

Abstract
Elastomeric, fully degradable and biocompatible biomaterials are rare, with current options presenting significant limitations in terms of ease of functionalization and tunable mechanical and degradation properties. We report a new method for covalently crosslinking tyrosine residues in silk proteins, via horseradish peroxidase and hydrogen peroxide, to generate highly elastic hydrogels with tunable properties. The tunable mechanical properties, gelation kinetics and swelling properties of these new protein polymers, in addition to their ability to withstand shear strains on the order of 100%, compressive strains greater than 70% and display stiffness between 200 – 10,000 Pa, covering a significant portion of the properties of native soft tissues. Molecular weight and solvent composition allowed control of material mechanical properties over several orders of magnitude while maintaining high resilience and resistance to fatigue. Encapsulation of human bone marrow derived mesenchymal stem cells (hMSC) showed long term survival and exhibited cell-matrix interactions reflective of both silk concentration and gelation conditions. Further biocompatibility of these materials were demonstrated with in vivo evaluation. These new protein-based elastomeric and degradable hydrogels represent an exciting new biomaterials option, with a unique combination of properties, for tissue engineering and regenerative medicine.
Keywords: silk, hydrogels, elastomers, biomaterials, biopolymers
1. Introduction
Matching mechanical properties to native tissue extracellular matrix (ECM) is a primary consideration during the selection of biomaterials for tissue engineering or regenerative medicine.[1] In particular, stiffness has a significant impact on the differentiation of stem cells and maintenance of cell phenotype during cell culture.[2] Hydrogels are particularly attractive for these applications, as their stiffness can be fine tuned and they can incorporate growth factors and other cell signaling factors to optimize cell functions.[3] Due to this versatility, numerous synthetic and natural polymer hydrogels have been studied, including those based on polyesters,[4] polyurethanes,[5] polyethers,[6, 7] elastin,[8] resilin[9] and collagen.[10] However, limited options remain for biomaterials with tunable mechanical properties that can match the resilience and elasticity of native tissues, while also being biocompatible, degradable and allow for the incorporation of cells, proteins and signaling factors.
Several biocompatible and biodegradable synthetic polymers have been developed that exhibit high resilience and recovery from both applied tensile and compressive forces.[11] Poly(glycerol sebacate) (PGS)[12] has shown utility as a scaffold for engineering vascular,[13] cardiac,[14] and nerve[15] tissues, but has several significant drawbacks including rapid degradation limiting its use to short term scaffolding.[16] Moreover, the requirement for organic solvents during processing prevents cell or protein encapsulation.[17] Synthetic bioelastomers based on polyurethanes modified with degradable segments have also been developed[5] and used for soft tissue,[18] bone,[19] and myocardial [20] repairs. While these materials have shown promise, concerns remain over potential toxicity of degradation products and the ability of these materials to resist long term strains in vivo.[21] Additional polymers suitable for use as elastomeric biomaterials include variants of poly(ethylene glycol),[6] poly(ε-caprolactone),[7] and poly(vinyl alcohol).[22] However, all of these polymers typically require organic solvents for processing, limiting incorporation of cells and bioactive molecules and many present issues with acidic degradation products.[23]
ECM proteins, purified from native tissues or produced via recombinant DNA methodologies have also been used for tissue engineering scaffolds. For example, elastin proteins, which provide elastic behavior in many native tissues, have been explored for tissue engineering of soft, elastic matrices.[8, 24] Recombinant resilin sequences have shown promising mechanical properties.[9, 25] Despite the unique properties and biomimetic nature of these protein biomaterials, they are limited in supply and are currently cost prohibitive for larger scale applications. In order to gain the benefits of proteins as biomaterial matrices, due to their diverse amino acid chemistries for functionalization and biodegradability, recent studies have explored blending with synthetic polymers or other readily available proteins or ECM components. For example, elastin was alloyed with silk,[26] collagen,[27] and poly(lactide-co-glycolide),[28] while resilin was mixed with poly(ethylene glycol)[29] in order to extend utility.
With the prior studies above, there remains a persistent need for a versatile, elastomeric, degradable, accessible and tunable biomaterial. Silk fibroin, a natural biopolymer that is extracted from the cocoons of the Bombyx mori silkworm, has been explored extensively for use in tissue engineering and regenerative medicine.[30] Silk is non-immunogenic[31] and is FDA approved for some medical devices including sutures and as a support structure during reconstructive surgery. Silk can be solubilized and used to form different material formats including films, foams, electrospun mats and hydrogels.[32] The versatility of silks and the large body of previous work showing the safety and efficacy of silk in tissue engineering, combined with its natural resilience, make silk protein a good candidate for elastomeric biomaterials.
Silk hydrogels can be formed by the addition of energy to an aqueous solution of the protein to drive chain mobility, or via dehydration of the solution; sonication,[33] vortexing,[34] decreased pH,[35] surfactants,[36] or electric fields[37] have been reported. Sonicated and vortexed silk hydrogels were useful for cell encapsulation, drug release and neuron growth, but did not exhibit elasticity; plastically deforming at strains greater than about 10%.[33, 34] Silk electrogels have shown remarkable elasticity and adhesion, withstanding strains of 1,000%, but require the application of an electric field and low pH.[37] One common factor between these hydrogels is that they are formed through physical entanglements and hydrogen bonding between hydrophobic domains, often resulting in β-sheet formation. These β-sheet crystals are responsible for the structure, strength and long term stability of the hydrogels, but also result in brittle behavior, as the crystals prevent long range displacements.[38]
In order to overcome the brittle nature of the β-sheet induced silk hydrogels, we report on enzyme catalyzed crosslinking of amino acid phenolic groups, which account for more than 5% of the amino acids in silk,[39] by reactions with horseradish peroxidase (HRP) and hydrogen peroxide. This method has been shown to be effective to form hydrogels from gelatin,[40] hyaluronic acid – tyramine conjugates,[41] and mixtures of tyramine substituted hyaluronic acid and alginate.[42] Since silk has the requisite phenol groups in tyrosine side chains, this approach was exploited as a crosslinking mechanism. Andersen (1964) found that silk solutions acted on by horse radish peroxidase and hydrogen peroxide formed gels that were stable in water and were highly elastic.[43] Aeschbach et al. (1976) also referenced enzymatic crosslinking of silks in a fundamental study on the formation of dityrosine crosslinks by tyrosine oxidation.[44] More recently similar reactions were used to graft chitosan onto silk using tyrosinases in an effort to form novel conjugates of the two proteins.[45] However, no systematic study of peroxidase-induced crosslinked silk hydrogels have been reported in terms of reaction conditions related to elastomeric mechanical properties, or in the context of cell and tissue culture.
The goal of the present study was to characterize the gelation, material properties and cytotoxicity of enzymatically crosslinked silk hydrogels. This technique results in highly tunable elastomeric silk hydrogels capable of supporting cell encapsulation. These enzymatically crosslinked hydrogels have controllable gelation kinetics, mechanical properties and are produced using all aqueous processing, offering a broad platform for biomaterial functionalization and utility. Furthermore the silk hydrogels are amenable to stiffening by induction of β-sheet while maintaining a considerable degree of elasticity. These highly tunable protein elastomers offer a new opportunity for degradable, biocompatible and useful polymers for biomaterials and regenerative medicine needs.
2. Results
2.1. Mechanism of HRP-mediated silk gelation and structural characterization
The addition of HRP to a silk solution in the presence of H2O2, results in the formation of a stable, highly elastic, transparent gel. This is due to the well known reaction whereby HRP facilitates crosslinking of the tyrosines in the silk fibroin via the formation of free radical species in the presence of hydrogen peroxide. H2O2 forms an oxyferryl center and a porphyrin-based cation radical at the active site of HRP, resulting in an activated enzyme which is a powerful reducing agent.[46] HRP then undergoes two single electron oxidation reactions in the presence of a phenolic oxidizing agent to return to its ground state. The overall reaction results in the formation of two water molecules and two phenolic radicals. In this reaction, tyrosine radicals formed through the HRP catalyzed reaction can react with one another to form covalent bonds (Figure 1a).[47] The formation of the dityrosine bonds and the resultant protein secondary structures were assessed by fluorescence spectroscopy and by Circular Dichroism (CD). The CD spectra (Figure 1b) of the silk solution with HRP revealed random coil conformations with a minima near 200 nm and no distinct peaks. Following gelation, a more ordered conformation was evident with a pronounced minimum around 200 nm and a peak near 195 nm, confirming that the gelation reaction was not due to typical anti-parallel β-sheet formation, which would have a distinct minimum at 218 nm. Dityrosine formation was confirmed by comparison of the fluorescence excitation-emission matrix (EEM) for the silk and HRP solutions and the fully formed hydrogel (Figure 1c, d). These data show a distinct shift in the fluorescence maxima from the solution with an excitation of 290 nm and an emission of 350 nm to the gel with a peak excitation of 300 nm with an emission at 425 nm. The shift in the fluorescence indicates the formation of the covalent crosslinks[48] and the intensity of the fluorescence may be correlated to crosslink density as it increased as a function of silk concentration (Figure S1a). Furthermore, enzymatic crosslinking resulted in the formation of optically clear gels with negligible absorbance above 350 nm (Figure S1b), and emission of blue fluorescence when irradiated under UV, that was not present in the precursor solution (Figure 1e).
Figure 1. Chemistry and structural characterization.
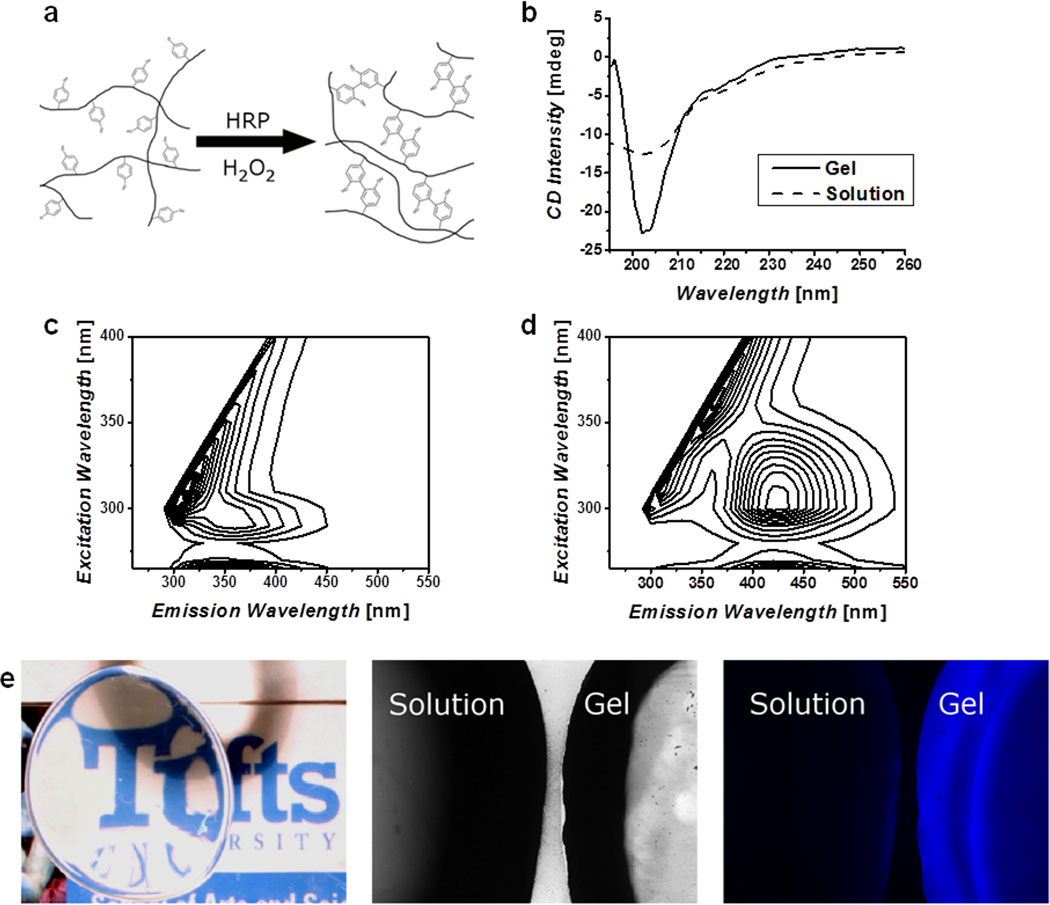
a) Schematic representation of the crosslinking of tyrosine residues on silk molecules, these covalent bonds allow for chain extension creating highly elastic hydrogels. b) Circular Dichroism (CD) spectra of silk solution and enzymatically formed hydrogels, show a change to a helical structure and not β-sheet as found in other silk materials. Fluorescence excitation-emission spectra of solution (c) and gel (d) confirm the formation of dityrosine bonds. e) The resultant hydrogels are optically clear and exhibit a blue fluorescence when irradiated with UV that is not present in the precursor solution.
2.2. Rheological properties of HRP silk hydrogels
The kinetics of gelation and shear properties of the enzymatically crosslinked silk hydrogels were assessed using rheology. Hydrogels of varying molecular weights and protein concentrations were formed in situ in a cone and plate geometry maintained at 37°C. By controlling the solution concentration as well as the molecular weight of the silk fibroin chains the rate at which the gels formed as well as the final mechanical properties were tunable. The degumming time of the silk fibers was inversely related to the molecular weight with 10mb (10 minutes of boiling to remove the sericin) generating the highest molecular weight. The gelation kinetics, frequency and strain responses for solutions of 5% w/v protein are shown in Figure 2a, b, and c, respectively. These tests indicated that increasing both the molecular weight and the protein concentration resulted in significantly stiffer gels. However, it was found that all gels tested, regardless of molecular weight and protein concentration were frequency independent and were able to withstand shear strains of at least 100%, above which they plastically deformed resulting in a rapid decrease in storage modulus. Based on the conditions studied stiffness values between 200 – 10,000 Pa were obtained, which covers three orders of magnitude and a significant portion of native tissues properties (Figure 3d). In all cases the loss modulus (data not shown) was below the lower limit of the rheometer, indicating that these were highly elastic materials with a negligible viscous component.
Figure 2. Rheological properties of hydrogels.
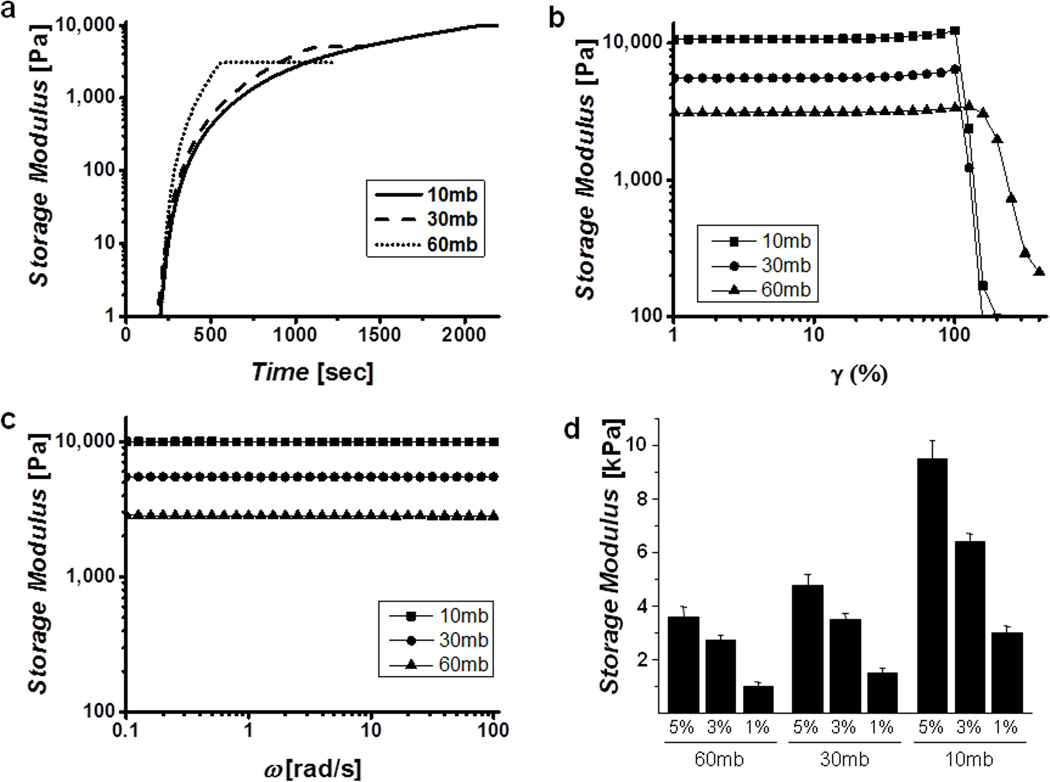
Representative gelation kinetics (a), strain sweeps (b) and frequency sweeps (c) of different molecular weight gels show controllable kinetics and final mechanical properties in highly resilient, frequency independent gels. Curves indicated were performed at a protein concentration of 5% w/v in DI water. d) Final storage modulus as a function of molecular weight and concentration, demonstrate the wide range of stiffness achievable.
Figure 3. Compressive properties of hydrogels.
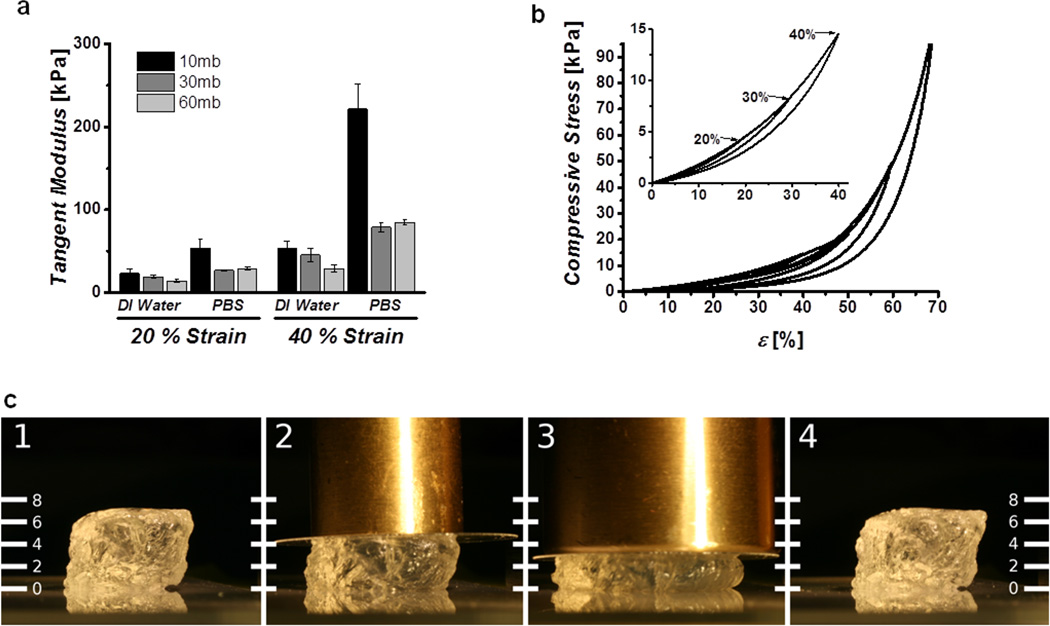
a) Tangent moduli of 5% w/v cast gels swelled in water and PBS show strain and molecular weight dependence. b) Cyclic compression curves of gels immersed in PBS showing excellent recovery below 70% strain, inset highlights complete recovery below 40% strain. c) Image of hydrogel undergoing ~50% compression, under 50 g (2) and 100 g (3) brass weights and showing complete recovery after removal (4). Scale units are in milimeters.
2.3. Evaluation of stiffness and recovery in unconfined compression
Hydrated unconfined compression studies were carried out to determine the compressive properties of the covalently crosslinked hydrogels. Preformed gels (8 mm ∅, 3 mm height, 5% w/v silk concentration) were equilibrated in either DI water or PBS for 12 hours and the final diameter measured. The samples were then placed under a tare load of 3 grams and subjected to 5 compressive cycles to 40% strain to eliminate loading artifacts. The sixth cycle was recorded and the compressive modulus, quantified as the tangent of the stress-strain curve at 20 and 40% strains, was calculated. The stiffness of the hydrogels increased with the magnitude of the compression and increasing molecular weight and were stiffer when immersed in PBS than in DI water (Figure 3a). The solvent effect was a result of significant swelling behavior in DI water, and deswelling in PBS, likely due to electrostatic shielding, which resulted in different absolute protein concentrations (Figure S2). Despite this swelling or contraction, the gels recovered from compressive strains of over 70% with minimal hysteresis as indicated by representative cyclic loading curves (Figure 3b) and pictorial representations (Figure 3c). While these physiologically extreme strains highlight the excellent resilience of the hydrogels, fatigue resistance of the hydrogel to high cycle moderate strains was more relevant to their use in biomedical applications. To evaluate the fatigue properties of the hydrogels, the dynamic modulus was monitored over 3,600 cycles to a strain magnitude of 10% (Figure S3). The samples showed excellent recovery and resistance to fatigue, with minimal changes in their dynamic compressive moduli. These analyses indicate that molecular weight and swelling solvent of the hydrogels allowed control of material properties over several orders of magnitude while maintaining high resilience and resistance to fatigue, even after large numbers of cycles at relatively large strains.
2.6. Cytotoxicity and cell encapsulation
To verify that the hydrogels were viable substrates for cell growth and encapsulation, hMSCs were cultured on the surface of preformed gels as well as encapsulated during the gelation process. Cells seeded onto preformed gels showed similar adhesion as the control TCP surface (Figure 4a) and supported cell growth and proliferation for greater than 3 weeks (Figure 4b). Live/dead staining and confocal imaging confirmed that the cells proliferated until confluence (Figure 4c). The influence of the gelation reaction and hydrogel network on cell-matrix interactions was further explored by encapsulating hMSC's in 3% w/v gels formed in sterile water. An Alamar blue assay one day following encapsulation indicated that 52.9 ± 2.3% of the cells survived the gelation versus those plated on TCP (Figure 5a). Live/dead staining indicated that the surviving cells were well distributed throughout the thickness of the hydrogel (Figure 5b). Subsequently, the hydrogels supported survival and proliferation for at least 29 days, reaching a plateau of 4.2 ± 0.3-fold increase of over day 1 values (Figure 5c). Confocal imaging confirmed viability, but showed that the cells exhibited minimal interaction with the matrix and retained a spherical morphology with few extensions (Figure 5d). To avoid cell mortality during the encapsulation process cells were also tested in 3% w/v silk gels formed in half concentration DMEM solution. This method did not significantly impact initial cell survival (Figure 5e), however, the cells began proliferating immediately and showed a significantly faster growth rate (p<0.001) when compared to those encapsulated in water (Figure 5f). More interestingly, the morphology of the cells encapsulated in DMEM was strikingly different with greater cell spreading and cell extensions on the order of several hundred microns (Figure 5g).
Figure 4. Human mesenchymal stem cell interactions on silk gel surfaces.
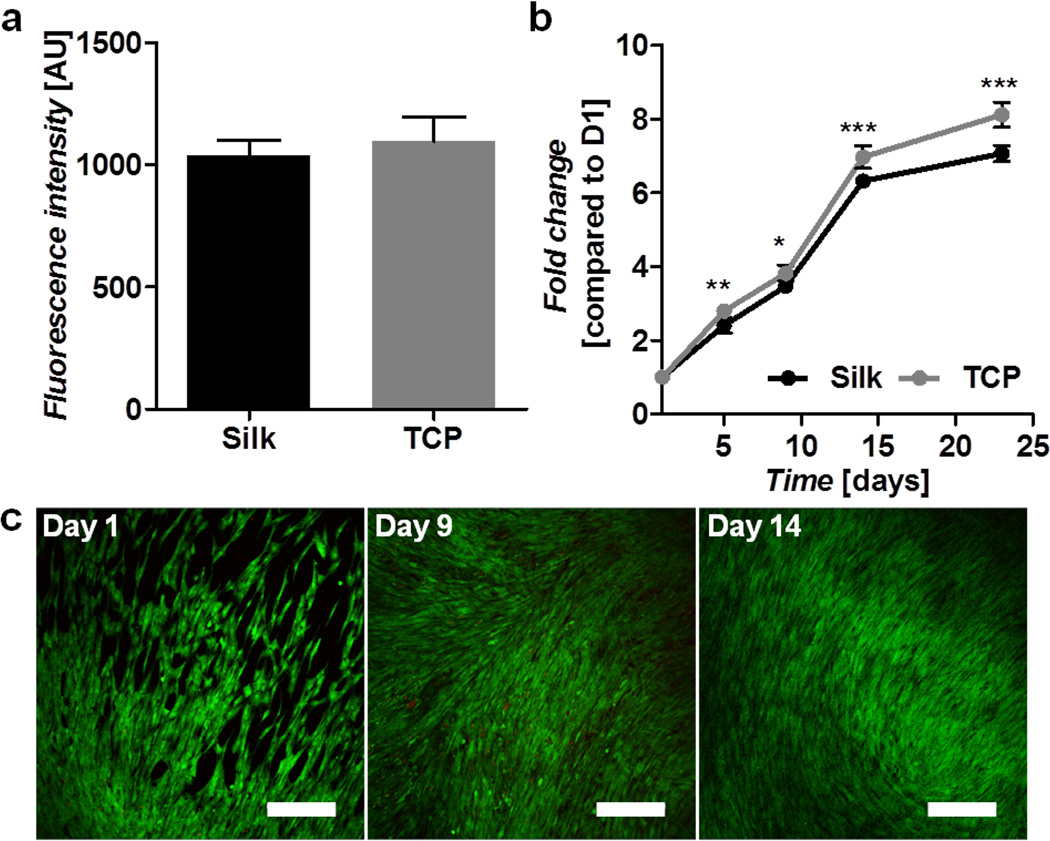
a) Cell attachment on silk and TCP at day 1 post-seeding as determined by Alamar blue. b) Cell proliferation on silk gels and TCP over a 24-day period as determined by Alamar blue and presented as fold change compared to day 1. c) Live (green) and dead (red) cell staining on silk gels over a 14-day period. Scale bars are 300 µm.
Figure 5. Human mesenchymal stem cells encapsulated in silk gels.
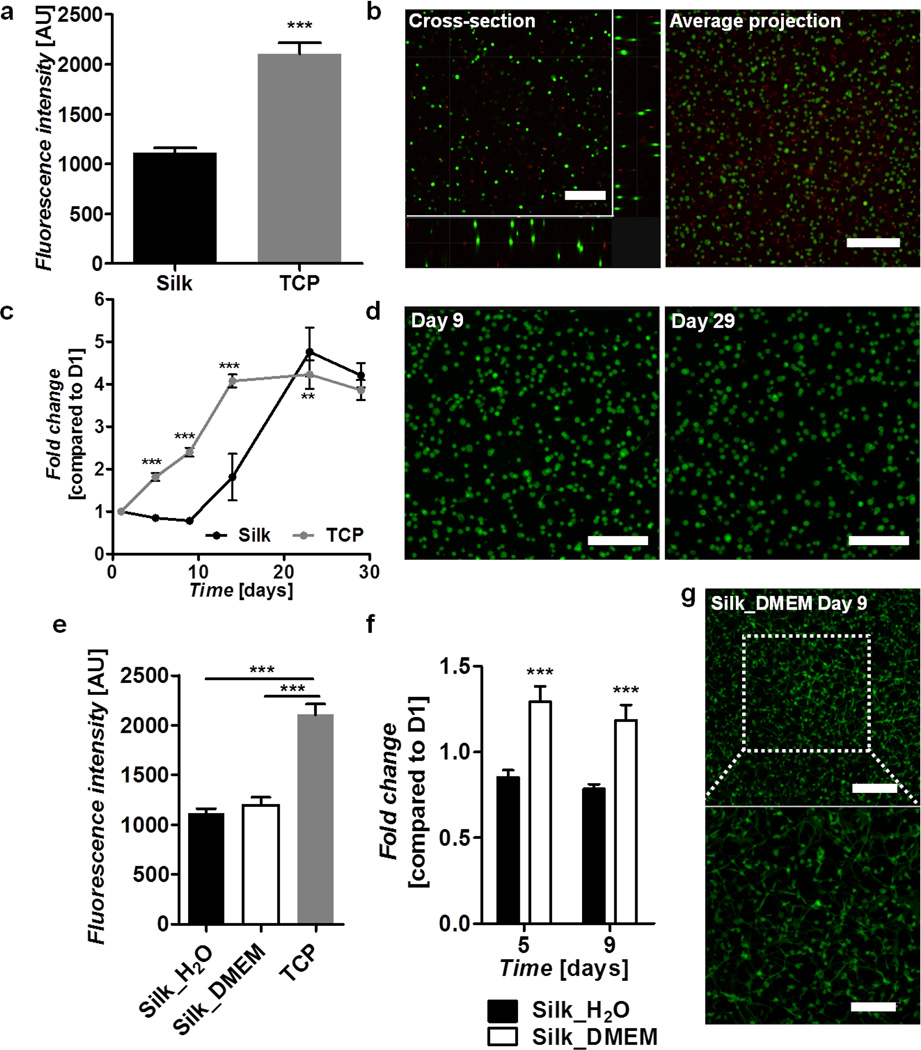
Silk gels were formed in water (a–d) or DMEM (e–g). a) Cell survival following encapsulation in silk gels compared to cells seeded on TCP as determined by Alamar blue at day 1 post-encapsulation. b) Live (green) and dead (red) staining on cells encapsulated in silk gels at day 1 post-encapsulation showing a cross-section and an average projection of a 352 µm thick image stack. c) Cell proliferation on silk gels and TCP over a 29 day period as determined by Alamar blue and presented as fold change compared to day 1. d) Live (green) and dead (red) staining of cells encapsulated in silk gels at days 9 and 29 post-seeding showing an average projection of 212 µm and 212 µm thick image stacks respectively. Scale bars are 300 µm. e) Cell survival following encapsulation in silk gels formed in water and DMEM compared to cells seeded on TCP as determined by Alamar blue at day 1 post-encapsulation. f) Comparison of cell proliferation in silk gels formed in water and DMEM as determined by Alamar blue at days 5 and 9 post-encapsulation and presented as fold change compared to day 1. g) Live (green) and dead (red) staining on cells encapsulated in a silk gel formed in DMEM at day 9 post-encapsulation showing an average projection of a 180 µm thick mage stack. Scale bars are 300 µm (top) and 175 µm (bottom).
2.6. In vivo biocompatibility of enzymatic hydrogels
Preformed hydrogels (5 mm ∅, 3 mm height) of 2% w/v and 6% w/v silk protein were implanted subcutaneously in mice and examined histologically at one, two and four weeks post-implantation. Explanted gels formed from 2% w/v solution contracted appreciably while the 6% w/v hydrogels maintained their initial volume (Figure S4). H&E staining showed that the silk gels elicited a typical foreign body response at respective harvest time points, without excessive inflammation. Despite the contraction of the 2% gels after implantation there were no significant differences in host response to the hydrogels and only the 6% w/v gels are shown for clarity. After one week of implantation (Figure 6a) the gels had retained their shape, with immune cells at the interface of the gel and tissue, with minimal infiltration into the bulk of the gel. Sectioning of explants after two weeks (Figure 6b) showed a less distinct interface between the gel and adjacent tissues as the silk gel became integrated. This trend continued at four weeks (Figure 6c) with further erosion of the gel periphery and evidence of gel fragments that had been broken from the bulk and were fully surrounded by cells. The degradation of the gel was accompanied by an apparent decrease in the number of adjacent immune cells.
Figure 6. In vivo interactions with implanted silk gels.
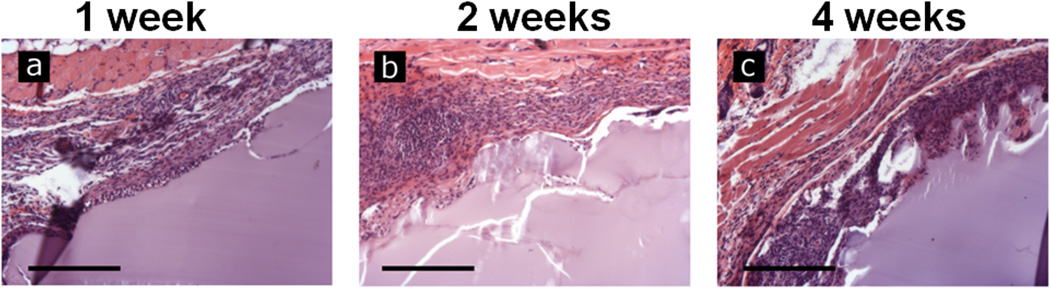
Preformed hydrogels of 6% w/v were implanted subcutaneously in a mouse model. Gels were explanted and examined histologically by H&E staining 1 (a), 2 (b) and 4 (c) weeks following implantation. The hydrogels showed progressive cell infiltration and degradation as the duration was increased. Scale bars are 250 µm.
3. Discussion
Advancements in tissue engineering and regenerative medicine are dependent on the development of easily prepared, inexpensive, biocompatible and biodegradable materials that can match the resilience, durability and mechanical properties found in native tissues. The covalent dityrosine bonds of the hydrogels presented here result in a robust hydrogel network to achieve high stiffness, while the lack of rigid crystalline segments imbues them with excellent elasticity and resilience. Furthermore, silk precursor materials have been well-studied, can be prepared in bulk and have been shown to be both biocompatible and biodegradable. The wide range of properties and high resilience of these hydrogels suggest that they may be useful for numerous in vivo and in vitro applications where soft, tunable and elastomeric substrates are required.
The bonding of adjacent tyrosine residues has been implicated as a fundamental component in several elastic and structural proteins including resilin,[9, 43] elastin,[49] and keratin.[50] Dityrosine residues have also been found to occur in wild type Tussah silk fibroin, but were not present in the domesticated B. mori fibroin investigated here.[50] The widespread existence of dityrosines in structural proteins combined with the well-studied peroxidase-mediated oxidative coupling of accessible tyrosines in the presence of hydrogen peroxide has led to the development of several hydrogel systems based on this reaction, including hyaluronic acid,[51] gelatin,[40] and alginate.[42] In addition to providing mild, physiologically relevant reaction conditions, the peroxidase reaction allows for significant tunability by varying the solution and reagent parameters, including tyrosine concentration and accessibility, HRP and H2O2 concentrations and substrate molecular weight. As demonstrated in this study, the same mechanism is also applicable to regenerated silk fibroin solutions, where the formation of the tyrosine crosslinked networks results in an optically clear, highly elastic protein hydrogel (Figure 1). This suggests that in addition to the variables explored here, control over HRP and H2O2 may serve as additional modes to further tune the hydrogels.
While many of the hydrogels developed in the biomaterials field to date have shown tunable mechanics, cell encapsulation features, biocompatibility, biodegradability or elasticity, none of the current options in the field offers the combination of all of these characteristics. The enzymatically crosslinked hydrogels presented here meet all of these criteria with shear moduli from the hundreds of Pascals to over ten kilopascals and tangent moduli from fifteen to four hundred kilopascals. Further, these new gels are not cytotoxic and they support cell encapsulation, are biocompatible and biodegradable and are capable of recovering from extreme compressive strains. Additionally, the sensitivity of the gels to swelling in low ionic strength solvents and contraction in high salt solution (Figure S2) is governed by both the initial protein concentration and molecular weight, allowing for selection of both modulus and degree of contraction for a given application.
Most importantly these hydrogels represent excellent candidates as biomaterials for applications that require the ability to fully recover from large strains or long term cyclic compression. It has been shown in numerous cases that scaffold modulus and mechanical stimulation are critical for proper development of tissues.[52] Similarly, excessive local stress and strain have been implicated in pathological remodeling of tissues.[53] Thus, it is imperative that a material for use in engineering or replacing these dynamic tissues support long term strains without appreciable changes in mechanical properties. As demonstrated in the present study, these enzymatically formed silk hydrogels are capable of withstanding 3,600 cycles of 10% strain with negligible changes in modulus, suggesting that they are a likely platform for use in dynamic tissue engineering such as cardiac and skeletal muscle.
In addition to their superior resilience, these hydrogels also allow for long term hMSC survival both on the surface and encapsulated within the matrix. Adhesion of cells onto the surface of gels was not significantly different than on TCP and while proliferation was slower than on TCP, the fold change was of the same order of magnitude. This suggests that the hydrogels are not cytotoxic and warrant further investigation as elastomeric biomaterials. While surface seeding of cells is an important screen, numerous synthetic elastomers that require toxic crosslinking agents and solvents are also capable of supporting cell growth after proper curing and removal or inactivation of the deleterious substances. Therefore, to advance the elastomeric biomaterials field, new materials must also allow for the direct encapsulation of cells in addition to withstanding repeated strains. Due to retardation of the gelling reaction in a solution of physiologic salt concentration, cells were initially encapsulated in hydrogels formed in water or cell culture media at reduced concentrations. Despite cell losses during the encapsulation process, the cells that survived were robust and capable of long term proliferation and survival. These results are promising and show the potential of the hydrogels for three dimensional cell culture, either to avoid cell-specific morphologies (as in the case of the water), or different morphologies (depending on the concentration of DMEM used during crosslinking). With suitable gel functionalization or inclusion of ECM components, cell responses should be tunable to specific tissue needs.
The dramatically different cell morphologies formed in DMEM indicated that network strength was weaker than those formed in water. The weaker network formation in media was confirmed using rheology, where gels formed in media were less stiff than those crosslinked in water (Figure S5). Thus, it seems that the cells were able to exploit weaker networks to spread and form extensions that were visible in the confocal images (Figure 5g). The hypothesis that network strength was responsible for the differences in cell morphology was further supported by experiments at lower protein concentrations (1.5% w/v vs. 3% w/v). Cells grown in the lower concentration gels, with weaker mechanics and a higher water content, had similar morphologies to those formed in the DMEM-based hydrogels with a flattened appearance and distinct extensions (Figure S6). These experiments show that in addition to tunable mechanics these hydrogels may allow for control of cell-matrix interactions and the ability to influence cell shape. This may serve as an independent means of manipulating cell differentiation, as cell shape has been previously implicated as a factor in commitment of a stem cell to a given lineage.[54] While similar properties have been shown in synthetic hydrogels with highly controlled chemistries,[55] the ability to incorporate this functionality into a protein based hydrogel provides a more natural platform to study these interactions. Previous work has already shown the utility of silk in controlling neuron outgrowth,[56] hMSC differentiation and survival,[33, 57] and development of adipose like tissue,[58] the present hydrogels allows for the further expansion of these diverse applications and signaling cues.
The success of the hydrogels for cell encapsulation and utility in vivo may also be enhanced by recent developments based on silk systems. The molding of channels into scaffolds to enhance diffusion was found to be advantageous for cell infiltration and soft tissue repair.[59] Additionally, the mild crosslinking reaction, makes the technique amenable to incorporation, delivery, and release of bioactive molecules including, growth factors,[60] antibodies,[61] and antibiotics.[62] Silk has also enjoyed utility as a platform for electronics, optics and related technological applications.[63] The hydrogels described here have potential as substrates for these technologies where the elastomeric nature can be exploited to create flexible, biocompatible, three dimensional optical and electronic devices. The combination of these attributes, in addition to their high resilience, makes this silk gelation system promising for numerous applications in regenerative medicine.
4. Conclusions
The present work demonstrates the formation of a new family of natural, biocompatible, biodegradable elastomeric hydrogels for use in tissue engineering and regenerative medicine. The gels have exceptional resilience, highly tunable properties, support cell survival and proliferation and were well tolerated when implanted in vivo. Additionally, the all aqueous processing makes the process amenable to incorporation of other ECM proteins and growth factors, allowing for the control of cell-matrix interactions and increasing the versatility of this new biomaterials platform. This unique combination of characteristics, in a protein based hydrogel, eliminates many of the disadvantages inherent in current elastomeric biomaterials. The development of an elastomeric biomaterial that can faithfully recapitulate numerous properties of native extracellular matrices, while retaining a high level of versatility, will allow the engineering of more natural and functional tissues.
5. Experimental
Preparation of Aqueous Silk Solution
Silk solutions were prepared using our previously established procedures.[32] Briefly, 5 grams of B. mori silkworm cocoons were immersed in 2L of boiling 0.02 M Na2CO3 solution (Sigma-Aldrich, St. Louis, MO) for 10, 30 or 60 minutes, subsequently referred to as 10 mb, 30 mb and 60 mb respectively, to remove the sericin protein coating. Degummed fibers were collected and rinsed with distilled water three times, then air-dried. The fibers were solubilized in 9.3 M LiBr (Sigma-Aldrich, St. Louis, MO) at 60°C for 4 hours. 15 mL of this solution was then dialyzed against 1 L of distilled water (water changes after 1, 3, 6, 24, 36, and 48 hours) with a regenerated cellulose membrane (3,500 MWCO, Thermo Scientific, Rockford, IL). The solubilized silk protein solution was then centrifuged twice (9700 RPM, 20 min., 4°C) to remove insoluble particulates. Protein concentration was determined by drying a known mass of the silk solution under a hood for 12 hours and assessing the mass of the remaining solids.
Preparation of Enzymatically Crosslinked Silk Hydrogels
Horseradish peroxidase (HRP), type VI (Sigma-Aldrich, St. Louis, MO) lyophilized powder was mixed with deionized water to form a stock solution with a concentration of 1,000 U/mL. The HRP solution was added to the silk solution in a ratio of 10 Units of HRP to 1 mL of silk solution. To initiate gelation, 10µL of 165 mM hydrogen peroxide (Sigma Aldrich, St. Louis, MO) solution were added per mL of silk solution, for a final concentration of 1.65 mM, and mixed by gentle pipetting prior to setting.
Circular Dichroism (CD)
To perform CD, a 100 µl aliquot of 2% w/v silk solution, 1 µl of HRP and 1 µl of hydrogen peroxide was thoroughly mixed and immediately loaded into 0.01 mm path length demountable quartz cuvette (Starna Cells, Atascadero, CA). The sample was then loaded into the spectrophotometer and allowed to gel at 37°C for 1 hour at which time the spectra were obtained from 260 to 190 nm at a resolution of 0.5 nm. CD measurements were acquired using an Aviv model 62DS spectrophotometer equipped with a temperature controller (AVIV Biomedical, Inc., Lakewood, NJ). CD spectra presented are the average of three measurements and were smoothed using 9 point Savitzky-Golay smoothing algorithm in OriginPro 9.1 (Origin Lab, Northampton, MA).
Fluorescence Spectroscopy
To assess the intrinsic fluorescence of the solution and hydrogels, 1.5 ml of 1% w/v silk solution was pipetted into a 3 mm pathlength quartz cuvette (Starna Cells, Atascadero, CA) and excitation-emission was recorded using a Hitachi F4500 Spectrofluorometer (Hitachi, Schaumburg, IL) from 250 nm to 500 nm in 10 nm increments at an intensity of 700 V using a 2.5 nm slit width to avoid saturation. The silk solution was then rinsed from the cuvette and 1.5 mL of new solution, with HRP and H2O2 were mixed and pipetted into the cuvette. The solution, with initiator, was allowed to gel for 2 hours at room temperature and another excitation-emission spectrum was collected. Spectra were then processed to subtract background fluorescence from the solvent and cuvette and normalized to account for differences in the xenon lamp output spectrum.
Rheology
Rheological measurements were conducted on the enzymatically crosslinked hydrogels to determine the gelation time and mechanical strength. All rheology was carried out on a TA Instruments ARES-LS2 rheometer (TA Instruments, New Castle, DE) using a 25mm stainless steel conical plate (angle: 0.0994 rad) and a temperature controlled Peltier plate set to 37°C. Briefly, gels were mixed by gentle pipetting and 420 µL of solution was loaded under the geometry. Prior to gelation, the geometry was lowered to the specified gap and low viscosity oil was placed around the outside edge of the cone to prevent water evaporation. A dynamic time sweep was conducted at a frequency of 1 Hz (6.283 rad/sec) and 1% strain for 4,000 seconds or until the sample had reached a plateau modulus, whichever occurred first. Following gelation, dynamic strain and frequency sweeps were carried out on the gels to ensure that measurements occurred within the viscoelastic regime and to assess the elasticity and relaxation times of the hydrogels. Silk solutions degummed for 10, 30, and 60 minutes were tested at concentrations of 1%, 3% and 5%.
Hydrogel Cyclic Compression Testing
Unconfined compression tests were conducted to assess the mechanical properties of the silk hydrogels as well as the recovery following multiple compression cycles. Five percent solutions of silk, degummed for 10, 30 and 60 minutes were used for the cyclic compression testing. Preformed enzymatically crosslinked hydrogels were biopsy punched into cylinders (8 mm ∅, 3 mm height) and allowed to fully swell in DI water or contract in PBS for approximately 12 hours. The final diameter of the samples were measured and used for calculation of compressive stresses. Samples were loaded in a TA Instruments RSA3 Dynamic Mechanical Analyzer (TA Instruments, New Castle, DE) between stainless steel parallel plates in an immersion bath. The upper plate was lowered until a compression force of ~3 g was registered. Samples were subjected to five preloading cycles to 40% strain in order to eliminate artifacts. The sixth cycle was recorded and tangent moduli were calculated at 20% and 40% strains for comparison. All testing was conducted at a constant crosshead speed of 1 mm/min. Sequential strain testing until failure was conducted in 10% strain increments from 20% to 80% strain following five preconditioning cycles to 40% strain. Fatigue stability of the gels was tested by monitoring the dynamic modulus at a frequency of 0.5 Hz and 10% strain over the course of 3,600 cycles.
Cell survival and proliferation
Human mesenchymal stem cells (hMSC's) were isolated from fresh bone marrow aspirate (Lonza, Basel, Switzerland) as previously described[64] and cultured in Dulbecco's Modified Eagle Medium (supplemented with 10% fetal bovine serum and 1% antibiotic/antimycotic) (Life Technologies, Grand Island, NY) and seeded at passage 3–4. For surface seeding, gels of roughly 400 µm thickness, of 60mb silk solution at a concentration of 3% wt/v were prepared as described above and allowed to cure for 1 hour, at 37 °C in an incubator. Cells were seeded at 210 cells per mm2, and allowed to adhere for 150 minutes prior to flooding with media. In order to minimize the time the cells were exposed to sub-physiological osmolarity, encapsulation was accomplished by partially gelling the silk solutions. The 60mb silk solution HRP and H2O2 were gently mixed allowed to gel for approximately 10 minutes, at which time cells at 500 or 1,000 cells per mm3 were added to the solution and 100 µl per well was added to 48 well plates or glass bottom petri dishes. The plates were placed into an incubator and allowed to gel for an additional 10 minutes at which time the wells were flooded with media. All cell culture was performed in an incubator maintained at 37°C and 5% CO2.
Metabolic Activity
The relative metabolic activity of the cells in each scaffold was determined by AlamarBlue assay (Life Technologies, Grand Island, NY) according to the manufacturer's directions. Scaffolds were rinsed with Phosphate Buffered Saline (PBS) and incubated in DMEM medium with 10% AlamarBlue reagent for 4 hours at 37°C with 5% CO2. Following incubation with the reagent, aliquots (100 µl) were placed into black 96 well plates and the fluorescence quantified using a plate reader with an excitation wavelength of 550 nm and an emission wavelength of 590 nm. Cells plated in tissue culture wells were maintained as above and utilized as controls, while acellular hydrogels were used to adjust for background fluorescence.
Cell Imaging with Confocal Laser Scanning Microscopy
Imaging of the surface seeded and encapsulated cells was performed by staining with a LIVE/DEAD Viability/Cytotoxicity Kit (Life Technologies, Grand Island, NY) according to the manufacturer's instructions. Briefly, cells were incubated with calcein AM and ethidium homodimer-1 (EthD-1) for 60 minutes to stain live (green) and dead cells (red) respectively. After staining the gels were washed three times with PBS and imaged using a Leica DMIRE2 confocal laser scanning microscope (CLSM) (Wetzlar, Germany) with excitation at 488 nm and emission at 499–537 nm for live cells and excitation at 543 and emission at 620–650 nm for dead cells.
Subcutaneous Implantation
All in vivo experimentation was conducted under protocols approved by the Tufts Institutional Animal Care and Use Committee. Mice used in this study were 4–6 week old BALBc female mice (Charles River Laboratories, Wilmington, MA). The mice were randomly assigned into three timepoints with three animals per group. Each animal had two preformed gels (5 mm ∅, 3 mm height) of 2% w/v and 6% w/v protein concentration, subcutaneously implanted in lateral pockets under general anesthesia of oxygen and isofluorane. At week 1, 2 or 4 post-implantation, animals were euthanized and the specimens along with the adjacent tissues were collected for histological examination.
Histochemistry
Explants were fixed in 10% neutral buffered formalin (NBF) and embedded in paraffin following xylene and graded ethanol incubation. Samples were sectioned to 6 µm thickness, placed on glass slides and paraffin was removed. Sections were then stained with hematoxylin and eosin (H&E) (Sigma-Aldrich, St. Louis, MO) to visualize cell nuclei and cytoplasm. Following staining samples were imaged using a Zeiss Axiovert 40 CFL microscope and a 10× objective (Carl Zeiss AG, Germany).
Statistical Analyses
Data are expressed as mean ± standard deviation. One or two-way analysis of variance (ANOVA) and Tukey post-hoc analysis were used to determine statistically significant differences. Statistical significance was accepted at the p < 0.05 level and indicated in figures as *p<0.05, **p<0.01, ***p<0.001.
Supplementary Material
Acknowledgements
B.P.P. was supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. This work was supported by the NIH (EB002520, EY020856, EB014283) and the AFOSR (FA9550-10-1-1-0172). The authors thank Prof. Gary Leisk for assistance with mechanical characterization and Dr. Bruce Panilaitis for assistance interpreting histology.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Seliktar D. Science. 2012;336:1124. doi: 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]
- 2.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman AS. Advanced Drug Delivery Reviews. 2012 [Google Scholar]; Peppas N, Huang Y, Torres-Lugo M, Ward J, Zhang J. Annual Review of Biomedical Engineering. 2000;2:9. doi: 10.1146/annurev.bioeng.2.1.9. [DOI] [PubMed] [Google Scholar]
- 4.Rai R, Tallawi M, Grigore A, Boccaccini AR. Progress in polymer science. 2012;37:1051. [Google Scholar]
- 5.Zdrahala RJ, Zdrahala IJ. Journal of biomaterials applications. 1999;14:67. doi: 10.1177/088532829901400104. [DOI] [PubMed] [Google Scholar]
- 6.Gaharwar AK, Dammu SA, Canter C-J, Wu JM, Schmidt G. Biomacromolecules. 2011;12:1641. doi: 10.1021/bm200027z. [DOI] [PubMed] [Google Scholar]
- 7.Amsden B. Soft Matter. 2007;3:1335. doi: 10.1039/b707472g. [DOI] [PubMed] [Google Scholar]
- 8.Mithieux SM, Rasko JE, Weiss AS. Biomaterials. 2004;25:4921. doi: 10.1016/j.biomaterials.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 9.Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DC, Merritt DJ, Dixon NE. Nature. 2005;437:999. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 10.Kang H-W, Tabata Y, Ikada Y. Biomaterials. 1999;20:1339. doi: 10.1016/s0142-9612(99)00036-8. [DOI] [PubMed] [Google Scholar]
- 11.Shi R, Chen D, Liu Q, Wu Y, Xu X, Zhang L, Tian W. International journal of molecular sciences. 2009;10:4223. doi: 10.3390/ijms10104223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Ameer GA, Sheppard BJ, Langer R. Nature biotechnology. 2002;20:602. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 13.Motlagh D, Yang J, Lui KY, Webb AR, Ameer GA. Biomaterials. 2006;27:4315. doi: 10.1016/j.biomaterials.2006.04.010. [DOI] [PubMed] [Google Scholar]; Fidkowski C, Kaazempur-Mofrad MR, Borenstein J, Vacanti JP, Langer R, Wang Y. Tissue engineering. 2005;11:302. doi: 10.1089/ten.2005.11.302. [DOI] [PubMed] [Google Scholar]
- 14.Chen Q-Z, Bismarck A, Hansen U, Junaid S, Tran MQ, Harding SE, Ali NN, Boccaccini AR. Biomaterials. 2008;29:47. doi: 10.1016/j.biomaterials.2007.09.010. [DOI] [PubMed] [Google Scholar]; Ravichandran R, Venugopal JR, Sundarrajan S, Mukherjee S, Ramakrishna S. Tissue Engineering Part A. 2011;17:1363. doi: 10.1089/ten.TEA.2010.0441. [DOI] [PubMed] [Google Scholar]
- 15.Sundback CA, Shyu JY, Wang Y, Faquin WC, Langer RS, Vacanti JP, Hadlock TA. Biomaterials. 2005;26:5454. doi: 10.1016/j.biomaterials.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Pomerantseva I, Krebs N, Hart A, Neville CM, Huang AY, Sundback CA. Journal of Biomedical Materials Research Part A. 2009;91:1038. doi: 10.1002/jbm.a.32327. [DOI] [PubMed] [Google Scholar]
- 17.Chen Q-Z, Ishii H, Thouas GA, Lyon AR, Wright JS, Blaker JJ, Chrzanowski W, Boccaccini AR, Ali NN, Knowles JC. Biomaterials. 2010;31:3885. doi: 10.1016/j.biomaterials.2010.01.108. [DOI] [PubMed] [Google Scholar]
- 18.Guan J, Fujimoto KL, Sacks MS, Wagner WR. Biomaterials. 2005;26:3961. doi: 10.1016/j.biomaterials.2004.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fromstein J, Woodhouse K. Journal of Biomaterials Science, Polymer Edition. 2002;13:391. doi: 10.1163/156856202320253929. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen FF, Karring T, Gogolewski S. Acta Orthopaedica. 1992;63:66. doi: 10.3109/17453679209154853. [DOI] [PubMed] [Google Scholar]; Gorna K, Gogolewski S. Journal of Biomedical Materials Research Part A. 2003;67:813. doi: 10.1002/jbm.a.10148. [DOI] [PubMed] [Google Scholar]
- 20.McDevitt TC, Woodhouse KA, Hauschka SD, Murry CE, Stayton PS. Journal of Biomedical Materials Research Part A. 2003;66:586. doi: 10.1002/jbm.a.10504. [DOI] [PubMed] [Google Scholar]
- 21.Santerre J, Woodhouse K, Laroche G, Labow R. Biomaterials. 2005;26:7457. doi: 10.1016/j.biomaterials.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 22.Nuttelman CR, Henry SM, Anseth KS. Biomaterials. 2002;23:3617. doi: 10.1016/s0142-9612(02)00093-5. [DOI] [PubMed] [Google Scholar]
- 23.Metters A, Anseth K, Bowman C. Polymer. 2000;41:3993. [Google Scholar]
- 24.Daamen WF, Veerkamp J, Van Hest J, Van Kuppevelt T. Biomaterials. 2007;28:4378. doi: 10.1016/j.biomaterials.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 25.Qin G, Rivkin A, Lapidot S, Hu X, Preis I, Arinus SB, Dgany O, Shoseyov O, Kaplan DL. Biomaterials. 2011;32:9231. doi: 10.1016/j.biomaterials.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu X, Wang X, Rnjak J, Weiss AS, Kaplan DL. Biomaterials. 2010;31:8121. doi: 10.1016/j.biomaterials.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buttafoco L, Kolkman N, Engbers-Buijtenhuijs P, Poot A, Dijkstra P, Vermes I, Feijen J. Biomaterials. 2006;27:724. doi: 10.1016/j.biomaterials.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Li M, Mondrinos MJ, Chen X, Gandhi MR, Ko FK, Lelkes PI. Journal of Biomedical Materials Research Part A. 2006;79:963. doi: 10.1002/jbm.a.30833. [DOI] [PubMed] [Google Scholar]
- 29.McGann CL, Levenson EA, Kiick KL. Macromolecular Chemistry and Physics. 2013;214:203. doi: 10.1002/macp.201200412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vepari C, Kaplan DL. Progress in polymer science. 2007;32:991. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meinel L, Hofmann S, Karageorgiou V, Kirker-Head C, McCool J, Gronowicz G, Zichner L, Langer R, Vunjak-Novakovic G, Kaplan DL. Biomaterials. 2005;26:147. doi: 10.1016/j.biomaterials.2004.02.047. [DOI] [PubMed] [Google Scholar]
- 32.Rockwood DN, Preda RC, Yücel T, Wang X, Lovett ML, Kaplan DL. Nature protocols. 2011;6:1612. doi: 10.1038/nprot.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Kluge JA, Leisk GG, Kaplan DL. Biomaterials. 2008;29:1054. doi: 10.1016/j.biomaterials.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yucel T, Cebe P, Kaplan DL. Biophysical journal. 2009;97:2044. doi: 10.1016/j.bpj.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim U-J, Park J, Li C, Jin H-J, Valluzzi R, Kaplan DL. Biomacromolecules. 2004;5:786. doi: 10.1021/bm0345460. [DOI] [PubMed] [Google Scholar]
- 36.Kang GD, Nahm JH, Park JS, Moon JY, Cho CS, Yeo JH. Macromolecular rapid communications. 2000;21:788. [Google Scholar]
- 37.Leisk GG, Lo TJ, Yucel T, Lu Q, Kaplan DL. Advanced Materials. 2010;22:711. doi: 10.1002/adma.200902643. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto A, Chen J, Collette AL, Kim U-J, Altman GH, Cebe P, Kaplan DL. The Journal of Physical Chemistry B. 2006;110:21630. doi: 10.1021/jp056350v. [DOI] [PubMed] [Google Scholar]
- 39.Wray LS, Hu X, Gallego J, Georgakoudi I, Omenetto FG, Schmidt D, Kaplan DL. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2011;99:89. doi: 10.1002/jbm.b.31875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amini AA, Nair LS. Journal of Bioactive and Compatible Polymers. 2012;27:342. [Google Scholar]
- 41.Kurisawa M, Chung JE, Yang YY, Gao SJ, Uyama H. Chemical communications. 2005:4312. doi: 10.1039/b506989k. [DOI] [PubMed] [Google Scholar]
- 42.Ganesh N, Hanna C, Nair SV, Nair LS. International Journal of Biological Macromolecules. 2013;55:289. doi: 10.1016/j.ijbiomac.2012.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen SO. Biochimica et Biophysica Acta (BBA)-General Subjects. 1964;93:213. doi: 10.1016/0304-4165(64)90289-2. [DOI] [PubMed] [Google Scholar]
- 44.Aeschbach R, Amadoò R, Neukom H. Biochimica et Biophysica Acta (BBA) - Protein Structure. 1976;439:292. doi: 10.1016/0005-2795(76)90064-7. [DOI] [PubMed] [Google Scholar]
- 45.Kang G, Lee K, Ki C, Nahm J, Park Y. Macromol. Res. 2004;12:534. [Google Scholar]; Freddi G, Anghileri A, Sampaio S, Buchert J, Monti P, Taddei P. Journal of Biotechnology. 2006;125:281. doi: 10.1016/j.jbiotec.2006.03.003. [DOI] [PubMed] [Google Scholar]; Sampaio S, Taddei P, Monti P, Buchert J, Freddi G. Journal of Biotechnology. 2005;116:21. doi: 10.1016/j.jbiotec.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Rodríguez-López JN, Lowe DJ, Hernández-Ruiz J, Hiner AN, García-Cánovas F, Thorneley RN. Journal of the American Chemical Society. 2001;123:11838. doi: 10.1021/ja011853+. [DOI] [PubMed] [Google Scholar]
- 47.Veitch NC. Phytochemistry. 2004;65:249. doi: 10.1016/j.phytochem.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 48.Harms GS, Pauls SW, Hedstrom JF, Johnson CK. Journal of Fluorescence. 1997;7:283. [Google Scholar]
- 49.LaBella F, Keeley F, Vivian S, Thornhill D. Biochemical and biophysical research communications. 1967;26:748. doi: 10.1016/s0006-291x(67)80137-2. [DOI] [PubMed] [Google Scholar]
- 50.Raven DJ, Earland C, Little M. Biochimica et Biophysica Acta (BBA)-Protein Structure. 1971;251:96. doi: 10.1016/0005-2795(71)90065-1. [DOI] [PubMed] [Google Scholar]
- 51.Darr A, Calabro A. J Mater Sci: Mater Med. 2009;20:33. doi: 10.1007/s10856-008-3540-0. [DOI] [PubMed] [Google Scholar]
- 52.Altman GH, Horan RL, Lu HH, Moreau J, Martin I, Richmond JC, Kaplan DL. Biomaterials. 2002;23:4131. doi: 10.1016/s0142-9612(02)00156-4. [DOI] [PubMed] [Google Scholar]
- 53.Balachandran K, Sucosky P, Jo H, Yoganathan AP. American Journal of Physiology-Heart and Circulatory Physiology. 2009;296:H756. doi: 10.1152/ajpheart.00900.2008. [DOI] [PubMed] [Google Scholar]; Grodzinsky AJ, Levenston ME, Jin M, Frank EH. Annual Review of Biomedical Engineering. 2000;2:691. doi: 10.1146/annurev.bioeng.2.1.691. [DOI] [PubMed] [Google Scholar]
- 54.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Developmental cell. 2004;6:483. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 55.DeForest CA, Anseth KS. Annual review of chemical and biomolecular engineering. 2012;3:421. doi: 10.1146/annurev-chembioeng-062011-080945. [DOI] [PubMed] [Google Scholar]
- 56.Hopkins AM, De Laporte L, Tortelli F, Spedden E, Staii C, Atherton TJ, Hubbell JA, Kaplan DL. Advanced Functional Materials. 2013;23:5140. [Google Scholar]
- 57.Murphy AR, John PS, Kaplan DL. Biomaterials. 2008;29:2829. doi: 10.1016/j.biomaterials.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mauney JR, Nguyen T, Gillen K, Kirker-Head C, Gimble JM, Kaplan DL. Biomaterials. 2007;28:5280. doi: 10.1016/j.biomaterials.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rnjak-Kovacina J, Wray LS, Golinski JM, Kaplan DL. Advanced Functional Materials. 2013 doi: 10.1002/adfm.201302901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Numata K, Kaplan DL. Advanced Drug Delivery Reviews. 2010;62:1497. doi: 10.1016/j.addr.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guziewicz N, Best A, Perez-Ramirez B, Kaplan DL. Biomaterials. 2011;32:2642. doi: 10.1016/j.biomaterials.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pritchard EM, Valentin T, Panilaitis B, Omenetto F, Kaplan DL. Advanced Functional Materials. 2012 doi: 10.1002/adfm.201201636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D-H, Viventi J, Amsden JJ, Xiao J, Vigeland L, Kim Y-S, Blanco JA, Panilaitis B, Frechette ES, Contreras D. Nature materials. 2010;9:511. doi: 10.1038/nmat2745. [DOI] [PMC free article] [PubMed] [Google Scholar]; Omenetto FG, Kaplan DL. Science. 2010;329:528. doi: 10.1126/science.1188936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Altman GH, Horan RL, Martin I, Farhadi J, Stark P, Volloch V, Richmond JC, Vunjak-Novakovic G, Kaplan DL. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2002;16:270. doi: 10.1096/fj.01-0656fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.