

Abstract
Intellectual disability and cerebellar atrophy occur together in a large number of genetic conditions and are frequently associated with microcephaly and/or epilepsy. Here we report the identification of causal mutations in Sorting Nexin 14 (SNX14) found in seven affected individuals from three unrelated consanguineous families who presented with recessively inherited moderate-severe intellectual disability, cerebellar ataxia, early-onset cerebellar atrophy, sensorineural hearing loss, and the distinctive association of progressively coarsening facial features, relative macrocephaly, and the absence of seizures. We used homozygosity mapping and whole-exome sequencing to identify a homozygous nonsense mutation and an in-frame multiexon deletion in two families. A homozygous splice site mutation was identified by Sanger sequencing of SNX14 in a third family, selected purely by phenotypic similarity. This discovery confirms that these characteristic features represent a distinct and recognizable syndrome. SNX14 encodes a cellular protein containing Phox (PX) and regulator of G protein signaling (RGS) domains. Weighted gene coexpression network analysis predicts that SNX14 is highly coexpressed with genes involved in cellular protein metabolism and vesicle-mediated transport. All three mutations either directly affected the PX domain or diminished SNX14 levels, implicating a loss of normal cellular function. This manifested as increased cytoplasmic vacuolation as observed in cultured fibroblasts. Our findings indicate an essential role for SNX14 in neural development and function, particularly in development and maturation of the cerebellum.
Main Text
Intellectual disability (ID) syndromes with a small cerebellum constitute a clinically and genetically heterogeneous group of neurological disorders for which the underlying molecular etiology is diverse and established in only a small subset. Several different cellular mechanisms have been implicated including mutations in SIL1, coding for an endoplasmic reticulum resident cochaperone, which cause Marinesco-Sjogren syndrome (MSS [MIM 248800]);1 sialic acid disorders such as Salla disease (MIM 604369);2 disorders of peroxisome biogenesis in atypical Refsum disease (MIM 614879);3 congenital disorders of glycosylation, especially type 1a caused by mutations in PMM2 (MIM 212065);4 and the X-linked ID-small cerebellum syndrome caused by mutations in OPHN1 (MIM 300486), a Rho-GTPase-activating protein (GAP).5 Related phenotypes include the group designated as pontocerebellar hypoplasias,6 within which causal mutations have been found in a number of genes involved in tRNA biogenesis (including RARS2 [MIM 611523], TSEN54 [MIM 225753], TSEN34 [MIM 612390], TSEN2 [MIM 612389], and CLP1 [MIM 615803]),6–10 in rRNA processing (the exosomal genes EXOSC3 [MIM 614678] and EXOSC8 [MIM 606019]),11,12 and another (CHMP1A [MIM 614961]), with a dual role in protein sorting at the endosome and chromatin modification at the nucleus.13 Other cellular processes include synaptic and cell junction function (CASK [MIM 300749]),14 cell cycle progression, and cell division (the serine-threonine kinase VRK1 [MIM 607596]).15 At the biochemical level, it is not clear how disruption of many of these genes leads to hindbrain hypoplasia or atrophy. The classification and diagnostic approach of cerebellar disease associated with ID in childhood is complex and challenging, often depending on the careful identification and assessment of neuroradiological and other clinical findings.15–18
We recently described an autosomal-recessive condition in a consanguineous Portuguese family (family 1) in which two sisters (Figures 1 and 2A) share a similar phenotype, characterized by severe cerebellar ataxia, severe intellectual disability (ID), absent speech, coarse facial features, relative macrocephaly, brachycamptodactyly of fifth fingers, and early-onset cerebellar atrophy (Table 1).21 To perform genetic analysis aimed at identifying the causal mutation, informed consents were obtained for all of the parents, probands, and siblings according to protocols approved by the ethical review committees at Great Ormond Street Hospital and Coimbra Hospital Centre. Specific parental consent was also given for the use of all of the clinical data and facial photographs included in this manuscript. The family was then investigated by first delineating regions of shared homozygosity followed by whole-exome sequencing to identify variants in the implicated regions. Homozygosity mapping was performed on the two parents (III.1 and III.2), two affected siblings (IV.3 and IV.6), and four unaffected siblings (IV.1, IV.4, IV.5, and IV.7) using the Infinium HD HumanCytoSNP-12 BeadChip (Illumina). This revealed 20 candidate regions spanning a total of 35,846,704 bp, which contained 450 RefSeq and 886 UCSC transcripts (Table S1 available online). Haplotype analysis of the SNP data defined the largest homozygous region of ∼18 Mb on 6q13-q14 (hg19; chr6: 70,500,118–88,497,536). Exome sequencing of the proband (IV.3) from family 1 was performed using Agilent SureSelect v.4 and Illumina TruSeq. Enriched libraries were sequenced on an Illumina HiSeq2000 by Perkin Elmer, resulting in a mean of 66× read depth with 68% of targeted bases covered at least 1×. We sequenced one sample per lane, aligning the resulting reads to the reference genome build GRCh37/hg19 using Burrows-Wheeler Aligner (v.0.5.7) and for variant calling we applied GATK base quality score recalibration,22 INDEL realignment, and duplicate removal and performed SNP and INDEL discovery and genotyping across all samples simultaneously using variant quality score recalibration.23 Variant annotation and interpretation analysis was generated through the use of Ingenuity Variant Analysis software v.3.0.20140422 from Ingenuity Systems. With the use of filters outlined in Table S2 designed to pinpoint novel or rare homozygous damaging variants, we reduced the number of variants from an initial 159,274 genome wide, to a single likely causal mutation. This was a unique, homozygous nonsense variant (c.2596C>T [p.Gln866∗]) within SNX14, located at 6q14.3, which is within the largest autozygous interval (Figures 2B and 2C; Table S3). Sanger sequencing using Big Dye Terminator v.1.1 (Life Technologies) on an ABI 3730 sequencer (Applied Biosystems) confirmed that the mutation was homozygous in both affected sisters and segregated as an autosomal recessive, being heterozygous in both parents and absent or heterozygous in four unaffected siblings (Figure 2A). The SNX14 locus generates two transcripts consisting of either 29 exons encoding the 946 amino acid isoform a (RefSeq NM_153816.2) or 26 exons, lacking exons 14, 23, and 24, encoding a shorter protein of 893 amino acids known as isoform b (RefSeq NM_020468.3). Both isoforms share the same four conserved domains: the PX (phosphoinositide binding, Phox homology), RGS (regulator of G protein signaling), PXA (PX-associated domain A), and PXC (PX-associated domain C) (Figure 2C). The c.2596C>T mutation was identified within an exon that codes for both transcripts (exon 26, based on the longer transcript) and was predicted to result in a protein truncation that would either remove the last 81 amino acids, including part of the PXC domain or, alternatively, trigger nonsense-mediated decay (Figure 2C).
Figure 1.
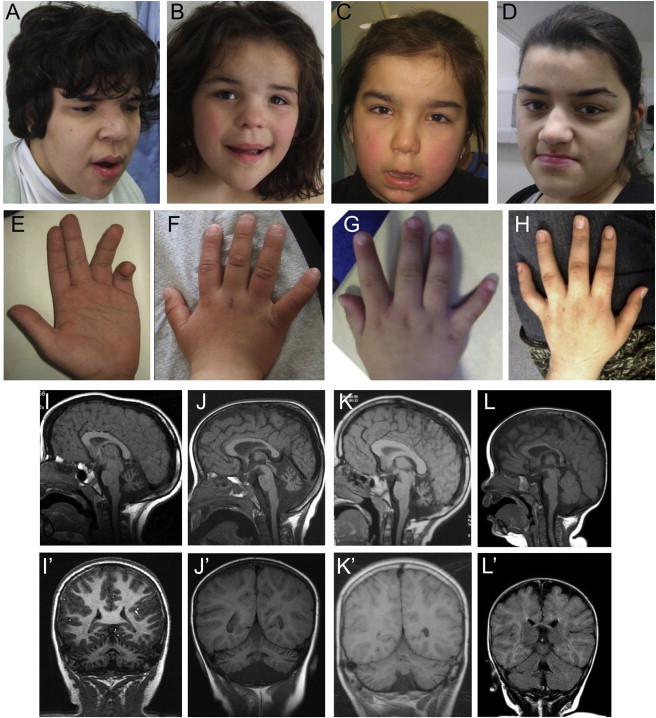
Phenotype of Affected Individuals from the Three Families Presented
Photographs and brain MRI scans from family 1 individual IV.3 aged 19 years (A) and 22 years (E); family 1 individual IV.6 aged 6 years (B, F) and 7 years (I, I′); family 2 individual V.1 aged 4.5 years (C, G, J, J′); family 3 individual III.2 aged 22 years (D, H) and 10 years (K, K′); and family 2 individual V.2 aged 9 months (L, L′). Notice the similar facial features mainly characterized by broad face, fullness of the upper eyelid, broad nasal base and slight underdevelopment of the alae, broad and long philtrum, thick lower lip vermillion, and fifth finger brachycamptodactyly. In the first years of life, no neuroradiological anomalies were observed in affected individuals, as depicted here by the normal T1-weighted mid-sagittal (L) and coronal (L′) MRI sections, which have not yet been repeated for this individual. MRI images performed for other individuals during infancy are unavailable but were reported to be normal. At older ages, affected individuals have a small cerebellum with thin folia and enlarged fissures, suggestive of global cerebellar atrophy as shown here in the T1-weighted MRI images from three of the children, one from each family (mid-sagittal sections I–K; coronal sections I′–K′). The pons appear small but in comparison are well preserved.
Figure 2.
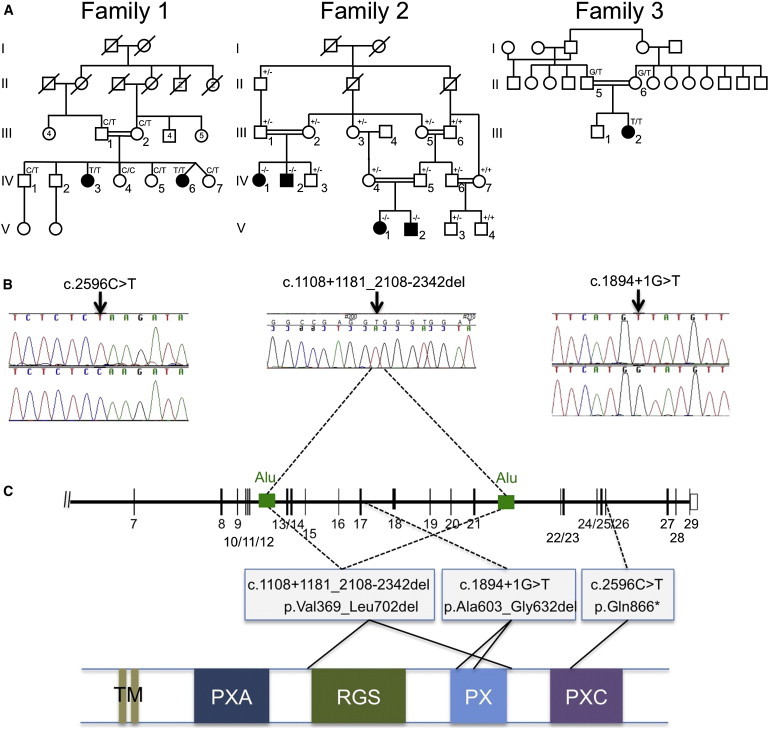
Identification of SNX14 Mutations in Three Affected Families
(A) Pedigrees of the three families showing genotypes in tested individuals.
(B) Sequence traces for families 1 and 3 show point mutations in genomic DNA (top trace, mutant; bottom trace, wild-type). For family 2, the sequence trace spans the deletion breakpoint, in genomic DNA, and indicates the location of the breakpoints within two Alu repeats in the schematic diagram of the SNX14 locus shown in (C) (see also Figure S1 for further details).
(C) Schematic representation of part of the SNX14 genomic locus (top) and the SNX14 protein (bottom) indicating the location and effect of the mutations detected in the three families. The protein consists of two predicted transmembrane domains (TM) at the N terminus, followed by the PXA domain, RGS domain, conserved PX phosphoinositide binding domain, and PXC domain situated toward the C terminus. The deletion in family 2 is predicted to remove the RGS and PX domains, whereas the splice site mutation in family 3 removes part of the PX domain.
Table 1.
Clinical Findings in the Affected Individuals
| Mutation in SNX14 (RefSeq NM_153816.3) |
Family 1 |
Family 2 |
Family 3 |
Total | ||||
|---|---|---|---|---|---|---|---|---|
| homoz p.Gln866∗ (c.2596C>T) | homoz p.Val369_Leu702del (c.1108+1181_2108−2342del) | homoz p.Ala603_Gly632del (c.1894+1G>A) | ||||||
| Subject ID | IV.3 | IV.6 | IV.1 | IV.2 | V.1 | V.2 | III.2 | |
| Sex | F | F | F | M | F | M | F | 5F, 2M |
| Present age (years) | 26 | 14 | 32 | 16 | 10 | 3 | 23 | |
| Growth at Birtha | ||||||||
| Gestational age (weeks) | 40 | 34 (twin) | U | U | 40 | 40 | U | |
| Length, cm (centile) | 49 (50th) | 42 (10th) | U | U | U | U | U | |
| Weight, g (centile) | 3,600 (50th) | 2,100 (25th) | U | U | 2,650 (3–10th) | 2,800 (10–25th) | U | |
| Head circumference, cm (centile) | 35.5 (50th) | 32 (50th) | U | U | U | 35.5 (90th) | U | |
| Growth, Postnatala | ||||||||
| Age (years) | 22 | 14.5 | 29 | 14 | 6.7 | 1.0 | 22 | |
| Height, cm (centile) | 155 (10th) | 140 (<3rd) | U | U | (91st) | 76.1 (<75th) | 157.7 (9–25th) | |
| Head circumference (centile) | 56.4 (75–90th) | 57.5 (90–97th) | 59 (>97th) | 55 (50th) | 55 (>97th) | 49.5 (97th) | 56.5 (75–90th) | |
| Neurodevelopment | ||||||||
| Intellectual disability | severe | severe | severe | severe | severe | NA | moderate | |
| Speech (1st words, years) | absent | sev impaired (13) | absent | absent | absent | absent | impaired (3) | 5/7 |
| Hypotonia | + | + | - | + | + | + | + | 6/7 |
| Sitting (age, months) | 18 | 16 | very late | >36 | 8 | >12 | 12 | 18b |
| Walking with help (age, months) | + (24) | + (20) | crawls | crawls | + (24) | − | + (18)c | 4/7 |
| Ataxia | + | + | wheelchair | wheelchair | + | NA | + | 5/6 |
| Talipes equino-varum | + | + | + | U | − | − | − | 3/6 |
| Hypo/areflexia | + | + | + | + | − | NA | + | 5/6 |
| Craniofacial Features | ||||||||
| Coarse features | + | + | + | + | + | + | + | 7/7 |
| Short palpebral fissures | + | + | + | + | + | + | − | 6/7 |
| Fullness of the upper eyelid | + | + | + | + | + | + | − | 6/7 |
| Broad/bulbous nose | + | + | + | + | + | + | + | 7/7 |
| Broad deep long philtrum | + | + | + | + | + | + | − | 6/7 |
| Thick lip vermilions (upper + lower) | + | + | + | + | + | + | + (lower) | 7/7 |
| Skeleton and Limbs | ||||||||
| Scoliosis/kyphosis | − | − | + | + | − | − | − | 2/7 |
| Brachy/camptodactyly of 5th fingers | + | + | + | + | + | − | + | 6/7 |
| Short and broad finger/toes | + | + | + | + | + | + | (+) | 7/7 |
| Elbow motion limitation | + | + | U | − | − | − | + | 3/6 |
| Hearing loss | SN | + | − | SN | SN | − | SN | 5/7 |
| Brain Imaging | MRI | MRI | CT | MRI | MRI | MRI | MRI | |
| Age at last evaluation | 20 years | 7 years | U | 10 months | 4.5 years | 9 months | 10 years | |
| Cerebellar atrophy | + P | + P | + | − | + P | − | + P | 5/7 |
| Pontine thinning | + | − | U | − | + | − | (+) | 4/7 |
Abbreviations and symbols are as follows: +, positive; −, negative/normal; U, data unknown; P, progressive: i.e., two or more sequential scans showed development/progression of the cerebellar atrophy; SN, sensorineural, moderate-severe, bilateral; NA, not applicable.
Growth measurements: For the Turkish individuals, the charts used for head circumference measurements were described by Evereklioglu et al.19 and Elmali et al.20 The Portuguese individuals were compared to national charts except for head circumference >36 cm, or where not available, the charts described by Evereklioglu et al.19 were used.
Median for sitting age.
Subject III.2 from family 3 was the only individual who progressed to independent ambulation, which was reached at age 3 years.
We identified a Turkish consanguineous family (family 2) with four affected individuals sharing similar clinical features to family 1 (Table 1; Figures 1 and 2A). SNP genotyping (as above), revealed 11 regions of homozygosity shared between all three of the affected individuals available at the time of mapping (IV.1, IV.2, and V.1), with the largest (8,056,114 bp) including the interval containing SNX14 in 6q14.3, being the only autozygous region in common with family 1 (Table S1). Individual V.1 underwent exome sequencing at Dasman Diabetes Institute (Kuwait City, Kuwait) using methodologies as described for family 1, with 89% of target bases covered at least 1× with an average depth of 33× per base. Using the same filtering parameters as those for family 1, we were able to reduce the number of variants from 199,920 to 18 (Table S2). As examination of these failed to identify any of these within the regions of shared homozygosity, we decided to look in more detail at SNX14. Specifically, we viewed the reads across SNX14 using Integrative Genomics Viewer (IGV) software (Broad Institute),24 which revealed a homozygous deletion of 9 consecutive exons from exon 13 to 21 (with respect to isoform a) (Figure S1A and Table S4), while all DNAs sequenced at the same time from unaffected subjects had good read depth for this interval (Figure S1A). The deletion was confirmed, first noting the lack of PCR amplification of individual exons from genomic DNAs, then by testing a series of primers flanking the approximate breakpoint position (Figure S1B). The latter strategy allowed determination of the precise sequence of the junction fragment. The deletion most likely occurred as a consequence of illegitimate recombination between two Alu-repeat sequences found in intron 12 and intron 21. This resulted in a deletion spanning 25,640 bases, described as c.1108+1181_2108−2342del with reference to isoform a cDNA or hg19 coordinates chr6: 86,255,692–86,230,052 (Figure S1C). In addition, Affymetrix CytoScan 750K array analysis of the youngest affected individual (V.2) revealed a homozygous deletion spanning five consecutive probes within SNX14, corresponding to the interval described above (Figure S2). Sanger sequencing of cDNA synthesized from fibroblast mRNA (V.1) confirmed that there was in-frame splicing between exon 12 and exon 22 (Figure S1D), which is predicted to remove 334 amino acids (p.Val369_Leu702del) from the full-length protein, including the entire RGS and PX domains (Figure 2C). We genotyped all of the available family members using a PCR-based approach to confirm segregation of the deletion. This revealed that only the four affected individuals were homozygous for the deletion, although eight unaffected individuals were heterozygous and three were homozygous for the reference allele (Figure 2A).
We next searched the Great Ormond Street Hospital Clinical Genetics records to identify affected individuals based solely on similar features. We performed text searches of the electronic medical records of all children known to the Clinical Genetics department using the terms “pontocerebellar hypoplasia,” “cerebellar hypoplasia,” OR “cerebellar atrophy” AND “hearing loss” and manually examined all sets of notes and clinical photographs, excluding all affected individuals with an alternative diagnosis or with microcephaly. This strategy identified family 2 (above) and a single further individual in an unrelated Turkish consanguineous family (family 3) among more than 50 case subjects matching the pontocerebellar or cerebellar search terms. The individual III.1 from family 3 (Figure 2A) had cerebellar ataxia, sensorineural hearing loss, slightly coarse facial features, relative macrocephaly, and bilateral fifth finger camptodactyly but generally had milder features than the other affected individuals in terms of the radiological degree of cerebellar atrophy and her motor, intellectual, and speech development (Table 1). Sanger sequencing of SNX14 exons and exon-intron boundaries in III.1 identified a homozygous canonical splice site mutation (c.1894+1G>A), which was heterozygous in both parents (Figure 2A). Using mRNA isolated from peripheral blood lymphocytes, we confirmed in-frame splicing of exon 18 to exon 20 (isoform a), completely skipping exon 19 (Figure S3). This was predicted to result in the deletion of 30 amino acids (p.Ala603_Gly632del) from within the PX domain (Figure 2C).
Using fibroblast cDNA, we compared mutant and wild-type transcript levels using quantitative RT-PCR with Power SYBR green PCR Master Mix on StepOnePlus Real-Time PCR Systems (Life Technologies). Compared to controls, we noted significantly reduced levels of expression in families 1 and 3, whereas levels in family 2 were similar (Figure 3). To investigate protein levels, SNX14 was analyzed by immunoblotting (Figure S4). A single band of ∼110 kDa was reproducibly obtained in control fibroblasts, but no band was detected in either family 1:IV.6 or 2:V.1. For family 1:IV.6, only a low level of mRNA was present, and this may produce an unstable or subdetectable level of protein. This also ties in with the possibility of activation of the nonsense-mediated decay pathway. For family 2:V.1, the deletion removes 112 of the 131 amino acid peptides used to raise the anti-SNX14 antibody. Therefore, it is conceivable that a truncated protein is present but not detectable with this assay. For family 3:III.2, a protein of slightly lower molecular weight (∼107 kDa) is detected, approximating to the loss of amino acids due to skipping of exon 19. Collectively, the mutation data are supportive of a loss of normal biological function for SNX14.
Figure 3.
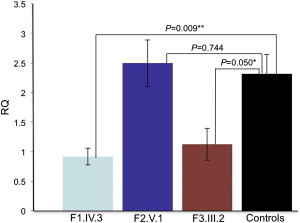
SNX14 RNA Expression in Fibroblasts from Affected and Control Individuals
qRT-PCR showing relative quantification (RQ ΔΔCT method) of SNX14 expression in affected fibroblasts samples from each family compared with controls. Both GAPDH and HPRT1 were used as combined endogenous controls, with RQ calculated using StepOne analysis software v.2.1 (Life Technologies). Each experiment was biologically replicated three times with each sample analyzed in triplicate. Using a two-tailed t test, family 1 individual IV.3 shows a 60% decrease in expression compared to combined controls, where fibroblasts from five different control individuals have been analyzed (N = 5), ∗∗p = 0.009. Family 3 individual III.2 shows a 51% drop in expression compared to controls, ∗p = 0.05. However, family 2 individual V.1 shows no significant difference in expression compared to controls, p = 0.74. Error bars correspond to mean ± SEM.
Therefore, we identified a total of seven affected individuals from three unrelated families who share a distinct rare syndrome resulting from SNX14 mutations. Neither point mutation was present in the NHLBI-ESP-EVS 4,870 exomes or in our internal database of 358 exomes. The Database of Genomic Variants lists 13 unaffected individuals who are heterozygous for 7 different but overlapping microdeletions of ∼31–130 kb (n = 1/2,026; 1/2,026; 7/2026; 1/1,557; 1/443; 1/17,421; 1/17,421)25–28 and a single 40 kb microduplication (n = 1/2,026),25 all involving parts of the SNX14 locus. However, a review of 4,500 GOSH children screened using the Affymetrix 750K array did not identify any further individuals, other than from family 2, who carry deletions or duplications that included SNX14.
Only five individuals with microdeletions involving SNX14 are well described in the literature.29–32 Two of these are included in DECIPHER, which lists seven individuals with intellectual disability and other various defects who have large (∼5 Mb) heterozygous deletions, and a further two with duplications that encompass SNX14 and other nearby genes. Of these, only the two unrelated individuals described by Wentzel et al.,31 who have large interstitial deletions (8.7 Mb and 4.5 Mb) containing SNX14 and three neighboring genes, have some phenotypic similarity to our affected individuals. In particular this includes the presence of ID, similar facial dysmorphic features, hearing loss, and macrocephaly. Additionally, one of these individuals had camptodactyly and limited movement of the elbows. None of them were reported to have cerebellar atrophy or ataxia, but both of them had walking difficulties and one was reported to have dyspraxia. Currently, we cannot exclude the presence of a second, SNX14-specific point mutation on the non-CNV-carrying chromosome in these cases.
In an attempt to further extend the phenotypic spectrum associated with SNX14 mutations, we investigated whole-exome sequencing data obtained for a series of 36 individuals with idiopathic pontocerebellar hypoplasia, and 168 from dominant and recessive families with idiopathic cerebellar ataxia, of which 138 were recessive/early onset or no family history. However, no likely causal variants or CNVs affecting SNX14 were identified. These data suggest that autosomal-recessive SNX14 mutations are associated with a narrow clinical spectrum. The presence of coarse faces and the absence of microcephaly and epilepsy are distinctive features within this group of conditions, confirming our findings reported in the description of the first family.21 Additionally, sensorineural hearing loss and camptodactyly of fifth fingers seem also to be prevalent (Table 1). Nevertheless, it should be noted that clinical recognition is challenging in infants because dysmorphic features, cerebellar involvement, ID, speech impairment, and ataxia are progressive and absent at an early age. Neuroradiological scans performed in the first years of life appeared normal but later were characterized by a globally small cerebellum (Table 1, Figure 1). Both the hemispheres and vermis are affected and there is separation of the folia indicating atrophy rather than hypoplasia (Figure 1). Evidence for slight pontine thinning was seen in older patients. This phenotype is significantly different from the pontocerebellar hypoplasias group of conditions, which are usually more severe, with prenatal-onset hypoplasia/atrophy of cerebellum and pons, associated with progressive microcephaly and seizures.
SNX14 is a member of the large family of sorting nexin proteins but it has only recently been investigated for its tissue distribution and cellular function. The earliest report describes Snx14 mRNA expression using an in vitro motoneuron selection method.33 Mice in this study were found to have the highest levels of Snx14 mRNA expression at E12.5, restricted to neuronal lineages such as the spinal cord. Expression in the brain was seen in the ventral ventricular region, the floor plate, V (trigeminal) and VIII (vestibulocochlear) cranial ganglia, the saccule of the inner ear, the developing pituitary gland, and eye. In general, mRNA distribution was described as colocalizing with Islet-1 expression, including Islet-1-positive motoneurons.33 Furthermore, Northern blot analysis in different adult mouse tissues revealed Snx14 expression in cerebellum and hippocampus and at much lower levels in cortex, muscle, liver, lung, and heart.33
We investigated SNX14 mRNA expression in human fetal tissue using RT-PCR and found it to be ubiquitously present in all tissues analyzed, including heart, skin, brain, kidney, bone, liver, eye, and placenta (Figure S5). The Allen Brain Atlas reports in situ hybridization for the p56 mouse brain, where the highest levels of Snx14 expression were found in the granule and Purkinje cell layers of the cerebellar cortex but also in the hippocampus (granule layers and dentate gyrus) and the piriform. This was supported by data from both the human UK Brain Expression Consortium (UKBEC) and the Human Brain Transcriptome database.34,35 SNX14 is expressed in all brain regions, with generally increasing levels during prenatal development and then plateauing. Interestingly, the pattern the cerebellum is the region where SNX14 is most highly expressed and transcript levels continue to increase through postnatal life until adult (UKBEC data, Figure S6). Most recently, Huang et al.36 described mouse SNX14 levels as being high in brain, testis, and lung, similarly showing an increase in cerebellar levels from embryonic to postnatal stages (E16.5–p63). Despite a broadly distributed spatiotemporal pattern, which might suggest function in many tissues, high levels in the central nervous system and particularly cerebellum correspond to the described phenotype (with internal organs spared), particularly the cerebellar atrophy seen in affected individuals from all three families presented here.
To date there are 49 mammalian proteins known to contain a PX domain and the majority of these are classified as sorting nexin proteins.37 The PX domain binds to phosphoinositides (PtdIns3P) on the cytoplasmic leaflets of various organelles and it defines both subcellular localization and function of different PX domain-containing proteins including endosomal sorting and trafficking.37 Mutations in the p47phox subunit of NADPH oxidase are known to cause autosomal-recessive chronic granulomatous disease (MIM 233700),38 whereas SNX27 has been indirectly linked to synaptic dysfunction in Down syndrome through impaired transcriptional regulation,39 and recently SNX10 mutations have been demonstrated to cause a nonsyndromic autosomal-recessive form of osteopetrosis (MIM 615085), an osteoclast-related bone disease.40
Based on predicted domain structure, SNX14 is classified within the PXA-RGS-PX-PXC subfamily along with SNX13, SNX19, and SNX25.41 Like SNX13, SNX14 also contains a putative double transmembrane domain including a short cytoplasmic leader sequence and an RGS domain, implying they share a similar function (Figure 2C). The RGS domain of SNX13 can bind to and increase the GTPase activity of the G-alphas (Gαs) subunit of G-protein coupled receptors (GPCRs). Mediated by the PX domain, this activity can occur at the endosome, allowing Gαs signal attenuation at the interface with this protein sorting and degradation pathway.42,43 Loss of functional SNX13 in the mouse resulted in dramatically altered endocytic/lysosomal compartmentalization in visceral yolk sac endoderm, with abnormal localization of several endocytic markers and with the appearance of abundant autophagic vacuoles.44
A role for SNX14 in endosomal sorting and the regulation of protein degradation is supported by recent analyses of the prenatal human brain transcriptome.45 Weighted gene coexpression network analysis (WGCNA) was used to group genes expressed within mid-fetal human neocortex into modules in an unsupervised manner.46 This approach is extremely useful in identifying modules of biologically related genes that are not just coexpressed, but coregulated.47 This method was applied to predict that SNX14 is highly coexpressed within a module (C25, black),45 which is significantly enriched for genes involved in cellular protein metabolism (Gene ontology term GO:0044267∼cellular protein metabolic process, Bonferroni-corrected p value = 9.18 × 10−5) and vesicle-mediated transport between the ER and Golgi (GO:0006888∼ER to Golgi vesicle-mediated transport, Bonferroni-corrected p value = 7.59 × 10−4).
This prompted us to examine cellular ultrastructural morphology in skin biopsy material and fibroblast cell lines, using a JEOL 1400 transmission electron microscope (Figure 4). Compared to control samples, the skin biopsy showed an adequate epithelial layer with a moderate degree of hyperkeratosis and occasional apoptotic bodies. Vacuoles with fine nonspecific granular material were identified in keratinocytes (Figure 4B). In the dermis, collagen and elastic tissue had a normal appearance and distribution. Fibroblasts demonstrated infrequent vacuoles with granular material and rare electron-dense laminated inclusions suggestive of lipid degeneration. No sweat glands were available for assessment and small myelinated and unmyelinated nerves were unremarkable. Next, we investigated ultrathin sections from cultured skin fibroblasts from one affected individual from each of the three families and control subjects. Affected fibroblasts were found to have frequent cytoplasmic vacuolation with electron-dense material of variable morphology including some with evidence of lamella structure as seen in multilamellar bodies (Figures 4D–4F). Interestingly, a proportion of fibroblasts from affected individuals but not control subjects immunostain positive for p62 in a granular pattern (Figure S7), which might suggest a defect in the autophagy pathway, as seen in many neurodegenerative and neurodevelopmental disorders.48 Interestingly, cultured fibroblasts from individuals with MSS similarly contain numerous cytoplasmic electron-dense, sometimes multilamellar inclusion, bodies in both individuals with and without SIL1 mutations.1 MSS and the SNX14 phenotype share the presence of ID, cerebellar atrophy, hypotonia, and ataxia. Therefore SNX14 should be screened in MSS-like individuals not carrying an SIL1 mutation, especially in those without microcephaly, cataracts, or myopathy (creatinine kinase levels were in the normal range for the four individuals tested who have SNX14 mutations).
Figure 4.
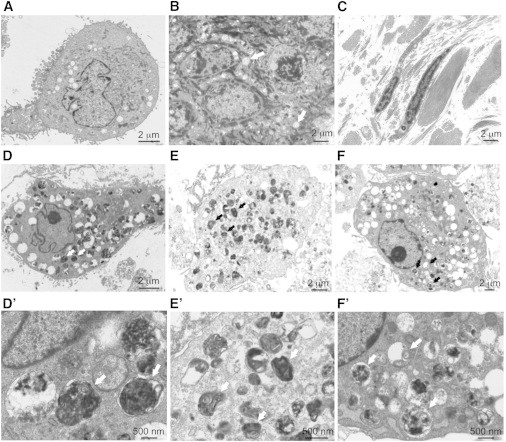
Electron Micrographs of Skin Biopsy and Cultured Fibroblasts
(A) Control cultured fibroblast at 800× magnification.
(B and C) Skin section from family 3 individual III.2 at 800× magnification.
(B) In the epidermis, there was mild hyperkeratosis with keratinocytes showing increased vacuolation, containing fine nonspecific granular material (arrows).
(C) In the dermis, collagen and elastic tissue had a normal appearance and distribution with infrequent vacuoles in the fibroblasts.
(D–F) Cultured fibroblasts from family 1 individual IV.6, family 2 individual V.1, and family 3 individual III.2, respectively, at 800× magnification. The cells showed numerous cytoplasmic vacuoles, often containing dense staining material suggestive of lipid degeneration. Same cells at 3,000× magnification shown in (D′)–(F′). Vacuoles contained granular material or multilamellar bodies or were empty (see white and black arrows for examples).
Zheng et al.44 reported that heterozygous Snx13+/− mice do not have an obvious phenotype, whereas Snx13−/− mice die between E10.5 and E14.5. Mutants are small and have exencephaly and abnormal cephalic vascularization, presumably in response to defective nutrient uptake and transport, particularly in the yolk sac endoderm, which could account for embryonic developmental delay. Snx14 knockdown studies using lentiviral shRNA, specifically in mouse cortical pyramidal neurons, were recently reported.36 Despite incomplete (60%) knockdown, significantly reduced intrinsic excitability and synaptic function was recorded. The study of Huang et al.36 also reported Snx14 to be maternally imprinted but suggest that this might be mouse specific. Our data support this because unaffected heterozygous individuals inherit the mutation on either the maternal or paternal allele (Figure 2A).
In conclusion, in terms of a potential mechanism there is a compelling case for a role of SNX14 in synaptic transmission.40 Synaptic dysfunction is well known to be implicated in neurodevelopmental disorders associated with ID49 and this may underlie the pathogenesis of SNX14 mutations. Other genes coding RGS proteins, especially of the Rho GTPase family, cause ID syndromes and have a crucial role in synaptic structure/function.50 Interestingly, a significant phenotypic overlap is seen with OPHN1 encoding a Rho-GAP protein, in which mutations cause an X-linked form of syndromic ID typically associated with cerebellar abnormalities, dysmorphic (often coarse) facial features, and sometimes macrocephaly.5,50 Overall, our results implicate an essential role for SNX14 with the breakdown and recycling of cellular components in human cerebellar development and maintenance.
Acknowledgments
We would like to thank the families who contributed to this study. We would also like to thank the expert assistance of Kerra Pearce (UCL Genomics) for running the SNP genotyping arrays, Osama Alsmadi and Dinu Antony for exome sequencing at The Dasman Diabetes Institute Genome Centre (Kuwait City), and Manuela Grazina (Faculty of Medicine, University of Coimbra) and Biljana Lukovic (Chemical Pathology, Great Ormond Street Hospital for Children) for providing an affected individual’s fibroblast culture. P.S. is supported by the Great Ormond Street Hospital Children’s Charity. S.B.S. was supported by Fundação para a Ciência e Tecnologia (SFRH/BD/46778/2008). A.C.T. is a Wellbeing of Women Research Associate. G.E.M.’s Fetal Growth and Development research group is supported by Wellbeing of Women, MRC, Sparks and the National Institute for Health Research Biomedical Research Centre (NIHR BRC) at Great Ormond Street Hospital for Children NHS Foundation Trust, and UCL Institute of Child Health. F.B. is supported by the Joshua Deeth foundation. We thank the additional members of the GOSgene Scientific Advisory Board (Philip Beales, Robert Kleta, Horia Stanesku, Nicholas Lench, Bobby Gaspar, Michael Hubank, and Elia Stupka). GOSgene is supported by the NIHR BRC at Great Ormond Street Hospital for Children NHS Foundation Trust and UCL Institute of Child Health. This report is independent research by the NIHR BRC Funding Scheme. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Footnotes
This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/3.0/).
Contributor Information
Sérgio B. Sousa, Email: sbsousa@chc.min-saude.pt.
Philip Stanier, Email: p.stanier@ucl.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Allen Mouse Brain Atlas, http://mouse.brain-map.org
Database of Genomic Variants (DGV), http://dgv.tcag.ca/dgv/app/home
Ingenuity Variant Analysis, http://www.ingenuity.com/products/variant-analysis
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Ezgu F., Krejci P., Li S., de Sousa C., Graham J.M., Jr., Hansmann I., He W., Porpora K., Wand D., Wertelecki W. Phenotype-genotype correlations in patients with Marinesco-Sjögren syndrome. Clin. Genet. 2014;86:74–84. doi: 10.1111/cge.12230. [DOI] [PubMed] [Google Scholar]
- 2.Strehle E.M. Sialic acid storage disease and related disorders. Genet. Test. 2003;7:113–121. doi: 10.1089/109065703322146795. [DOI] [PubMed] [Google Scholar]
- 3.Waterham H.R., Ebberink M.S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta. 2012;1822:1430–1441. doi: 10.1016/j.bbadis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Matthijs G., Schollen E., Pardon E., Veiga-Da-Cunha M., Jaeken J., Cassiman J.J., Van Schaftingen E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat. Genet. 1997;16:88–92. doi: 10.1038/ng0597-88. [DOI] [PubMed] [Google Scholar]
- 5.Philip N., Chabrol B., Lossi A.M., Cardoso C., Guerrini R., Dobyns W.B., Raybaud C., Villard L. Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J. Med. Genet. 2003;40:441–446. doi: 10.1136/jmg.40.6.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Namavar Y., Chitayat D., Barth P.G., van Ruissen F., de Wissel M.B., Poll-The B.T., Silver R., Baas F. TSEN54 mutations cause pontocerebellar hypoplasia type 5. Eur. J. Hum. Genet. 2011;19:724–726. doi: 10.1038/ejhg.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budde B.S., Namavar Y., Barth P.G., Poll-The B.T., Nürnberg G., Becker C., van Ruissen F., Weterman M.A., Fluiter K., te Beek E.T. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 2008;40:1113–1118. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- 9.Karaca E., Weitzer S., Pehlivan D., Shiraishi H., Gogakos T., Hanada T., Jhangiani S.N., Wiszniewski W., Withers M., Campbell I.M., Baylor Hopkins Center for Mendelian Genomics Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaffer A.E., Eggens V.R., Caglayan A.O., Reuter M.S., Scott E., Coufal N.G., Silhavy J.L., Xue Y., Kayserili H., Yasuno K. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014;157:651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan J., Yourshaw M., Mamsa H., Rudnik-Schöneborn S., Menezes M.P., Hong J.E., Leong D.W., Senderek J., Salman M.S., Chitayat D. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boczonadi V., Müller J.S., Pyle A., Munkley J., Dor T., Quartararo J., Ferrero I., Karcagi V., Giunta M., Polvikoski T. EXOSC8 mutations alter mRNA metabolism and cause hypomyelination with spinal muscular atrophy and cerebellar hypoplasia. Nat. Commun. 2014;5:4287. doi: 10.1038/ncomms5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mochida G.H., Ganesh V.S., de Michelena M.I., Dias H., Atabay K.D., Kathrein K.L., Huang H.T., Hill R.S., Felie J.M., Rakiec D. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat. Genet. 2012;44:1260–1264. doi: 10.1038/ng.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Najm J., Horn D., Wimplinger I., Golden J.A., Chizhikov V.V., Sudi J., Christian S.L., Ullmann R., Kuechler A., Haas C.A. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008;40:1065–1067. doi: 10.1038/ng.194. [DOI] [PubMed] [Google Scholar]
- 15.Renbaum P., Kellerman E., Jaron R., Geiger D., Segel R., Lee M., King M.C., Levy-Lahad E. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am. J. Hum. Genet. 2009;85:281–289. doi: 10.1016/j.ajhg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Arrigo S., Viganò L., Grazia Bruzzone M., Marzaroli M., Nikas I., Riva D., Pantaleoni C. Diagnostic approach to cerebellar disease in children. J. Child Neurol. 2005;20:859–866. doi: 10.1177/08830738050200110101. [DOI] [PubMed] [Google Scholar]
- 17.Anheim M., Fleury M., Monga B., Laugel V., Chaigne D., Rodier G., Ginglinger E., Boulay C., Courtois S., Drouot N. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11:1–12. doi: 10.1007/s10048-009-0196-y. [DOI] [PubMed] [Google Scholar]
- 18.Poretti A., Wolf N.I., Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood. Eur. J. Paediatr. Neurol. 2008;12:155–167. doi: 10.1016/j.ejpn.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Evereklioglu C., Doganay S., Er H., Gunduz A., Tercan M., Balat A., Cumurcu T. Craniofacial anthropometry in a Turkish population. Cleft Palate Craniofac. J. 2002;39:208–218. doi: 10.1597/1545-1569_2002_039_0208_caiatp_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 20.Elmali F., Altunay C., Mazicioglu M.M., Kondolot M., Ozturk A., Kurtoglu S. Head circumference growth reference charts for Turkish children aged 0-84 months. Pediatr. Neurol. 2012;46:307–311. doi: 10.1016/j.pediatrneurol.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 21.Sousa S.B., Ramos F., Garcia P., Pais R.P., Paiva C., Beales P.L., Moore G.E., Saraiva J.M., Hennekam R.C.M. Intellectual disability, coarse face, relative macrocephaly, and cerebellar hypotrophy in two sisters. Am. J. Med. Genet. A. 2014;164A:10–14. doi: 10.1002/ajmg.a.36235. [DOI] [PubMed] [Google Scholar]
- 22.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O’Hara R., Casalunovo T., Conlin L.K., D’Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 28.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turleau C., Demay G., Cabanis M.O., Lenoir G., de Grouchy J. 6q1 monosomy: a distinctive syndrome. Clin. Genet. 1988;34:38–42. doi: 10.1111/j.1399-0004.1988.tb02613.x. [DOI] [PubMed] [Google Scholar]
- 30.Van Esch H., Rosser E.M., Janssens S., Van Ingelghem I., Loeys B., Menten B. Developmental delay and connective tissue disorder in four patients sharing a common microdeletion at 6q13-14. J. Med. Genet. 2010;47:717–720. doi: 10.1136/jmg.2010.077586. [DOI] [PubMed] [Google Scholar]
- 31.Wentzel C., Lynch S.A., Stattin E.L., Sharkey F.H., Annerén G., Thuresson A.C. Interstitial deletions at 6q14.1-q15 associated with obesity, developmental delay and a distinct clinical phenotype. Mol. Syndromol. 2010;1:75–81. doi: 10.1159/000314025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowry R.B., Chernos J.E., Connelly M.S., Wyse J.P. Interstitial deletions at 6q14.1q15 associated with developmental delay and a marfanoid phenotype. Mol. Syndromol. 2013;4:280–284. doi: 10.1159/000354038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carroll P., Renoncourt Y., Gayet O., De Bovis B., Alonso S. Sorting nexin-14, a gene expressed in motoneurons trapped by an in vitro preselection method. Dev. Dyn. 2001;221:431–442. doi: 10.1002/dvdy.1163. [DOI] [PubMed] [Google Scholar]
- 34.Trabzuni D., Ryten M., Walker R., Smith C., Imran S., Ramasamy A., Weale M.E., Hardy J. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J. Neurochem. 2011;119:275–282. doi: 10.1111/j.1471-4159.2011.07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang H.-S., Yoon B.-J., Brooks S., Bakal R., Berrios J., Larsen R.S., Wallace M.L., Han J.E., Chung E.H., Zylka M.J., Philpot B.D. Snx14 regulates neuronal excitability, promotes synaptic transmission, and is imprinted in the brain of mice. PLoS ONE. 2014;9:e98383. doi: 10.1371/journal.pone.0098383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teasdale R.D., Collins B.M. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease. Biochem. J. 2012;441:39–59. doi: 10.1042/BJ20111226. [DOI] [PubMed] [Google Scholar]
- 38.Casimir C.M., Bu-Ghanim H.N., Rodaway A.R., Bentley D.L., Rowe P., Segal A.W. Autosomal recessive chronic granulomatous disease caused by deletion at a dinucleotide repeat. Proc. Natl. Acad. Sci. USA. 1991;88:2753–2757. doi: 10.1073/pnas.88.7.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X., Zhao Y., Zhang X., Badie H., Zhou Y., Mu Y., Loo L.S., Cai L., Thompson R.C., Yang B. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat. Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pangrazio A., Fasth A., Sbardellati A., Orchard P.J., Kasow K.A., Raza J., Albayrak C., Albayrak D., Vanakker O.M., De Moerloose B. SNX10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity. J. Bone Miner. Res. 2013;28:1041–1049. doi: 10.1002/jbmr.1849. [DOI] [PubMed] [Google Scholar]
- 41.Worby C.A., Dixon J.E. Sorting out the cellular functions of sorting nexins. Nat. Rev. Mol. Cell Biol. 2002;3:919–931. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 42.Zheng B., Ma Y.-C., Ostrom R.S., Lavoie C., Gill G.N., Insel P.A., Huang X.-Y., Farquhar M.G. RGS-PX1, a GAP for GalphaS and sorting nexin in vesicular trafficking. Science. 2001;294:1939–1942. doi: 10.1126/science.1064757. [DOI] [PubMed] [Google Scholar]
- 43.Zheng B., Lavoie C., Tang T.-D., Ma P., Meerloo T., Beas A., Farquhar M.G. Regulation of epidermal growth factor receptor degradation by heterotrimeric Galphas protein. Mol. Biol. Cell. 2004;15:5538–5550. doi: 10.1091/mbc.E04-06-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng B., Tang T., Tang N., Kudlicka K., Ohtsubo K., Ma P., Marth J.D., Farquhar M.G., Lehtonen E. Essential role of RGS-PX1/sorting nexin 13 in mouse development and regulation of endocytosis dynamics. Proc. Natl. Acad. Sci. USA. 2006;103:16776–16781. doi: 10.1073/pnas.0607974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller J.A., Ding S.L., Sunkin S.M., Smith K.A., Ng L., Szafer A., Ebbert A., Riley Z.L., Royall J.J., Aiona K. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konopka G. Functional genomics of the brain: uncovering networks in the CNS using a systems approach. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011;3:628–648. doi: 10.1002/wsbm.139. [DOI] [PubMed] [Google Scholar]
- 48.Lee K.M., Hwang S.K., Lee J.A. Neuronal autophagy and neurodevelopmental disorders. Exp. Neurobiol. 2013;22:133–142. doi: 10.5607/en.2013.22.3.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zoghbi H.Y., Bear M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012;4:a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ba W., van der Raadt J., Nadif Kasri N. Rho GTPase signaling at the synapse: implications for intellectual disability. Exp. Cell Res. 2013;319:2368–2374. doi: 10.1016/j.yexcr.2013.05.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.