

Abstract
The 17p13.1 microdeletion syndrome is a recently described genomic disorder with a core clinical phenotype of intellectual disability, poor to absent speech, dysmorphic features, and a constellation of more variable clinical features, most prominently microcephaly. We identified five subjects with copy-number variants (CNVs) on 17p13.1 for whom we performed detailed clinical and molecular studies. Breakpoint mapping and retrospective analysis of published cases refined the smallest region of overlap (SRO) for microcephaly to a genomic interval containing nine genes. Dissection of this phenotype in zebrafish embryos revealed a complex genetic architecture: dosage perturbation of four genes (ASGR1, ACADVL, DVL2, and GABARAP) impeded neurodevelopment and decreased dosage of the same loci caused a reduced mitotic index in vitro. Moreover, epistatic analyses in vivo showed that dosage perturbations of discrete gene pairings induce microcephaly. Taken together, these studies support a model in which concomitant dosage perturbation of multiple genes within the CNV drive the microcephaly and possibly other neurodevelopmental phenotypes associated with rearrangements in the 17p13.1 SRO.
Introduction
Copy-number losses and gains involving different segments of the chromosome 17 short arm lead to a number of well-characterized genomic disorders. These include chromosome 17p13.3 duplication syndrome (MIM 613215),1 Miller-Dieker lissencephaly syndrome (MDLS [MIM 247200]),2 Charcot-Marie-Tooth disease type 1A (CMT1A [MIM 118220]),3 hereditary neuropathy with liability to pressure palsies (HNPP [MIM 162500]),4 Potocki-Lupski microduplication syndrome (PTLS [MIM 610883]),5 and Smith-Magenis microdeletion syndrome (SMS [MIM 182290]).6
The first clinical cases with 17p13.1 microdeletion syndrome (MIM 613776) were reported in 2009 and 20107–11 and were delineated further by Zeesman et al.,12 who described two individuals with similar clinical phenotypes carrying de novo deletions. This study helped narrow the critical genomic interval to <250 kb. Duplications have been observed rarely and are associated with moderate intellectual disability and mild dysmorphic features;13,14 clinical similarity with Silver-Russell syndrome (SRS [MIM 180860]) was reported in one case.14 In contrast, subjects with 17p13.1 microdeletion syndrome present marked phenotypic variability that includes severe intellectual disability, poor to absent speech, dysmorphic features, and microcephaly. The variable clinical presentation may correlate with gene content and with size of the deleted segments, which, in the literature, reveals nonrecurrent events that ranged from 245 kb to 4.4 Mb. Based on this observation, Zeesman et al.12 suggested that two or more separate microdeletion syndromes may exist within that entire region and that 17p13.1 microdeletion syndrome may constitute a “contiguous gene microdeletion syndrome” as originally proposed by Schmickel.15 This is an attractive hypothesis not easy to test due to the uniqueness of each rearrangement size as well as the rarity of individuals with such disorders reported to date.
We identified five subjects with 17p13.1 copy-number variants (CNVs) and performed detailed clinical evaluation and molecular analyses of the genomic rearrangements. Retrospective analysis of these combined data sets narrowed the smallest region of overlap (SRO) associated with microcephaly to a genomic interval containing nine annotated loci. Functional studies in which the dosage of each of these genes was manipulated in zebrafish embryos provided evidence for genetic complexities. Not only did we observe that multiple individual genes can induce microcephaly with concomitant cell cycling defects, but we also observed strong genetic interactions among discrete gene sets. In fact, the genetic complexities revealed from studies of 17p13.1 microdeletion syndrome have similarities to observations for a nonrecurrent CNV on 8q24.16
These data stand in contrast to the current models for several CNVs, the phenotype of which is driven by dosage perturbation of a single major driver: e.g., PMP22 mutations (MIM 601097) can lead to CMT1A,3,17 subjects with TBX1 mutations (MIM 602054) can reproduce some of the phenotypes observed in DiGeorge/velocardiofacial syndromes (MIM 188400/192430),18 CREBBP mutations (MIM 600140) can reproduce Rubinstein-Taybi syndrome (MIM 180894),19 and similarly for EHMT1 (MIM 607001) in Kleefstra syndrome (MIM 610253)20 and for NSD1 (MIM 606681) in Sotos syndrome (MIM 117550).21 Here we provide evidence that dosage alteration of any one of four genes—ASGR1(MIM 108360), ACADVL (MIM 609575), DVL2 (MIM 602151), and GABARAP (MIM 605125)—mapping within the SRO in humans is sufficient to cause head size defects in zebrafish embryos potentially due to an increased apoptosis. Furthermore, epistatic interactions between them and nearby gene products exert a contributory effect that is likely partially responsible for the incomplete penetrance and variable expressivity of the phenotype.
Subjects and Methods
Subjects
Subjects BAB3036, BAB3045, BAB3277, and BAB3302 carrying genomic rearrangements in 17p13.1 were identified at the Baylor College of Medicine Medical Genetics Laboratories as part of their clinical diagnostic evaluation. DECIPHER4155 was identified at the Clinical Genetics Department, Vejle Hospital (Vejle, Denmark). Informed consent was obtained for participation in the research study. This research was performed with the approval of the Baylor College of Medicine Institutional Review Board (H-27609). Clinical phenotype for each subject is described in Table 1.
Table 1.
Clinical Presentation of Subjects with Rearrangements Spanning 17p13.1
| Subject ID | BAB3045 | BAB3036 | BAB3277 | BAB3302 | DECIPHER4155 |
|---|---|---|---|---|---|
| CNV | triplication | deletion | deletion | deletion | deletion |
| Inheritance | de novo | unknown (adopted child) | unknown | de novo | de novo |
| Size | 1.25 Mb | 0.157 Mb | 0.241 Mb | 0.94 Mb | 1.15 Mb |
| Gender | F | F | M | F | F |
| Age | 9 months | 12 years 6 months | 2 years 10 months | 7 years 11 months | 13 years |
| Cognitive/psych | GDD | GDD, nonverbal, profound ID, ASD, severe behavior disorder | GDD, nonverbal | GDD, ID, uses a few words | GDD, profound ID, nonverbal |
| Absolute/relative microcephaly | +/− | +/+ | +/+ | −/− | abnormal head shape |
| Dysmorphic facies | + | + | + | - | + |
| Cardiac | VSD, PDA | unremarkable | NA | NA | NA |
| MRI brain | NA | unremarkable | borderline to thin corpus callosum | delayed myelination | cerebral atrophy, reduced white matter, small cerebellum |
| Other | proximally flexed halluces, hypoplastic nails, unilateral transverse palmar crease, sensorineural hearing loss | dysplastic toenails, ligamentous laxity, inverted nipples, congenital entropion, bilateral exotropia | café au lait macule, esotropia | thoracolumbar scoliosis, small syrinx | joint laxity, multiple joint dislocation, myopia |
Abbreviations are as follows: NA, not available; GDD, global developmental delay; ID, intellectual disabilities; ASD, autism spectrum disorder; VSD, ventricular septal defect; PDA, patent ductus arteriosus.
We also studied further the rearrangements involving 17p13.1 in individuals DECIPHER2009 and DECIPHER2173 who were identified in a Brazilian survey of subjects with intellectual disability.8 From that cohort, we excluded DECIPHER2203 due to the presence of mosaicism in 50% of white blood cells, which could confound statistical analysis. To enlarge our sampling, we performed a literature search for clinical reports of individuals carrying delineated genomic alterations spanning 17p13.1. For instance, (1) samples that were analyzed by oligonucleotide array and/or had breakpoint junctions sequenced so that only those with genomic data comparable in resolution to our molecular data were utilized and (2) subjects for whom complete growth parameters (occipitofrontal head circumference, height, weight, and age at the time of clinical ascertainment) were available were included here for further analysis.
Clinical Laboratory Studies
Biological samples from subjects BAB3036, BAB3045, BAB3277, and BAB3302 were studied using chromosomal microarray analysis (CMA), GTG-banded chromosome analysis, and fluorescence in situ hybridization (FISH) at the Baylor College of Medicine Medical Genetics Laboratories as described.22 FISH to examine for the presence of deletion or duplication involving the 17p13.1 region was performed using BAC probes RP11-599B13 (test) and RP11-601N13 (control).
Head Circumference Analysis
Head circumference was measured as the maximal occipitofrontal head circumference (OFC), using standard techniques. Age- and sex-matched head circumference (OFC), weight, and height (HT) centiles were obtained using Abase, a PalmOS based calculator, and converted into Z scores. References for height, weight, and OFC were based on official standards for the United States/Canada populations.23 Absolute microcephaly was defined as OFC ≤ 1.88 SD below the mean for age and sex.24 Relative microcephaly was calculated by adjusting the OFC to the HT (zOFC-zHT).24,25 Analysis and graphing were implemented using GraphPad Prism 5.
Refining of CNV Size and Content
To determine size, genomic extent, and gene content for each rearrangement, we used a customized tiling-path oligonucleotide microarray spanning the entire 17p chromosome that was designed using the Agilent e-array website. We selected 42,000 probes covering chr17: 29,000–22,100,000 (hg18), which represents an average distribution of 1 probe per 526 bp. Probe labeling and hybridization were performed as described.25
Breakpoint Junction Sequencing and Analysis
Sample-specific primers for PCR were designed at the apparent boundaries of the deleted or amplified segments as inferred from the aCGH result marked by a transition from normal copy number to copy number loss or gain. Standard PCR reactions were performed using HotStartTaq Polymerase (QIAGEN), and long-range PCR reactions were performed using TaKaRa LA Taq (Clontech). Sequences of the primers that successfully produced PCR sample-specific bands are shown in Table S1. PCR products were sequenced by Sanger sequencing methodology.
Analysis of DNA sequencing was performed using UCSC Genome Browser; similarity analysis was performed using Needleman-Wunsch Global Sequence Alignment Tool.
Modeling Gene-Dosage Defects in Zebrafish Embryos
Capped human mRNA corresponding to the nine genes present within the defined SRO (ASGR1, DLG4 [MIM 602887], ACADVL, DVL2, GABARAP, CTDNEP1 [MIM 610684], CLDN7 [MIM 609131], SLC2A4 [MIM 138190], and EIF5A [MIM 600187]) was generated in vitro with the mMessage mMachine SP6 kit (Life Technologies) from linearized gateway compatible vector pCS2+ containing the cloned cDNA of interest as described.26 Capped mRNA (100 ng) was injected into 1- to 4-cell zebrafish embryos and scored for microcephaly at 4 dpf. At 4 dpf, embryos were fixed in 4% paraformaldehyde and head size was measured as the distance between eyes as described.27 Splice blocking morpholinos (sb-MOs) were designed and obtained from Gene Tools Inc.; the sequence of each MO is listed in Table S2; 0.5 nl of diluted MO corresponding to 3 ng was injected in zebrafish embryos at 1- to 4-cell stage and scored for microcephaly at 4 dpf. Graphs representing the probability distribution of head size measurements generated with the mean and standard deviation calculated from approximately 50 embryos from each condition. The Z score was calculated for the mean value for each condition compared to the mean and standard deviation of control embryos. To test the knockdown of gene expression, RNA was isolated using Trizol (Life Technologies) from zebrafish embryos injected with the morpholino at 1 dpf, cDNA prepared with Superscript III (Invitrogen), and RT-PCR carried out with primers listed in Table S3.
Immunohistochemistry of Zebrafish Embryos
Zebrafish embryos at 4 dpf were fixed in freshly prepared 4% paraformaldehyde for 6 hr and transferred to ice-cold methanol for 24 hr. The embryos were rehydrated to 100% PBS by a step-wise increasing concentration of PBS with 5 min incubations followed by bleaching in 3% hydrogen peroxide and 0.5% potassium hydroxide in PBS for 30 min. Embryos were blocked in 2% BSA and 10% FBS in PBS for 30 min and incubated with anti-phospho Histone H3 (Santa Cruz Biotechnology) at a dilution of 1:750 overnight at 4°C and secondary anti-rabbit Alexa488 at room temperature for 30 min. Both antibody incubations were followed by three washes in PBS-Tween. Zebrafish embryos were visualized using Nikon AZ100 microscope and representative images were generated by capturing a z stack of each embryo and merging them. The total numbers of phospho-Histone H3-positive cells were counted by ImageJ software using a cell counter plugin. The number of phospho-histone-positive cells was calculated by subtracting the number of cells in the eye from the total number of cells obtained by tracing the head of the embryos. TUNEL assays were carried out in 3 dpf embryos. In brief, zebrafish embryos injected with 3 ng of MO were fixed in 4% PFA, bleached, treated with proteinase K for 5 min, and post-fixed again in 4% PFA. The TUNEL staining kit (ApopTag, Millipore) was used as per manufacturer’s protocol; embryos were visualized with Nikon AZ100. Images were processed in Adobe Photoshop and analyses were carried out using ImageJ.
Cell Culture, shRNA, Transfection, and Cell Cycle Analysis
Neuro2A cells were grown in 10% FBS, DMEM medium. To test the expression of the nine genes in the SRO, RT-PCR was carried out from RNA isolated from Neuro2A cells as described above, with primers listed in Table S4. To analyze the knockdown efficiency of each expressed gene, four to five different shRNAs expressing pLKO.1 vector for each gene (Table S5) were obtained from Sigma. Neuro2A cells were transfected with either nontargeting scrambled siRNA (Dharmacon) or a pool consisting of four targets of siRNA against Asgr1 (Dharmacon) at a final concentration of 20 nM with Dharmafect I (Dharmacon). The remaining seven genes in the SRO were knocked down by transfecting a pool of 0.5 μg of each pLKO.1 vector expressing shRNA targeting the gene of interest with Dharmafect I (Dharmacon) as per manufacturer’s protocol. The siRNA and the shRNA sequences used for the knockdown experiments have been listed in Table S5. At 48 hr after transfection, RNA was isolated and real-time PCR analyses were carried out to test the knockdown efficiency with primers listed in Table S6. Cell cycle analysis was carried out by transfecting pLKO.1 shRNA constructs as described above; 72 hr after transfection, cells were trypsinized, resuspended in growth medium, washed in PBS, and fixed in 4% PFA for 10 min with 0.1% TX-100 and propidium iodide (10 μg/ml) in PBS. The staining procedure was carried out on ice. Cell cycle profile was analyzed by flow cytometry; we calculated the percentage of S/G2/M versus G0/G1 in a population of 10,000 cells.
Results
High-Resolution aCGH and Breakpoint Junction Sequence Analysis
We studied the 17p13.1 molecular rearrangement of seven subjects (BAB3036, BAB3045, BAB3277, BAB3302, DECIPHER2009, DECIPHER2173, and DECIPHER4155) with rearrangements varying in size from ∼157 kb to 2.7 Mb; five subjects (BAB3036, BAB3045, BAB3277, BAB3302, and DECIPHER4155) are being reported for the first time. Individual BAB3036 carries the smallest deletion (157 kb) reported thus far for the 17p13.1 microdeletion syndrome (Figures 1 and 2A). Six affected individuals carry nonrecurrent copy-number losses. One subject (BAB3045) presents with a copy-number gain encompassing ∼1.25 Mb; normalized log2 (Cy5/Cy3) ratio of the CGH signal reached mean thresholds of +1.0 indicating copy-number state (CN) = 4 consistent with a triplication (Figure 1B). The presence of four red signals corresponding to the tested 17p13.1 region in interphase cells supports the presence of a triplicated segment by FISH (Figure S1 available online).
Figure 1.
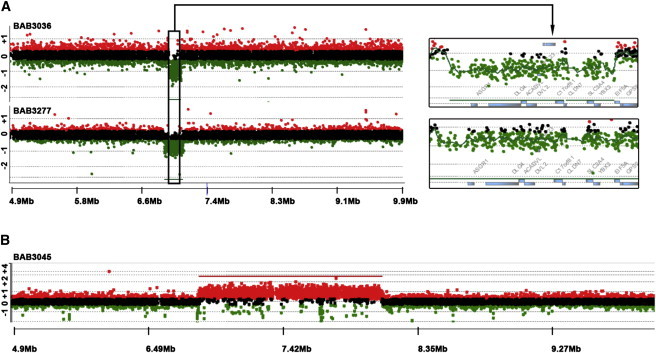
High-Resolution aCGH Plot for Individuals BAB3036, BAB3277, and BAB3045
(A) Individuals with small deletions of 17p13.1 region. Black box delimits the smallest region of overlap deleted in subjects with microcephaly.
(B) Individual with triplication involving 17p13.1 region.
CNVs were detected by oligonucleotide probes for which the mean normalized log2 (Cy5/Cy3) ratio of the CGH signal reached mean thresholds of −1 (indicating a heterozygous deletion [CN = 1]), 0.6 (indicating a duplication [CN = 3]), or 1.0 (indicating a triplication [CN = 4]).
Figure 2.
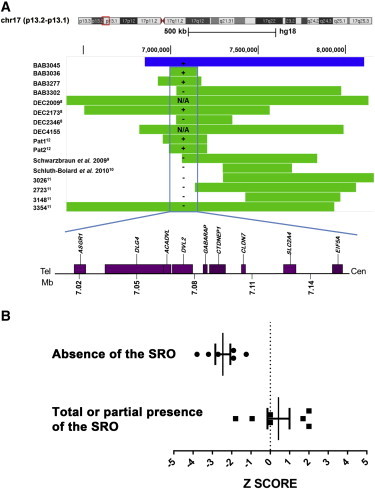
Representative Summary of CNVs Involving 17p13.1
(A) Top: Individuals with deletions are represented by green rectangles and the individual with a triplication is represented by blue rectangle. The graphical normalized data for each individual was obtained by applying the most distal and proximal oligonucleotide genomic probe coordinates to the custom track at UCSC Genome Browser website. Positions are given relative to build hg18. Vertical blue rectangle delimits the smallest region of overlap (SRO) for the microcephaly phenotype in this cohort. Plus sign indicates presence of absolute microcephaly; minus sign indicates absence of absolute microcephaly (refer to Table 2 for Z score values). N/A indicates information not available. Bottom: Dosage-sensitive region associated with small head size (157 kb) chr17: 6,996,378–7,152,828 (hg18).
(B) Graph of Z scores of head circumferences in subjects with 17p13.1 deletions. Black dots: individual Z score of subjects who harbor CNVs encompassing entirely predefined SRO of 157 kb. Black squares: individual Z scores of subjects who harbor CNVs that do not encompass predefined SRO or encompass it partially. All measurements are plotted as age- and sex-matched Z scores. Bars indicate mean and 95% confidence intervals.
We designed a variety of primer pairs to obtain the breakpoint junction for each of the nonrecurrent 17p13.1 rearrangements. We sequenced the junction fragment of deletions present in four individuals (BAB3036, BAB3277, DECIPHER2009, and DECIPHER2173; Figure S2). In two cases, BAB3277 and DECIPHER2173, deletions were mediated by repetitive elements. The deletion in BAB3277 involved a pair of directly oriented LTR13A located 250 kb apart; each of those LTRs spans ∼963 nt and shares 96% of nucleotide sequence similarity. The deletion in DECIPHER2173 was mediated by two directly oriented Alu elements, constituents of different families (AluSg and AluY) and located 1 Mb apart in the haploid reference genome; they share 82% nucleotide sequence identity. The deletion observed in DECIPHER2009 also occurred between two repetitive elements (L1PA13 and AluJb), but there is no significant nucleotide similarity between them; microhomology is not apparent at the junction, which is characterized by a CCT insertion of unknown origin. BAB3036 presents a complex breakpoint junction that incorporated 76 nt of an AluY element located 760 kb telomeric to the rearrangement. In addition, two point mutations within that Alu element are present: a G>A at the breakpoint junction and a G>C located 19 nt away from the junction (Figure S2). Mutation G>A is not present in dbSNP; therefore, it can be either a rare mutation present in the ancestral chromosome from which the 76 nt insertion originated or it can be a mutation generated concomitantly with the formation of the genomic rearrangement;28 G>C is deemed to be a single-nucleotide polymorphism (rs9894826). There is no apparent microhomology at the breakpoint junctions of that Alu insertion, which may be concealing polymerase slippages in both distal and proximal strands upon the Alu insertion (Figure S2). Replication-based mechanisms for formation of genomic rearrangements can be error prone and polymerase slippage can be observed at or near breakpoint junctions.28–30
Small Head Size Associated with Rearrangements Involving a Specific 157 kb Region
Comparative analysis of the gene content in subjects reported here and in the literature narrowed the dosage-sensitive SRO associated with a reduced head size or microcephaly to 157 kb, spanning chr17: 6,996,378–7,152,828 (Figure 2, Table 2); six individuals presented with absolute microcephaly (see Subjects and Methods for definitions), including BAB3045, who carries a triplication. Remarkably, the mean Z score for the head circumference (mean zOFC) of those individuals whose deletions include the SRO is −2.46 ± 0.39 (mean ± SEM). In contrast, in subjects carrying CNVs that do not encompass the SRO, we observed a greater variation in the head size with a mean zOFC of +0.42 ± 0.57. Comparison of the two groups, using an unpaired two-tailed t test with Welch’s correction for unequal variances, showed that they differ significantly (p = 0.0018). These data strongly suggest that the genomic SRO is significantly associated with microcephaly (Figure 2B), arguing that dosage-sensitive genes inside the genetically refined and mapped SRO contribute to head size development.
Table 2.
Growth Parameters for Subjects with Rearrangements at 17p13.1
| Subjects | Gender | Age | OFC (Z) | HT (Z) | WT (Z) | zOFC-zHT | Breakpoint Junctions Chr17 (hg18) | CNV |
|---|---|---|---|---|---|---|---|---|
| BAB3045 | F | 9 months | −3.1∗ | −4.2 | −5 | +1.1 | 6,853,665–8,107,394 | TRP |
| BAB3036 | F | 12 years 6 months | −3.8∗ | −1.25 | +2.5 | −2.55∗ | 6,996,378–7,152,828a | DEL |
| BAB3277 | M | 2 years 10 months | −2.7∗ | −0.65 | +0.7 | −2.05∗ | 6,936,148–7,177,360 a | DEL |
| BAB3302 | F | 7 years 11 months | 0 | −0.45 | +0.7 | +0.45 | 7,033,635–7,977,678 | DEL |
| DECIPHER20098 | M | NA | NA | NA | NA | NA | 5,415,715–8,132,381 a | DEL |
| DECIPHER21738 | M | 8 years 8 months | −1.9∗ | −0.94 | −0.8 | −0.96 | 6,512,788–7,567,293 a | DEL |
| DECIPHER23468 | F | 5 years | −1.8 | −1.8 | −1.8 | 0 | 7,033,251–7,354,034 | DEL |
| DECIPHER4155 | F | 13 years | NA | NA | NA | NA | 6,838,295–7,991,364 | DEL |
| Pt 112 | M | 2 years | −1.9∗ | −0.1 | −1 | −1.8 | 6,957,207–7,209,585 | DEL |
| Pt 212 | F | 8 years | −3.2∗ | −0.5 | −1.5 | −2.7∗ | 6,991,049–7,209,612 | DEL |
| Schwarzbraun et al.9 | F | at term birth | +1.7 | +1 | +1.7 | 0 | 7,066,210–7,840,187 | DEL |
| Schluth-Bolard et al.10 | M | at term birth | +2 | +0.95 | +0.95 | +1.05 | 7,299,400–7,694,600 | DEL |
| 302611 | F | 33 years | 0 | −2 | −2 | +2 | 7,300,398–8,273,016 | DEL |
| 272311 | F | 7 years 7 months | −0.94 | −0.94 | +0.90 | 0 | 7,140,464–8,061,771 | DEL |
| 314811 | F | 5 years 9 months | +2 | −1.25 | +1.25 | +3.25 | 7,429,371–7,972,019 | DEL |
| 335411 | M | 3 years 6 months | −1.25 | +0.5 | +0.5 | −1.75 | 5,500,927–7,937,620 | DEL |
Abbreviations are as follows: OFC, occipito-frontal head circumference; HT, height; WT, weight; zOFC-zHT, Z score after adjusting the OFC to the HT; DEL: deletion; TRP: triplication. Asterisk (∗) indicates values that fit absolute microcephaly (zOFC ≤ −1.88) and relative microcephaly (zOFC-zHT ≤ −1.88).
Breakpoint junction coordinates based on Sanger sequencing data.
In Vivo Testing of Potential Microcephaly Drivers
To identify dosage-sensitive genes that might contribute to microcephaly within the predefined SRO, we used overexpression and suppression of the human genes in zebrafish embryos. Recent studies have shown that the systematic assessment of dosage-sensitive genes within a CNV using a surrogate head size assay in zebrafish embryos can potentially inform the contribution of transcripts within the CNV to head size defects in individuals with microcephaly.16,27 In contrast to prior studies on the 16p11.2 CNV, in which duplication and deletion produce mirror microcephalic and macrocephalic phenotypes, respectively,25,27 both gain and loss of genetic material within the SRO on 17p13.1 is associated with absolute microcephaly (OFC ≤ 1.88 SD; Tables 1 and 2). As a first test, we injected 100 pg of capped human mRNA for each of the nine human genes in the SRO into ∼100 embryos at the 1- to 4-cell stage. We then measured the distance between the eyes as a surrogate for the human microcephaly trait25,27 at 4 days postfertilization (4 dpf), a time assessed to be optimal for fish head size scoring.26 We set a Z score of ≤−1.88 as a minimum criterion for microcephaly to mirror the clinical criteria in humans. We found that overexpression of 7/9 genes (except DLG4 and SLC2A4) induced significant microcephaly (Figures 3A–3C); importantly, we did not observe any significant changes in either body length, number of somites, or the development of the swim bladder, supporting the contention that this phenotype was unlikely to be the result of overall developmental delay.
Figure 3.
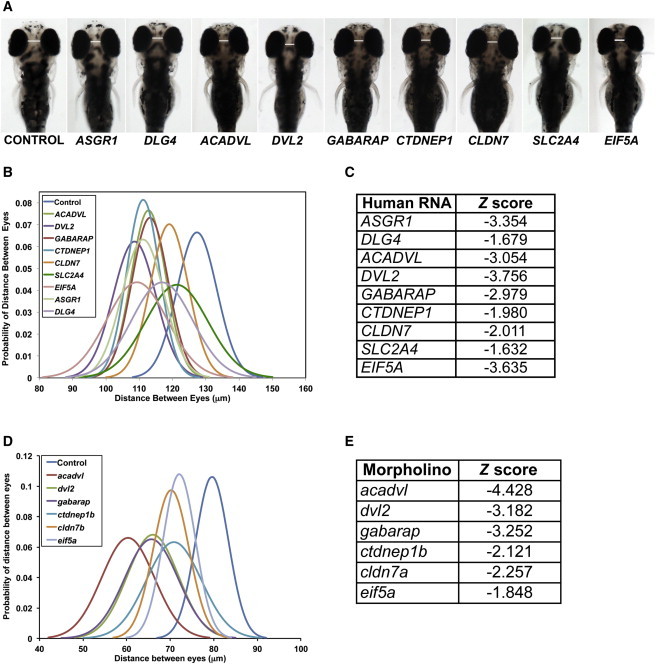
Delimited 17p13.1 Region Harbors Dosage-Sensitive Genes as Experimentally Assayed in Zebrafish
(A) Overexpression of capped human mRNA corresponding to nine loci in the 17p13.1 region in zebrafish embryos leads to microcephaly at 4 dpf. Representative dorsal images of zebrafish embryos injected with the indicated human mRNA scored for microcephaly at 4 dpf.
(B) Graph represents the probability distribution curve of distance between eyes in zebrafish embryos upon overexpression injection of individual mRNAs.
(C) List of individual capped human mRNAs and corresponding Z score measured as distance between eyes in zebrafish at 4 dpf (B).
(D) Graph represents the probability distribution curve of distance between eyes in zebrafish embryos injected with splice blocker morpholinos of the indicated genes and scored for microcephaly at 4 dpf.
(E) The corresponding Z scores of the probability distribution curves in (D).
Next, we asked whether the seven genes that induced microcephaly when overexpressed could also give rise to the same phenotype when suppressed. Reciprocal BLAST of the zebrafish genome identified a direct ortholog for 6/7 genes. ASGR1 could not be detected in the zebrafish genome and thus could not be modeled. Two other genes, CTDNEP1 and CLDN7, have two copies in the zebrafish genome; we focused on the respective paralogs with the highest identity to the human protein, namely ctdnep1b and cldn7a. For each available zebrafish orthologous gene, we first tested the expression during development by analysis of our in-house RNAseq data from 3 dpf and 5 dpf wild-type embryos. We detected 5–20 copies per million for each of the six genes, with cldn7a expressed at the lowest level among this gene set (5 copies/million); we therefore proceeded to design splice-blocking morpholinos (sbMOs), which we then titrated to establish optimal dosage, based on monitoring efficiency (by RT-PCR) and on scoring for toxicity and nonspecific phenotypes such as cardiac edema and gross developmental delay (Figure S3, Tables S2 and S3).
We then injected each sbMO and tested for microcephaly using the same criteria as our overexpression studies. Suppression of 5/6 genes (acavdl, cldn7a, ctdnep1, dvl2, and gabarap) induced significant microcephaly (Z < −2.0), whereas eif5a was marginal (Z −1.848; Figures 3D and 3E). Three genes in particular (acadvl, dvl2, and gabarap) had the most striking phenotype (Z scores between −3.1 and −4.4). The phenotype was dose dependent and significant pathologies were not observed in other organs that might suggest nonspecific effects. We attempted rescue experiments for the three genes associated with the strongest phenotypes with human mRNA. Dvl2 was intractable due to the severe phenotype induced by the mRNA itself. For the other two genes, head size defects were rescued (Figure S4). As an additional measure of specificity, we suppressed two genes outside the SRO, GPS2 and CD68, both of which are expressed during early zebrafish development and have a single direct ortholog; we did not observe microcephaly at three different MO injection concentrations (Figure S5).
Previous studies have shown that head size defects are associated with a reduced number of dividing cells in the developing brain of zebrafish embryos.27,31 To ask whether this was the case here, we stained embryos suppressed for each of acadvl, dvl2, and gabarap (the three genes yielding the most severe phenotypes) with anti-phospho-histone H3, a marker of dividing cells. Counting of pH3-positive cells in 10–20 embryos masked to injection cocktail showed a significant reduction for each of acadvl, dvl2, and gabarap (Figures 4A and 4B). To test whether these changes were a result of apoptosis, we performed TUNEL staining on the embryos suppressed for those same three loci. Suppression of both acadvl and dvl2 MOs resulted in increased apoptosis as quantified by comparing TUNEL-positive cells between morphants and controls; likewise, suppression of gabarap induced a comparatively more modest but still significant increase (Figures 4C and 4D).
Figure 4.
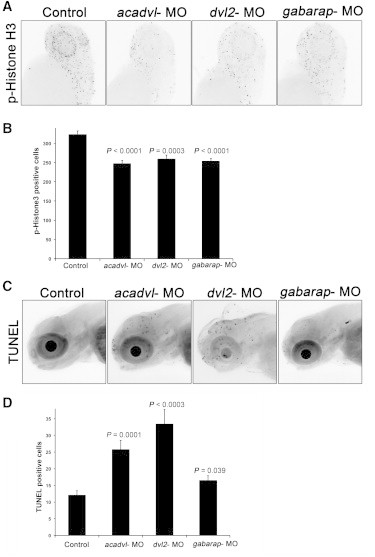
Loss of the Primary Drivers of Microcephaly Lead to Reduced Cell Division in Zebrafish
(A) Zebrafish embryos were injected with the morpholinos as indicated and stained for phospho-histone H3 at 2 dpf. Representative images are shown for control and morpholino suppression. The dashed lines in the controls indicate the regions used for calculating the number of histone-positive cells.
(B) Graph represents average and standard error of histone-positive cells in each sample from 20 embryos.
(C) Representative images of TUNEL staining of 3 dpf zebrafish embryos either uninjected or injected with 3 ng of acadvl, dvl2, or gabarap MO.
(D) Graph represents the average number of TUNEL-positive cells and standard error for each condition.
Intrigued by this observation, and mindful of the caution necessary when interpreting zebrafish data in the context of mammalian pathology, we asked whether the changes in the phospho-Histone H3 and TUNEL staining observed in the developing brain might be reflective of mitotic defects in mammalian neuronal cells. We therefore tested for possible effects of RNAi-mediated silencing of the genes in the SRO on the mitotic index of Neuro2A cells, which partially retain the characteristics of neurons.32 RT-PCR detected message for 8/9 genes in the SRO; we could not detect Cldn7a expression (Figure S6). The siRNA pool targeting asgr1 and the predesigned and cloned shRNA targeting the remaining seven genes in the SRO were able to efficiently knockdown the expression of all eight genes by real-time qPCR, 48 hr posttransfection (Figure S7). Subsequently, when we performed cell cycle analysis 3 days posttransfection, when cells were targeted with shRNA against Acadvl, Dvl2, and Gabarap (the three genes shown in our in vivo studies to give rise to severe microcephaly), significant reduction in the percentage of cells at S/G2/M phases of the cell cycle was observed; Asgr1, which was intractable to in vivo suppression, gave a similar pattern (Figure 5), whereas the remaining testable genes not associated with overt microcephaly did not exhibit obvious cell cycle alterations.
Figure 5.
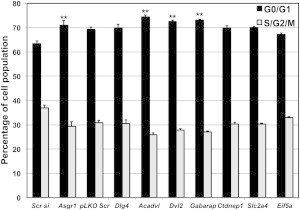
Loss of the Primary Drivers Result in Decreased S/G2/M Population Neuro2A Cells
Neuro2A cells were transfected with a pool of four different siRNAs targeting Asgr1 and compared with cells transfected with scrambled siRNA (Scr si). The remaining seven genes were suppressed with a combination of five shRNAs in pLKO.1 vector for each gene and compared against the control cells transfected with scrambled shRNA (pLKO.1 scr). Three days after transfection, cells were harvested, fixed, permeabilized, and stained with propidium iodide (PI). Cell cycle analysis was carried by flow cytometry and the average percentage of G0/G1 and S/G2/M population of each condition from three experiments is represented and the error bars indicate the standard deviation. ∗∗p < 0.005.
Genetic Interaction between Loci within the SRO
Taken together, our overexpression and suppression data in zebrafish embryos and the cell cycle phenotypes in mammalian neuronal cells intimated that dosage perturbation of multiple genes within the 17p13.1 predefined human SRO could induce head size defects of a magnitude analogous to what was observed in the subjects reported here. This raised the question whether these genes might each be sufficient to induce the phenotype, or whether there might be potential gene-gene interactions within this CNV. We therefore performed pairwise interaction experiments with the six genes that gave tractable, reproducible microcephaly phenotypes in either overexpression or suppression assays in zebrafish embryos (acadvl, dvl2, gabarap, cldn7a, ctdnep1b, and eif5a). Each transcript was titrated so that subeffective injection of each single MO did not yield microcephaly; we then added MOs together and scored 50–100 embryos per condition, masked to injection cocktail. Genetic interactions that were unique for each transcript were observed (Table 3). For example, subeffective cosuppression of acadvl and dvl2, which by themselves yielded Z scores > −1.0, induced Z scores of −1.91, possibly indicative of an additive effect, whereas acadvl+gabarap gave rise to Z scores of −3.5, suggesting a multiplicative effect. By contrast, acadvl+ctdnep1b gave no appreciable phenotype (Z −0.06), whereas ctdnep1b+dvl2 gave the most severe phenotype (Z −5.0).
Table 3.
Analysis of Pairwise Genetic Interactions for Microcephaly Reveal Specific Loci that Sensitize the Phenotype in Zebrafish Embryos
| Gene 1 | Gene 2 | Z Score |
|---|---|---|
| acadvl | – | −0.98 |
| cldn7a | – | −0.75 |
| ctdnep1b | – | −0.35 |
| dvl2 | – | −0.41 |
| eif5a | – | −0.62 |
| gabarap | – | 0.61 |
| acadvl | cldn7a | −0.85 |
| acadvl | ctdnep1b | −0.06 |
| acadvl | dvl2 | −1.91∗ |
| acadvl | eif5a | −0.07 |
| acadvl | gabarap | −3.5∗ |
| cldn7a | ctdnep1b | −1.6 |
| cldn7a | dvl2 | −2.02∗ |
| cldn7a | eif5a | −1.3 |
| cldn7a | gabarap | −3.5∗ |
| ctdnep1b | dvl2 | −5.0∗ |
| ctdnep1b | eif5a | −1.3 |
| ctdnep1b | gabarap | −4.2∗ |
| dvl2 | eif5a | −1.1 |
| dvl2 | gabarap | −1.1 |
| eif5a | gabarap | −0.8 |
Zebrafish embryos were injected with suboptimal doses (1 ng) of morpholino and 50–100 embryos were scored at 4 dpf for the distance between eyes. The Z scores represent the difference in distance between eyes with respect to the control population; asterisk (∗) indicates Z scores demonstrating interaction that lead to microcephaly.
Discussion
Microcephaly can have an environmental or genetic etiology. Impaired neurogenesis seems to be the main underlying cause and includes defects in the mitotic apparatus, extended or shortened cell cycle that causes disturbance of the delicate symmetric-asymmetric division balance, defects in DNA replication or genome maintenance, and elevated rate of cell apoptosis (reviewed in Alcantara and O’Driscoll33). Over the last two decades several genetic disorders have been reported, the majority of which include microcephaly as one of the main clinical phenotypes. This observation supports the contention that microcephaly can be an indication of perturbed cell maintenance and that important gene(s) product(s) or cell processes are profoundly deregulated.
Here we present clinical and molecular studies of five individuals with nonrecurrent copy-number variation encompassing the 17p13.1 region; their core clinical phenotype includes microcephaly, dysmorphic features, intellectual disability, and global developmental delay. Four out of five subjects present 17p13.1 copy-number loss whereas one subject presents 17p13.1 copy-number gain consistent with triplication. Breakpoint analysis and clinical assessment of newly identified subjects, combined with published cases, identified a minimal dosage-sensitive region of ∼157 kb that is likely the major driver of head size defects for CNVs involving 17p13.1. This region contains nine annotated genes, which were further modeled in vitro and in vivo. Those assays revealed that as many as seven dosage-sensitive transcripts exist within the SRO segment that likely contribute to the microcephaly phenotype: ASGR1, ACADVL, DVL2, GABARAP, CTDNEP1, CLDN7, and EIF5A. Among these, our in vivo modeling data intimate at least three major genes, ACADVL, DVL2, and GABARAP, as the primary drivers of the microcephaly phenotype; overexpression in zebrafish and in vitro modeling indicates that ASGR1 might be a fourth one. In fact, the silencing of all four orthologous genes in a model of mammalian neuronal cells led to reduced mitotic index. Further TUNEL assay of cells derived from developing embryos knocked down for acadvl, dvl2, and gabarap shown an increased number of apoptotic cells.
These findings are likely to indicate the pathomechanism(s) that drive clinical pathology in subjects carrying this CNV. ASGR1 encodes the most abundant major subunit of the hetero-oligomeric asialoglycoprotein receptor (ASGPR). The asialoglycoprotein receptor is the major lectin of hepatocytes, an integral membrane protein that mediates endocytosis and lysosomal degradation of glycoproteins with exposed terminal galactose or N-acetylgalactosamine residues.34 There is no obvious link between dosage changes in ASGR1 and brain size, but our analysis of RNA expression data of ASGR1 from 20 neuroanatomic human brain regions available at the Allen Human Brain Atlas indicates that this gene is expressed in several brain regions. Furthermore, ASGR1 shows expression in fetal brain (Human Gene Expression Atlas 2, Genomics Institute of the Novartis Research Foundation). Remarkably, mutations in STAMBP (MIM 606247) that lead to diminished expression of the encoded protein deubiquinating (DUB) isopeptidase STAMBP cause microcephaly-capillary malformation syndrome (MIC-CAP [MIM 614261]);35 STAMBP has a key role in the regulation of cell surface receptor-mediated endocytosis and ubiquitin-dependent sorting of receptors to lysosomes, thereby providing a precedent for dysregulation of the endocytosis pathway in the causation of severe microcephaly.35 ACADVL encodes very long chain acyl CoA dehydrogenase (VLCAD), an inner mitochondrial membrane protein involved in fatty acid β-oxidation pathway.36 Recessive mutations in ACADVL result in VLCAD deficiency (MIM 201475), a disease characterized by cardiomyopathy due the dependence of energy of cardiomyocytes on fatty acid oxidation, and rare cases of neonatal and early childhood pathogenesis have resulted in death.37 Obligate carriers of ACADVL mutations do not present microcephaly and although there is a minimal dependence of neurons on fatty acid oxidation as an energy resource, it is intriguing that 1% to 5% of individuals with autism spectrum disorders have mitochondrial disease, highlighting the potential significance of mitochondrial function in brain physiology.38 DVL2 is a component of the planar cell polarity pathway (β-catenin-independent Wnt signaling). Loss of Dvl2−/− in mice leads to significant lethality with increased neural crest defects.39 Of interest, the expression of DVL2 is increased significantly in subjects with glioblastoma multiforme (GBM), a common form of malignant glioma in adults, suggesting a role for DVL2 in uncontrolled self-renewal of neural progenitor cells. The depletion of DVL2 in glioblastoma cell lines induced mitotic arrest and differentiation in vitro and inhibited the formation of tumors in immunodeficient mice due to intracranial injection of glioblastoma cells.40 Finally, GABARAP is a Type A γ-aminobutyric acid receptor (GABAAR)-associated protein that binds the γ2 subunit of the receptor.41 These receptors belong to ligand-gated ion channel family and are activated by GABA released by inhibitory axonal terminals (reviewed in Fritschy and Brunig42). GABARAP is involved in GABAA receptor trafficking43 and increases channel conductance44 and clustering on the membrane.45 In the context of the observed defects in cell cycle profiles and increased apoptosis in the zebrafish developing brain, we do not yet know the precise contribution of the three major driver genes but the contribution of dosage imbalance of each transcript/protein will likely be different. For example, Dvl2 is involved in the mitotic spindle rotation, attachment to the kinetochore, and spindle assembly checkpoint.46 By contrast, GABARAP is an integral part of the autophagosome compartment and recently has been shown to interact with the antiapoptotic protein Bcl-2.47 Although further studies are required to delineate the functional significance of this interaction, it is possible that our increased TUNEL-positive cells observed in embryos targeted with gabarap MO might suggest a deregulation of the apoptotic-autophagy pathway in the brain. Finally, suppression of the enzyme activity of ACADVL results in increased generation of reactive oxygen species (ROS).48 Therefore, the increased apoptosis observed due to the loss of acadvl might be a result of ROS generation.
Komoike et al.49 analyzed an individual with intellectual disability, intractable epilepsy, and dysmorphic features who carried a 2.3 Mb deletion in the 17p13.2p13.1 region that overlaps with the previously identified ∼180 kb SRO.49 Based on these data, DLG4 and GABARAP were tested as potential dosage-sensitive genes associated with microcephaly by morpholino suppression of the orthologs in zebrafish. Suppression of dlg4 did not yield any appreciable phenotype, whereas gabarap MO resulted in microcephaly and hypoplastic mandible in 3 dpf zebrafish embryos, a result fully concordant with our studies. These results increase the confidence in our data; in our unbiased approach we eliminated DLG4 as a possible candidate for microcephaly, whereas suppression of GABARAP resulted in severe microcephaly and constituted one of the sensitizing loci for this phenotype.
CNVs associated with abnormal head size were reported previously in genomic disorders such as those involving 16p11.2 and 1q21.1;25,50 interestingly, deletions and duplication involving those loci lead to mirror phenotypes (i.e., microcephaly versus macrocephaly). Nonetheless, triplication encompassing the 17p13.1 region did not lead to a mirror trait (i.e., macrocephaly) in the subject reported here; in fact, BAB3045 presented with absolute microcephaly (Z score −3.1) but normocephaly when head circumference was adjusted to the height (zOFC-zHT +1.1). Two subjects carrying 17p13.1 deletions, DECIPHER2173 and one individual described by Zeesman et al.,12 also presented with absolute microcephaly, which was less significant when adjusted to body height, suggesting that subjects with copy-number alterations involving the 17p13.1 might present with small body size (Table 2) in some cases. Our in vivo studies indicate that apoptosis might play an important role as the underlying disease mechanism for this CNV. Interestingly, the list of genes that can cause microcephaly due to excessive apoptosis when mutated in humans seems to be growing fast. Individuals with homozygous mutation affecting the RNA kinase involved in tRNA splicing, CLP1 (MIM 608757), show brain malformation and microcephaly; and neuronal progenitor cells from CLP1 kinase-defective mice showed increased cell death despite normal cell proliferation.51 Recently, dAnkle2, the Drosophila ortholog of human ANKLE2, was identified in an X chromosome forward screen for fly lethal recessive mutations and further observed in a homozygous state in a subject with very severe microcephaly and brain dysgenesis.52
Furthermore, our data indicate that microcephaly associated with CNV spanning the 17p13.1 is subject to variable expression (Tables 1 and 2), suggesting that genes present in that SRO are potentially affected by epistasis, a hypothesis tested directly in zebrafish embryos. Our analyses revealed discrete pairing of genes that can interact to drive microcephaly and suggest that the clinical pathology is more likely to be driven by contributory effects of each of seven genes (Figure 6). Therefore, for this CNV, we propose a model in which epistasis within the genomic region modulates the clinical presentation, as opposed to a model where each gene contributes to a discrete component of the syndromic phenotype.
Figure 6.
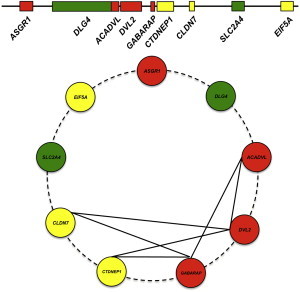
Schematic Model for Dosage-Sensitive Region at 17p13.1 Associated with Small Head Size in Human
Top: Model representing loci that leads to severe (red), moderate (yellow), and no scored phenotype (green).
Bottom: Summary of the binary genetic interactions tested between loci for microcephaly in zebrafish embryos.
Most 17p13.1 deletions are deemed to arise by recombination between repetitive elements, especially Alu-Alu elements, in direct orientation. A previous study reported that 67% of deletions involving that region originated by recombination between Alu elements sharing 81%–84% of nucleotide similarity.11 In our cohort, rearrangements generated by recombination between repetitive elements predominate: repetitive elements were present at both breakpoints in three out of four deletions (75%) that had breakpoints sequenced; two out of four (50%) deletions were mediated by these elements (DECIPHER2173, AluSg-AluY with 82% of nucleotide similarity and BAB3277, LTR13A-LTR13A with 96% of nucleotide similarity). Deletions mediated by repetitive elements have been observed for a number of genomic disorder-associated loci, often nonrecurrent rearrangements: for example, deletions observed in rare pathogenic CNVs,53 deletions involving FOXL2 (MIM 605597) causing blepharophimosis-ptosis-epicanthus inversus syndrome (BPES [MIM 110100]),54 deletions leading to autosomal-dominant spastic paraplegia-4 (SPG4 [MIM 182601]),55 although recurrent rearrangements are also observed, as the 3.4 Mb HERV-H-mediated 3q13.2-q13.31 deletions that lead to a syndrome marked by motor and language delay,56 and the HERV-I-mediated AZFa deletion that causes Y-linked spermatogenic failure (SPGFY2 [MIM 415000]).57,58 In addition to repetitive-element-mediated events, the complex rearrangement including the segmental insertion of 76 nt from a nearby AluY element in BAB3036 suggests that replication-based mechanisms, such as fork stalling and template switching (FoSTeS) or microhomology-mediated-break-induced replication (MMBIR),28,59,60 also underlie rearrangements involving 17p13.1.
In conclusion, our data support an oligogenic model for microcephaly driven by dosage alteration of four major genes located in a small region of 17p13.1. This contrasts sharply with the model originally proposed by Roy Schmickel of “contiguous gene deletion syndrome,” wherein each dosage-sensitive gene contributes independently to the development of a particular clinical phenotype. These new data add further complexity to the comprehension of phenotypic effects of CNVs in humans. We recognize that our study is limited by our inability to perturb reliably more than two genes at a time and we cannot exclude the possibility that more complex interactions exert an effect. Likewise, we do not have the dynamic range to determine whether these interactions are additive or multiplicative, nor did we test for potential positional effects of genes mapping outside the SRO. Additionally, it is possible that other genes encompassed by larger 17p13.1 CNVs or even located elsewhere in the genome are relevant to this phenotype; in fact, modifier genes likely have a role in defining penetrance because our data indicate that lack of the SRO 157 kb segment is necessary but not enough to lead to microcephaly (Figure 2). Further, we are also cautious in interpreting functional data from model organisms as surrogates of clinical phenotypes in humans, especially because, for both our in vitro and in vivo systems, it is not possible to recapitulate accurately the haploinsufficient genotype found in humans; indeed, our suppression studies likely exceed haploinsufficiency and thus exaggerate the phenotypic impact of each gene within this CNV. Nonetheless, the zebrafish model has been shown to be useful in recapitulating microcephaly,27,31,49 an observation consistent with the analogous Z scores seen in embryos and in subjects carrying 17p13.1 CNVs.
Acknowledgments
We thank the families for their participation in the study. This work was supported in part by US National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS058529 to J.R.L. and 5K08NS062711 to M.B.R.; US National Human Genome Research Institute/National Heart Blood and Lung Institute (NHGRI/NHBLI) grant U54HG006542 to J.R.L.; by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), a Young Investigator fellowship from the Science without Borders Program grant 402520/2012-2 to C.M.B.C.; and a grant from the Simons Foundation to N.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NINDS, NHGRI, NIH, or CNPq. J.R.L. holds stock ownership in 23andMe, Inc., and is a coinventor on multiple United States and European patents related to molecular diagnostics. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Medical Genetics Laboratories.
Contributor Information
Nicholas Katsanis, Email: nicholas.katsanis@dm.duke.edu.
James R. Lupski, Email: jlupski@bcm.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent eArray, https://earray.chem.agilent.com
Allen Human Brain Atlas, http://human.brain-map.org/
Baylor College of Medicine Medical Genetics Laboratories, https://www.bcm.edu/research/medical-genetics-labs/
Database of Genomic Variants (DGV), http://dgv.tcag.ca/dgv/app/home
DECIPHER, http://decipher.sanger.ac.uk/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
Arrays have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE62004.
References
- 1.Bi W., Sapir T., Shchelochkov O.A., Zhang F., Withers M.A., Hunter J.V., Levy T., Shinder V., Peiffer D.A., Gunderson K.L. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.vanTuinen P., Dobyns W.B., Rich D.C., Summers K.M., Robinson T.J., Nakamura Y., Ledbetter D.H. Molecular detection of microscopic and submicroscopic deletions associated with Miller-Dieker syndrome. Am. J. Hum. Genet. 1988;43:587–596. [PMC free article] [PubMed] [Google Scholar]
- 3.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 4.Chance P.F., Alderson M.K., Leppig K.A., Lensch M.W., Matsunami N., Smith B., Swanson P.D., Odelberg S.J., Disteche C.M., Bird T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 5.Potocki L., Bi W., Treadwell-Deering D., Carvalho C.M., Eifert A., Friedman E.M., Glaze D., Krull K., Lee J.A., Lewis R.A. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen K.S., Manian P., Koeuth T., Potocki L., Zhao Q., Chinault A.C., Lee C.C., Lupski J.R. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 7.Adam M.P., Justice A.N., Schelley S., Kwan A., Hudgins L., Martin C.L. Clinical utility of array comparative genomic hybridization: uncovering tumor susceptibility in individuals with developmental delay. J. Pediatr. 2009;154:143–146. doi: 10.1016/j.jpeds.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krepischi-Santos A.C., Rajan D., Temple I.K., Shrubb V., Crolla J.A., Huang S., Beal S., Otto P.A., Carter N.P., Vianna-Morgante A.M., Rosenberg C. Constitutional haploinsufficiency of tumor suppressor genes in mentally retarded patients with microdeletions in 17p13.1. Cytogenet. Genome Res. 2009;125:1–7. doi: 10.1159/000218743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarzbraun T., Obenauf A.C., Langmann A., Gruber-Sedlmayr U., Wagner K., Speicher M.R., Kroisel P.M. Predictive diagnosis of the cancer prone Li-Fraumeni syndrome by accident: new challenges through whole genome array testing. J. Med. Genet. 2009;46:341–344. doi: 10.1136/jmg.2008.064972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schluth-Bolard C., Sanlaville D., Labalme A., Till M., Morin I., Touraine R., Edery P. 17p13.1 microdeletion involving the TP53 gene in a boy presenting with mental retardation but no tumor. Am. J. Med. Genet. A. 2010;152A:1278–1282. doi: 10.1002/ajmg.a.33316. [DOI] [PubMed] [Google Scholar]
- 11.Shlien A., Baskin B., Achatz M.I., Stavropoulos D.J., Nichols K.E., Hudgins L., Morel C.F., Adam M.P., Zhukova N., Rotin L. A common molecular mechanism underlies two phenotypically distinct 17p13.1 microdeletion syndromes. Am. J. Hum. Genet. 2010;87:631–642. doi: 10.1016/j.ajhg.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeesman S., Kjaergaard S., Hove H.D., Kirchhoff M., Stevens J.M., Nowaczyk M.J. Microdeletion in distal 17p13.1: a recognizable phenotype with microcephaly, distinctive facial features, and intellectual disability. Am. J. Med. Genet. A. 2012;158A:1832–1836. doi: 10.1002/ajmg.a.35508. [DOI] [PubMed] [Google Scholar]
- 13.Belligni E.F., Di Gregorio E., Biamino E., Calcia A., Molinatto C., Talarico F., Ferrero G.B., Brusco A., Silengo M.C. 790 Kb microduplication in chromosome band 17p13.1 associated with intellectual disability, afebrile seizures, dysmorphic features, diabetes, and hypothyroidism. Eur. J. Med. Genet. 2012;55:222–224. doi: 10.1016/j.ejmg.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Coutton C., Devillard F., Vieville G., Amblard F., Lopez G., Jouk P.S., Satre V. 17p13.1 microduplication in a boy with Silver-Russell syndrome features and intellectual disability. Am. J. Med. Genet. A. 2012;158A:2564–2570. doi: 10.1002/ajmg.a.35553. [DOI] [PubMed] [Google Scholar]
- 15.Schmickel R.D. Contiguous gene syndromes: a component of recognizable syndromes. J. Pediatr. 1986;109:231–241. doi: 10.1016/s0022-3476(86)80377-8. [DOI] [PubMed] [Google Scholar]
- 16.Dauber A., Golzio C., Guenot C., Jodelka F.M., Kibaek M., Kjaergaard S., Leheup B., Martinet D., Nowaczyk M.J., Rosenfeld J.A. SCRIB and PUF60 are primary drivers of the multisystemic phenotypes of the 8q24.3 copy-number variant. Am. J. Hum. Genet. 2013;93:798–811. doi: 10.1016/j.ajhg.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sereda M., Griffiths I., Pühlhofer A., Stewart H., Rossner M.J., Zimmerman F., Magyar J.P., Schneider A., Hund E., Meinck H.M. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 18.Yagi H., Furutani Y., Hamada H., Sasaki T., Asakawa S., Minoshima S., Ichida F., Joo K., Kimura M., Imamura S. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 19.Petrij F., Giles R.H., Dauwerse H.G., Saris J.J., Hennekam R.C., Masuno M., Tommerup N., van Ommen G.J., Goodman R.H., Peters D.J. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 20.Kleefstra T., Brunner H.G., Amiel J., Oudakker A.R., Nillesen W.M., Magee A., Geneviève D., Cormier-Daire V., van Esch H., Fryns J.P. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurotaki N., Imaizumi K., Harada N., Masuno M., Kondoh T., Nagai T., Ohashi H., Naritomi K., Tsukahara M., Makita Y. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 22.Boone P.M., Bacino C.A., Shaw C.A., Eng P.A., Hixson P.M., Pursley A.N., Kang S.H., Yang Y., Wiszniewska J., Nowakowska B.A. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zankl A., Molinari L. ABase-a tool for the rapid assessment of anthropometric measurements on handheld computers. Am. J. Med. Genet. A. 2003;121A:146–150. doi: 10.1002/ajmg.a.20185. [DOI] [PubMed] [Google Scholar]
- 24.Lainhart J.E., Bigler E.D., Bocian M., Coon H., Dinh E., Dawson G., Deutsch C.K., Dunn M., Estes A., Tager-Flusberg H. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am. J. Med. Genet. A. 2006;140:2257–2274. doi: 10.1002/ajmg.a.31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinawi M., Liu P., Kang S.H., Shen J., Belmont J.W., Scott D.A., Probst F.J., Craigen W.J., Graham B.H., Pursley A. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niederriter A.R., Davis E.E., Golzio C., Oh E.C., Tsai I.C., Katsanis N. In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp. 2013:e50338. doi: 10.3791/50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho C.M., Pehlivan D., Ramocki M.B., Fang P., Alleva B., Franco L.M., Belmont J.W., Hastings P.J., Lupski J.R. Replicative mechanisms for CNV formation are error prone. Nat. Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hicks W.M., Kim M., Haber J.E. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010;329:82–85. doi: 10.1126/science.1191125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deem A., Keszthelyi A., Blackgrove T., Vayl A., Coffey B., Mathur R., Chabes A., Malkova A. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9:e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beunders G., Voorhoeve E., Golzio C., Pardo L.M., Rosenfeld J.A., Talkowski M.E., Simonic I., Lionel A.C., Vergult S., Pyatt R.E. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013;92:210–220. doi: 10.1016/j.ajhg.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tremblay R.G., Sikorska M., Sandhu J.K., Lanthier P., Ribecco-Lutkiewicz M., Bani-Yaghoub M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods. 2010;186:60–67. doi: 10.1016/j.jneumeth.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Alcantara D., O’Driscoll M. Congenital microcephaly. Am. J. Med. Genet. C. Semin. Med. Genet. 2014;166C:124–139. doi: 10.1002/ajmg.c.31397. [DOI] [PubMed] [Google Scholar]
- 34.Stockert R.J. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol. Rev. 1995;75:591–609. doi: 10.1152/physrev.1995.75.3.591. [DOI] [PubMed] [Google Scholar]
- 35.McDonell L.M., Mirzaa G.M., Alcantara D., Schwartzentruber J., Carter M.T., Lee L.J., Clericuzio C.L., Graham J.M., Jr., Morris-Rosendahl D.J., Polster T., FORGE Canada Consortium Mutations in STAMBP, encoding a deubiquitinating enzyme, cause microcephaly-capillary malformation syndrome. Nat. Genet. 2013;45:556–562. doi: 10.1038/ng.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoyama T., Ueno I., Kamijo T., Hashimoto T. Rat very-long-chain acyl-CoA dehydrogenase, a novel mitochondrial acyl-CoA dehydrogenase gene product, is a rate-limiting enzyme in long-chain fatty acid beta-oxidation system. cDNA and deduced amino acid sequence and distinct specificities of the cDNA-expressed protein. J. Biol. Chem. 1994;269:19088–19094. [PubMed] [Google Scholar]
- 37.Strauss A.W., Powell C.K., Hale D.E., Anderson M.M., Ahuja A., Brackett J.C., Sims H.F. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc. Natl. Acad. Sci. USA. 1995;92:10496–10500. doi: 10.1073/pnas.92.23.10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Legido A., Jethva R., Goldenthal M.J. Mitochondrial dysfunction in autism. Semin. Pediatr. Neurol. 2013;20:163–175. doi: 10.1016/j.spen.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Hamblet N.S., Lijam N., Ruiz-Lozano P., Wang J., Yang Y., Luo Z., Mei L., Chien K.R., Sussman D.J., Wynshaw-Boris A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- 40.Pulvirenti T., Van Der Heijden M., Droms L.A., Huse J.T., Tabar V., Hall A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res. 2011;71:7280–7290. doi: 10.1158/0008-5472.CAN-11-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H., Bedford F.K., Brandon N.J., Moss S.J., Olsen R.W. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- 42.Fritschy J.M., Brünig I. Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol. Ther. 2003;98:299–323. doi: 10.1016/s0163-7258(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z.W., Olsen R.W. GABAA receptor associated proteins: a key factor regulating GABAA receptor function. J. Neurochem. 2007;100:279–294. doi: 10.1111/j.1471-4159.2006.04206.x. [DOI] [PubMed] [Google Scholar]
- 44.Everitt A.B., Luu T., Cromer B., Tierney M.L., Birnir B., Olsen R.W., Gage P.W. Conductance of recombinant GABA (A) channels is increased in cells co-expressing GABA(A) receptor-associated protein. J. Biol. Chem. 2004;279:21701–21706. doi: 10.1074/jbc.M312806200. [DOI] [PubMed] [Google Scholar]
- 45.Chen L., Wang H., Vicini S., Olsen R.W. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc. Natl. Acad. Sci. USA. 2000;97:11557–11562. doi: 10.1073/pnas.190133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kikuchi K., Niikura Y., Kitagawa K., Kikuchi A. Dishevelled, a Wnt signalling component, is involved in mitotic progression in cooperation with Plk1. EMBO J. 2010;29:3470–3483. doi: 10.1038/emboj.2010.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma P., Schwarten M., Schneider L., Boeske A., Henke N., Lisak D., Weber S., Mohrlüder J., Stoldt M., Strodel B. Interaction of Bcl-2 with the autophagy-related GABAA receptor-associated protein (GABARAP): biophysical characterization and functional implications. J. Biol. Chem. 2013;288:37204–37215. doi: 10.1074/jbc.M113.528067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabuyama Y., Suzuki T., Nakazawa N., Yamaki J., Homma M.K., Homma Y. Dysregulation of very long chain acyl-CoA dehydrogenase coupled with lipid peroxidation. Am. J. Physiol. Cell Physiol. 2010;298:C107–C113. doi: 10.1152/ajpcell.00231.2009. [DOI] [PubMed] [Google Scholar]
- 49.Komoike Y., Shimojima K., Liang J.S., Fujii H., Maegaki Y., Osawa M., Fujii S., Higashinakagawa T., Yamamoto T. A functional analysis of GABARAP on 17p13.1 by knockdown zebrafish. J. Hum. Genet. 2010;55:155–162. doi: 10.1038/jhg.2010.1. [DOI] [PubMed] [Google Scholar]
- 50.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karaca E., Weitzer S., Pehlivan D., Shiraishi H., Gogakos T., Hanada T., Jhangiani S.N., Wiszniewski W., Withers M., Campbell I.M., Baylor Hopkins Center for Mendelian Genomics Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamamoto S., Jaiswal M., Charng W.L., Gambin T., Karaca E., Mirzaa G., Wiszniewski W., Sandoval H., Haelterman N.A., Xiong B. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159:200–214. doi: 10.1016/j.cell.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vissers L.E., Bhatt S.S., Janssen I.M., Xia Z., Lalani S.R., Pfundt R., Derwinska K., de Vries B.B., Gilissen C., Hoischen A. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 54.Verdin H., D’haene B., Beysen D., Novikova Y., Menten B., Sante T., Lapunzina P., Nevado J., Carvalho C.M., Lupski J.R., De Baere E. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet. 2013;9:e1003358. doi: 10.1371/journal.pgen.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boone P.M., Yuan B., Campbell I.M., Scull J.C., Withers M.A., Baggett B.C., Beck C.R., Shaw C.J., Stankiewicz P., Moretti P. The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am. J. Hum. Genet. 2014;95:143–161. doi: 10.1016/j.ajhg.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shuvarikov A., Campbell I.M., Dittwald P., Neill N.J., Bialer M.G., Moore C., Wheeler P.G., Wallace S.E., Hannibal M.C., Murray M.F. Recurrent HERV-H-mediated 3q13.2-q13.31 deletions cause a syndrome of hypotonia and motor, language, and cognitive delays. Hum. Mutat. 2013;34:1415–1423. doi: 10.1002/humu.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamp C., Hirschmann P., Voss H., Huellen K., Vogt P.H. Two long homologous retroviral sequence blocks in proximal Yq11 cause AZFa microdeletions as a result of intrachromosomal recombination events. Hum. Mol. Genet. 2000;9:2563–2572. doi: 10.1093/hmg/9.17.2563. [DOI] [PubMed] [Google Scholar]
- 58.Sun C., Skaletsky H., Rozen S., Gromoll J., Nieschlag E., Oates R., Page D.C. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum. Mol. Genet. 2000;9:2291–2296. doi: 10.1093/oxfordjournals.hmg.a018920. [DOI] [PubMed] [Google Scholar]
- 59.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 60.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.