

Abstract
Rare-variant association studies in common, complex diseases are customarily conducted under an additive risk model in both single-variant and burden testing. Here, we describe a method to improve detection of rare recessive variants in complex diseases termed RAFT (recessive-allele-frequency-based test). We found that RAFT outperforms existing approaches when the variant influences disease risk in a recessive manner on simulated data. We then applied our method to 1,791 Finnish individuals with type 2 diabetes (T2D) and 2,657 matched control subjects. In BBS10, we discovered a rare variant (c.1189A>G [p.Ile397Val]; rs202042386) that confers risk of T2D in a recessive state (p = 1.38 × 10−6) and would be missed by conventional methods. Testing of this variant in an established in vivo zebrafish model confirmed the variant to be pathogenic. Taken together, these data suggest that RAFT can effectively reveal rare recessive contributions to complex diseases overlooked by conventional association tests.
Introduction
Genome-wide association studies (GWASs) have identified numerous additive genomic variants associated with complex disorders,1 which appear to impact liability to disease (or affect a normally distributed trait) linearly with an effect size of a when heterozygous and 2a when homozygous. Most of these variants are common (≥5% frequency) and have only small effect sizes (odds ratio [OR] < 1.2) on complex disease classifications. However, such variants do not explain the full heritability of disease. As rare variants have been comparatively less well studied, attention has shifted to exome- or genome-sequence-based approaches to identifying additional risk factors. Another less-well-explored hypothesis is that there might be variants that influence disease susceptibility in a nonadditive fashion, for instance, if the variants influence disease risk recessively, that is, only when the minor allele is found in a homozygous state, or in trans with another minor allele to form compound heterozygotes. However, most widely used tests for disease association, such as allele-based regression tests or burden tests for aggregation of rare alleles, assume an additive model. Such approaches are well powered to detect common alleles even if they influence disease risk in a nonadditive fashion. Common variants in genes such as FUT2 (MIM 182100) and TYRP1 (MIM 115501) have also been demonstrated to influence complex diseases and traits in a recessive manner;2,3 these loci were detected by the typical additive approaches because there are many homozygotes observed for these common variants and therefore strong signal exists even under the additive model.4
In contrast, the power of the additive model to detect recessive alleles diminishes greatly at lower frequencies, since the numbers of homozygotes observed are far fewer. Consequently, if there are rare variants that confer significant risk of the phenotype in a recessive manner, conventional tests are underpowered to detect such variants in complex diseases. However, extensive runs of homozygosity have been associated with complex diseases and traits such as height5 (MIM 606255), schizophrenia6 (MIM 181500), and autism spectrum disorders7,8 (MIM 209850), suggesting that rare recessive variants in these regions might be driving disease risk. Specifically for autism spectrum disorders, further studies have shown that there is a strong signal from recessively acting rare variants involved in the disease etiology.9,10 Moreover, since a high proportion of rare Mendelian disease alleles confer risk in a strictly dominant or recessive fashion, testing for nonadditive effects of rare variants in common, complex diseases might well yield fruitful associations missed by conventional association tests.
We describe a statistical methodology (termed RAFT for recessive allele frequency-based test) designed specifically for detecting variants with recessive effects on a dichotomous phenotype. We demonstrate that RAFT has considerably more power to detect disease-associated rare variants than conventional approaches. In applying RAFT to an exome chip data set comprising 4,448 individuals, 1,791 of whom are case subjects with type 2 diabetes (T2D [MIM 125853]), we detected a rare variant in Bardet-Biedl syndrome 10 (BBS10 [MIM 610148]) that confers significant risk of T2D in a recessive manner (p = 1.38 × 10−6). Functional testing of this allele in vivo confirmed its effect on protein function, providing further support for the genetic association. Thus, increasing power for specific modes of inheritance offers the potential to discover novel associations in common complex diseases (as it clearly has in rare diseases), particularly for rare recessive variants for which conventional statistical methods are bereft of power.
Material and Methods
Exome Chip Genotyping and Quality Control
The DNA samples were obtained from the Botnia and Direva Studies. For genotyping, the samples were sent to the Broad Institute and prepared for genetic analysis with two quality-control measures. First, DNA quantity was measured by Picogreen, and then all samples with sufficient total DNA and minimum concentrations for downstream activities were genotyped for a set of 24 SNPs with the Sequenom iPLEX Assay. These 24 validated markers include 1 gender assay and 23 SNPs located across the autosomes. The genotypes for these SNPs were used as a quality filter to advance samples, as well as a technical fingerprint validation (when applicable) for array genotypes. This study was approved by the MGH/Broad/HMS institutional review board, and appropriate informed consent was obtained from human subjects.
All genotyping was performed at the Broad Institute Genetic Analysis Platform. DNA samples were placed on 96-well plates and genotyped with the Illumina HumanExome v.1.1 SNP array. Genotypes were assigned with GenomeStudio v.2010.3 and the calling algorithm/genotyping module v.1.8.4 with the custom cluster file HumanExomev1_1_CEPH_A.egt. Subsequent processing of genotype calling was done by zCall.11 Samples with two or more discordant fingerprint genotypes and/or call rates below 97% were excluded from data analysis. Individuals with call rates below 99% were excluded from analysis, as were SNVs with call rates below 80% or with extreme deviations (p < 1 × 10−8) from Hardy-Weinberg equilibrium (HWE). The accuracy of genotypes for the BBS10 c.1189A>G variant was assessed via visual inspection of intensity plots (Figure S1, available online).
Case-Control Principal-Component Analysis and Matching
Principal-component analysis was performed on a set of linkage-disequilibrium-independent SNPs with PLINK12 and EIGENSTRAT.13 In addition, to reduce heterogeneity between the case and control subjects, we performed one-to-one matching for each case-control pair by using the first ten principal components and selecting the closest matched control for each case. Case-control pairs with a sum of the absolute differences in the first ten principal components of >0.06 were removed from the analyses. In addition, cases with excessive relatedness (identity by descent > 0.2) were removed from the analyses.
Data Filtering and Annotation
In addition, all variants with heterozygous counts of ≤3 (allele frequency of ∼0.025%) in case and control subjects were removed to reduce the number of variants that were miscalled as rare homozygotes instead of heterozygotes. To further reduce genotyping errors, variants with HWE p values of ≤1 × 10−3 in the controls were removed. The variants were then annotated with the Variant Effect Predictor.
Estimating the Probability of Observing Homozygotes
The key factor in any homozygosity analysis is the probability of minor allele (a) being homozygous P(aa), which in theory is simply P(a)2 and is estimated directly from the observed allele counts. Two scenarios exist when this expectation may not be met and can be readily accommodated up front in this analysis framework. Substructure or consanguinity in a population can cause global departure from HWE, resulting in systematic genome-wide excess homozygosity. In such cases, we can fit a regression to all variants in the data set to estimate a substructure or homozygosity by descent factor F by using the standard model P(aacorrected) = FP(a) + (1 − F)P(a)2 and can then utilize this corrected value as P(aa) below. This might be important for analysis of populations with high inbreeding.
Additionally, in actual genotyping and sequencing data, we encounter occasions where local departures from HWE in control subjects (as well as case subjects) are observed as a result of hemizygous deletions or systematic genotyping errors. While such local genotyping artifacts can be detected and avoided in many ways, in cases where the observed rate of homozygous genotypes in control subjects exceeds the expected corrected P(aa), we can conservatively set the P(aa) used below to the higher observed rate in control subjects, thereby insuring that the probability of homozygosity that the test statistic relies on is insulated from both global and local sources of inflation.
RAFT Statistics
Under the alternative hypothesis described below, the maximum-likelihood estimate of P(aa) is altered slightly (since excess homozygosity in cases is presumed to arise from case ascertainment), but this difference is generally negligible and not tested as a parameter in the association test below. Under the alternative hypothesis of recessive association, the probability of the minor allele being homozygous in a case P(aa | case, γ), given a genotypic relative risk to homozygotes (γ), is
And the probability of not observing a homozygote for the minor allele in a case subject is
The alternative calculation for homozygotes for the minor allele in control subjects P(aa | control, γ) is similar. However, for selected control subjects, the formulation includes the estimated prevalence of the disease (ϕ), which we have fixed as a constant to 1 in 100,000. For convenience, we use the variable x to represent the prevalence of disease in control individuals where , which is the prevalence under the null. As such, P(aa | control, γ) and can be calculated by
and .
And the resulting likelihood of the observation of number of alternate homozygotes in the case subjects, number of heterozygotes and reference homozygotes in the case subjects, number of alternate homozygotes in the control subjects, number of heterozygotes and reference homozygotes in the control subjects, is proportional to
or
where ncase is the total number of case subjects and ncontrol is the total number of control subjects. We next performed an expectation maximization (EM) step to maximize the log likelihood ratios between the alternate model L1 and the null model L0, to obtain the following RAFT statistic:
or
where γcase is equivalent to the OR of the alternate homozygotes for the variant. To calculate a p value, we can estimate the following to a chi-square distribution with 1 degree of freedom ():
Simulations Using Whole-Exome Sequencing Data for Finns and NFEs
We used two different populations—Finns and non-Finnish Europeans (NFEs)—to simulate a scenario where the samples used in the association are not homogeneously from a single population even if case and control subjects are well matched. Using whole-exome sequencing data available for Finns and NFEs,14 we randomly sampled genotypes from 2,500 Finns and 2,500 NFEs and assigned them as case subjects. Similarly, we randomly sampled genotypes for another 2,500 Finns and 2,500 NFEs and assigned them as control subjects. For each variant, we calculated the number of individuals that were heterozygous or homozygous for the minor allele in both case and control subjects. To test the RAFT statistic in a simulated data set without population substructure, we simulated genotypes of 5,000 Finns as case subjects and 5,000 Finns as control subjects. These simulations were repeated 100 times to obtain the 95% confidence interval for the distribution of p values calculated with the RAFT statistic.
Microinjection and Morphometric Analysis of Zebrafish Embryos
Translation blocker (TB) morpholinos against zebrafish bbs10 were synthesized by Gene Tools (5′-CGTTAAACCTCTTCTGTGAACCAGC-3′). Human BBS10 mRNA was in vitro transcribed with mMESSAGE mMACHINE SP6 Kit (Ambion). A volume of 0.5 nl mixture of 5 ng bbs10 TB and/or 75 pg mRNA was microinjected into the yolk of embryos at the 1- to 8-cell stage. At the 9- to 10-somite stage, embryos were analyzed for convergent extension defects according to previously established phenotypic criteria.15–17
Glucose Tolerance Test
At 1 day postfertilization (dpf), zebrafish embryos were dechorionated and transferred into egg water (recipe in Westerfield18) with 1% D-glucose. At 4 dpf, total RNA was extracted from zebrafish embryos with Trizol (Invitrogen) and then reverse transcripted into cDNA with SuperScript III (Invitrogen), to provide template for real-time qPCR. The expression levels of pdk2 and runx1, as well as gapdh for internal control, were examined with the following primers: dr-pdk2-qF, 5′-CGAATTAGCCAATAAACCAACAAA-3′; dr-pdk2-qR, 5′-CACACTTCACCTGCATTTCCA-3′; dr-runx1-qF, 5′-CGTCTTCACAAACCCTCCTCAA-3′; dr-runx1-qR, 5′-GCTTTACTGCTTCATCCGGCT-3′; dr-gapdh-qF, 5′-TTGTAAGCAATGCCTCCTGC-3′; dr-gapdh-qR, 5′-CTGTGTTGCTGTGATGGCAT-3′.
Results
RAFT Statistic
Instead of directly comparing homozygous counts in case and control subjects, the RAFT statistic evaluates the likelihood of observing the number of homozygotes in the cases (Ncases) compared to the expected number of homozygotes given the allele frequency of the variant and normalizes this by the same statistic for observing Ncontrols, the number of homozygotes in the controls compared to the expected number of homozygotes. For instance, if there is a variant (variant A) with 0.5% allele frequency, we would expect to observe 0.05 homozygotes in 2,000 individuals (0.005 × 0.005 × 2,000) for this variant. Similarly, if there is another variant (variant B) with 5% allele frequency, we would expect to observe 5 homozygotes in 2,000 individuals (0.05 × 0.05 × 2,000). However, if we observed five homozygotes who are case subjects and zero homozygotes who are control subjects for both variants A and B, the RAFT statistic will assign a higher LOD score to variant A than to variant B because it is more unusual to observe five homozygotes in the case group for a 0.5% variant than for a 5% variant. The normalization of the log likelihood ratio with the observation in the control subjects brings in the case-control comparison and, more importantly, ensures that the results are adjusted for regions where there is excessive homozygosity in the control subjects (such as hemizygous copy-number polymorphic regions, regions with genotyping errors or arising from unusual substructure) that might not be truly associated with disease risk. A conventional test such as Fisher’s exact test will assign the same probabilities for both variants based on the observed numbers of homozygotes. However, the RAFT statistic essentially compares the observed five homozygotes in the case subjects with the expected number of homozygotes (0.05 for variant A), resulting in increased power.
Greater Power with RAFT for Detecting Rare Variants under a Recessive Model
To compare RAFT to existing approaches, we performed simulations to evaluate the power for detecting variants with allele frequency ranging from 0.1% to 20% that confer a modest risk (OR = 10) of disease under a recessive mode of inheritance (Figure 1). We observed that there might greater than 80% power to detect common recessive variants (allele frequency > 10%) by using the standard additive tests (chi-square and transmission disequilibrium test [TDT]) and that RAFT or a conventional recessive test (Fisher’s exact test on the homozygous counts [Hom-FET]) did not confer much advantage over these additive tests (Figure 2). In contrast, RAFT provides significantly more power to detect lower-frequency recessive variants (allele frequency ≤ 5%) than do all the other tests.
Figure 1.
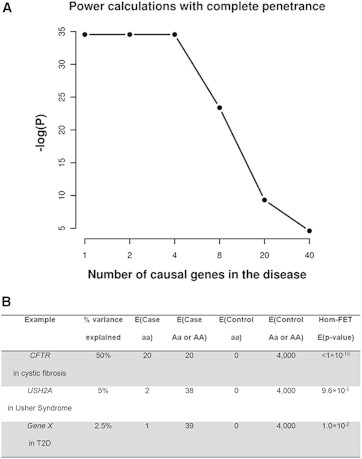
Scenarios to Illustrate p Values Obtained with Fisher’s Exact Test on the Homozygous Counts
(A) p values obtained if there are 1, 2, 4, 8, 20, or 40 genes with complete penetrance that are causal for the disease.
(B) p values with different percentages of variance explained by the homozygotes (aa) in the cases, assuming complete penetrance. This is an ideal scenario and the power for detection will be lower if the variants exhibit incomplete penetrance or if there are fewer controls available.
Figure 2.
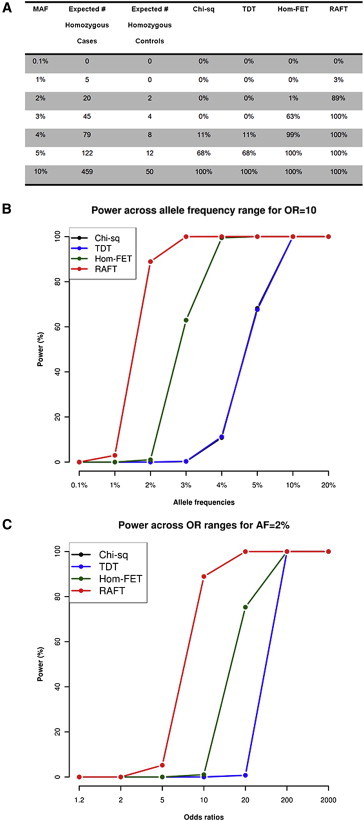
Power Calculations for Various Tests in Detecting Recessive Variants with OR = 10 and Disease Prevalence of 1% in 5,000 Case Subjects and 5,000 Control Subjects
(A) Power calculations for detecting recessive variants across different allele frequencies by using two allelic tests (chi-square and TDT), as well as two recessive tests (Fisher’s exact test on the homozygotes [Hom-FET] and RAFT).
(B) Graphical representation for the power calculations for chi-square, TDT, Hom-FET, and RAFT across various allele-frequency ranges from 0.1% to 20% with the same parameters.
(C) Power calculations for the same scenario but a variant with 2% allele frequency across various recessive ORs from 1.2 to 2,000.
Applying RAFT on Previously Reported Recessive Gene MKKS
We applied the RAFT statistic on a previously published instance where 2 out of 17 independent families affected by Bardet-Biedl syndrome (BBS [MIM 209900]) were reported to harbor the same homozygous c.280delT frameshift in MKKS19 (MIM 604896). The c.280delT frameshift was shown to be pathogenic and causal for BBS in these two families. However, the p value for this observation calculated by a conventional recessive test such as Hom-FET is 1.46 × 10−5, which is shy of the exome-wide significance threshold of 2.5 × 10−6 after correcting for 20,000 genes. Instead, by incorporating the allele frequency of the variant (the c.280delT variant was found to be heterozygous in 1 out of 4,300 individuals of European ancestry from the NHLBI Exome Variant Server and was found to be heterozygous in another 2 out of the 17 families affected by BBS), the p value calculated with RAFT for this observation is highly significant at 2.26 × 10−9. In an exome-wide screen involving several more families and many more variants, it is thus possible that a conventional test might miss the association between the MKKS c.280delT variant and BBS while the RAFT statistic flags this variant as highly significant.
The RAFT Statistic Conforms to the Null Hypothesis when Tested on a Simulated Finnish Data Set
To evaluate the null behavior of our test statistic, we performed RAFT analysis on simulated data by using allele frequencies from a whole-exome sequencing data set composed of 3,000 individuals of Finnish ancestry and 3,000 NFEs14 and consisting of 590,003 coding region variants. For each variant, we randomly simulated 5,000 case subjects and 5,000 control subjects by using the Finnish allele frequencies and applied the RAFT analysis. We found that across all allele frequency bins (common ≥ 5%, low-frequency 1%–5%, or rare < 1%), we obtained similar numbers of observed and expected variants in the various p value bins (Table S1), suggesting that the RAFT statistic conforms to the null hypothesis when the individuals are drawn from a homogenous population and there is no underlying signal of any type.
Nonhomogeneous Ancestry Can Cause Inflation to the RAFT Statistic
Given that RAFT is a method designed to test for an excess of homozygous variants given the allele frequency, artifacts in the data that cause deviation from HWE could inflate the test statistics calculated by RAFT. One such factor is population stratification. However, similar to existing genome-wide association studies, this can be detected by existing methods such as genomic control to identify whether the case subjects are well-matched to the control subjects in terms of ancestry. Another more subtle and pernicious factor for RAFT is population substructure, where the case subjects and control subjects may be sampled equally from a heterogeneous population that consists of two or more distinct population ancestries. For instance, if the case subjects and control subjects are derived from a mixture of Finns and NFEs, this might result in the Wahlund effect where there is excessive homozygosity and reduced heterozygosity. Unlike direct case-control comparisons, where balanced mixtures would not inflate allele frequency comparisons, such underlying heterogeneity can inflate the RAFT statistic.
To evaluate further the effect of population substructure on RAFT (where the case and control subjects are well matched for ancestry but contain ethnically different subpopulations in both case and control subjects), we randomly generated 5,000 case subjects and 5,000 control subjects by using equal proportions of Finns and NFEs in the cases and controls. When we ran the test statistic on the simulated data, we indeed observed inflation among the common variants (Table S2). When we performed additional simulations on just the Finns alone, we did not observe any significant amount of inflation (Table S3). Such inflation, if observed, is fully managed by the inclusion of the F term in the determination of the background probability of homozygosity as described in the Material and Methods.
RAFT Discovers a Rare BBS10 Variant Associated with T2D from T2D Exome Chip Data
We next asked whether we can use RAFT to discover disease-associated variants by applying existing tests to a set of exome chip genotyping data for 1,791 T2D case subjects and 2,657 control subjects of Finnish ancestry matched by principal-component analysis (Figure S2). A series of quality control checks were performed to test whether there is any major population stratification between the case and control subjects, which will result in global inflation when running an additive test on the low-frequency and common variants (≥1% allele frequency). However, Fisher’s exact test on the allele counts (FET) did not show any evidence for inflation (genomic control λ = 1.06, Figure S3). We note that even though the current sample size is underpowered to detect the previously published T2D common variants with genome-wide significance, the top hit is an intronic variant found in a reported T2D locus (CDKAL1 [MIM 611259], OR = 1.28, p = 7.75 × 10−6). Likewise, a conventional recessive test such as Hom-FET on the common variants detected CDKAL1 (homozygous OR = 1.59, p = 1.36 × 10−6), reinforcing the fact that both additive and recessive tests are capable of detecting the same common variants, regardless of the true underlying mode of inheritance (Figure S4). The results from both tests did not show any inflation from the null distribution, suggesting that the case and control subjects were well matched for ancestry and that there was no major excessive homozygosity found in the cases. We further applied FET on the allele counts for rare variants (<1% allele frequency) but did not observe any inflation, further suggesting that there is no major stratification between case and control subjects, even in the rare variants (Figure S5). Next, we applied RAFT to the data set and observed departure from the null distribution only among the common variants (≥5% allele frequencies) and rare variants (<1% allele frequencies), but not the low-frequency variants (1%–5% allele frequencies).
The top hit from applying the RAFT approach (excluding common variants—the strongest of which, as noted, flag known associations with T2D) was a rare missense variant (rs202042386; c.1189A>G [p.Ile397Val]) with 0.4% allele frequency in the overall sample in BBS10 (p = 1.38 × 10−6) with three homozygotes found in case subjects and none in control subjects (Table 1). Given that we tested 34,673 variants with <1% allele frequency, the study-wide significance threshold is approximately 0.05/34,673 = 1.4 × 10−6, and the c.1189A>G missense variant in BBS10 is thus study-wide significant.
Table 1.
Top Results from RAFT on the T2D Exome Chip Data Set, with the OR and P Calculated with RAFT and the p Values from Fisher’s Exact Test on the Allelic Counts and Fisher’s Exact Test on the Homozygous Counts
| Chr | Pos | Ref | Alt | Gene | AA | PolyPhen2 | Case AA | Case Aa | Case aa | Con AA | Con Aa | Con aa | AF Case | AF Con | OR | p Value | FET p Value | Hom-FET p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 76740576 | T | C | BBS10 | p.Ile397Val | benign | 1775 | 13 | 3 | 2638 | 19 | 0 | 0.005 | 0.004 | 131 | 1.38 × 10−6 | 0.25 | 0.07 |
| 19 | 39669106 | C | G | PAK4 | p.Arg555Gly | probably damaging | 1725 | 13 | 2 | 2581 | 6 | 0 | 0.005 | 0.001 | 856 | 1.61 × 10−6 | 0.002 | 0.16 |
| 2 | 218762553 | T | C | TNS1 | p.Met46Val | probably damaging | 1769 | 19 | 3 | 2635 | 22 | 0 | 0.007 | 0.004 | 98 | 3.43 × 10−6 | 0.07 | 0.07 |
| 22 | 19502488 | G | A | CDC45 | p.Gly485Ser | probably damaging | 1783 | 6 | 2 | 2647 | 10 | 0 | 0.003 | 0.002 | 316 | 1.29 × 10−5 | 0.37 | 0.16 |
| 1 | 230979511 | C | A | C1orf198 | p.Arg42Ser | benign | 1784 | 5 | 1 | 2656 | 1 | 0 | 0.002 | 0.0002 | 10000 | 3.45 × 10−5 | 0.009 | 0.40 |
| 1 | 17326989 | G | A | ATP13A2 | p.Ala244Val | benign | 1790 | 0 | 1 | 2656 | 1 | 0 | 0.0006 | 0.0002 | 10000 | 3.45 × 10−5 | 0.57 | 0.40 |
| 12 | 50529528 | T | C | CERS5 | p.Ile287Val | benign | 1790 | 0 | 1 | 2656 | 1 | 0 | 0.0006 | 0.0002 | 10000 | 3.45 × 10−5 | 0.57 | 0.40 |
| 6 | 106553606 | C | G | PRDM1 | p.Thr524Arg | benign | 1790 | 0 | 1 | 2654 | 1 | 0 | 0.0006 | 0.0002 | 10000 | 3.45 × 10−5 | 0.57 | 0.40 |
| 2 | 30974108 | C | T | CAPN13 | p.Arg366Gln | benign | 1788 | 1 | 1 | 2656 | 1 | 0 | 0.0008 | 0.0002 | 10000 | 3.45 × 10−5 | 0.31 | 0.40 |
| 2 | 75278553 | C | T | TACR1 | p.Val253Met | probably damaging | 1789 | 1 | 1 | 2656 | 1 | 0 | 0.008 | 0.0002 | 10000 | 3.45 × 10−5 | 0.31 | 0.40 |
| 1 | 230927671 | G | A | CAPN9 | p.Met547Ile | possibly damaging | 1789 | 1 | 1 | 2656 | 1 | 0 | 0.0008 | 0.0002 | 10000 | 3.45 × 10−5 | 0.31 | 0.40 |
| 17 | 72368372 | G | A | GPR142 | p.Arg341Gln | benign | 1789 | 1 | 1 | 2656 | 1 | 0 | 0.008 | 0.0002 | 10000 | 3.45 × 10−5 | 0.31 | 0.40 |
Of note, this allele appears to be enriched in Finns, appearing at 0.4% in this sample, and is vanishingly rare outside of Finland—appearing at only one copy in more than 13,000 chromosomes in the Europeans and African Americans from the Exome Variant Server despite excellent average coverage (∼92×) in the data. We explored the region of homozygosity around the BBS10 c.1189A>G variant in the three cases and found that they shared a 7 Mbp homozygous region in common. In addition, we found that the three homozygotes have longer runs of homozygosity than most other individuals in the data set (Figure S6), but they are not outliers in terms of inbreeding coefficients (Figure S7), suggesting that the homozygosity for the BBS10 c.1189A>G variant in these three individuals arose from a single founder haplotype.
In Silico Prediction for BBS10 c.1189A>G
BBS10 is a small 2-exon gene located on the q-arm of chromosome 12. It encodes a protein that is 723 amino acids long (∼81 kDa). Recessive mutations in BBS10 have been reported to result in BBS,15 a multisystemic ciliopathy often characterized by vision or hearing loss, polydactyly (additional fingers), autism, and obesity. The BBS10 c.1189A>G variant was predicted by in silico tools such as PolyPhen-2,20 SIFT,21 GERP,22 and phastCons23 to be benign or tolerated. Multiple sequence alignment across 46 vertebrates showed that the variant is not conserved and that there are several vertebrates with the same mutant valine amino acid as the T2D homozygotes, such as the rhesus, baboon, rat, pika, dog, hedgehog, elephant, and rock hyrax (Figure 3). However, we note that these vertebrates, with the exception of the rock hyrax, had a cysteine amino acid prior to the valine amino acid. In human, chimp, orangutan, and kangaroo rat, the isoleucine at position 397 was preceded by a serine amino acid. It is possible that the valine amino acid found in the three homozygotes with T2D results in disease in humans when it is preceded by the serine amino acid.
Figure 3.
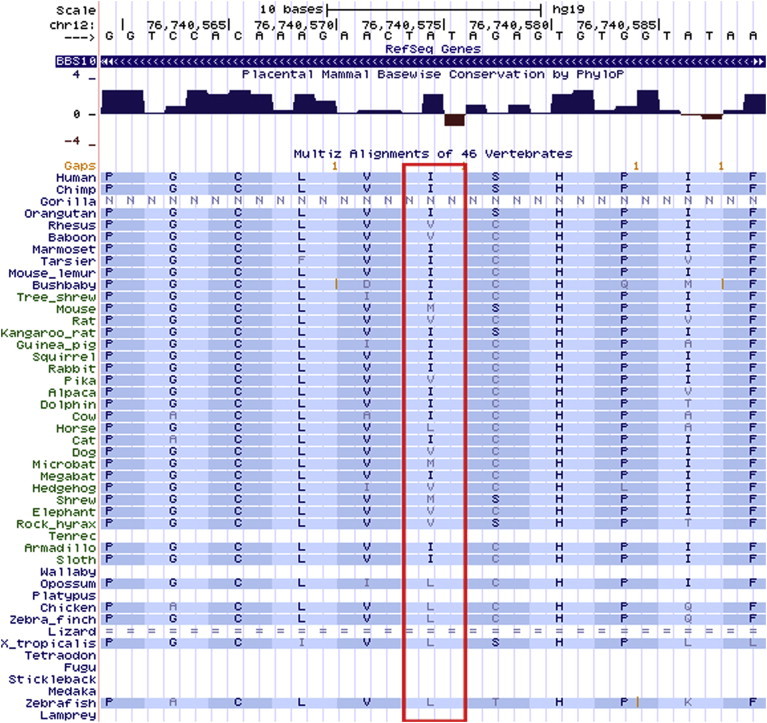
The p.Ile397Val Residue Is Not Conserved across 46 Vertebrates, and the Valine Residue Is Found in Other Vertebrates
Detailed Phenotyping for the Three Homozygotes
Given the known relevance of recessive-acting mutations in BBS10 to diabetes-related phenotypes, the borderline study-wide significant finding at BBS10 c.1189A>G was sufficient to evaluate this variant further. Since manifestations of BBS include not only T2D but also obesity24 (MIM 601665), we surveyed the phenotypic data available for these three individuals. Upon re-examination of their medical records, we found that one of the individuals had bilateral hypoacusis or partial hearing loss, a characteristic often associated with BBS. Two of the homozygotes are obese (body mass index [BMI] = 39.2, age = 70; BMI = 37.73, age = 67), and one of them is overweight (BMI = 28.83, age = 69). The lipid levels for one of the individuals (BMI = 37.7) showed that most of her lipid measurements are in the normal range (total cholesterol = 22nd percentile, low-density lipoprotein = 19th percentile, high-density lipoprotein = 14th percentile), but she has elevated levels of triglycerides in the 91st percentile, consistent with the obesity status. None of the three individuals were reported to have cardinal phenotypes associated with BBS such as polydactyly, intellectual disability, cystic kidney disease, or retinal dystrophy.
Functional Evaluation of BBS10 c.1189A>G
We have shown previously that suppression of most BBS-associated genes during zebrafish embryogenesis leads to convergent extension defects indicative of defective noncanonical Wnt signaling, likely through compromised proteasomal activity.15–17,25,26 This tool has been used previously to annotate systematically nonsynonymous changes by using in vivo complementation, where candidate alleles are introduced into human mRNA and are then tested by their ability to rescue morphant phenotypes.17 Microinjection of translation blocker morpholino to knockdown bbs10 in zebrafish embryos gave rise to convergent extension defects (Figure 4A), reproducing previously reported phenotypes.15 Although coinjection of human wild-type BBS10 mRNA reduced significantly the proportion of zebrafish embryos with convergent extension defects, essentially to the level of control embryos, coinjection of human BBS10 mRNA encoding the c.1189A>G variant failed to rescue the phenotype, suggesting that c.1189A>G variant is a loss-of-function allele (Figure 4B). Therefore, in vivo functional testing supports the pathogenicity of the c.1189A>G variant as discovered by the RAFT analysis.
Figure 4.
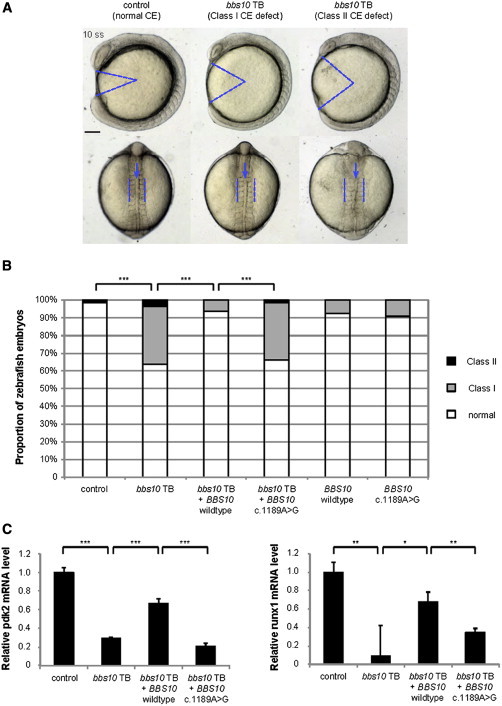
In Vivo Knockdown and Rescue Experiments in Zebrafish for BBS10 c.1189A>G
(A) Lateral view (top) and dorsal view (bottom) of representative zebrafish embryos at the 10-somite stage. In comparison with control embryos, embryos injected with bbs10 translation blocker (TB) morpholino exhibited an expanded body gap angle (marked with blue lines in lateral view) and elongated somites (marked with blue lines in dorsal view) and kinked notochord (marked with blue arrows in dorsal view), indicating convergent extension (CE) defects. Scale bar represents 100 μM.
(B) Distribution of zebrafish embryos with different CE defect severity levels based on previously established criteria (class I, mild; class II, severe15–17 in each condition). Coinjection of human wild-type human BBS10 mRNA (bbs10 TB + BBS10 wild-type) rescues CE defect in bbs10 TB-injected embryos, while coinjection of human BBS10 mRNA with c.1189A>G mutation (bbs10 TB + BBS10 c.1189A>G) does not. Chi-square test was used to test significance; ∗∗∗p < 0.001.
(C) The relative mRNA levels of the genes pdk2 and runx1 in 4 days postfertilization (dpf) zebrafish embryos treated with 1% D-glucose at 1 dpf. Decreased mRNA levels in bbs10 TB-injected embryos were significantly rescued by coinjection of human wild-type human BBS10 mRNA (bbs10 TB + BBS10 wild-type), but not coinjection of human BBS10 mRNA encoding the c.1189A>G mutation (bbs10 TB + BBS10 c.1189A>G). t test was used to test significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Previous glucose tolerance test studies in this model organism have shown that glucose treatment leads to increased mRNA level of several genes, including pdk2 and runx1,27 suggesting that these genes participate in the pathway that functions to tolerate high glucose. To further test this hypothesis and evaluate the role of the c.1189A>G variant, we coinjected the zebrafish embryos with bbs10 morpholino and human BBS10 mRNA. We observed that suppression of bbs10 in embryos treated with glucose results in decreased expression of pdk2 and runx1 and that these phenotypes could be rescued by coinjection of human BBS10 wild-type mRNA, but not mRNA encoding the BBS10 c.1189A>G variant (Figure 4C). These observations suggest that bbs10 might be involved in the glucose tolerance pathway in the zebrafish embryos, and that the c.1189A>G variant in human BBS10 is a loss-of-function allele with regards to this assay, further supporting the pathogenicity of the c.1189A>G allele.
Screening for Homozygotes in Other BBS-Associated Genes in T2D Case and Control Subjects
Given our observation that a rare variant in BBS10 contributes to T2D risk in a recessive manner, we screened for additional rare homozygotes (found at <1% allele frequency in Europeans from the Exome Variant Server) in the other 17 genes that have been associated with BBS28 in a larger superset of 4,947 T2D case subjects and 9,201 control subjects with exome chip genotyping data available (Table S4). Including the BBS10 c.1189A>G variant, we observed a total of 11 homozygotes in the case subjects, compared to 6 in the control subjects (OR = 3.41, 95% confidence interval = [2.41, 4.41], Fisher’s exact test p = 0.019). Excluding the three homozygotes for the BBS10 c.1189A>G variant, there is a still modest excess of homozygotes observed in the case subjects (OR = 2.48, 95% confidence interval = [1.42, 3.54], Fisher’s exact test p = 0.095). It might be possible to identify which variants in these additional BBS-associated genes might also be associated with T2D in future studies involving larger sample sizes.
Discussion
In this study, we describe an approach to detect rare complex-disease-associated recessive alleles that might be missed by conventional methods. The application of this approach on a T2D exome chip data set consisting of ∼4,500 Finnish individuals revealed one such significant finding, c.1189A>G in BBS10 (p = 1.38 × 10−6), while a conventional analysis using Fisher’s exact test yielded insignificant p values for both allelic (p = 0.25) and homozygous (p = 0.07) counts. While there are confounding issues in applying RAFT to a case-control data set, such as population stratification and substructure, we have addressed these both conventionally (by matching case and control subjects for ancestry by using principal-component analysis, as well as by confirming the null distribution of standard association tests) and with elements unique to the RAFT analysis. Specifically, miscalled hemizygosity arising from common deletions in the genome uncovering a rare variant (as well as general genotyping errors) that result in site-specific excesses of homozygotes are handled by the simultaneous identical analysis in control subjects and genome-wide inflation due to substructure managed by a specific correction to the expected homozygosity as a function of frequency.
Previously, we have shown that in an ascertained case-control cohort, there is more power to detect rare risk variants compared to rare protective variants.29 Similarly in this study, the case subjects were selected for genotyping because of their T2D status, whereas the control subjects were drawn from a larger population of individuals without T2D. As such, we have applied the RAFT method in a one-tailed test to screen for only rare recessive variants that confer risk of T2D, that is, if there are more rare homozygotes in T2D cases than expected. We note that under a different study design, it might be worthwhile testing and screening for rare protective variants as well.
Consistent with a role of the c.1189A>G allele in the phenotype tested, functional studies indicated this allele to encode a loss-of-function change. BBS-associated genes such as BBS4 (MIM 600374) have been implicated in GWASs on BMI,30 and while it is plausible that BBS-associated genes might influence T2D through obesity-induced mechanisms, it has also been described that 6% of BBS subjects have T2D,31 and impaired glucose tolerance has been observed in young BBS subjects,32 suggesting that a fraction of BBS subjects might potentially have T2D as a result of severe insulin resistance33 and that there might be other obesity-independent mechanisms involved as well.
Given that many genes such as the BBS-associated genes can result in complex diseases such as obesity-induced T2D, it will be interesting to systematically test and evaluate the extent to which rare recessive variants in BBS-associated genes influence obesity and T2D. While our functional studies in zebrafish embryos are compelling, further studies to design and optimize glucose tolerance assays to systematically evaluate whether these BBS-associated genes can affect glucose tolerance in adult zebrafish will be required. It will be interesting to understand whether this pathway is dependent on obesity-associated factors, such as adipocyte dysfunction, and the disease mechanisms behind the BBS mutations.
The discovery of rare recessive variants in critical genes associated with complex diseases has broader implications in terms of genetic discoveries for complex diseases as well. For instance, rare recessive mutations in TREM2 (MIM 605086) were initially described to cause a rare disease named the Nasu-Hakola disease34,35 (MIM 221770), characterized by early-onset and severe dementia. More recently, the region encompassing TREM2 was implicated in GWASs on Alzheimer disease36 (MIM 104300), and rare heterozygous variants in TREM2 were also found to confer risk of Alzheimer disease.37,38 Similarly, further studies will be needed for understanding whether rare heterozygous variants in BBS10 might result in increased risk of T2D and obesity.
There are several potential extensions to the approach we have described in this work. For instance, RAFT can be modified to evaluate the significance of genes rather than variants by estimating the frequency of rare variants (such as loss-of-function variants or rare deleterious nonsynonymous variants) across a gene, similar to estimating the allele frequencies for variants. For instance, the “composite allele frequency” of loss-of-function variants within a gene can be estimated with the number of individuals who have one loss-of-function variant on the same copy of the gene. And similar to the single-variant version of RAFT described in this manuscript, we can evaluate whether the number of individuals who are recessive (harbor homozygous or compound-heterozygous loss-of-function variants within the same gene) are enriched beyond the squared of the composite allele frequency. However, one potential drawback with adopting a gene-based test is that the high rate of benign variants can dilute any potential signal in the gene.
In addition, the approach can also be extended and modified for evaluating epistatic interactions across genes and loci. To date, there are 17 genes reported to be involved in BBS,28 and mutations in different genes can interact in an oligogenic mode of disease transmission.39 It will be interesting, by using methods such as RAFT and in vivo complementation assay in zebrafish, to test whether certain combinations of rare variants in different BBS-associated genes can similarly confer risk of T2D or obesity in an oligogenic manner.
Understanding the role of BBS-associated variants in T2D and obesity can aid in genetic testing of family members for early intervention. In addition, rare recessive variants such as BBS10 c.1189A>G can contribute to various complex diseases, although the extent of the contribution from such recessive variants in complex diseases is not clear. Given the reported roles of rare recessive variation in Mendelian diseases, it seems plausible there may be many such variants yet to be identified in complex diseases. As such, targeted methods such as RAFT may contribute more broadly to whole-exome or whole-genome sequencing efforts in identifying rare recessive variants associated with complex diseases.
Acknowledgments
The Botnia PPP and Direva studies are supported by grants from Academy of Finland and the Juselius and Folkhälsan Foundations. The in vivo functional study is supported by NIH grants R37GM04360, R01HD04260, R01DK072301, and R01DK075972 (N.K.). The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). Finally, the authors would also like to thank anonymous reviewers 1 and 2 for their words of encouragement and insightful, detailed feedback, which have helped improve this manuscript substantially.
Contributor Information
Elaine T. Lim, Email: tengting.lim@childrens.harvard.edu.
Mark J. Daly, Email: mjdaly@atgu.mgh.harvard.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindesmith L., Moe C., Marionneau S., Ruvoen N., Jiang X., Lindblad L., Stewart P., LePendu J., Baric R. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003;9:548–553. doi: 10.1038/nm860. [DOI] [PubMed] [Google Scholar]
- 3.Kenny E.E., Timpson N.J., Sikora M., Yee M.C., Moreno-Estrada A., Eng C., Huntsman S., Burchard E.G., Stoneking M., Bustamante C.D., Myles S. Melanesian blond hair is caused by an amino acid change in TYRP1. Science. 2012;336:554. doi: 10.1126/science.1217849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vukcevic D., Hechter E., Spencer C., Donnelly P. Disease model distortion in association studies. Genet. Epidemiol. 2011 doi: 10.1002/gepi.20576. Published online March 17, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McQuillan R., Eklund N., Pirastu N., Kuningas M., McEvoy B.P., Esko T., Corre T., Davies G., Kaakinen M., Lyytikäinen L.P., ROHgen Consortium Evidence of inbreeding depression on human height. PLoS Genet. 2012;8:e1002655. doi: 10.1371/journal.pgen.1002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller M.C., Simonson M.A., Ripke S., Neale B.M., Gejman P.V., Howrigan D.P., Lee S.H., Lencz T., Levinson D.F., Sullivan P.F., Schizophrenia Psychiatric Genome-Wide Association Study Consortium Runs of homozygosity implicate autozygosity as a schizophrenia risk factor. PLoS Genet. 2012;8:e1002656. doi: 10.1371/journal.pgen.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gamsiz E.D., Viscidi E.W., Frederick A.M., Nagpal S., Sanders S.J., Murtha M.T., Schmidt M., Triche E.W., Geschwind D.H., State M.W., Simons Simplex Collection Genetics Consortium Intellectual disability is associated with increased runs of homozygosity in simplex autism. Am. J. Hum. Genet. 2013;93:103–109. doi: 10.1016/j.ajhg.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chahrour M.H., Yu T.W., Lim E.T., Ataman B., Coulter M.E., Hill R.S., Stevens C.R., Schubert C.R., Greenberg M.E., Gabriel S.B., Walsh C.A., ARRA Autism Sequencing Collaboration Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu T.W., Chahrour M.H., Coulter M.E., Jiralerspong S., Okamura-Ikeda K., Ataman B., Schmitz-Abe K., Harmin D.A., Adli M., Malik A.N. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim E.T., Raychaudhuri S., Sanders S.J., Stevens C., Sabo A., MacArthur D.G., Neale B.M., Kirby A., Ruderfer D.M., Fromer M., NHLBI Exome Sequencing Project Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein J.I., Crenshaw A., Carey J., Grant G.B., Maguire J., Fromer M., O’Dushlaine C., Moran J.L., Chambert K., Stevens C., Swedish Schizophrenia Consortium. ARRA Autism Sequencing Consortium zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics. 2012;28:2543–2545. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 14.Lim E.T., Würtz P., Havulinna A.S., Palta P., Tukiainen T., Rehnström K., Esko T., Mägi R., Inouye M., Lappalainen T., Sequencing Initiative Suomi (SISu) Project Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoetzel C., Laurier V., Davis E.E., Muller J., Rix S., Badano J.L., Leitch C.C., Salem N., Chouery E., Corbani S. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006;38:521–524. doi: 10.1038/ng1771. [DOI] [PubMed] [Google Scholar]
- 16.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 17.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westerfield M. Fifth Edition. University of Oregon Press; Eugene: 2007. THE ZEBRAFISH BOOK: A Guide for the Laboratory Use of Zebrafish (Danio rerio) [Google Scholar]
- 19.Katsanis N., Beales P.L., Woods M.O., Lewis R.A., Green J.S., Parfrey P.S., Ansley S.J., Davidson W.S., Lupski J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000;26:67–70. doi: 10.1038/79201. [DOI] [PubMed] [Google Scholar]
- 20.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margulies E.H., Blanchette M., Haussler D., Green E.D., NISC Comparative Sequencing Program Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaghloul N.A., Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y.P., Tsai I.C., Morleo M., Oh E.C., Leitch C.C., Massa F., Lee B.H., Parker D.S., Finley D., Zaghloul N.A. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Invest. 2014;124:2059–2070. doi: 10.1172/JCI71898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris J.M., Esain V., Frechette G.M., Harris L.J., Cox A.G., Cortes M., Garnaas M.K., Carroll K.J., Cutting C.C., Khan T. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood. 2013;121:2483–2493. doi: 10.1182/blood-2012-12-471201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forsythe E., Beales P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013;21:8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan Y., Lim E.T., Sandholm N., Wang S.R., McKnight A.J., Ripke S., Daly M.J., Neale B.M., Salem R.M., Hirschhorn J.N., DIAGRAM Consortium. GENIE Consortium. GIANT Consortium. IIBDGC Consortium. PGC Consortium An excess of risk-increasing low-frequency variants can be a signal of polygenic inheritance in complex diseases. Am. J. Hum. Genet. 2014;94:437–452. doi: 10.1016/j.ajhg.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monda K.L., Chen G.K., Taylor K.C., Palmer C., Edwards T.L., Lange L.A., Ng M.C., Adeyemo A.A., Allison M.A., Bielak L.F., NABEC Consortium. UKBEC Consortium. BioBank Japan Project. AGEN Consortium A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat. Genet. 2013;45:690–696. doi: 10.1038/ng.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imhoff O., Marion V., Stoetzel C., Durand M., Holder M., Sigaudy S., Sarda P., Hamel C.P., Brandt C., Dollfus H., Moulin B. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin. J. Am. Soc. Nephrol. 2011;6:22–29. doi: 10.2215/CJN.03320410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green J.S., Parfrey P.S., Harnett J.D., Farid N.R., Cramer B.C., Johnson G., Heath O., McManamon P.J., O’Leary E., Pryse-Phillips W. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N. Engl. J. Med. 1989;321:1002–1009. doi: 10.1056/NEJM198910123211503. [DOI] [PubMed] [Google Scholar]
- 33.Beales P.L., Elcioglu N., Woolf A.S., Parker D., Flinter F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med. Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 34.Numasawa Y., Yamaura C., Ishihara S., Shintani S., Yamazaki M., Tabunoki H., Satoh J.I. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur. J. Neurol. 2011;18:1179–1183. doi: 10.1111/j.1468-1331.2010.03311.x. [DOI] [PubMed] [Google Scholar]
- 35.Kaneko M., Sano K., Nakayama J., Amano N. Nasu-Hakola disease: the first case reported by Nasu and review. Neuropathology. 2010 doi: 10.1111/j.1440-1789.2010.01127.x. Published online May 24, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Cruchaga C., Kauwe J.S., Harari O., Jin S.C., Cai Y., Karch C.M., Benitez B.A., Jeng A.T., Skorupa T., Carrell D., GERAD Consortium. Alzheimer’s Disease Neuroimaging Initiative (ADNI) Alzheimer Disease Genetic Consortium (ADGC) GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78:256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P.V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A.I., Lah J.J. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J.S., Younkin S., Alzheimer Genetic Analysis Group TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 2004;13(Spec No 1):R65–R71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.