

Abstract
Using a combination of exome sequencing and linkage analysis, we investigated an English family with two affected siblings in their 40s with recessive Charcot-Marie Tooth disease type 2 (CMT2). Compound heterozygous mutations in the immunoglobulin-helicase-μ-binding protein 2 (IGHMBP2) gene were identified. Further sequencing revealed a total of 11 CMT2 families with recessively inherited IGHMBP2 gene mutations. IGHMBP2 mutations usually lead to spinal muscular atrophy with respiratory distress type 1 (SMARD1), where most infants die before 1 year of age. The individuals with CMT2 described here, have slowly progressive weakness, wasting and sensory loss, with an axonal neuropathy typical of CMT2, but no significant respiratory compromise. Segregating IGHMBP2 mutations in CMT2 were mainly loss-of-function nonsense in the 5′ region of the gene in combination with a truncating frameshift, missense, or homozygous frameshift mutations in the last exon. Mutations in CMT2 were predicted to be less aggressive as compared to those in SMARD1, and fibroblast and lymphoblast studies indicate that the IGHMBP2 protein levels are significantly higher in CMT2 than SMARD1, but lower than controls, suggesting that the clinical phenotype differences are related to the IGHMBP2 protein levels.
Main Text
Charcot-Marie-Tooth disease (CMT) is a genetically heterogeneous disorder of the peripheral nervous system with an estimated prevalence of 1 in 2,500 individuals.1 Clinical manifestations of CMT include slowly progressive distal weakness, wasting, and sensory loss, which spreads proximally as the disease progresses. Clinically, CMT can be divided into two major phenotypic types: a demyelinating form (CMT type 1 [CMT1]) and an axonal form (CMT type 2 [CMT2]).2–6 Mutations in 15 unique genes have so far been identified as causing CMT2. Despite this significant progress, about 70% of people with CMT2 do not have a genetic diagnosis.2–9 The identification of the remaining CMT2 genes is expected to yield important insights into the disease pathways and pathophysiology associated with axonal degeneration. In addition, it is becoming evident that the phenotypic and genotypic intersection of CMT2 with related motor neuron disorders of axonal degeneration and other neuromuscular diseases is more extensive than previously thought, increasing the importance of gene identification and characterization in this area.10,11
We initially studied a family where two siblings were affected with CMT2. The onset was in late childhood, with slowly progressive disease and parents that were clinically and electrically unaffected (family A). The proband is currently 43 (family A, individual 1) and her sister is 40 years of age (family A, individual 2), both work, are able to drive, and use a stick to walk with silicon ankle foot orthosis. Examination of the index case at 43 years of age revealed bilateral foot drop, distal weakness, and wasting in the upper and lower limbs, with mild proximal lower limb weakness (Figure 1). Reflexes were absent and there was sensory loss in the feet and hands. Cranial nerves were normal apart from a trombone-shaped tongue (Figure 1). There were no respiratory problems. Chest X-ray and sleep study was normal; nerve conduction studies and sural nerve biopsy indicated an axonal neuropathy (Figure 2). Her sister had milder clinical features, and examination findings at the age of 40 years revealed bilateral foot drop, distal weakness, and wasting in the upper and lower limbs and areflexia. There were no respiratory problems and an axonal neuropathy was seen on nerve conduction studies (Table 1, Table 2, Table 3; see also Table S1 available online).
Figure 1.
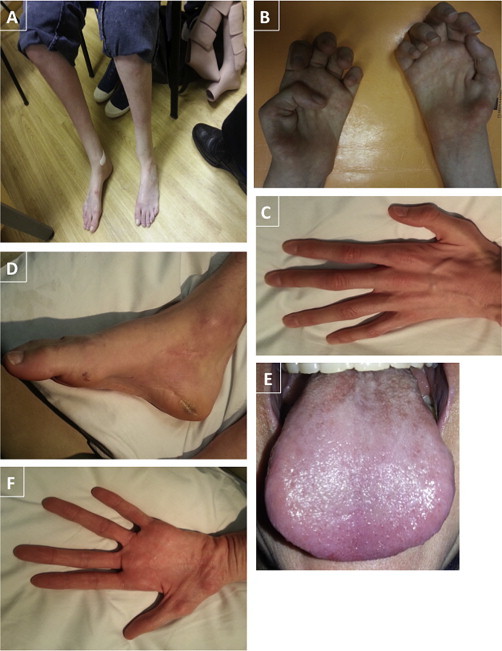
Photographs of CMT2 Individuals with IGHMBP2 Mutations
(A) Legs and feet of family A, with individual II.1 also showing silicon ankle foot orthosis.
(B) Hands of family A, individual II.1.
(C) Left hand of family B, individual II.2.
(D) Right foot of family B, individual II.1.
(E) Trombone-shaped tongue of family A, individual II.1.
(F) Left hand of family B, individual II.1.
Figure 2.
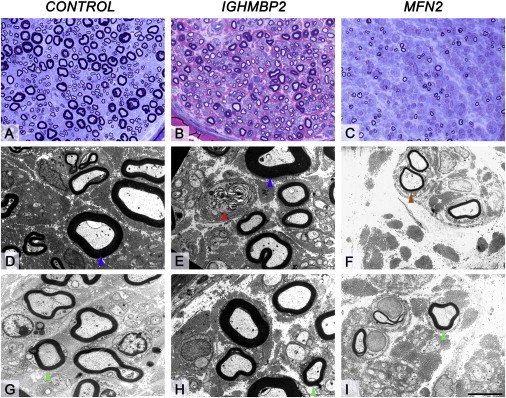
Morphological Appearances of the Sural Nerve Biopsy in the Individual with IGHMBP2 Mutation, Healthy Age-Matched Control and Individual with MFN2 Mutation
(A, D, and G) Sural nerve biopsy of a healthy age-matched control.
(B, E, and H) Sural nerve biopsy of a patient with IGHMBP2 mutation.
(C, F, and I) Sural nerve biopsy of an individual with known MFN2 mutation. Semithin resin sections stained with toluidine blue (A, healthy age-matched control; C, individual with known MFN2 mutation) and methylene blue azure–basic fuchsin (MBA-BF) (B, individual with IGHMBP2 mutation). When compared with the control (A), the biopsy of the individual with IGHMBP2 mutation (B) shows a moderate reduction in density of the large myelinated fibers, whereas the small myelinated fibers are well preserved and regeneration clusters is not a feature. In contrast, in the individual with MFN2 mutation (C), there is near complete loss of large fibers and severe widespread loss of small myelinated fibers. Ultrastructural assessment reveals occasional actively degenerating axonal profiles (E, red arrowhead) in the individual with IGHMBP2 mutation. In the individual with MFN2 mutation rare regeneration clusters are seen (F, brown arrowhead). The thickness and configuration of the myelin sheaths of remaining large (D and E, blue arrowheads) and small myelinated fibers (G, H, and I, green arrowheads) are similar to that seen in a healthy age-matched control.
Scale bar represents 35 μm in (A)–(C) and 5 μm in (D)–(I).
Table 1.
List of IGHMBP2 Mutations Found in Individuals with Axonal Neuropathy
| Family | Ethnicity | Sex | Diagnosis | Age at Onset | Current Age | Protein Change | Nucleotide Change |
|---|---|---|---|---|---|---|---|
| A | English | Female | CMT2 | 7 years | 43 years | p.Cys46∗ + p.Arg971Glufs∗4 | c.138T>A + c.2911_2912delAG |
| A | English | Female | CMT2 | 6 years | 40 years | p.Cys46∗ + p.Arg971Glufs∗4 | c.138T>A + c.2911_2912delAG |
| B | English | Male | CMT2 | 5 years | 23 years | p.Cys46∗ + p.Arg971Glufs∗4 | c.138T>A + c.2911_2912delAG |
| C | Serbian | Male | CMT2 | 2 years | 14 years | p.Cys46∗ + p.Phe202Val | c.138T>A + c.604T>G |
| C | Serbian | Female | CMT2 | 2 years | 15 years | p.Cys46∗ + p.Phe202Val | c.138T>A + c.604T>G |
| D | Pakistani | Female | CMT2 + Down Syndrome | 7 years | 20 years | p.Pro531Thr + p.Val580Ile | c.1591C>A + c.1738G>A |
| E | Vietnamese | Female | CMT2 | 3 years | 39 years | p.Arg605∗ + p.His924YTyr | c.1813C>T + c.2770C>T |
| F | English | Male | CMT2 | 4 years | 15 years | p.Ser80Gly + p.Cys496∗ | c.238A>G + c.1488C>A |
| G | USA | Female | CMT2 | 6 years | 10 years | p.Trp386Arg + p.Arg971Glufs∗4 | c.1156T>C + c.2911_2912delAG |
| H | Polish | Female | CMT2 | 4 years | 28 years | p.990_994del (Hom) | c.2968_2980del (Hom) |
| I | Italian | Female | CMT2 | 1 years | 12 years | p.Val373Gly + p.Ala528Thr | c.1118T>G + c.1582G>A |
| I | Italian | Male | CMT2 | 1 years | 6 years | p.Val373Gly + p.Ala528Thr | c.1118T>G + c. 1582G>A |
| J | Korean | Male | CMT2 | 5 years | 41 years | p.Asn245Ser (Het) | c.734A>G (Het) |
| K | English | Male | CMT2 | 7 years | 20 years | p.Arg605∗ (Het) + deletion | c.1813C>T (Het) + deletion |
| K | English | Female | CMT2 | 10 years | 18 years | p.Arg605∗ (Het) + deletion | c.1813C>T (Het) + deletion |
Hom, homozygous; Het, Heterozygous.
Table 2.
Electrophysiology Data for the Individuals with CMT2
| Individual | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 12 | 13 | 11 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family no. | A | A | B | C | C | D | E | F | G | H | I | I | J | K | K |
| Sex/age (y) | F/43 | F/40 | M/23 | M/14 | F/15 | F/19 | F/39 | M/15 | F/10 | F/28 | F/12 | M/6 | M/41 | M/20 | F/18 |
| Ethnicity | English | English | English | Serbian | Serbian | Pakistani | Vietnam | English | USA | Polish | Italian | Italian | Korean | English | English |
| Age at first symptoms | 7 years | 6 years | <5 years | <2 years | <2 years | <10 years | <3 years | 4 years | 6 years | 4 years | 1 years | 1 years | 5 years | 7 years | 10 years |
| First symptoms | Toe walking | Toe walking | Difficulty walking | Delayed milestones | Delayed walking | hypotonia, foot drop | Delayed milestones | Foot drop | Foot drop | Hand weakness | Limb weakness | equino-varus | Gait difficulty | Foot drop | Feet deformity |
| Weaknessa | |||||||||||||||
| UL | +++ | +++ | ++ | +++ | +++ | +++ | +++ | ++ | N | ++ | +++ | + | + | ++ | + |
| LL | +++ | +++ | ++ | +++ | +++ | +++ | +++ | ++ | ++ | +++ | +++ | ++ | ++ | ++ | + |
| Pinprickb | |||||||||||||||
| UL | N | N | + | N | N | n/a | N | n/a | N | + | + | n/a | + | N | N |
| LL | + | N | + | N | N | n/a | N | n/a | N | n/a | + | n/a | + | N | N |
| Vibrationc | |||||||||||||||
| UL | N | N | N | N | N | n/a | N | n/a | N | n/a | n/a | n/a | ++ | N | N |
| LL | + | N | + | N | N | n/a | N | n/a | N | n/a | n/a | n/a | ++ | N | N |
| Reflexes | |||||||||||||||
| UL | Abs | Abs | Abs | Abs | Abs | Abs | Abs | n/a | + | +/− | Abs | abs | abs | N | N |
| LL | Abs | Abs | Abs | Abs | Abs | Abs | Abs | n/a | Abs (AJ) | Abs | Abs | abs | abs | AJ | +/− |
| Bulbar | Rhomboid tongue | Wasted tongue | No | No | No | Wasted tongue | No | n/a | No | No | No | No | No | No | No |
| Respiratory support | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No |
| Overall maximal function | Independent ambulation | Independent ambulation | Independent ambulation | n/a | n/a | Independent ambulation | n/a | n/a | n/a | n/a | Independent ambulation | Walking with stick | Independent ambulation | Independent ambulation | Independent ambulation |
| Walking aids | AFO | AFO (past) | n/a | WC | WC | WC | WC since 16 | AFO | AFO | WC | WC since age 5 years | Bilateral support | AFO | AFO+Crutches | No |
AFO, ankle-foot orthosis; n/a, not available; LL, lower limbs; UL, upper limbs; WC, wheelchair.
Weakness: N, normal; + > 4, distal muscles, ++ < 4, distal muscles, +++, proximal weakness (knee flexion and extension, elbow flexion and extension or above).
Pinprick and vibration sensation: N, normal; +, reduced below wrist/ankle; ++, reduced below knee/elbow; +++, reduced at or above elbow/knee.
Reflexes: N, normal/present; ++, brisk; +++, brisk with extensor plantars; +/−, present with reinforcement; abs, absent; abs (AJ), absent ankle jerks only.
Table 3.
Electrophysiology Data from the Individuals from Our CMT2 Cohort
| Individual | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family no. | A | A | B | C | D | D | E | I | I | J | K | K |
| Age at examination (y) | 17 | 25 | 16 | 20 | 13 | 7 | 32 | 7 | 1.5 | 40 | 12 | 10 |
| Radial n. | ||||||||||||
| Sensory Amp | 2 μV | Abs | Abs | 13 μV | NT | NT | n/a | Abs | n/a | NT | 16 μV | n/a |
| Sensory CV | 50 m/s | Abs | Abs | 69 m/s | NT | NT | n/a | Abs | n/a | NT | 63 m/s | n/a |
| Median n. | ||||||||||||
| Motor DML | NT | Abs | Abs | 3.5 ms | 5.1 ms | 3.1 ms | Abs | Abs | n/a | 6 ms | 2.8 ms | 3.2 ms |
| Motor Amp | NT | Abs | Abs | 5.7 mV | 0.02 mV | 2.8 mV | Abs | Abs | n/a | 0.7 mV | 18.8 mV | 21.8 mV |
| Motor CV | NT | Abs | Abs | 46 m/s | 30 m/s | 42 m/s | Abs | Abs | n/a | 33.6 m/s | 58 m/s | 58 m/s |
| Sensory Amp | Abs | Abs | Abs | 6 μV | Abs | 20 μV | 2.2 uV | Abs | Abs | Abs | 32 μV | 26 μV |
| Sensory CV | Abs | Abs | Abs | 45 m/s | Abs | 49 m/s | 59.8 m/s | Abs | Abs | Abs | 60 m/s | 52 m/s |
| Ulnar n. | ||||||||||||
| Motor DML | 3.8 ms | 3.3 ms | 4.3 ms | 3.5 ms | NT | NT | Abs | n/a | n/a | 3.1 | 2.8 ms | 3.2 ms |
| Motor Amp | 0.8 mV | 3.7 mV | 5.7 mV | 2.9 mV | NT | NT | Abs | n/a | n/a | 14.3 | 8.9 mV | 12.8 mV |
| Motor CV | 51 m/s | 51 m/s | 45 m/s | 46 m/s | NT | NT | Abs | n/a | 55 m/s | 41.1 | 58 m/s | 62 m/s |
| Sensory Amp | NT | Abs | Abs | Abs | Abs | 12 μV | 2.0 uV | n/a | n/a | Abs | 16 μV | 14 μV |
| Sensory CV | NT | Abs | Abs | Abs | Abs | 48 m/s | 50.3 m/s | n/a | n/a | Abs | 67 m/s | 53 m/s |
| Peroneal n. | ||||||||||||
| Motor DML | NT | Abs | Abs | NT | NT | Abs | n/a | Abs | Abs | Abs | Abs | 4.9 ms |
| Motor Amp | NT | Abs | Abs | NT | NT | Abs | n/a | Abs | Abs | Abs | Abs | 4.6 mV |
| Motor CV | NT | Abs | Abs | NT | NT | Abs | n/a | Abs | Abs | Abs | Abs | 51 m/s |
| Tibial n. | ||||||||||||
| Motor DML | 9.3 ms | Abs | Abs | NT | Abs | Abs | n/a | n/a | Abs | Abs | 6.3 ms | 4.3 ms |
| Motor Amp | 0.08 mV | Abs | Abs | NT | Abs | Abs | n/a | n/a | Abs | Abs | 2 mV | 8.2 mV |
| Motor CV | 34 m/s | Abs | Abs | NT | Abs | Abs | n/a | n/a | Abs | Abs | 46 m/s | 50 m/s |
| Sural n. | ||||||||||||
| Sensory Amp | Abs | Abs | Abs | Abs | Abs | Abs | n/a | Abs | Abs | Abs | 38 μV | 35 μV |
| Sensory CV | Abs | Abs | Abs | Abs | Abs | Abs | n/a | Abs | Abs | Abs | 59 m/s | 49 m/s |
Abs, absent; NT, not tested.
Known mutations in genes implicated in CMT2 were excluded by Sanger sequencing and whole-exome sequencing and linkage analysis were carried out, with informed consent and IRB ethics approval UCL/UCLH 99/N103. Exome sequencing was performed as previously described12 using the Agilent SureSelect kit and run on the Illumina HiSeq 2500. Sequences were aligned with the Burrows-Wheeler Aligner, duplicates were removed with Picard, indels aligned and base quality scores recalibrated with the Genome Analysis Toolkit (GATK). The average sequencing depth was 55-fold with variants being filtered according to pathogenicity, inheritance pattern, and segregation in the family.
Two compound heterozygous mutations were identified in the affected individuals in immunoglobulin helicase μ-binding protein 2 (IGHMBP2 [MIM 600502]; RefSeq: NM_002180.2), a nonsense 5′ mutation (c.138T>A: p.Cys46∗) and a 3′ frameshift mutation in the last exon of the gene (c.2911_2912delAG: p.Arg971Glufs∗4). The mother and father were heterozygous for the c.138T>A and c.2911_2912delAG mutations, respectively. These mutations were absent from the 1000 Genomes database (healthy controls) and our in-house exome database of 480 clinically and neuropathologically normal controls. Mutations in IGHMBP2 have previously been associated with a different phenotype, spinal muscular atrophy with respiratory distress type 1 (SMARD1 [MIM: 604320]), a devastating neuromuscular disorder with muscle weakness and atrophy severely affecting the diaphragm.13–16 SMARD1 mutations are typically missense in the helicase domain or mutations where both alleles are loss-of-function, usually in the 5′ region of the gene.14,17 The onset of this condition is usually in the first few weeks of life with early respiratory failure and death in infancy, typically before 12 months of age.14–39 The longest surviving children reported were 13 and 15 years of age, they had profound upper and lower limb muscle and trunk weakness and respiratory compromise.18,30 Three children have been reported with delayed onset of respiratory distress of between 4 and 10 years old and designated juvenile SMARD1.18,35,39
IGHMBP2 was Sanger sequenced in a cohort of 85 likely recessive CMT2 families, and CMT exome sequence data was analyzed from the Hussman Institute for Human Genomics. A total of 11 CMT2 families with IGHMBP2 mutations were identified (Table 1). All families with CMT2 and IGHMBP2 mutations showed an autosomal recessive pattern of inheritance but in two families only one heterozygous pathogenic mutation was identified (Table 1; Figure 3). The phenotype consisted of childhood onset, progressive CMT2 with mild proximal weakness and scoliosis in some cases. Sensory involvement was mild glove and stocking and electrophysiology indicated a sensory and motor axonal neuropathy in all cases (Tables 2 and 3). Two further cases had unusually shaped tongues (Figure 1); none of the cases had significant respiratory compromise, recurrent chest infections or previous ventilator assistance or sleep apnea. One case had trisomy 21 and Down syndrome (Tables 2 and 3).
Figure 3.
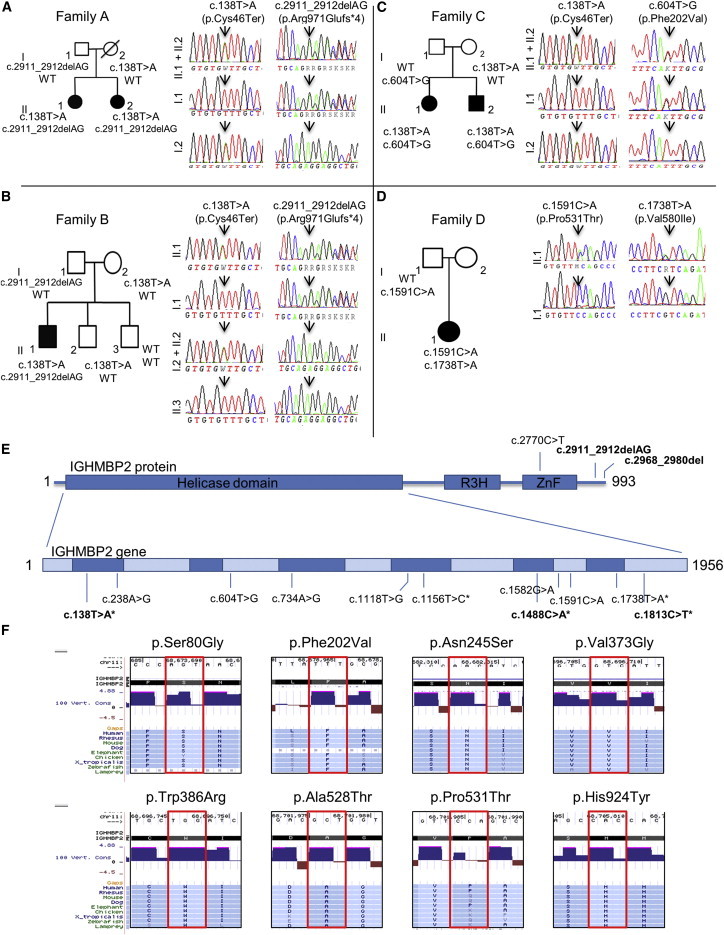
Pedigrees of Four CMT2 Families Affected by IGHMBP2 Mutations and Chromatograms of These Mutations
(A–D) Pedigrees of family A (A), family B (B), family C (C), and family D (D).
(E) A schematic of IGHMBP2 (993 amino acids) showing the helicase, R3H, and ZnF domains. The relative base pair positions of the identified mutations are located. Mutations in bold are nonsense or frameshift and result in an altered transcript. ∗ = pathogenic mutations found before in SMARD1 patients.
(F) Conservation of the missense mutations found in IGHMBP2. A selected subset of 9 species were chosen, representing the 100 species available at the USCS browser. Red boxes indicate the location of the amino acid changed due to the mutation.
Three families (five individuals) carried the p.Cys46∗ nonsense variant in combination with either an AG deletion, causing a p.Arg971Glufs∗4 frameshift in the last exon (family A and B), or a novel p.Phe202Val variant (family C). Haplotype analysis indicated that a common founder was unlikely in the individuals with p.Cys46∗ variants (Figure S1, Table S2). In the IGHMBP2 helicase domain40 (PDB code 4B3G), Cys46 is located in the β-barrel of domain 1B and the side chain does not interact with any neighboring residues. Phe202 is part of an α-helix in domain 1A and is pathogenic, but not central to the protein structure, suggesting a milder phenotype. Family 4 has compound heterozygous missense mutations and presents with a known severe pathogenic variant p. Val580Ile and a novel variant p.Pro531Thr. Pro531 lies in a loop region and is exposed to the solvent region on the protein surface. The side chain of the residue does not interact with neighboring residues and will likely have a milder phenotype (Figure S2). Val580 lies near a β strand in the core of domain 2A and interacts with Ser539 on a neighboring strand to stabilize the RecA-like fold. Mutating Val580 to isoleucine, which has a longer side chain, likely disrupts the formation of the β sheet and hence destabilizes domain 2A (Figure S2). Similarly, in family G (p.Trp386Arg), mutating a hydrophobic residue to a positively charged residue can result in protein instability due to the loss of some favorable van der Waals contacts with neighboring hydrophobic residues. The other missense mutations at Asn245, Val373, and Ala528 (families I and J) are also predicted to cause protein instability, resulting in loss of functional protein40 (Figure S2). In the two families with a single IGHMBP2 mutation and recessive CMT2 phenotype, we additionally analyzed the 5′ promoter region and the exome BAM files for sequencing coverage and carried out IGHMBP2 cDNA sequencing in the two affected individuals from family K. The cDNA analysis identified that the stop mutation was hemizygous, suggesting a deletion on the other allele (Figure S3; Table 1).
The neurophysiological pattern in individuals with CMT2 and IGHMBP2 mutations was consistent with a mild motor and sensory axonal polyneuropathy (velocities 40–50 m/s) (Table 3). This is in contrast to SMARD1 with markedly reduced motor conduction velocities (around 20 m/s), particularly in the legs, and a very marked reduction or loss of the compound muscle action potential.41 Nerve biopsy in CMT2 family A, individual 1 showed a moderate reduction in density of the large myelinated fibers, whereas the small myelinated fibers are well preserved. This is in contrast with the individual with a MFN2 mutation where there is near complete loss of large fibers and severe widespread loss of small myelinated fibers. Ultrastructural assessment revealed occasional actively degenerating axonal profiles in CMT2 with an IGHMBP2 mutation, but these were rare in MFN2 patients (Figure 2).
Fibroblast and lymphoblastoid cell lines from families 1 and 2 were used to investigate whether the c.138T>A mutation resulted in nonsense mediated decay. The presence of both wild-type (WT) and mutant mRNA persisted in carriers and affected individuals, indicating that NMD has not been activated (Figure S3). Because the c.2911_2912del mutation is located in the last exon of the gene, we would not expect nonsense-mediated decay to occur. Fibroblasts from individuals with SMARD1 with heterozygous or homozygous frameshift mutations also failed to show NMD (Figure S3), suggesting that IGHMBP2 is protected from NMD and likely produces truncated protein products.
Considering the presumed existence of a truncated protein in the CMT2 cell lines, and for the missense mutations, immunofluorescence experiments were performed to detect changes in the localization or potential clustering of the truncated protein. Misfolded or mislocalized proteins can interact inappropriately with other cellular factors to cause toxicity. However, results show no difference between fibroblast lines of individuals with SMARD1 or CMT2 in comparison with controls and carriers (Figure 4; Figure S4).
Figure 4.
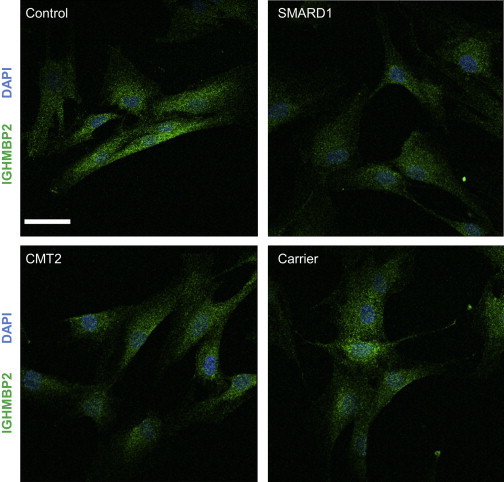
Localization of the IGHMBP2 Protein in Fibroblasts
Scale bar represents 44.00 μm. Green represents IGHMBP2; blue represents 4’,6-diamidino-2-phenylindole (DAPI) staining for the nucleus. No difference in clustering of the truncated protein is found between the control and both the affected individuals and the carrier. SMARD1 = p.Gly98Fs; CMT2 = p.Cys46∗ + p.Arg971Glufs∗4; Carrier = p.Arg971Glufs∗4. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.05% Triton X-100 and blocked in 10% FBS. Coverslips were incubated with a 1:1000 dilution of primary antibody (Millipore) for 60 min, washed with PBS and incubated with a 1:2000 dilution of goat anti-mouse immunoglobulin G Alexa Fluor 488-A11008 secondary antibody (Invitrogen) for 60 min. Following, the coverslips were washed with PBS and mounted on microscope slides with Prolong Gold Antifade with DAPI and imaged using a Zeiss 710 confocal microscope (Carl Zeiss AG) with the 63× oil immersion objective.
Protein quantification was estimated in both fibroblast and lymphoblastoid cell lines from IGHMBP2-associated CMT2, SMARD1, carriers, and controls to investigate whether abundance of residual protein correlates with the severity of the disease (Table S3). When comparing the fibroblast lines of six CMT2 and two SMARD1 individuals against controls, a clear difference in protein levels can be observed (Figure 5). Looking at the levels of the protein in all fibroblast and lymphoblastoid cell lines, single heterozygous carriers of IGHMBP2-associated CMT2 mutations were found to have intermediate IGHMBP2 protein levels in between affected and control individuals (Figure 5). Interestingly, in the three individuals with the p.Cys46∗ and p.Arg971Glufs∗4 combination of variants, a band was detected between 70–80 kDa. This band was not observed in any other affected individuals, carriers or controls (Figure 5). Using online tools, we estimated the molecular weight of the truncated protein resulting from the p.Cys46∗ variant at 52 kDa, whereas the p.Arg971Glufs∗4 frameshift results in a protein of 109 kDa. In previous experiments, physicochemical properties of the WT protein in comparison with the p.Thr493Ile variant, known to cause SMARD1, have been investigated. Results showed a degradation band at 75 kDa that was primarily present in the p.Thr493Ile transfected cells and comprises the N-terminal amino acid residues 1–674.8 Similar to this variant, the p.Cys46∗ variant or the p.Arg971Glufs∗4 frameshift in these individuals could alter the physicochemical properties of the protein and results in a degradation product at 75 kDa. Because neither of the carriers with either the p.Cys46∗ and p.Arg971Glufs∗4 variant show a band at this molecular weight, it could be hypothesized that the lower levels of functioning protein in the compound heterozygous individuals activate a feedback mechanism that preserved any residual truncated protein.
Figure 5.
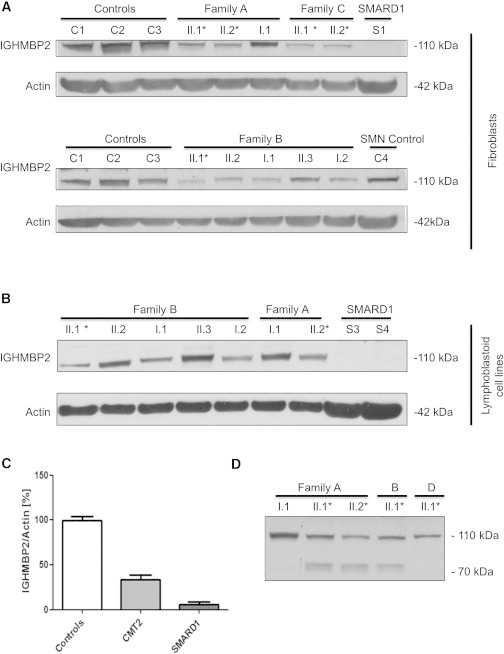
Protein Levels of IGHMBP2 Normalized Against an Actin Control in All Individuals for Both Fibroblasts and Lymphoblastoid Cell Lines
(A) Protein levels in fibroblast cell lines of families A, B, and C.
(B) Protein levels in lymphoblastoid cell lines of families A and B. Family A consists of two affected individuals (II.1 + II.2) with lower levels of the protein in fibroblasts in comparison with the carrier of the p.Arg971Glufs∗4 mutation (I.1). This is consistent in lymphoblasts, where individual II.2 has lower levels than the carrier (I.1). Family B consists of one affected individual (I.1) with lower levels of the IGHMBP2 protein in comparison with the carriers of the p.Cys46∗ mutation (II.2, I.1) or the p.Arg971Glufs∗4 mutation (I.2). These all have lower levels than the unaffected sibling of the patient (II.3). This is consistent in the lymphoblasts. For family C, only two patient fibroblasts cell lines were available, both showing reduced levels in comparison with controls. All SMARD1 fibroblasts and lymphoblasts have lower levels than any of the individuals. ∗ = individuals with CMT2.
(C) Protein levels of the IGHMBP2 protein normalized against actin in controls, CMT2 individuals, and SMARD1 individuals. There is a significant difference between all groups. All samples were standardized against two controls: C1 and C2. Data are presented as mean ± SEM. Statistical analysis was performed with Bonferroni’s multiple comparison test. ∗p < 0.05; ∗∗∗p < 0.0001.
(D) Existence of a degradation band around 70–80 kDa in individuals with CMT2 and a combination of the p.Cys46∗ and p.Arg971Glufs∗4 mutations.
Cells were lysed in 50 μl of NP40 buffer (150 mM Tris (pH 8), 1 mM EDTA, 150 mM NaCl, 0.5% NP40) containing 1× complete protease inhibitor cocktail (Roche). 80 μg of protein was run on a 4%–12% Bis-Tris gel, blocked in 5% (w/v) milk for 1 hr at room temperature. Membranes were incubated overnight with the primary antibody (Millipore) at 4°C. β-actin (Sigma) was used as a loading control.
The mRNA expression of IGHMBP2 in six brain regions was assessed in humans during development.42 After birth, the expression of IGHMBP2 shows an increase in the cerebellar cortex, whereas in other brain regions there is a small decrease. IGHMBP2 expression levels seem to be constant throughout adult life (Figure S5). In adults, using data from ten regions of normal human postmortem brain tissue,42 the highest IGHMBP2 expression levels were also in the cerebellum. Expression in other body tissues was ubiquitous, with moderate expression in fibroblasts and lymphoblastoid cell lines (Figure S5). These data indicate the importance of the IGHMBP2 protein in the peripheral nerve but suggest that in other tissues with high expression, such as the cerebellum, the protein has a less important function as individuals with IGHMBP2 mutations do not have cerebellar signs. CMT2 is characterized by a highly heterogeneous genotype, with mutations in several unique genes being responsible for disease. The genetic background plays an important role in the classification of the disease and is crucial to find common pathways to explain the characteristic features seen in most affected individuals. No direct interactions between IGHMBP2 and any of the CMT2 proteins have been found so far. However, with the GeneMANIA prediction server,43 the presence of a network of interacting proteins known in CMT2 with IGHMBP2 can be observed (Figure S6). Given that many people with CMT2 are genetically undefined, and with the increasing amount of genetic data available, network analysis will be important in identifying causative and modifying gene pathways.
Together, our studies indicate that autosomal recessive IGHMBP2 mutations are a cause of CMT2. The clinical presentation is of a relatively pure form of CMT2, some more severe than others as seen in Tables 2 and 3, and typically what is seen in a number of the other genetic causes of CMT2, such as those individual with defects in MFN2, MPZ, MED25, and Lamin A/C genes. In contrast, SMARD1 usually presents in the first few days or weeks of life and children usually die before they are 1 year old. In addition, neurophysiology is much milder in CMT2 as compared to SMARD1 (Table 3), and the CMT2 sural nerve biopsy shows similar mild features (Figure 2).
Previous work by Guenther and Grohmann and colleagues also quantified the residual IGHMBP2 protein levels in a mouse model of SMARD1 and in lymphoblastoid cell lines from children with SMARD1. They found significant differences in the IGHMBP2 protein levels of individuals with typical congenital SMARD1, juvenile SMARD1 (respiratory distress at 3.5 months), and controls. Despite the reduction in protein levels, IGHMBP2 mRNA levels were not decreased in individuals with SMARD1 and IGHMBP2 mutations, an identical result which we also found in individuals with CMT2 (data not shown). This suggests that defective or truncated proteins undergo posttranslational degradation. Although we have found a number of IGHMBP2 mutations associated with CMT2, and mutations are usually different to SMARD1 in type and combination and result in higher residual protein levels in CMT2 as compared with SMARD1 and controls, we are cautious whether this always correlates with the onset of disease and phenotype. Protein levels are reduced in missense and nonsense or frameshift mutations, but the numbers are too few to correlate exact figures and there might be differences between mutation types. In addition, these experiments were carried out on material such as fibroblast and lymphoblastoid cell lines, which are not primarily affected in CMT2 or SMARD1. However, IGHMBP2 mRNA is widely expressed throughout the body and it is likely that these tissues might reflect the consequences of mutations. A further genetic factor that might modify the phenotype was identified in the IGHMBP2 mouse model (nmd) and was contained within the BAC-27k3 transgene. Expression of this transgene completely rescued the reduction in the total number of myelinated axons in the nmd femoral motor nerves. The syntenic genomic area in humans contains four tRNATyr genes and the activator of basal transcription 1 (ABT1) gene;29 no variations were found in these genes in the index cases with CMT2 studied here.
IGHMBP2 consists of 993 amino acids, 7 putative helicase motifs,44 and a DEAD box-like motif, typical for RNA helicases.45 IGHMBP2 contains a DNA-binding domain at position 638−786, including the helicase motifs V and VI44,46,47 and the nucleic acid-binding R3H motif48 is involved in immunoglobulin switching,47 in pre-mRNA processing,45 and in regulation of transcription by DNA binding.49 In this respect, IGHMBP2 resembles the SMN protein, which binds directly to DNA and RNA.50,51 Senataxin (SETX [MIM 608465]) encodes an 302.8 kD protein that contains a DNA/RNA helicase domain with strong homology to human UPF1 regulator of nonsense transcripts homolog (UPF1 [MIM 601430]) and IGHMBP2. Heterozygous mutations in SETX cause a type of motor neuronopathy called ALS4 and different mutations, inherited in a recessive fashion cause an ataxia with neuropathy called ataxia-oculomotor apraxia type 2 (AOA2 [MIM 606002]). The overlap in homology suggests that DNA/RNA helicase dysfunction might play an important role in the development of different types of neuropathy. The helicase domain of SETX also showed strong homology with UPF1, which, like IGHMBP2, also plays a role in producing mature mRNA.52 We suggest that mutations in IGHMBP2 may lead to the dysfunction of the helicase activity of this protein. It is conceivable that the reduced protein levels or the abnormal IGHMBP2 protein in SMARD1 and CMT2 impair the capacity of neurons to produce error-free mature mRNA, thus leading to neuronal degeneration. The overlap in gene type and function with the identification of different phenotypes illustrate the increasingly recognized shared molecular mechanisms that underlie the inherited neuropathies.
Acknowledgments
The authors would like to thank all the participants of the study for their essential help with this work. This study was supported by the Medical Research Council (MRC UK), The Wellcome Trust, The Brain Research Trust (BRT), The French Muscular Dystrophy Association (AFM). We are also supported by the MRC Neuromuscular Diseases Centre grant (G0601943) and we thank the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) for their support. This work was also partially funded by the University of Antwerp (TOP BOF 29069 to A.J.), the Fund for Scientific Research-Flanders (FWO G054313N to A.J.), The Ministry of Science and Technological Development, Republic of Serbia (project No. 17 3016 and 17 508), NIH (R01NS075764 and U54NS065712 S.Z.), the CMT Association (S.Z.), the Muscular Dystrophy Campaign (MDC), the Muscular Dystrophy Association (MDA), The European Research Council (309548), the Randerson Foundation, the Association Belge contre les Maladies Neuromusculaires (ABMM) and the EU FP7/2007-2013 under grant agreement number 2012-305121 (NEUROMICS). This study was also supported by the National Institute for Health Research (NIHR) University College London Hospitals (UCLH) Biomedical Research Centre (BRC).
Footnotes
This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/3.0/).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Database of Genomic Variants (DGV), http://dgv.tcag.ca/dgv/app/home
Ingenuity Variant Analysis, http://www.ingenuity.com/products/variant-analysis
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Harel T., Lupski J.R. Charcot-Marie-Tooth disease and pathways to molecular based therapies. Clin Genet. 2014 doi: 10.1111/cge.12393. Published online May 9, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Reilly M.M., Shy M.E. Diagnosis and new treatments in genetic neuropathies. J. Neurol. Neurosurg. Psychiatry. 2009;80:1304–1314. doi: 10.1136/jnnp.2008.158295. [DOI] [PubMed] [Google Scholar]
- 4.Saporta M.A., Shy M.E. Inherited peripheral neuropathies. Neurol. Clin. 2013;31:597–619. doi: 10.1016/j.ncl.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shy M.E. Inherited peripheral neuropathies. Continuum (Minneap Minn). 2011;17:294–315. doi: 10.1212/01.CON.0000396963.75069.66. [DOI] [PubMed] [Google Scholar]
- 6.Timmerman V., Strickland A.V., Züchner S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes (Basel) 2014;5:13–32. doi: 10.3390/genes5010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bombelli F., Stojkovic T., Dubourg O., Echaniz-Laguna A., Tardieu S., Larcher K., Amati-Bonneau P., Latour P., Vignal O., Cazeneuve C. Charcot-Marie-Tooth Disease Type 2A: From Typical to Rare Phenotypic and Genotypic Features. J. Am. Med. Assoc. Neurol. 2014;71:1036–1042. doi: 10.1001/jamaneurol.2014.629. [DOI] [PubMed] [Google Scholar]
- 8.Wee C.D., Kong L., Sumner C.J. The genetics of spinal muscular atrophies. Curr. Opin. Neurol. 2010;23:450–458. doi: 10.1097/WCO.0b013e32833e1765. [DOI] [PubMed] [Google Scholar]
- 9.Shy M.E., Patzkó A. Axonal Charcot-Marie-Tooth disease. Curr. Opin. Neurol. 2011;24:475–483. doi: 10.1097/WCO.0b013e32834aa331. [DOI] [PubMed] [Google Scholar]
- 10.Timmerman V., Clowes V.E., Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp. Neurol. 2013;246:14–25. doi: 10.1016/j.expneurol.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Roberts R.C. The Charcot-Marie-Tooth diseases: how can we identify and develop novel therapeutic targets? Brain. 2012;135:3527–3528. doi: 10.1093/brain/aws311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumner C.J., d’Ydewalle C., Wooley J., Fawcett K.A., Hernandez D., Gardiner A.R., Kalmar B., Baloh R.H., Gonzalez M., Züchner S. A dominant mutation in FBXO38 causes distal spinal muscular atrophy with calf predominance. Am. J. Hum. Genet. 2013;93:976–983. doi: 10.1016/j.ajhg.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertini E., Gadisseux J.L., Palmieri G., Ricci E., Di Capua M., Ferriere G., Lyon G. Distal infantile spinal muscular atrophy associated with paralysis of the diaphragm: a variant of infantile spinal muscular atrophy. Am. J. Med. Genet. 1989;33:328–335. doi: 10.1002/ajmg.1320330309. [DOI] [PubMed] [Google Scholar]
- 14.Grohmann K., Schuelke M., Diers A., Hoffmann K., Lucke B., Adams C., Bertini E., Leonhardt-Horti H., Muntoni F., Ouvrier R. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- 15.Grohmann K., Varon R., Stolz P., Schuelke M., Janetzki C., Bertini E., Bushby K., Muntoni F., Ouvrier R., Van Maldergem L. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann. Neurol. 2003;54:719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- 16.Kaindl A.M., Guenther U.P., Rudnik-Schöneborn S., Varon R., Zerres K., Schuelke M., Hübner C., von Au K.J. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) J. Child Neurol. 2008;23:199–204. doi: 10.1177/0883073807310989. [DOI] [PubMed] [Google Scholar]
- 17.Grohmann K., Rossoll W., Kobsar I., Holtmann B., Jablonka S., Wessig C., Stoltenburg-Didinger G., Fischer U., Hübner C., Martini R., Sendtner M. Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1) Hum. Mol. Genet. 2004;13:2031–2042. doi: 10.1093/hmg/ddh222. [DOI] [PubMed] [Google Scholar]
- 18.Rudnik-Schöneborn S., Stolz P., Varon R., Grohmann K., Schächtele M., Ketelsen U.P., Stavrou D., Kurz H., Hübner C., Zerres K. Long-term observations of patients with infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Neuropediatrics. 2004;35:174–182. doi: 10.1055/s-2004-820994. [DOI] [PubMed] [Google Scholar]
- 19.Jędrzejowska M., Madej-Pilarczyk A., Fidziańska A., Mierzewska H., Pronicka E., Obersztyn E., Gos M., Pronicki M., Kmieć T., Migdał M. Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur. J. Paediatr. Neurol. 2014;18:183–192. doi: 10.1016/j.ejpn.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 20.van der Heijde D., Calin A., Dougados M., Khan M.A., van der Linden S., Bellamy N.J. Selection of instruments in the core set for DC-ART, SMARD, physical therapy, and clinical record keeping in ankylosing spondylitis. Progress report of the ASAS Working Group. Assessments in Ankylosing Spondylitis. J. Rheumatol. 1999;26:951–954. [PubMed] [Google Scholar]
- 21.Guenther U.P., Schuelke M., Bertini E., D’Amico A., Goemans N., Grohmann K., Hübner C., Varon R. Genomic rearrangements at the IGHMBP2 gene locus in two patients with SMARD1. Hum. Genet. 2004;115:319–326. doi: 10.1007/s00439-004-1156-0. [DOI] [PubMed] [Google Scholar]
- 22.Maystadt I., Zarhrate M., Landrieu P., Boespflug-Tanguy O., Sukno S., Collignon P., Melki J., Verellen-Dumoulin C., Munnich A., Viollet L. Allelic heterogeneity of SMARD1 at the IGHMBP2 locus. Hum. Mutat. 2004;23:525–526. doi: 10.1002/humu.9241. [DOI] [PubMed] [Google Scholar]
- 23.Diers A., Kaczinski M., Grohmann K., Hübner C., Stoltenburg-Didinger G. The ultrastructure of peripheral nerve, motor end-plate and skeletal muscle in patients suffering from spinal muscular atrophy with respiratory distress type 1 (SMARD1) Acta Neuropathol. 2005;110:289–297. doi: 10.1007/s00401-005-1056-y. [DOI] [PubMed] [Google Scholar]
- 24.Corti S., Locatelli F., Papadimitriou D., Donadoni C., Del Bo R., Crimi M., Bordoni A., Fortunato F., Strazzer S., Menozzi G., Salani S., Bresolin N., Comi G.P. Transplanted ALDHhiSSClo neural stem cells generate motor neurons and delay disease progression of nmd mice, an animal model of SMARD1. Hum Mol Genet. 2006;15:167–187. doi: 10.1093/hmg/ddi446. [DOI] [PubMed] [Google Scholar]
- 25.Guenther U.P., Varon R., Schlicke M., Dutrannoy V., Volk A., Hübner C., von Au K., Schuelke M. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum. Mutat. 2007;28:808–815. doi: 10.1002/humu.20525. [DOI] [PubMed] [Google Scholar]
- 26.Hartley L., Kinali M., Knight R., Mercuri E., Hubner C., Bertini E., Manzur A.Y., Jimenez-Mallebrera C., Sewry C.A., Muntoni F. A congenital myopathy with diaphragmatic weakness not linked to the SMARD1 locus. Neuromuscul. Disord. 2007;17:174–179. doi: 10.1016/j.nmd.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Kaindl A.M., Guenther U.P., Rudnik-Schöneborn S., Varon R., Zerres K., Gressens P., Schuelke M., Hubner C., von Au K. [Distal spinal-muscular atrophy 1 (DSMA1 or SMARD1)] Arch. Pediatr. 2008;15:1568–1572. doi: 10.1016/j.arcped.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 28.Corti S., Nizzardo M., Nardini M., Donadoni C., Salani S., Del Bo R., Papadimitriou D., Locatelli F., Mezzina N., Gianni F. Motoneuron transplantation rescues the phenotype of SMARD1 (spinal muscular atrophy with respiratory distress type 1) J. Neurosci. 2009;29:11761–11771. doi: 10.1523/JNEUROSCI.2734-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Planell-Saguer M., Schroeder D.G., Rodicio M.C., Cox G.A., Mourelatos Z. Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet. 2009;18:2115–2126. doi: 10.1093/hmg/ddp134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joseph S., Robb S.A., Mohammed S., Lillis S., Simonds A., Manzur A.Y., Walter S., Wraige E. Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul. Disord. 2009;19:193–195. doi: 10.1016/j.nmd.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 31.Fanos V., Cuccu A., Nemolato S., Marinelli V., Faa G. A new nonsense mutation of the IGHMBP2 gene responsible for the first case of SMARD1 in a Sardinian patient with giant cell hepatitis. Neuropediatrics. 2010;41:132–134. doi: 10.1055/s-0030-1262852. [DOI] [PubMed] [Google Scholar]
- 32.Uchiumi F., Enokida K., Shiraishi T., Masumi A., Tanuma S. Characterization of the promoter region of the human IGHMBP2 (Smubp-2) gene and its response to TPA in HL-60 cells. Gene. 2010;463:8–17. doi: 10.1016/j.gene.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 33.Chalançon M., Debillon T., Dieterich K., Commare M.C. [A rare cause of respiratory failure in infants: distal spinal-muscular atrophy 1 (DSMA1 or SMARD1)] Arch. Pediatr. 2012;19:1082–1085. doi: 10.1016/j.arcped.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Eckart M., Guenther U.P., Idkowiak J., Varon R., Grolle B., Boffi P., Van Maldergem L., Hübner C., Schuelke M., von Au K. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Pediatrics. 2012;129:e148–e156. doi: 10.1542/peds.2011-0544. [DOI] [PubMed] [Google Scholar]
- 35.Messina M.F., Messina S., Gaeta M., Rodolico C., Salpietro Damiano A.M., Lombardo F., Crisafulli G., De Luca F. Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature. Eur J Paediatr Neurol. 2012;16:90–94. doi: 10.1016/j.ejpn.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Krieger F., Elflein N., Ruiz R., Guerra J., Serrano A.L., Asan E., Tabares L., Jablonka S. Fast motor axon loss in SMARD1 does not correspond to morphological and functional alterations of the NMJ. Neurobiol. Dis. 2013;54:169–182. doi: 10.1016/j.nbd.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 37.Lin X., Zhang Q.J., He J., Lin M.T., Murong S.X., Wang N., Chen W.J. Variations of IGHMBP2 Gene Was Not the Major Cause of Han Chinese Patients With Non-5q-Spinal Muscular Atrophies. J. Child Neurol. 2013;29:NP35–NP39. doi: 10.1177/0883073813497827. [DOI] [PubMed] [Google Scholar]
- 38.Litvinenko I., Kirov A.V., Georgieva R., Todorov T., Malinova Z., Mitev V., Todorova A. One Novel and One Recurrent Mutation in IGHMBP2 Gene, Causing Severe Spinal Muscular Atrophy Respiratory Distress 1 With Onset Soon After Birth. J. Child Neurol. 2013;29:799–802. doi: 10.1177/0883073813477203. [DOI] [PubMed] [Google Scholar]
- 39.Guenther U.P., Handoko L., Varon R., Stephani U., Tsao C.Y., Mendell J.R., Lützkendorf S., Hübner C., von Au K., Jablonka S. Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J. Mol. Med. 2009;87:31–41. doi: 10.1007/s00109-008-0402-7. [DOI] [PubMed] [Google Scholar]
- 40.Lim S.C., Bowler M.W., Lai T.F., Song H. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res. 2012;40:11009–11022. doi: 10.1093/nar/gks792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pitt M., Houlden H., Jacobs J., Mok Q., Harding B., Reilly M., Surtees R. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain. 2003;126:2682–2692. doi: 10.1093/brain/awg278. [DOI] [PubMed] [Google Scholar]
- 42.Trabzuni D., Ryten M., Walker R., Smith C., Imran S., Ramasamy A., Weale M.E., Hardy J. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J. Neurochem. 2011;119:275–282. doi: 10.1111/j.1471-4159.2011.07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warde-Farley D., Donaldson S.L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C.T. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Suppl):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuta T.R., Fukita Y., Miyoshi T., Shimizu A., Honjo T. Isolation of cDNA encoding a binding protein specific to 5′-phosphorylated single-stranded DNA with G-rich sequences. Nucleic Acids Res. 1993;21:1761–1766. doi: 10.1093/nar/21.8.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molnar G.M., Crozat A., Kraeft S.K., Dou Q.P., Chen L.B., Pardee A.B. Association of the mammalian helicase MAH with the pre-mRNA splicing complex. Proc. Natl. Acad. Sci. USA. 1997;94:7831–7836. doi: 10.1073/pnas.94.15.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miao M., Chan S.L., Fletcher G.L., Hew C.L. The rat ortholog of the presumptive flounder antifreeze enhancer-binding protein is a helicase domain-containing protein. Eur. J. Biochem. 2000;267:7237–7246. doi: 10.1046/j.1432-1327.2000.01836.x. [DOI] [PubMed] [Google Scholar]
- 47.Fukita Y., Mizuta T.R., Shirozu M., Ozawa K., Shimizu A., Honjo T. The human S mu bp-2, a DNA-binding protein specific to the single-stranded guanine-rich sequence related to the immunoglobulin mu chain switch region. J. Biol. Chem. 1993;268:17463–17470. [PubMed] [Google Scholar]
- 48.Grishin N.V. The R3H motif: a domain that binds single-stranded nucleic acids. Trends Biochem. Sci. 1998;23:329–330. doi: 10.1016/s0968-0004(98)01258-4. [DOI] [PubMed] [Google Scholar]
- 49.Chen N.N., Kerr D., Chang C.F., Honjo T., Khalili K. Evidence for regulation of transcription and replication of the human neurotropic virus JCV genome by the human S(mu)bp-2 protein in glial cells. Gene. 1997;185:55–62. doi: 10.1016/s0378-1119(96)00630-0. [DOI] [PubMed] [Google Scholar]
- 50.Lorson C.L., Androphy E.J. The domain encoded by exon 2 of the survival motor neuron protein mediates nucleic acid binding. Hum. Mol. Genet. 1998;7:1269–1275. doi: 10.1093/hmg/7.8.1269. [DOI] [PubMed] [Google Scholar]
- 51.Meister G., Bühler D., Laggerbauer B., Zobawa M., Lottspeich F., Fischer U. Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum. Mol. Genet. 2000;9:1977–1986. doi: 10.1093/hmg/9.13.1977. [DOI] [PubMed] [Google Scholar]
- 52.Mendell J.T., ap Rhys C.M., Dietz H.C. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science. 2002;298:419–422. doi: 10.1126/science.1074428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.