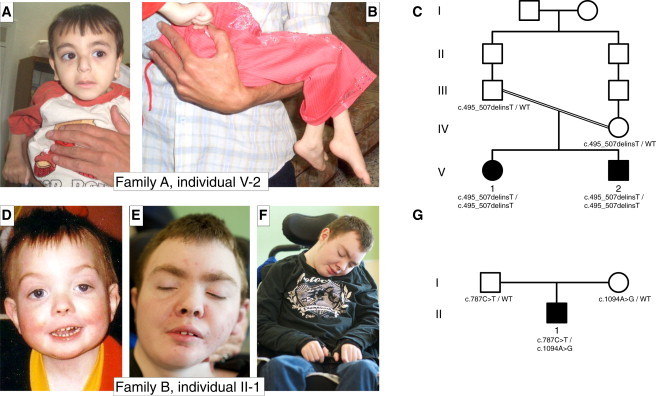
Pedigrees and Pictures of Affected Individuals
(A and B) Individual V-2 from family A suffers from severe intellectual disability, early-onset epilepsy, spasticity, and microcephaly and is slightly dysmorphic with a long philtrum, high-arched eyebrows, large ears, and a mildly hypoplastic and flattened nasal root.
(C) Pedigree of family A. The parents are first-degree cousins once removed.
(D–F) Individual II-1 from family B is shown at 2 years of age (D) and at 19 years of age (E and F). He has growth failure, seizures, developmental delay, and only minor dysmorphic features of hypertelorism, ptosis, facial asymmetry, a short nose, a long smooth philtrum, high-arched eyebrows (evident at the younger age), and a thin upper lip. By 19 years of age, he has some coarsening of his facial features and has developed seizures that are progressively difficult to control and spastic quadriparesis with neuroregression and encephalopathy.
(G) Pedigree of family B. The parents are not related.
Family B has a single affected male (individual II-1; Figure 1) born to a healthy 32-year-old female after an uncomplicated pregnancy. His birth weight was 2.865 kg, and apart from being born with cryptorchidism, he had no major medical problems. This individual is now 19 years of age and has a complex medical and neurodevelopmental history. He developed early-onset bilateral nuclear cataracts requiring surgery at 2 years of age. By 6 months of age, he had started to show significant growth failure with microcephaly. Currently, his height, weight, and head circumference are well below the third percentile (−4 to −5 SDs). At 10 months of age, he developed generalized tonic-clonic seizures, which progressed to a more complex semiology including myoclonic and complex partial seizures and features of atypical Lennox-Gastaut syndrome. His seizures have become more difficult to control with time, and he is now on polytherapy and a ketogenic diet. Despite these therapies, he still has daily clinical seizures, and his electroencephalogram shows subclinical seizures with background slowing consistent with epileptic encephalopathy. He has developed progressive spastic quadriparesis requiring hamstring and tendon releases, bilateral hip osteotomies, and internal fixation of his spine for scoliosis. He has no history of rhizomelia, and at age 19 he has symmetric short stature without rhizomelia. He has never been able to walk or stand unsupported. Developmentally, he is nonverbal and has a history of regression in that he has lost the ability to sit, to use augmentative communication devices or gaze for communication, and to hold a cup. At 19 years of age, he is drowsy and difficult to arouse and has profound intellectual disability. When awake, he shows social interaction and is calm and gentle. On physical examination, he has severe growth retardation, microcephaly, ptosis, coarse facial features, and generalized spasticity with large- and small-joint contractures (Figure 1). His MRI shows cerebellar hemispheric and vermian atrophy with progressive multiple punctate white-matter lesions throughout the cerebral cortices bilaterally, as well as more confluent white-matter changes in both subcortical and deep white-matter regions (Figure 2). There is no evidence of cervical spinal stenosis, although given the associations between low plasmalogen levels and multilevel cervical spinal stenosis, repeat and serial imaging for surveillance purposes are being arranged. A recent skeletal survey confirmed the absence of rhizomelia, and there was no significant evidence of an underlying bone dysplasia: it showed normal morphology of the long bones, a mild increase in vertebral body height of the lumbosacral spine, and a normal pelvis except for mild coxa valga (Figure 2). Extensive genetic and metabolic investigations were all normal.
Figure 2.
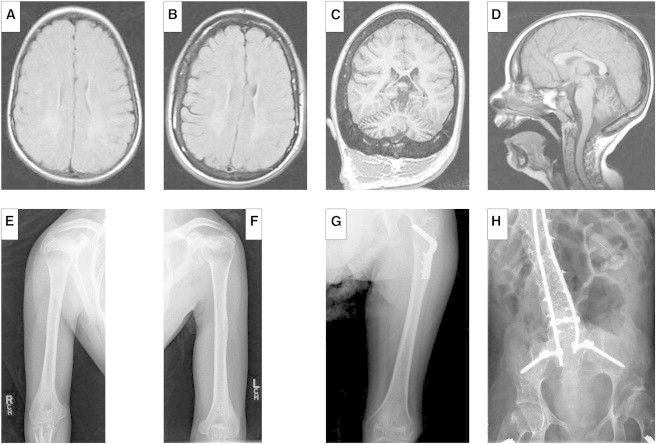
MRI Pictures and X-Rays of Individual II-1 from Family B
(A–D) MRI of individual II-1. Axial T2-weighted images obtained at 9 years of age (A) and again at 15 years of age (B) show increased signal intensity diffusely but more prominently in the posterior periventricular and peritrigonal regions bilaterally. There is subtle evidence of progression of these white-matter changes over time. (C) A coronal MP-RAGE-weighted image at 15 years of age shows prominence of the cerebellar folia consistent with mild to moderate cerebellar atrophy. (D) A sagittal T1-weighted image at 9 years of age shows mild atrophy of the cerebellum and no evidence of cervical spinal stenosis.
(E–H) Skeletal X-rays of individual II-1 from family B. Skeletal X-rays of individual II-1’s humeri (E and F), left femur (G), and lumbosacral spine and pelvis (H) were taken at 19 years of age. There is no evidence of rhizomelic shortening of the humeri or femurs. The spine has decreased intervertebral spaces and tall vertebral bodies. Internal instrumentation was used to treat his scoliosis. There is mild coxa valga of the femoral necks, and the internal fixation is from his bilateral hip osteotomies.
Genotyping and mapping analyses were performed in family A as previously reported.26 Four candidate regions (chr4: 111,364,166–113,972,855 bp; chr6: 138,484,928–144,671,770 bp; chr11: 7,712,660–19,368,171 bp, and chr15: 75,083,493–79,273,984 bp) homozygous in both affected children were identified for a total length of 24.6 Mb. DNA from individual V-1 from family A underwent exome capture and next-generation sequencing on the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies) and a SOLiD4 instrument (Life Technologies) as described previously.27 An average target coverage of 56.7× was obtained, and 58.2% of reads were covered to a depth of at least 20× and 73.6% were covered to a depth of at least 5×. A total of 36,345 single-nucleotide variants (SNVs) and 2,563 insertions or deletions (indels) were identified, and 415 SNVs and 77 indels were neither annotated in dbSNP nor reported in 1000 Genomes, the NHLBI Exome Variant Server, or in-house control exomes. Of these, only one variant, c.495_507delinsT (p.Glu165_Pro169delinsAsp) in FAR1 (RefSeq accession number NM_032228.5), was located in a homozygous candidate region, involved conserved amino acids, and was predicted to affect the protein sequence. Because the depth of coverage was low, we repeated sequencing of the coding sequence (CDS) in the candidate regions (233 genes) with a custom targeted-capture kit (Agilent Technologies). We achieved at least 20× coverage for 85.4% of the target sequence and at least 5× coverage for 90.3% of the sequence. Combining reads from exome and CDS custom targeted sequencing, we achieved a mean coverage of 373× for the CDS within the regions of homozygosity, and 93.1% of the CDS target region was covered at least 5×. No additional candidate variants were identified with this approach. The FAR1 variant segregated with severe intellectual disability in the family and was absent in 280 ethnically matched control individuals.
To investigate the cause of sporadic intellectual disability, we performed trio-based whole-exome sequencing on individual II-1 from family B and his parents at the Alberta Children’s Hospital Research Institute Genomics and Bioinformatics Facility (University of Calgary). Genomic DNA from affected individual II-1 and his parents underwent exome enrichment with the Agilent SureSelect V5 Kit (Agilent Technologies) prior to sequencing with a 5500xl SOLiD sequencer (Life Technologies). A total of 31,044 coding variants were identified in the proband. The average target depth was 115×, and 88% of target bases were covered at least 20× and 92% were covered at least 10×. After failing to identify any pathogenic de novo variants, we examined genes in which two rare protein-coding variants were inherited, one from each parent. We identified biallelic variants in FAR1, c.787C>T (p.Arg263∗) and c.1094A>G (p.Asp365Gly), which were inherited paternally and maternally, respectively. Neither mutation has been reported in dbSNP or the NHLBI Exome Variant Server. The candidate variant p.Arg263∗ introduces a premature stop codon, and p.Asp365Gly affects a residue absolutely conserved in 100 vertebrate species and is predicted to be deleterious by in silico missense prediction tools SIFT,28 PolyPhen-2,29 and MutationTaster.30
To provide further evidence of pathogenicity for the variants detected, we sought to model the structure of the FAR1 reductase domain (residues 1–424), in which all of the identified variants resided. Molecular modeling of the p.Glu165_Pro169delinsAsp substitution in family A showed that amino acids Glu, Val, Val, Tyr, and Pro form a loop near the active site in the wild-type protein (Figure 3). A deletion at this position alters the geometry of the active-site pocket (Figure 3), most likely impairing enzyme activity. Modeling of p.Arg263∗ in family B showed that the alteration, if it does not result in nonsense-mediated decay, is predicted to affect the entire domain, including the active center, and is therefore expected to result in complete loss of enzymatic activity. Molecular modeling of the p.Asp365Gly substitution showed that asparagine at position 365 normally forms stabilizing salt bridges with arginines at positions 263 and 266 (Figure 3). The presence of the p.Asp365Gly substitution is predicted to disrupt these interactions and thereby result in destabilization of the tertiary structure of FAR1 (Figure 3).
Figure 3.
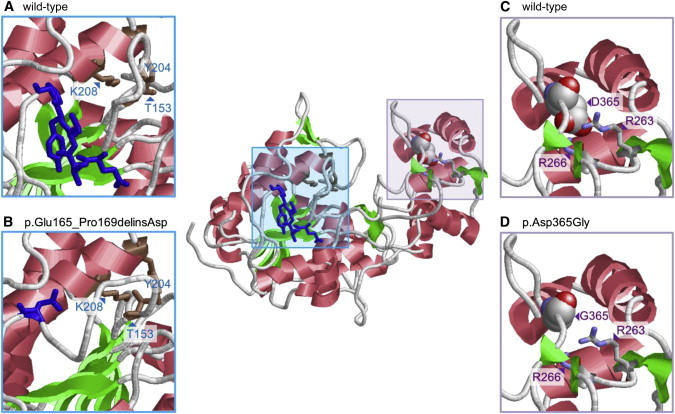
Molecular Modeling of Amino Acid Substitutions in FAR1
Molecular modeling of the FAR1 reductase domain is based on the crystal structure of the homologous reductase domain from a mycobacterial enzyme (Protein Data Bank ID 4DQV31). It was performed with the program Modeler 9.932 and resulted in a structure with good local geometry and no steric hindrance.
(Center) A model of wild-type FAR1 includes boxes at regions of amino acid substitutions.
(A) A model of wild-type FAR1 shows residues 165–169 (Glu-Val-Val-Tyr-Pro stretch) as blue sticks. The amino acids adopt an extended conformation and are located in a loop that connects the active-site residues of the enzyme (Thr153 [T153], Tyr204 [Y204], and Lys208 [K208], shown as brown sticks).
(B) The effect of the deletion was modeled with ModLoop,33 and RasMol34 was used for graphical presentation. An amino acid substitution in which the Glu-Val-Val-Tyr-Pro stretch is replaced by a single aspartate (Asp165, shown as a blue stick) is predicted to alter the geometry of the active-site pocket.
(C) Residue Asp365 (D365) is shown in a space-filled presentation and is colored according to the atom types. Asp365 interacts with Arg263 (R263) and Arg266 (R266), shown in stick presentation.
(D) Replacement of Asp365 with Gly365 (G365) is predicted to abolish salt bridges with Arg263 (R263) and Arg266 (R266) and thereby result in a destabilization of the FAR1 structure.
To provide a second line of evidence of the functional effects of the three identified mutations, we determined the effects of each mutation in a cell-based functional assay. Because FAR1 reduces both saturated and unsaturated fatty acyl-CoAs of 16 and 18 carbon atoms to their respective fatty alcohols,24,25 we sought to measure the enzymatic activity of wild-type and mutation-containing FAR1 in transfected human embryonic kidney 293 (HEK293) cell lines that were previously shown to have negligible FAR1 activity in comparison to cells transfected with plasmids encoding FAR1.24 We also confirmed this in our experiments (Figure 4). For this, we performed site-directed mutagenesis to introduce all three mutations—c.495_507delinsT (p.Glu165_Pro169delinsAsp), c.787C>T (p.Arg263∗), and c.1094A>G (p.Asp365Gly)—into human FAR1 cDNA (hFAR1) in the pCMV6-XL6 vector. Cells were transfected with 10 μg of pCMV6-XL6-hFAR1 containing wild-type or mutation-containing sequences. Forty-eight hours later, the cells were harvested, and lipids were extracted with chloroform according to the Bligh and Dyer method.35 The lipid extracts were silylated and separated by gas chromatography, and detection was carried out by electron-impact mass spectrometry.
Figure 4.
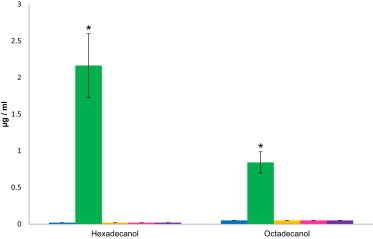
Results of the FAR1-Activity Assay
Quantification of hexadecanol and octadecanol in lipid extracts of HEK293 cells transfected with plasmids encoding wild-type FAR1 (green), plasmids encoding FAR1 with mutations c.495_507delinsT (p.Glu165_Pro169delinsAsp) (yellow), c.787C>T (p.Arg263∗) (magenta), or c.1094A>G (p.Asp365Gly) (purple), or an empty vector (blue). Only after transfection with plasmids encoding wild-type FAR1 could we measure hexadecanol and octadecanol; all other samples only showed traces. The decrease in hexadecanol and octadecanol levels was significant (p < 0.05). A detailed description of the methods is provided in the Figure S1 legend. Error bars represent the SEM.
After transfection with plasmids encoding wild-type FAR1, levels of hexadecanol and octadecanol in lipid extracts were elevated (Figure 4), which was expected given that these are the products of the enzyme after reduction of palmitic acid and stearic acid, respectively. Conversely, cells transfected with plasmids encoding FAR1 containing any of the mutations did not yield any significant levels of hexadecanol or octadecanol (p < 0.05; Figure 4; Figure S1, available online) and were thus similar to cells transfected with an empty vector. This result strongly suggests that the presence of any of these three mutations results in a complete loss of FAR1 activity. This is in accordance with the results of the measurement of red blood cell plasmalogens of individual II-1 from family B; they showed significantly decreased ratios of plasmalogen to fatty acids: 0.002 for C16 (normal range: 0.079–0.128) and 0.008 for C18 (normal range: 0.199–0.284) (Table 1).36,37
Table 1.
Ratios of Plasmalogen to Fatty Acids
|
Ratio of C16:0 DMA to C16:0 Fatty Acid |
Ratio of C18:0 DMA to C18:0 Fatty Acid |
|||
|---|---|---|---|---|
| Median | Range | Median | Range | |
| Individual II-1 from family B | 0.002 | – | 0.008 | – |
| Normal control individuals (n = 193) | 0.103 | 0.079–0.128 | 0.241 | 0.199–0.284 |
| Zellweger spectrum disorders (n = 125)28 | 0.014 | 0.001–0.085 | 0.042 | 0.001–0.189 |
| RCDP (n = 53)28 | 0.002 | 0.001–0.031 | 0.002 | 0.001–0.109 |
Ratios of plasmalogen to fatty acids were measured for individual II-1 from family B and are compared to ratios from healthy individuals and individuals with plasmalogen-synthesis disorders; the ratios are low and consistent with a defect in plasmalogen metabolism. DMA stands for dimethyl acetal (of plasmalogen).
FAR1 is a component of the plasmalogen-biosynthesis pathway and supplies fatty alcohols for processing by further enzymes in the peroxisome and ER (Figure 5).24 Impairment of plasmalogen synthesis is the primary biochemical deficiency present in individuals with RCDP.2,39 To date, mutations in three genes have been reported to cause RCDP. Specifically, these genes encode the transporter PEX7, involved in peroxisomal biogenesis, and the enzymes GNPAT and AGPS, which are both directly involved in plasmalogen synthesis (Figure 5).16–18,25,40 In individuals with RCDP, fatty alcohols supplied by FAR1 fail to be incorporated into ether lipids, leading to elevated fatty-alcohol levels and reduced plasmalogen levels.38 The three alterations we identified essentially abolish FAR1 activity and thus probably lead to lower levels of fatty alcohols and, as in RCDP, to low plasmalogen levels. This is supported by our observation that the ratios of plasmalogen to fatty acids in individual II-1 from family B were significantly decreased and comparable to those in persons affected by RCDP (Table 1).36,37
Figure 5.
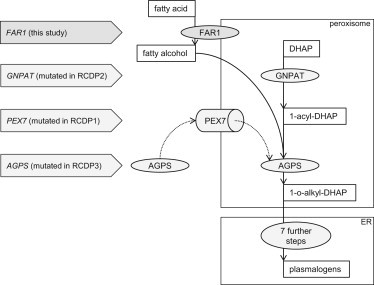
Plasmalogen-Biosynthesis Pathway and Associated Phenotypes
Plasmalogen synthesis is a multistep process that involves several enzymes. On the luminal side of the peroxisomal membrane, GNPAT acylates DHAP (dihydroxyacetone phosphate) at its sn-1 position. Then, AGPS is transported into the peroxisome by PEX7 and uses the 1-acyl-DHAP and the fatty alcohols, which are supplied by FAR1, as substrates and exchanges the acyl group for an alkyl group. The resulting 1-o-alkyl DHAP is then reduced to GPA (1-O-alkyl-2-hydroxy-sn-glycerophosphate) by an acyl/alkyl-DHAP reductase, which is located in both peroxisomal and ER membranes. Further processing is similar to the processing of diacyl glycerophospholipids and takes place in the ER.2,12,15 Mutations in GNPAT and AGPS cause a block in plasmalogen synthesis and lead to an accumulation of fatty alcohols that are no longer processed.38 Affected individuals develop RCDP.16,17 Mutations in PEX7 lead to an absence of peroxisomal AGPS and thus have similar effects on plasmalogen biosynthesis.18 In individuals V-1 and V-2 from family A and II-1 from family B, fatty acids are no longer metabolized to fatty alcohols by FAR1, likewise leading to a decrease in plasmalogen levels and an overlapping phenotype.
There is a remarkable similarity between the clinical features of individuals with RCDP and those of the persons reported here. These features include profound intellectual disability, congenital cataracts, growth restriction, spasticity, epilepsy, microcephaly, and cerebellar atrophy (Table 2). However, the most significant difference between individuals with classical RCDP and individuals V-1 and V-2 from family A and II-1 from family B is the absence of rhizomelia and skeletal dysplasia even by 19 years of age. Furthermore, individuals with classical RCDP usually do not survive the first decade of life. Thus, we were surprised by the extremely low plasmalogen levels (similar to those of classical RCDP) in 19-year-old individual II-1 from family B. The mechanisms underlying these phenotypic differences are not clear, but we expect that a kind of tissue-specific compensation mechanism exists. One possible explanation could be a compensating mechanism by FAR2 (fatty acyl-CoA reductase 2), a homolog of FAR1. FAR2 prefers only saturated C16 and C18 fatty acyl-CoAs, whereas FAR1 uses both saturated and unsaturated fatty acyl-CoAs as substrate.24 Nevertheless, given that the ratios of plasmalogen to fatty acids in red blood cells of individual II-1 from family B are similar to those of individuals with classic RCDP, this compensating mechanism would, if at all, be restricted to some tissues. One further argument against a compensation effect of FAR2 is the expression profile of FAR2, given that according to the data from GeneHub-GEPIS tissue,41 FAR2 is not expressed in bones or cartilage, whereas FAR1 is expressed in both. Another possible explanation is that it has been proposed that fatty alcohols might be alternatively supplied by β-oxidation.42 However, this is also less likely because individuals with deficits in peroxisomal β-oxidation metabolism, such as ACOX1 deficiency (MIM 264470) and HSD17B4 deficiency (MIM 261515), have normal plasmalogen levels.2,42 Dietary intake can also be a limited source of fatty alcohols and might help explain the phenotype differences between the individuals presented here and individuals with RCDP.2
Table 2.
Symptoms of RCDP and FAR1 Deficiency
| Symptoms |
Family A |
Family B |
Individuals with RCDP | |
|---|---|---|---|---|
| V-1 | V-2 | II-1 | ||
| Profound intellectual disability | + | + | + | + |
| Neuroregression | ? | ? | + | ++ |
| Early-onset seizures | + | + | + | + |
| Spastic quadriparesis | + | + | + | + |
| Microcephaly | + | + | + | + |
| Cataracts | − | + | + | + |
| Growth retardation | + | + | +++ | +++ |
| Chondrodysplasia punctata | − | − | − | + |
| Rhizomelia | − | − | − | ++ |
| Survival | alive at 5 years | alive at 3 years | alive at 19 years | demise by 10 years |
| Brain imaging | not available | Dandy-Walker malformation | cerebellar vermis atrophy, diffuse white-matter disease | cerebral and cerebellar atrophy, delayed myelination |
A comparison of symptoms from individuals with RCDP1, RCDP2, and RCDP3 and the individuals from our families A and B shows a significant overlap in phenotypes. Abbreviations are as follows: +, present; ++, moderate; +++, severe; −, not present; and ?, unknown.
We have identified three FAR1 mutations in two unrelated families sharing a number of clinical features that also clearly overlap those seen in RCDP. These findings provide evidence that FAR1 deficiency causes a peroxisomal disorder as a result of impaired plasmalogen biosynthesis. At least seven additional enzymes are involved in plasmalogens biosynthesis, and it is possible that other yet-to-be-identified mutations in these genes might account for some unexplained intellectual disability in combination with congenital cataracts in the general population. Further genetic analysis will allow for further delineation of this group of peroxisomal disorders with disturbed plasmalogen synthesis.
Acknowledgments
We are grateful to the families who participated in this study. We thank David Russell (Dallas) for providing us with the pCMV6-XL6-hFAR1 vector. We thank André Reis (Erlangen) for continued support. We thank Petra Rothe, Angelika Diem, and Farah Radwan from Erlangen for assistance with SNP array genotyping, Sanger sequencing, and next-generation sequencing. This study was supported by Deutsche Forschungsgemeinschaft through grants AB393/2-1 and AB393/2-2 to R.A.J. and by Deutscher Akademischer Austauschdienst through a scholarship to H.T. We also thank the Alberta Children’s Hospital Research Institute Genomics and Bioinformatics Facility, Paul Gordon and Richard Pon for their expertise, the Alberta Children’s Hospital Foundation for their generous funding support to A.M.I., J.S.P., and F.P.B., and Brenda McInnes for clinical support. We acknowledge the expert reviews of the skeletal survey by Sarah Nikkel (University of Ottawa) and of the neuroimaging by Luis Bello-Espinos (University of Calgary).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, https://esp.gs.washington.edu/drupal/
NIST Standard Reference Databases: Analytical Chemistry, http://www.nist.gov/srd/analy.cfm
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Wanders R.J. Metabolic functions of peroxisomes in health and disease. Biochimie. 2014;98:36–44. doi: 10.1016/j.biochi.2013.08.022. [DOI] [PubMed] [Google Scholar]
- 2.Braverman N.E., Moser A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta. 2012;1822:1442–1452. doi: 10.1016/j.bbadis.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Heymans H.S., Oorthuys J.W., Nelck G., Wanders R.J., Schutgens R.B. Rhizomelic chondrodysplasia punctata: another peroxisomal disorder. N. Engl. J. Med. 1985;313:187–188. [PubMed] [Google Scholar]
- 4.Heikoop J.C., Wanders R.J., Strijland A., Purvis R., Schutgens R.B., Tager J.M. Genetic and biochemical heterogeneity in patients with the rhizomelic form of chondrodysplasia punctata—a complementation study. Hum. Genet. 1992;89:439–444. doi: 10.1007/BF00194319. [DOI] [PubMed] [Google Scholar]
- 5.Brites P., Mooyer P.A., El Mrabet L., Waterham H.R., Wanders R.J. Plasmalogens participate in very-long-chain fatty acid-induced pathology. Brain. 2009;132:482–492. doi: 10.1093/brain/awn295. [DOI] [PubMed] [Google Scholar]
- 6.Khan M., Singh J., Singh I. Plasmalogen deficiency in cerebral adrenoleukodystrophy and its modulation by lovastatin. J. Neurochem. 2008;106:1766–1779. doi: 10.1111/j.1471-4159.2008.05513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy E.J., Schapiro M.B., Rapoport S.I., Shetty H.U. Phospholipid composition and levels are altered in Down syndrome brain. Brain Res. 2000;867:9–18. doi: 10.1016/s0006-8993(00)02205-8. [DOI] [PubMed] [Google Scholar]
- 8.Grimm M.O., Kuchenbecker J., Rothhaar T.L., Grösgen S., Hundsdörfer B., Burg V.K., Friess P., Müller U., Grimm H.S., Riemenschneider M., Hartmann T. Plasmalogen synthesis is regulated via alkyl-dihydroxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in Alzheimer’s disease. J. Neurochem. 2011;116:916–925. doi: 10.1111/j.1471-4159.2010.07070.x. [DOI] [PubMed] [Google Scholar]
- 9.Heymans H.S., Schutgens R.B., Tan R., van den Bosch H., Borst P. Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome) Nature. 1983;306:69–70. doi: 10.1038/306069a0. [DOI] [PubMed] [Google Scholar]
- 10.Schrakamp G., Schutgens R.B., Wanders R.J., Heymans H.S., Tager J.M., Van den Bosch H. The cerebro-hepato-renal (Zellweger) syndrome. Impaired de novo biosynthesis of plasmalogens in cultured skin fibroblasts. Biochim. Biophys. Acta. 1985;833:170–174. doi: 10.1016/0005-2760(85)90266-8. [DOI] [PubMed] [Google Scholar]
- 11.Glaser P.E., Gross R.W. Plasmenylethanolamine facilitates rapid membrane fusion: a stopped-flow kinetic investigation correlating the propensity of a major plasma membrane constituent to adopt an HII phase with its ability to promote membrane fusion. Biochemistry. 1994;33:5805–5812. doi: 10.1021/bi00185a019. [DOI] [PubMed] [Google Scholar]
- 12.Brites P., Waterham H.R., Wanders R.J. Functions and biosynthesis of plasmalogens in health and disease. Biochim. Biophys. Acta. 2004;1636:219–231. doi: 10.1016/j.bbalip.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Nagan N., Zoeller R.A. Plasmalogens: biosynthesis and functions. Prog. Lipid Res. 2001;40:199–229. doi: 10.1016/s0163-7827(01)00003-0. [DOI] [PubMed] [Google Scholar]
- 14.Munn N.J., Arnio E., Liu D., Zoeller R.A., Liscum L. Deficiency in ethanolamine plasmalogen leads to altered cholesterol transport. J. Lipid Res. 2003;44:182–192. doi: 10.1194/jlr.m200363-jlr200. [DOI] [PubMed] [Google Scholar]
- 15.James P.F., Lake A.C., Hajra A.K., Larkins L.K., Robinson M., Buchanan F.G., Zoeller R.A. An animal cell mutant with a deficiency in acyl/alkyl-dihydroxyacetone-phosphate reductase activity. Effects on the biosynthesis of ether-linked and diacyl glycerolipids. J. Biol. Chem. 1997;272:23540–23546. doi: 10.1074/jbc.272.38.23540. [DOI] [PubMed] [Google Scholar]
- 16.Wanders R.J., Schumacher H., Heikoop J., Schutgens R.B., Tager J.M. Human dihydroxyacetonephosphate acyltransferase deficiency: a new peroxisomal disorder. J. Inherit. Metab. Dis. 1992;15:389–391. doi: 10.1007/BF02435984. [DOI] [PubMed] [Google Scholar]
- 17.Wanders R.J., Dekker C., Hovarth V.A., Schutgens R.B., Tager J.M., Van Laer P., Lecoutere D. Human alkyldihydroxyacetonephosphate synthase deficiency: a new peroxisomal disorder. J. Inherit. Metab. Dis. 1994;17:315–318. doi: 10.1007/BF00711817. [DOI] [PubMed] [Google Scholar]
- 18.Purdue P.E., Zhang J.W., Skoneczny M., Lazarow P.B. Rhizomelic chondrodysplasia punctata is caused by deficiency of human PEX7, a homologue of the yeast PTS2 receptor. Nat. Genet. 1997;15:381–384. doi: 10.1038/ng0497-381. [DOI] [PubMed] [Google Scholar]
- 19.Motley A.M., Hettema E.H., Hogenhout E.M., Brites P., ten Asbroek A.L., Wijburg F.A., Baas F., Heijmans H.S., Tabak H.F., Wanders R.J., Distel B. Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a non-functional PTS2 receptor. Nat. Genet. 1997;15:377–380. doi: 10.1038/ng0497-377. [DOI] [PubMed] [Google Scholar]
- 20.Braverman N., Steel G., Obie C., Moser A., Moser H., Gould S.J., Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 1997;15:369–376. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- 21.Braverman N., Chen L., Lin P., Obie C., Steel G., Douglas P., Chakraborty P.K., Clarke J.T., Boneh A., Moser A. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum. Mutat. 2002;20:284–297. doi: 10.1002/humu.10124. [DOI] [PubMed] [Google Scholar]
- 22.Bams-Mengerink A.M., Majoie C.B., Duran M., Wanders R.J., Van Hove J., Scheurer C.D., Barth P.G., Poll-The B.T. MRI of the brain and cervical spinal cord in rhizomelic chondrodysplasia punctata. Neurology. 2006;66:798–803. doi: 10.1212/01.wnl.0000205594.34647.d0. discussion 789. [DOI] [PubMed] [Google Scholar]
- 23.Motley A.M., Brites P., Gerez L., Hogenhout E., Haasjes J., Benne R., Tabak H.F., Wanders R.J., Waterham H.R. Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1. Am. J. Hum. Genet. 2002;70:612–624. doi: 10.1086/338998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng J.B., Russell D.W. Mammalian wax biosynthesis. I. Identification of two fatty acyl-Coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 2004;279:37789–37797. doi: 10.1074/jbc.M406225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honsho M., Asaoku S., Fukumoto K., Fujiki Y. Topogenesis and homeostasis of fatty acyl-CoA reductase 1. J. Biol. Chem. 2013;288:34588–34598. doi: 10.1074/jbc.M113.498345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abou Jamra R., Wohlfart S., Zweier M., Uebe S., Priebe L., Ekici A., Giesebrecht S., Abboud A., Al Khateeb M.A., Fakher M. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet. 2011;19:1161–1166. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murakami Y., Tawamie H., Maeda Y., Büttner C., Buchert R., Radwan F., Schaffer S., Sticht H., Aigner M., Reis A. Null mutation in PGAP1 impairing Gpi-anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet. 2014;10:e1004320. doi: 10.1371/journal.pgen.1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(Web Server issue):W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chhabra A., Haque A.S., Pal R.K., Goyal A., Rai R., Joshi S., Panjikar S., Pasha S., Sankaranarayanan R., Gokhale R.S. Nonprocessive [2 + 2]e- off-loading reductase domains from mycobacterial nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. USA. 2012;109:5681–5686. doi: 10.1073/pnas.1118680109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sánchez R., Sali A. Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol. Biol. 2000;143:97–129. doi: 10.1385/1-59259-368-2:97. [DOI] [PubMed] [Google Scholar]
- 33.Fiser A., Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 34.Sayle R.A., Milner-White E.J. RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 35.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 36.Björkhem I., Sisfontes L., Boström B., Kase B.F., Blomstrand R. Simple diagnosis of the Zellweger syndrome by gas-liquid chromatography of dimethylacetals. J. Lipid Res. 1986;27:786–791. [PubMed] [Google Scholar]
- 37.Moser A.B., Jones D.S., Raymond G.V., Moser H.W. Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem. Res. 1999;24:187–197. doi: 10.1023/a:1022549618333. [DOI] [PubMed] [Google Scholar]
- 38.Rizzo W.B., Craft D.A., Judd L.L., Moser H.W., Moser A.B. Fatty alcohol accumulation in the autosomal recessive form of rhizomelic chondrodysplasia punctata. Biochem. Med. Metab. Biol. 1993;50:93–102. doi: 10.1006/bmmb.1993.1050. [DOI] [PubMed] [Google Scholar]
- 39.Hoefler G., Hoefler S., Watkins P.A., Chen W.W., Moser A., Baldwin V., McGillivary B., Charrow J., Friedman J.M., Rutledge L. Biochemical abnormalities in rhizomelic chondrodysplasia punctata. J. Pediatr. 1988;112:726–733. doi: 10.1016/s0022-3476(88)80689-9. [DOI] [PubMed] [Google Scholar]
- 40.Honsho M., Asaoku S., Fujiki Y. Posttranslational regulation of fatty acyl-CoA reductase 1, Far1, controls ether glycerophospholipid synthesis. J. Biol. Chem. 2010;285:8537–8542. doi: 10.1074/jbc.M109.083311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Luoh S.M., Hon L.S., Baertsch R., Wood W.I., Zhang Z. GeneHub-GEPIS: digital expression profiling for normal and cancer tissues based on an integrated gene database. Nucleic Acids Res. 2007;35(Web Server issue):W152–W158. doi: 10.1093/nar/gkm381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hashimoto F., Furuya Y., Hayashi H. Accumulation of medium chain acyl-CoAs during beta-oxidation of long chain fatty acid by isolated peroxisomes from rat liver. Biol. Pharm. Bull. 2001;24:600–606. doi: 10.1248/bpb.24.600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.