

Abstract
Metastatic disease still lacks effective treatments, and remains the primary cause of cancer mortality. Therefore, there is a critical need to develop better strategies to inhibit metastatic cancer. The Rho family GTPase Rac is an ideal target for anti-metastatic cancer therapy, because Rac is a key molecular switch that is activated by a myriad of cell surface receptors to promote cancer cell migration/invasion and survival. Previously, we reported the design and development of EHop-016, a small molecule compound, which inhibits Rac activity of metastatic cancer cells with an IC50 of 1 μM. EHop-016 also inhibits the activity of the Rac downstream effector p21-activated kinase (PAK), lamellipodia extension, and cell migration in metastatic cancer cells. Herein, we tested the efficacy of EHop-016 in a nude mouse model of experimental metastasis, where EHop-016 administration at 25 mg/kg body weight (BW) significantly reduced mammary fat pad tumor growth, metastasis, and angiogenesis. As quantified by UPLC MS/MS, EHop-016 was detectable in the plasma of nude mice at 17 to 23 ng/ml levels at 12 h following intraperitoneal (i.p.) administration of 10 to 25 mg/kg BW EHop-016. The EHop-016 mediated inhibition of angiogenesis In Vivo was confirmed by immunohistochemistry of excised tumors and by In Vitro tube formation assays of endothelial cells. Moreover, EHop-016 affected cell viability by down-regulating Akt and Jun kinase activities and c-Myc and Cyclin D expression, as well as increasing caspase 3/7 activities in metastatic cancer cells. In conclusion, EHop-016 has potential as an anticancer compound to block cancer progression via multiple Rac-directed mechanisms.
Introduction
Cancer progression to metastasis contributes to the poor prognosis of cancer patients due to the aggressive and invasive behavior of cancer cells that evade the immune system and establish tumors at distant organs. Therefore, there is a critical need to design and develop therapeutics that can block cancer cell invasion and migration away from the primary tumor [1], [2]. The closely related members of the Rho family, Rac and Cdc42, have been extensively studied due to their pivotal roles in actin cytoskeleton organization, migration/invasion and metastasis, epithelial to mesenchymal transition, transcription, cell proliferation, cell cycle progression, apoptosis, vesicle trafficking, angiogenesis, and cell adhesions [3], [4], [5]. Indeed, studies from us and others have implicated hyperactive Rac1 and Rac3 with increased survival, proliferation, and invasion of many cancer types [6], [7], [8], [9], [10]. In addition to promoting cancer malignancy, Rac and Cdc42 have also been shown to be essential for Ras and other oncogene-mediated transformation [11], [12].
Racs [1], [2], [3] are activated by a myriad of cell surface receptors that include: integrins, G protein coupled receptors, growth factor receptors, and cytokine receptors. These cell surface receptors regulate cancer promoting signal cascades that have been implicated with Rac and its direct downstream effector p21-activated kinase (PAK) activity [13]. These pathways include: phosphoinositide 3-kinase (PI3-K)/Akt/mammalian target of Rapamycin (mTOR); signal transducer and activator of transcription (STATs); and the mitogen activated protein kinases (MAPKs): extracellular regulated kinase (ERK), jun kinase (JNK), and p38 MAPK [14], [15], [16], [17], [18]. Activated Rac has also been shown to affect cell proliferation via signaling to the oncogenes c-Myc and Cyclin D [19]. Therefore, Rac GTPases play a pivotal role in regulation of cancer malignancy, and targeting Racs appear to be a viable strategy to impede cancer metastasis [8], [15], [20], [21].
Unlike Ras, Rho GTPases are not mutated in disease but activated via the deregulation of expression and/or activity of their upstream regulators, guanine nucleotide exchange factors (GEFs) [22]. Accordingly, although ~ 9% of melanomas were recently found to contain an activating Rac mutation [23], and the hyperactive splice variant Rac1b is frequently overexpressed in cancer [24], a majority of the Rac proteins in human cancer are activated due to up-regulated GEFs [21], [25], [26]. So far, over 70 potential Rac GEFs are known; and many members of the largest family of Rac GEFs, the Dbl family, have been identified as oncogenes [22], [27], [28], [29]. Of the Rac GEFs, T-cell invasion and metastasis gene product (Tiam-1), Trio, Vav (1/2/3), and PIP3-dependent Rac exchanger (p-Rex1/2) have been implicated in the progression of metastatic breast and other cancers [30], [31], [32], [33], [34], [35]. Therefore, the binding of GEFs to Rac and Cdc42 has been targeted as a rational strategy to inhibit their activity; and thus, metastasis.
The Rac inhibitor NSC23766 was identified as a small molecule compound that inhibits the interaction of Rac with the GEFs Trio and Tiam1 [36], [37], [38]. NSC23766 has been used to demonstrate the significance of Rac activity in cancer cell proliferation, survival, migration, metastasis, and therapy resistance [10], [39], [40], [41], [42], [43]. However, the high effective concentrations (IC50 > 75 μM) of NSC23766 limit its use as a therapeutic agent [36]. Other known Rac inhibitors also have IC50s of 10 to 50 μM [44], [45]; including the recently published Rac inhibitors AZA1, ZINC69391, and IA-116 [46], [47]. At concentrations ranging from 5 to 20 μM, AZA1 acted as a dual inhibitor for Rac and Cdc42, and blocked prostate cancer cell proliferation, cell migration, and reduced Cyclin D1, and PAK and Akt activities [46]. Another compound ZINC69361, which inhibited Rac activity with an IC50 of 61 μM and reduced lung metastasis, was used as a lead to derive IA-116, which was selective for Rac and inhibited the interaction between Rac and the Rac GEF p-Rex1; albeit, at μM effective concentrations [47].
Recent studies have also shown the utility of the NSC23766 derivative AZA197, which was identified as a selective inhibitor for the closely related Rac homolog Cdc42. AZA197, at 1 to 10 μM, inhibited the Cdc42 GEF Dbs activity, PAK and ERK activities, and reduced Cyclin D levels, colon cancer cell proliferation, and cancer progression in a mouse model [48]. The potency of this inhibitor is similar to that of ML141 (CID2950007), another Cdc42 selective inhibitor with an IC50 ~ 3 to 5 μM [49], that was shown to inhibit melanoma cell migration [50]. These data demonstrate the utility of developing chemical probes to target both Rac and Cdc42 in malignant cancer.
To improve the efficacy of NSC23766 and its derivatives, we developed a panel of related compounds [51], and identified EHop-016 as a Rac inhibitor that is 100 times more potent than NSC23766, and binds to the effector domain of Rac1 with a tighter interaction [52]. To our knowledge, EHop-016 is one of the most potent Rac inhibitors that has been published, and is an effective tool for probing Rac function in cell and mouse models; as has been shown by us and others, in studies using breast cancer cell lines, leukemia, melanoma, and T lymphocytes [50], [52], [53], [54]. We reported that EHop-016 inhibits the Rac activity of metastatic cancer cells with an IC50 of 1 μM by blocking the specific interaction of Rac with the Rac GEF and oncogene Vav. EHop-016 also inhibits the activity of the Rac downstream effector PAK, lamellipodia extension, and cell migration in metastatic cancer cells at concentrations less than 10 μM, while concentrations ≥ 10 μM inhibits the activity of the close Rac homolog Cdc42, and cell viability [52], [53].
The aim of this study was to test the In Vivo effects of EHop-016 in cancer progression. We used metastatic cancer cell lines and a mouse model of experimental metastasis to demonstrate the efficacy of EHop-016 at reducing mammary fat pad tumor growth, metastasis, and angiogenesis. We also found that EHop-016 inhibited cell survival and proliferation regulators, and induced apoptosis in metastatic cancer cells; thus, highlighting its potential as an anticancer compound.
Materials and Methods
Cell Culture
The human metastatic breast cancer cell line MDA-MB-435 expressing green fluorescent protein (GFP) (kind gift of Dr. Danny Welch, The University of Alabama at Birmingham, AL, 2009) was cultured, as described in [55]. The prostate cancer cell line PC3 was purchased from American Type Culture Collection (ATCC, Manassas, VA) and was cultured in Roswell Park Memorial Institute (RPMI) Medium supplemented with 10% heat inactivated fetal bovine serum (FBS) at 37°C in 5% CO2. Primary Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from ATCC and were cultured at 37°C in 5% CO2 using the endothelial cell growth kit-BBE media (Vascular cell Basal media + added supplements) from ATCC, as per manufacturer instructions.
EHop-016 Synthesis
Ehop-016 was synthesized as previously described by us in [52]. Stock solutions were made in 10% dH2O and 90% DMSO.
Immunohistochemistry
Tumor specimens were embedded in optimal cutting temperature (OCT) medium. Sections (5 μm) were fixed for two minutes each in acetone, chloroform:acetone, and acetone at − 20°C. Washed slides were incubated in blocking buffer (3% horse serum, 3% goat serum) and then with anti-CD31 (1:50 dilution; Abcam, Cambridge, MA) overnight in a humid chamber at 4°C, followed by incubation with Alexa Fluor 594 goat anti-rabbit (1:2000; Life Technologies, Carlsbad, CA) for 1 h at room temperature. After washing with 1 × PBS, sections were counterstained with 49-6-diamidino-2-phenylindole (DAPI) (1:5,000; Santa Cruz Biotechnology, Santa Cruz, CA) and mounted. Digital photographs were obtained using a Nikon Eclipse TS 100 Inverted microscope (Nikon, Melville, NY) with the NIS-Elements F 3.0 software and a Zeiss AxiocamMRc (Carl Zeiss, Gottingen, Germany).
Capillary Tube Formation Assay
Capillary tube formation was analyzed using 1:5 Matrigel matrix (Corning, Tewksbury, MA) in ice-cold buffer (10 mM of Tris Base 0.7% NaCl, pH 8), solidified by incubation at 37°C for ~ 1.5 h. A total of 40,000 HUVECs/well, pre-treated with vehicle (0.1% DMSO) or 8 μM of Ehop-016 for 24 h, were seeded into Matrigel pre-coated (200 μl/well) 48-well plates. Fresh vehicle or 8 μM of Ehop-016 was added to the corresponding treatments during the assay. Tube formation was monitored following a 3 h incubation at 37°C and 5% CO2.
Rac Activity Assay
HUVECs or PC3 cells were treated with vehicle or 8 μM Ehop-016. After 24 h, cells were lysed and total protein was quantified using the Precision Red protein assay kit (Cytoskeleton, Inc., Denver, CO). Active Rac was pulled down using beads coupled to GST–p21-activated kinase (PAK)-Cdc42/Rac interactive binding (CRIB) motif (GST-PAK-PBD beads from Cytoskeleton, Denver, CO) as described in [7], [6]. Proteins were Western blotted using an anti-Rac antibody (Cell Signaling Technology, Inc., Danvers, MA). Positive bands were imaged using ChemiDoc MP system (Bio-Rad, Hercules, CA) and quantified using Image J software. Rac activity was determined as the Rac-GTP bound to the PAK–CRIB domain as a function of total Rac.
Apoptosis Assay
Apoptosis was measured by relative caspase 3/7 activity, as described in [56], using a Caspase-Glo3/7 Luminescence Assay Kit as per manufacturer’s instructions (Promega, Corp., Madison, WI, USA). Following treatment of MDA-MB-435 cells with vehicle or Ehop-016 at 5, 10, or 25 μM, 100 μl of Caspase-3/7 Glo reagent was added and incubated at room temperature for 60 minutes. Caspase-3/7 activities were determined by quantifying luminescence.
Western Blotting
MDA-MB-435 or PC3 cells were treated with vehicle, or 4 or 8 μM Ehop-016 for 24 h. Cells were immediately lysed as in [57] and total protein was quantified using the Precision Red protein assay kit (Cytoskeleton, Inc., Denver, CO). Equal total protein amounts were Western blotted using anti-Akt, anti-phospho AktThr308, anti-JNK, anti-phospho JNKThr183/Try185, anti-c-Myc, or anti-Cyclin D (Cell Signaling Technology, Inc., Danvers, MA) antibodies. The integrated density of positive bands was quantified using Image J software.
Animals
All animal studies were conducted under approved protocol #A8180112 by the University of Puerto Rico Medical Sciences Campus Institutional Animal Care and Use Committee, in accordance with the principles and procedures outlined in the NIH Guideline for the Care and Use of Laboratory Animals.
Female athymic nu/nu mice, 4 to 5 weeks old (Charles River Laboratories, Inc., Wilmington, MA) were maintained under pathogen-free conditions in HEPA-filtered cages (5 mice per cage) under controlled light (12 h light and dark cycle), temperature (22 to 24°C), and humidity (25%). The animals received autoclaved rodent diet (Tek Global, Harlan Teklad, Madison, WI) with 24.5% protein, 4.3% fat and 3.7% fiber and water ad libitum.
Tumor Establishment
GFP-MDA-MB-435 cells (~ 0.5 × 106) in Matrigel (BD Biosciences, San Jose, CA) were injected at the fourth right mammary fat pad under isofluorane inhalation (1% to 3% in oxygen using an inhalation chamber at 2 L/min) to produce orthotopic primary tumors as described in [57]. After tumor establishment (1 wk post-inoculation), the animals from the same litter with similar weight and tumor size were randomly divided into experimental treatment groups. The study was initiated with 10 mice/group. However, due to unforeseen mouse deaths (but not from EHop-016-mediated toxicity), the numbers on the last day were: Vehicle, N = 6; 10 mg/kg BW, N = 8; and 25 mg/kg BW, N = 4.
Administration of EHop-016
Mice were treated with vehicle (12.5% ethanol (Sigma-Aldrich, St. Louis, MO), 12.5% Cremophor (Sigma-Aldrich, St. Louis, MO), and 75% 1 × PBS pH 7.4), or 10 or 25 mg/kg BW Ehop-016 by intraperitoneal (i.p.) injection in a 100 μl volume every other day, 3 times a week. Treatments continued until sacrifice at day 55.
Whole Body Fluorescence Image Analysis
Mammary tumor growth was quantified as changes in the integrated density of GFP fluorescence, using methods developed by Hoffman and co-workers [58]. Mice were imaged one week following breast cancer cell inoculation (on day 1 of treatment administration) and once a week thereafter. The FluorVivo small animal In Vivo imaging system (INDEC Systems, Inc., Santa Clara, CA) was used for whole body imaging of GFP fluorescence.
Tumor fluorescence intensities were analyzed using Image J software (National Institutes of Health, Bethesda, MD). The final images were acquired on day 55. Relative tumor growth was calculated as the integrated density of fluorescence of each tumor on each day of imaging relative to the integrated density of fluorescence of the same tumor on day 1 of treatment administration, as described in [55], [57].
Analysis of Metastases
Following sacrifice, lungs, kidneys, livers, and spleens were excised and immediately stored in liquid N2. Stored organs were thawed and analyzed using an Olympus MV10 fluorescence macro zoom system microscope and images acquired with an Olympus DP71 digital camera, as described in [57]. Each organ was imaged on both sides. The fluorescent lesions (green component of RGB images) were quantified for integrated density of fluorescent pixels using Image J software.
Ehop-016 Detection by Ultra Performance Liquid Chromatography/Tandem Mass Spectrometry (UPLC-MS/MS)
Plasma Ehop-016 was quantified using an automated UPLC system coupled to a triple quadrupole tandem mass spectrometer (MS/MS) (Agilent Technologies, Santa Clara, CA). The data was collected and analyzed by the Agilent MassHunter software package (Version B.05.01). The UPLC separations were performed on a Poroshell 120 EC-C18 column (50 mm × 3.0 mm) with 2.7 μm particle size (Agilent, CA) under gradient conditions with a mobile phase of 1 mM ammonium fluoride aqueous solution (solution A) and 50% Acetonitrile/50% methanol/0.1% formic acid solution (solution B) at a flow rate of 0.5 ml/min at 40 °C. The initial mobile phase composition was 65% of solution A and 35% of solution B. The content of solution B was increased by a linear gradient to 98% from 2.5 minutes to 3.0 minutes. After 4.5 minutes, the content of solution B was decreased by a linear gradient to 35%. Finally, the column was equilibrated at the initial conditions for 1.5 minutes. The total run time for analysis was 6.5 minutes and the injection volume was 1 μl.
Statistical Analysis
Data are expressed as the mean ± SEM. Statistical analyses were done using Microsoft Excel and GraphPad Prism. Differences between groups were considered to be statistically significant at P ≤ .05. Differences between means for vehicle were compared with means for 10 mg/kg BW EHop-016 or 25 mg/kg BW Ehop-016 using Student’s t test. One-way ANOVAs were also performed for all 3 groups and the statistical significance determined by Kruskal–Wallis test and Dunn’s multiple comparisons test.
Results and Discussion
Metastasis, the migration of cancer cells away from the primary tumor to establish secondary tumors at distant sites, is a major cause of failure in cancer therapy and patient survival. Thus, there is an urgent need for strategies that specifically target migratory, and thus, metastatic cancer cells [2]. The Rho GTPases Rac and Cdc42 have been implicated in cancer progression via control of cancer cell proliferation, survival, migration/invasion, and metastasis [2], [59], [60], [61]. Therefore, we initiated the development of Rac and Cdc42 inhibitors as potential anti metastatic cancer therapeutics, using the established Rac inhibitor NSC23766 as a lead compound [51]. Recently, we disclosed the development of EHop-016, which inhibits Rac activity of metastatic cancer cells with an IC50 of 1 μM, and is the first compound reported to inhibit the activation of Rac by the oncogenic GEF Vav. EHop-016 inhibits the activity of the Rac downstream effector PAK, lamellipodia extension, and cell migration of metastatic cancer cells. At higher concentrations (≥ 10 μM) EHop-016 also inhibits Cdc42 activity and cell viability [52]. Herein, our objective was to test the feasibility of EHop-016 as a tool to inhibit metastatic cancer progression, using an athymic nude mouse model of experimental metastasis.
EHop-016 Reduces Mammary Tumor Growth
EHop-016 was administered by interperitoneal (i.p.) injection to nude mice with mammary tumors established from GFP-tagged MDA-MB-435 human metastatic cancer cells. Tumor growth was quantified as a measure of the fluorescence intensity of the primary mammary tumor of each mouse relative to day 1 from fluorescence images acquired once a week for 8 weeks. Administration of 25 mg/kg BW EHop-016 three times a week for 8 weeks resulted in a ~ 80% reduction in tumor growth compared to vehicle. As determined by Students t test, the decrease in tumor growth at 25 mg/kg BW EHop-016 was statistically significant when compared to vehicle or 10 mg/kg BW EHop-016 for the final four weeks of the study (Figure 1, A and B). On the final day of imaging, the comparison of tumor intensities between 0 and 10 mg/kg BW treatments with 25 mg/kg BW treatment was statistically significant when compared by the Kruskall–Wallis test. The Dunn’s multiple comparison test demonstrated statistical significance between 10 mg/kg BW treatment and the 25 mg/kg BW treatment, but not between 0 and the 25 mg/kg BW treatment. On the other hand, administration of 10 mg/kg BW EHop-016 did not cause significant changes in tumor growth when compared to the vehicle control (Figure 1B), as determined by the Students t test, as well as one-way ANOVA, using Kruskal-Wallis and Dunn’s multiple comparisons tests. These results demonstrate a concentration dependent effect of Ehop-016 on tumor growth.
Figure 1.
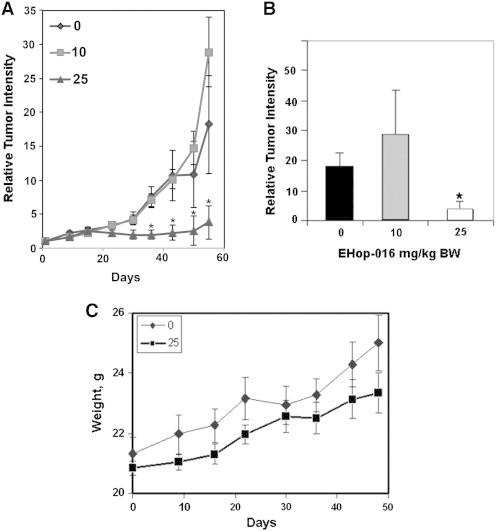
Effect of EHop-016 on mammary fat pad tumor growth. Athymic nude mice were inoculated at the mammary fat pad with GFP-MDA-MB-435 cells. Average relative tumor growth from fluorescence in situ images up to 55 days following 0, 10, or 25 mg/kg body weight (BW) EHop-016 (three times a week) was determined. (A) Relative tumor growth as a function of days following EHop-016 administration. (B) Average Relative tumor intensity on day 55 (N = 6, 0 mg/kg BW; N = 8,10 mg/kg BW; N = 4, 25 mg/kg BW). Error bars = SEM, * = p < 0.05. (C) Average mouse weight as a function of days following EHop-016 administration.
Figure 1C demonstrates that at 25 mg/kg BW, EHop-016 did not cause significant weight changes in the nude mice. Moreover, these animals did not demonstrate any gross phenotypical changes in skin color and malleability, or behavior. Alanine transaminase activity from liver lysates also demonstrated no change from vehicle controls (data not shown). Therefore, EHop-016 does not appear to be toxic to the animals at the effective concentration.
EHop-016 Levels in Mouse Plasma
To determine whether adequate EHop-016 levels were being delivered to the tumor tissue, we quantified the EHop-016 concentration in mouse plasma from blood collected at the end of the study. Mice were treated with EHop-016 three times a week for 55 days, with the final i.p. administration at 12 h prior to blood collection from cardiac puncture. A method was developed using UPLC-MS/MS to quantify EHop-016 from mouse plasma. EHop-016 was detected at ~ 17 ng/ml or 23 ng/ml (0.0395 or 0.0534 μM) in the mouse plasma, following 12 h administration of 4.65 mM EHop-016 (10 mg/kg BW EHop-016) or 11.61 mM EHop-016 (25 mg/kg BW EHop-016), respectively, in 100 μl in a 20 g mouse (Table 1). This low recovery rate in plasma may be due to a faster clearing rate of EHop-016 from the blood via high tissue absorbance of this highly lipophilic molecule. Alternatively, EHop-016 may become metabolized or become unavailable for detection due to sequestration by carrier proteins in the blood. These mechanisms of drug elimination from the mouse plasma are currently being explored.
Table 1.
EHop-016 levels in mouse plasma. EHop-016 was detected at ng/ml quantities from mouse plasma, using UPLC-MS/MS, following 10, or 25 mg/kg BW EHop-016 for 12 h. Data are expressed as the Mean ± SEM
| EHop-016 Administered (mg/kg BW) | Ehop-016 in plasma (ng/ml) |
|---|---|
| 10 | 17.1 ± 4.6 |
| 25 | 23.3 ± 6.5 |
EHop-016 Inhibits Metastasis
The In Vivo study was terminated at 55 days, and the distant organs were excised and quantified for fluorescent metastatic foci. As shown in Figure 2 and Table 2, EHop-016 at 25 mg/kg BW dramatically reduced metastasis to lung, liver, spleen, and kidneys.
Figure 2.
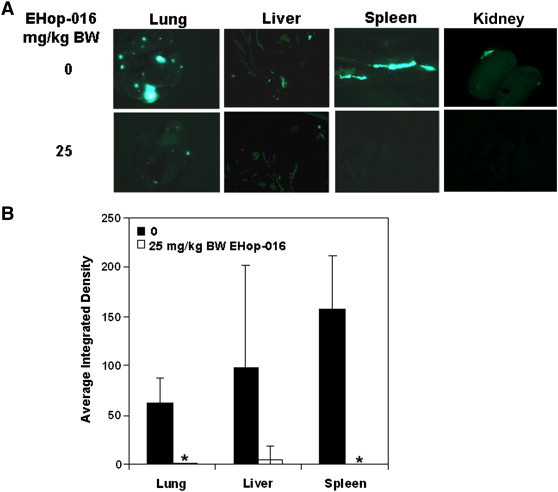
Effect of EHop-016 on metastasis. Mammary fat pad tumors were established in athymic nude mice using GFP-MDA-MB-435 cells. Mice were treated with 0, 10, or 25 mg/kg body weight (BW) EHop-016 (three times a week) (N = 5). Lungs, livers, kidneys, and spleens were removed at necropsy and imaged for fluorescent metastatic foci. (A) Representative organs under fluorescence microscopy for 25 mg/kg BW Ehop-016 treatment. (B) Average integrated intensity of fluorescent metastatic foci/organ. Error bars = SEM, * = p < 0.05.
Table 2.
Average integrated density (1 × 103) of metastatic foci
| Organ |
Lung | Spleen | Liver | Kidney |
|---|---|---|---|---|
| Treatment | ||||
| Veh | 61.4 | 157.4 | 97.6 | 7.4 |
| Ehop-016 (25 mg/kg BW) | 0.73 | 0 | 5.2 | 0 |
A number of studies have implicated Rac in cancer metastasis [15], [62]. Therefore, our results (Figure 1, Figure 2 and Table 2) further implicate Rac in the regulation of tumor growth and metastasis and demonstrate the utility of Rac inhibition as a strategy to block cancer progression. This study that demonstrates inhibition of metastasis, to all distant organs examined following EHop-016 treatment, may indicate that EHop-016 is inhibiting the intravasation step, when cancer cells migrate away from the primary mammary tumor to enter the circulation. Conversely, the marked reduction in tumor growth by EHop-016 may reduce the number of cells that are shed from the primary tumors and thus, effectively block metastasis. Future studies testing the effect of EHop-016 in spontaneous metastasis assays are expected to elucidate whether EHop-016 blocks the extravasation of metastatic cancer cells to establish metastases at distant sites.
EHop-016 Inhibits Angiogenesis
Angiogenesis is essential to cancer progression, where the endothelial cells in the tumor microenvironment form new blood vessels to sustain the growing tumor. A pertinent observation from our study was the relative lack of blood vessels at the surface of the mammary tumors from mice treated with 25 mg/kg BW EHop-016, compared to those treated with vehicle (Figure 3A). Therefore, tumor tissue was subjected to immunofluorescence for CD31, an endothelial cell marker. As expected, the tumors from mice that received vehicle controls demonstrated tubes arranged as capillaries. However, the tumors from mice that received EHop-016 demonstrated a ~ 85% decrease in capillary formation, as quantified from fluorescence micrographs (10 microscopic sections each for N = 2) of tissues stained for CD-31 for 0 and 25 mg/kg BW EHop-016 treatments (Figure 3A).
Figure 3.
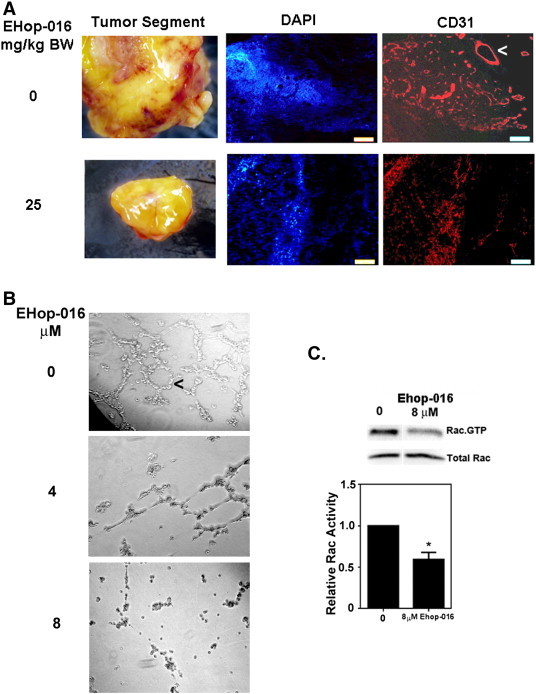
Effect of EHop-016 on angiogenesis. (A) Representative tumors from athymic nude mice inoculated at the mammary fat pad with GFP-MDA-MB-435 cells and treated with 0 or 25 mg/kg body weight (BW) EHop-016 (three times a week) (N = 5). Left, stereoscopic images of representative mammary tumors with blood vessels in red. Right, immunofluorescence micrographs of tissue sections with blood vessels stained for DAPI or CD31 (red fluorescence). (B) Representative micrographs following a tube formation assay using HUVEC cells treated with vehicle or 4 or 8 μM EHop-016 for 24 h (N = 4). Arrows indicate capillaries (tubes). (C) Representative Western blot and quantification of the Rac activity of HUVEC cells treated with 0 or 8 μM EHop-016 for 24 h from a pulldown assay for Rac.GTP (N = 2). Error bars = SEM, * = P < 0.05.
The ability of EHop-016 to inhibit Rac activity and capillary tube formation was also confirmed In Vitro using Human Umbilical Vein Endothelial Cells (HUVECs). As expected, EHop-016 inhibited the aggregation of endothelial cells into tubes. At 4 μM EHop-016, there was reduced tube formation, which was impaired at 8 μM, the concentration at which we observed a 50% reduction in Rac activity. (Figure 3B). Since Racs [1], [2] play an essential role in blood vessel morphogenesis via integrin signaling and endothelial cell proliferation/adhesion/migration mechanisms [63], [64], [65], we expect EHop-016 to additionally block tumor growth by reducing their blood supply via inhibition of the Rac activity of endothelial cells.
In this study, for the first time, we have shown that EHop-016 can be used effectively to block mammary tumor progression to metastasis. This anticancer activity of EHop-016 is predicted to be due to inhibition of Rac, and possibly Cdc42, activities in the human breast cancer cells as well as the endothelial cells in the tumor microenvironment. Therefore, EHop-016 may inhibit mammary tumor growth via multiple mechanisms of blocking the growth and migration of tumor cells and endothelial cells. Future studies will investigate the effect of EHop-016 on additional cells in the tumor microenvironment, such as macrophages and neutrophils as well as T and B lymphocytes that are regulated by Vav1/Rac2 signaling [66].
Recent studies have documented the utility of inhibiting Rac and Cdc42 to reduce tumor growth and metastasis in xenograft models. Another NSC23766 analog AZA1 (at 100 μg/day) was shown to inhibit Rac1 and Cdc42 in prostate cancer cells and reduce tumor growth via inhibition of Rac/Cdc42/PAK signaling to the actin cytoskeleton as well as Akt and Cyclin D to reduce cell survival and induce cell death [46]. The Rac GEF inhibitor ZINC639391 at 25 mg/kg BW, and its analog IA-116 at 3 mg/kg BW, resulted in reduced lung metastases from spontaneous metastases assays [47]. Similarly a Cdc42 specific inhibitor, AZA197, suppressed colon cancer growth via down-regulation of PAK and ERK activities, and Cyclin D1 expression [48]. Therefore, we expect EHop-016 to inhibit mammary tumor progression via multiple Rac/Cdc42/PAK-mediated signaling mechanisms.
EHop-016 Inhibits Rac/Cdc42/PAK-Mediated Cell Survival/Proliferation Signaling
To understand the mechanism by which EHop-016 reduces tumor growth, we investigated the effect of EHop-016 on apoptosis and cell survival signaling In Vitro. As previously shown by us, at concentrations ≥ 10 μM EHop-016 inhibits Rac and PAK activities by ~ 100% and Cdc42 activity by 75%, and reduces cell viability [52]. Figure 4 shows that in MDA-MB-435 metastatic cancer cells, at concentrations ≥ 10 μM, EHop-016 increases caspase 3/7 activity in a statistically significant (P < .05) and concentration-dependent manner with a maximum 1.6-fold induction at 25 μM, at concentrations that inhibit both Rac and Cdc42. This result indicates that EHop-016 may induce mitochondrial and death receptor-regulated apoptosis, and is consistent with a number of studies from various cancer types that have implicated a role for Rac/Cdc42/PAK signaling in cell survival and evasion of apoptosis [46], [53], [62].
Figure 4.
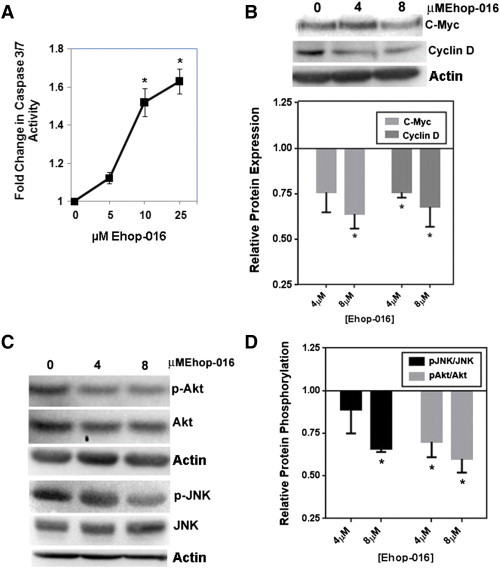
Effect of EHop-016 on apoptosis and Rac/Cdc42/PAK signaling in MDA-MB-435 cells. A) Fold change in caspase 3/7 activity in response to EHop-016. MDA-MB-435 cells treated for 24 h with 0, 5, 10, or 25 μM EHop-016 and subjected to caspase 3/7 activity assays. N = 3 ± SEM, Asterisk = P < .05. B-D. MDA-MB-435 cells treated with 0, 4, or 8 μM EHop-016 were lysed, and equal amounts of protein subjected to Western blotting followed by integrated density of positive bands. N = 2 to 5 ± SEM; Asterisk = P < .05. (B) Representative Western blot stained for c-Myc or Cyclin D and quantification of relative protein expression. (C) Representative Western blot stained for phospho (p)-Akt, Akt, p-JNK, or JNK. (D) Quantification of relative protein phosphorylation.
Our previous studies demonstrated that EHop-016, at concentrations < 10 μM, inhibits the Rac activity of metastatic breast cancer cells MDA-MB-435 and MDA-MB-231 [52], as well as the SKBR3 cell line (data not shown). To determine the potential of EHop-016 as a general Rac inhibitor, we also tested the effect of EHop-016 in the metastatic prostate cancer cell line PC3; a cell line that has been shown to be dependent on Rac/Vav signaling for migration/invasion [67]. Figure 5A demonstrates that 8 μM EHop-016 inhibits the Rac activity of PC3 cells by 50%.
Figure 5.
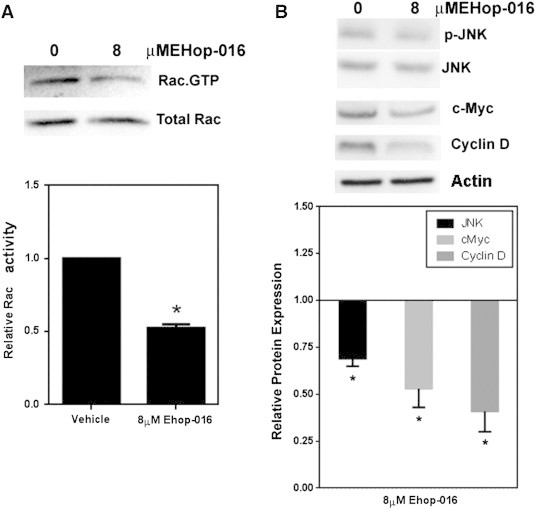
Rac activity and expression of cell proliferation regulators in response to EHop-016 in PC3 cells. PC3 cells treated for 24 h with 8 μM EHop-016 were subjected to the following assays. (A) Rac activity determined by a pulldown assay for Rac.GTP. Representative Western blot showing positive bands for Rac.GTP and total Rac. Relative Rac activity was quantified from the positive bands from Western blots and represented as the ratio of active Rac/Total Rac. (B) JNK, c-Myc and cyclin D expression in response to 8 μM EHop-016. Representative Western blots and quantification of positive bands for relative protein expression is shown. N = 2–4 ± SEM, Asterisk = P < .05.
To understand the mechanisms by which EHop-016 may reduce cell survival and induce apoptosis, we investigated the effect of EHop-016 on known Rac/Cdc42/PAK signaling pathway molecules, which have been implicated in controlling cell survival and proliferation. Activated Rac and Cdc42 may affect cell cycle progression via up-regulation of the oncogenes Cyclin D and c-Myc [10], [19], [68], [69], [70]. Rac/PAK signaling also regulates cell growth via signaling to Akt, ERK, JNK, and p38 MAPK [16], [71]. Figure 4, Figure 5 show that in both MDA-MB-435 and PC3 cells, EHop-016 significantly reduced the expression of the oncogenic cell cycle regulators c-Myc and Cyclin D expression by ~ 25% to 60%.
Next, we investigated the effect of EHop-016 on MAPK activity and expression. EHop-016 did not affect ERK activity or expression (data not shown). However, EHop-016 significantly reduced the JNK activity of MDA-MB-435 cells by ~ 30%. In PC3 prostate cancer cells, p-JNK levels were decreased but total JNK levels were also reduced to a similar extent, indicating that JNK expression is also down-regulated in this cell line. Moreover, in the MDA-MB-435 cells, EHop-016 reduced Akt activity by 40% at 4 and 8 μM, without affecting the Akt activity of PC3 cells (data not shown). These differences may be attributed to disparate cancer types of the two different cell lines. Studies have also linked Akt activity and thus, the regulation of the anti-apoptotic protein BAD with Rac action [72], and may account of the observed reduction in caspase activity in the MDA-MB-435 cell line, where a parallel 1.4-fold calculated increase in caspase activity is observed at 8 μM, when Akt activity is decreased by − 1.4-fold (Figure 4, A and B). Therefore, EHop-016 may reduce cell viability and tumor growth via a number of Rac-regulated pathways that control cell survival and death.
Conclusions
We have demonstrated that EHop-016 is a viable tool for blocking Rac activity via inhibition of the Vav/Rac interaction and thus, metastatic breast cancer cell migration In Vitro at μM concentrations [52]. Following this publication, the utility of EHop-016 as a Rac inhibitor has also been demonstrated in leukemia and melanoma cells [50], [53]. In this study, for the first time, we tested the efficacy of EHop-016 In Vivo. Our results, using a nude mouse model of experimental metastasis, demonstrate that EHop-016 significantly reduces mammary fat pad tumor growth and metastasis, as well as angiogenesis. The In Vitro assays with HUVEC cells, MDA-MB-435 cells, and PC3 cells further validate the use of EHop-016 to inhibit Rac, and thus, reduce cancer cell survival and proliferation, and inhibit metastatic cancer progression. Therefore, our data is significant for demonstrating the utility of developing chemical probes targeted at Rac, and the homolog Cdc42, as potential anti cancer therapeutics.
Acknowledgments
We wish to acknowledge Cristina Del Valle for excellent technical assistance. This study was supported by National Institute on Minority Health and Health Disparities of the National Institutes of Health (NIMHHD/NIH) U54MD008149, and Department of Defense/Breast Cancer Research Program (DoD/BCRP) W81XWH-07-1-0330 to SD; NIH/NIMHHD Research Centers in Minority Institutions (RCMI) 8G12MD007583, and Title V PPOHA P031S130068 from U.S. Department of Education to UCC; UPR RCM NIH/NIMHHD grants 5U54CA096297 and R25GM061838 to THB; and 2012 American Association of Colleges of Pharmacy (AACP) New Investigator Award to EH. The authors have no conflicts of interest to declare.
References
- 1.Condeelis J, Singer RH, Segall JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 3.Pai SY, Kim C, Williams DA. Rac GTPases in human diseases. Dis Markers. 2010;29:177–187. doi: 10.3233/DMA-2010-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 6.Azios NG, Krishnamoorthy L, Harris M, Cubano LA, Cammer M, Dharmawardhane SF. Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia. 2007;9:147–158. doi: 10.1593/neo.06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baugher PJ, Krishnamoorthy L, Price JE, Dharmawardhane SF. Rac1 and Rac3 isoform activation is involved in the invasive and metastatic phenotype of human breast cancer cells. Breast Cancer Res. 2005;7:R965–R974. doi: 10.1186/bcr1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan AY, Coniglio SJ, Chuang YY, Michaelson D, Knaus UG, Philips MR, Symons M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene. 2005;24:7821–7829. doi: 10.1038/sj.onc.1208909. [DOI] [PubMed] [Google Scholar]
- 9.Mira JP, Benard V, Groffen J, Sanders LC, Knaus UG. Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc Natl Acad Sci U S A. 2000;97:185–189. doi: 10.1073/pnas.97.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida T, Zhang Y, Rivera Rosado LA, Chen J, Khan T, Moon SY, Zhang B. Blockade of Rac1 activity induces G1 cell cycle arrest or apoptosis in breast cancer cells through downregulation of cyclin D1, survivin, and X-linked inhibitor of apoptosis protein. Mol Cancer Ther. 2010;9:1657–1668. doi: 10.1158/1535-7163.MCT-09-0906. [DOI] [PubMed] [Google Scholar]
- 11.Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Pedersen E, Basse A, Lefever T, Peyrollier K, Kapoor S, Mei Q, Karlsson R, Chrostek-Grashoff A, Brakebusch C. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation In Vivo. Oncogene. 2010;29:3362–3373. doi: 10.1038/onc.2010.95. [DOI] [PubMed] [Google Scholar]
- 13.Whale A, Hashim FN, Fram S, Jones GE, Wells CM. Signalling to cancer cell invasion through PAK family kinases. Front Biosci. 2011;16:849–864. doi: 10.2741/3724. [DOI] [PubMed] [Google Scholar]
- 14.Yang HW, Shin MG, Lee S, Kim JR, Park WS, Cho KH, Meyer T, Do HW. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol Cell. 2012;47:281–290. doi: 10.1016/j.molcel.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Soledad SM, Kazanietz MG. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal. 2012;24:353–362. doi: 10.1016/j.cellsig.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menges CW, Sementino E, Talarchek J, Xu J, Chernoff J, Peterson JR, Testa JR. Group I p21-activated kinases (PAKs) promote tumor cell proliferation and survival through the AKT1 and Raf-MAPK pathways. Mol Cancer Res. 2012;10:1178–1188. doi: 10.1158/1541-7786.MCR-12-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katz E, Sims AH, Sproul D, Caldwell H, Dixon MJ, Meehan RR, Harrison DJ. Targeting of Rac GTPases blocks the spread of intact human breast cancer. Oncotarget. 2012;3:608–619. doi: 10.18632/oncotarget.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell. 2011;42:50–61. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boettner B, Van Aelst L. The role of Rho GTPases in disease development. Gene. 2002;286:155–174. doi: 10.1016/s0378-1119(02)00426-2. [JID - 7706761] [DOI] [PubMed] [Google Scholar]
- 20.Mack NA, Whalley HJ, Castillo-Lluva S, Malliri A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle. 2011;10:1571–1581. doi: 10.4161/cc.10.10.15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33:4021–4035. doi: 10.1038/onc.2013.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23:9369–9380. doi: 10.1038/sj.onc.1208182. [DOI] [PubMed] [Google Scholar]
- 25.Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19:3013–3020. doi: 10.1038/sj.onc.1203621. [DOI] [PubMed] [Google Scholar]
- 26.Stengel K, Zheng Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell Signal. 2011;23:1415–1423. doi: 10.1016/j.cellsig.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 29.Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim Biophys Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 30.Adams HC, III, Chen R, Liu Z, Whitehead IP. Regulation of breast cancer cell motility by T-cell lymphoma invasion and metastasis-inducing protein. Breast Cancer Res. 2010;12:R69–R83. doi: 10.1186/bcr2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmby TR, Abe K, Karnoub AE, Der CJ. Vav transformation requires activation of multiple GTPases and regulation of gene expression. Mol Cancer Res. 2004;2:702–711. [PubMed] [Google Scholar]
- 32.Miller SL, DeMaria JE, Freier DO, Riegel AM, Clevenger CV. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol Endocrinol. 2005;19:939–949. doi: 10.1210/me.2004-0443. [DOI] [PubMed] [Google Scholar]
- 33.Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat. 2004;84:21–32. doi: 10.1023/B:BREA.0000018421.31632.e6. [DOI] [PubMed] [Google Scholar]
- 34.Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010;40:877–892. doi: 10.1016/j.molcel.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montero JC, Seoane S, Ocana A, Pandiella A. P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene. 2011;30:1059–1071. doi: 10.1038/onc.2010.489. [DOI] [PubMed] [Google Scholar]
- 36.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas EK, Cancelas JA, Chae HD, Cox AD, Keller PJ, Perrotti D, Neviani P, Druker BJ, Setchell KD, Zheng Y. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell. 2007;12:467–478. doi: 10.1016/j.ccr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Binker MG, Binker-Cosen AA, Gaisano HY, Cosen-Binker LI. Inhibition of Rac1 decreases the severity of pancreatitis and pancreatitis-associated lung injury in mice. Exp Physiol. 2008;93:1091–1103. doi: 10.1113/expphysiol.2008.043141. [DOI] [PubMed] [Google Scholar]
- 39.Gastonguay A, Berg T, Hauser AD, Schuld N, Lorimer E, Williams CL. The role of Rac1 in the regulation of NF-kB activity, cell proliferation, and cell migration in non-small cell lung carcinoma. Cancer Biol Ther. 2012;13:647–656. doi: 10.4161/cbt.20082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizukawa B, Wei J, Shrestha M, Wunderlich M, Chou FS, Griesinger A, Harris CE, Kumar AR, Zheng Y, Williams DA. Inhibition of Rac GTPase signaling and downstream prosurvival Bcl-2 proteins as combination targeted therapy in MLL-AF9 leukemia. Blood. 2011;118:5235–5245. doi: 10.1182/blood-2011-04-351817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen QY, Xu LQ, Jiao DM, Yao QH, Wang YY, Hu HZ, Wu YQ, Song J, Yan J, Wu LJ. Silencing of Rac1 modifies lung cancer cell migration, invasion and actin cytoskeleton rearrangements and enhances chemosensitivity to antitumor drugs. Int J Mol Med. 2011;28:769–776. doi: 10.3892/ijmm.2011.775. [DOI] [PubMed] [Google Scholar]
- 42.Hamalukic M, Huelsenbeck J, Schad A, Wirtz S, Kaina B, Fritz G. Rac1-regulated endothelial radiation response stimulates extravasation and metastasis that can be blocked by HMG-CoA reductase inhibitors. PLoS One. 2011;6:e26413–e26423. doi: 10.1371/journal.pone.0026413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, Wang Z, Jiang Y, Yang C. Inactivation of Rac1 reduces Trastuzumab resistance in PTEN deficient and insulin-like growth factor I receptor overexpressing human breast cancer SKBR3 cells. Cancer Lett. 2011;313:54–63. doi: 10.1016/j.canlet.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 44.Ferri N, Corsini A, Bottino P, Clerici F, Contini A. Virtual screening approach for the identification of new Rac1 inhibitors. J Med Chem. 2009;52:4087–4090. doi: 10.1021/jm8015987. [DOI] [PubMed] [Google Scholar]
- 45.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 46.Zins K, Lucas T, Reichl P, Abraham D, Aharinejad S. A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS One. 2013;8:e74924–e74936. doi: 10.1371/journal.pone.0074924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cardama GA, Comin MJ, Hornos L, Gonzalez N, Defelipe L, Turjanski AG, Alonso DF, Gomez DE, Menna LP. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem. 2013;14:840–851. doi: 10.2174/18715206113136660334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zins K, Gunawardhana S, Lucas T, Abraham D, Aharinejad S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of PAK1 activity. J Transl Med. 2013;11:295–310. doi: 10.1186/1479-5876-11-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Surviladze Z, Waller A, Strouse JJ, Bologa C, Ursu O, Salas V, Parkinson JF, Phillips GK, Romero E, Wandinger-Ness A. Anonymous. 2010. A potent and selective inhibitor of Cdc42 GTPase. [PubMed] [Google Scholar]
- 50.Maes H, Van ES, Krysko DV, Vandenabeele P, Nys K, Rillaerts K, Garg AD, Verfaillie T, Agostinis P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014;5:e1127–e1138. doi: 10.1038/cddis.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hernandez E, De LM-P, Dharmawardhane S, Vlaar CP. Novel inhibitors of Rac1 in metastatic breast cancer. P R Health Sci J. 2010;29:348–356. [PubMed] [Google Scholar]
- 52.Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De LM-P, Cubano LA, Vlaar CP, Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem. 2012;287:13228–13238. doi: 10.1074/jbc.M111.334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin H, Mali RS, Ma P, Chatterjee A, Ramdas B, Sims E, Munugalavadla V, Ghosh J, Mattingly RR, Visconte V. Pak and Rac GTPases promote oncogenic KIT-induced neoplasms. J Clin Invest. 2013;123:4449–4463. doi: 10.1172/JCI67509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manes TD, Pober JS. TCR-driven transendothelial migration of human effector memory CD4 T cells involves Vav, Rac, and myosin IIA. J Immunol. 2013;190:3079–3088. doi: 10.4049/jimmunol.1201817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlachterman A, Valle F, Wall KM, Azios NG, Castillo L, Morell L, Washington AV, Cubano LA, Dharmawardhane SF. Combined resveratrol, quercetin, and catechin treatment reduces breast tumor growth in a nude mouse model. Transl Oncol. 2008;1:19–27. doi: 10.1593/tlo.07100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong W, He L, Coppola M, Guo J, Esposito NN, Coppola D, Cheng JQ. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J Biol Chem. 2010;285:17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Castillo-Pichardo L, Martinez-Montemayor MM, Martinez JE, Wall KM, Cubano LA, Dharmawardhane S. Inhibition of mammary tumor growth and metastases to bone and liver by dietary grape polyphenols. Clin Exp Metastasis. 2009;26:505–516. doi: 10.1007/s10585-009-9250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang M, Baranov E, Jiang P, Sun FX, Li XM, Li L, Hasegawa S, Bouvet M, Al-Tuwaijri M, Chishima T. Whole-body optical imaging of green fluorescent protein-expressing tumors and metastases. Proc Natl Acad Sci U S A. 2000;97:1206–1211. doi: 10.1073/pnas.97.3.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 60.Chan SK, Hill ME, Gullick WJ. The role of the epidermal growth factor receptor in breast cancer. J Mammary Gland Biol Neoplasia. 2006;11:3–11. doi: 10.1007/s10911-006-9008-2. [DOI] [PubMed] [Google Scholar]
- 61.Arteaga CL, Truica CI. Challenges in the development of anti-epidermal growth factor receptor therapies in breast cancer. Semin Oncol. 2004;31:3–8. doi: 10.1053/j.seminoncol.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 62.King H, Nicholas NS, Wells CM. Role of p-21-activated kinases in cancer progression. Int Rev Cell Mol Biol. 2014;309:347–387. doi: 10.1016/B978-0-12-800255-1.00007-7. [DOI] [PubMed] [Google Scholar]
- 63.Galan Moya EM, Le GA, Gavard J. PAKing up to the endothelium. Cell Signal. 2009;21:1727–1737. doi: 10.1016/j.cellsig.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 64.Sandri C, Caccavari F, Valdembri D, Camillo C, Veltel S, Santambrogio M, Lanzetti L, Bussolino F, Ivaska J, Serini G. The R-Ras/RIN2/Rab5 complex controls endothelial cell adhesion and morphogenesis via active integrin endocytosis and Rac signaling. Cell Res. 2012;22:1479–1501. doi: 10.1038/cr.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Buul JD, Geerts D, Huveneers S. Rho GAPs and GEFs: Controling switches in endothelial cell adhesion. Cell Adh Migr. 2014;8:108–124. doi: 10.4161/cam.27599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 67.Valderrama F, Thevapala S, Ridley AJ. Radixin regulates cell migration and cell-cell adhesion through Rac1. J Cell Sci. 2012;125:3310–3319. doi: 10.1242/jcs.094383. [DOI] [PubMed] [Google Scholar]
- 68.Boureux A, Furstoss O, Simon V, Roche S. Abl tyrosine kinase regulates a Rac/JNK and a Rac/Nox pathway for DNA synthesis and Myc expression induced by growth factors. J Cell Sci. 2005;118:3717–3726. doi: 10.1242/jcs.02491. [DOI] [PubMed] [Google Scholar]
- 69.Fournier AK, Campbell LE, Castagnino P, Liu WF, Chung BM, Weaver VM, Chen CS, Assoian RK. Rac-dependent cyclin D1 gene expression regulated by cadherin- and integrin-mediated adhesion. J Cell Sci. 2008;121:226–233. doi: 10.1242/jcs.017012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito Y, Teitelbaum SL, Zou W, Zheng Y, Johnson JF, Chappel J, Ross FP, Zhao H. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J Clin Invest. 2010;120:1981–1993. doi: 10.1172/JCI39650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y, Rivera Rosado LA, Moon SY, Zhang B. Silencing of D4-GDI inhibits growth and invasive behavior in MDA-MB-231 cells by activation of Rac-dependent p38 and JNK signaling. J Biol Chem. 2009;284:12956–12965. doi: 10.1074/jbc.M807845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang FC, Kapur R, King AJ, Tao W, Kim C, Borneo J, Breese R, Marshall M, Dinauer MC, Williams DA. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity. 2000;12:557–568. doi: 10.1016/s1074-7613(00)80207-1. [DOI] [PubMed] [Google Scholar]