

Abstract
Inhibition of the proteasome offers many therapeutic possibilities in inflammation as well as in neoplastic diseases. However, clinical use of proteasome inhibitors is limited by the development of resistance or severe side effects. In our study we characterized the anti-tumor properties of the novel proteasome inhibitor BSc2118. The sensitivity of tumor lines to BSc2118 was analyzed in comparison to bortezomib using crystal violet staining in order to assess cell viability. The In Vivo distribution of BSc2118 in mouse tissues was tracked by a fluorescent-modified form of BSc2118 (BSc2118-FL) and visualized by confocal microscopy. Inhibition of the 20S proteasome was monitored both in cultured cell lines and in mice, respectively. Finally, safety and efficacy of BSc2118 was evaluated in a mouse melanoma model. BSc2118 inhibits proliferation of different tumor cell lines with a similar potency as compared with bortezomib. Systemic administration of BSc2118 in mice is well tolerated, even when given in a dose of 60 mg/kg body weight. After systemic injection of BSc2118 or bortezomib similar proteasome inhibition patterns are observed within the murine organs. Detection of BSc2118-FL revealed correlation of distribution pattern of BSc2118 with inhibition of proteasomal activity in cells or mouse tissues. Finally, administration of BSc2118 in a mouse melanoma model shows significant local anti-tumor effects. Concluding, BSc2118 represents a novel low-toxic agent that might be alternatively used for known proteasome inhibitors in anti-cancer treatment.
Introduction
The ubiquitin-proteasome system plays a crucial role in the maintenance of cellular homeostasis by participation in the degradation of the majority of cytosolic proteins. This system is involved in the regulation of the cell cycle, apoptosis, transcription, cell signaling, antigen presentation, inflammation and development [1]. This situation makes the ubiquitin-proteasome system to one of the most promising therapeutic targets for further drug screening and development.
The proteasome is an abundant cytosolic and nuclear protease complex, which contains a 20S proteasome core complex as central catalytic unit that harbors different proteolytic activities, i.e. a trypsin-like (T-L within the β2 subunit), a chymotrypsin-like (ChT-L within the β5 subunit) and a caspase-like (within the β1 subunit) [2]. Its activity within the cell is regulated by interaction of the 20S core with the regulatory 19S complex and with the PA28 complex at both ends of the proteasome cylinder [3]. The proteasome system is coupled with the ubiquitin system for controlled protein degradation [4], [5]. Therefore, inhibition of the proteasome leads in the first line to accumulation of polyubiquitinated proteins. Imbalance in cell cycle turn over and subsequent cell cycle arrest as well as the inhibition of NF-κB as a result from stabilization of IκBα are other hallmarks of proteasomal inhibition. Finally, inhibition of the 20S proteasome leads to induction of apoptosis that is a summary effect of the inability to degrade injurious substrates. In this context, the ChT-L activity is likely to be essential for most proteasomal functions and for the viability of cells. Irreversible inhibition or deletion of the β5 subunit carrying the ChT-L activity is therefore known to be lethal [6], [7].
Proteasome inhibition is an established therapeutic approach in anti-tumor drug development. In this context, proteasome inhibitors induce apoptosis more selectively in tumor than in normal cells, which is the most important rationale for application of these inhibitors in anti-tumor therapy. By stabilization of IκBα, proteasome inhibitors exert anti-inflammatory effects and promote death of tumor cells [8], [9], [10], [11], [12], [13].
Based on the catalytic specificity of the proteasome complex, a number of short peptide derived inhibitors (e.g., peptide boronic acids, vinyl sulfonates or peptide aldehydes) have been developed [14], [15], [16]. However, many of these were ultimately discarded from consideration for clinical use because of poor stability, low bioavailability and lack of specificity. The first drug applied in human diseases was bortezomib, a dipeptidyl boronic acid also known as PS-341 or Velcade (Millennium Pharmaceuticals, USA). Bortezomib selectively targets the catalytic β-subunits of the proteasome in a concentration dependent manner, thus inhibiting the chymotrypsin-like (β5/β5i) and to a lesser degree the caspase-like (β1/β1i) activity [17], [18]. The compound was initially approved for the treatment of drug-resistant multiple myeloma in 2003 [19]. Furthermore, this inhibitor was approved by the FDA for the treatment of previously untreated multiple myeloma as well as in Waldenström's macroglobulinemia and mantle cell lymphoma [20], [21], [22]. However, prolonged treatment with bortezomib induces substantial toxicity with frequent and potentially severe adverse events, such as peripheral neurotoxicity, cardiotoxicity, thrombocytopenia, anaemia, gastrointestinal symptoms, neuropathy, fever, fatigue, headache, arthralgia, rash as well as electrolyte disturbances and the development of drug resistance [23], [24], [25], [26]. Therefore, the search for new effective and low toxic inhibitors of the proteasome system is urgently needed.
Another proteasome inhibitor that has been frequently used in various experimental designs is MG132 (zLLL-CHO). In the present project, we characterized the novel inhibitor BSc2118 (patent no T30305), which is an analogue of MG132 with a better proteasome inhibition profile than MG132 [27]. Previously, BSc2118 has been shown to inhibit the ChTL activity of the proteasome, induce G2/M cell cycle arrest and promote apoptosis. BSc2118 further stabilizes IκBα and prevents NF-κB activation [27].
In the current work, we analyzed the distribution pattern of Bsc2118 both in various tumor cell lines and in mice as well as the inhibition profile in selected mice organs after intraperitoneal administration. We finally show that application of BSc2118 in mice induces local anti-tumor effects and is tolerated at higher doses as compared to bortezomib.
Material and Methods
Proteasome Inhibitors
BSc2118 and fluorescent Bodipy-BSc2118 was dissolved in DMSO at 20 mM and kept at − 20°C. Bortezomib (Lilly, Germany) was dissolved in distilled water at 4 mM and kept at − 20°C until usage.
Synthesis of BSc2118
BSc2118 was synthesized as described previously [28]. The fluorescent variant of BSc2118 (further named as BSc2118-FL) was synthesized as follows: a solution of 10 mg of the peptide NH2-Leu-Asp(tBu)-Leu-CHO (BSc2118) in 1 ml dimethylformamide DMF (pH = 8) was given to 10 mg of Bodipy-FL, SE (Molecular Probes, Germany). The reaction mixture was stirred overnight in darkness. Preparative purification by high-pressure liquid chromatography (HPLC) was carried out on a Shimadzu LC-8A system with an Agilent Zorbax, C18 SE column (250 × 21.2 mm), 7 μl, (buffer A: 0.2% trifluoroacetic acid TFA in water, buffer B: 0.2% TFA in water: acetonitrile, 1:4). The peptides were purified with a gradient of 60% B to 95% B in 50 min.
Cell Culture
HeLa cells and C26 murine colon cancer cells were cultured in RPMI-1640 (Biochrom AG, Germany) with stable glutamax, both supplemented with 10% heat-inactivated FBS (Invitrogen, Germany), 100 μg/ml streptomycin and 250 ng/ml amphotericin B (Invitrogen, Germany). For assessment of In Vitro cytostatic/cytotoxic activity of BSc2118, established human and mouse tumor cell lines were used. These cells were in particular: HCT116, PC3, LoVo, CaPan2, MDA435, Panc02, EMT6, C26, B16F10 (all ATCC) MeWo, MeWoCis, MeWoVin, MeWoEto, MeWoFote (obtained from Dr. H. Roekmann [29]), Mel-6, Mel-15, Mel-18, Mel-21, MaMel-63a (obtained from Dr. D. Schadendorf, Skin Cancer Unit Deutsches Krebsforschungszentrum, Heidelberg, Germany), WM35, WM902B, WM9 (obtained from Dr. M. Herlyn, Wistar Institute, Philadelphia, PA). All cells were grown either in DMEM or in RPMI-1640 and supplemented with 10% FCS plus antibiotics.
Estimation of GI50 (growth inhibition) of BSc2118 in a panel of tumor cell lines
The influence of BSc2118 on the growth of 22 tumor cell lines was analyzed using a crystal violet assay similarly as described for bortezomib by Adams et al [30]. GI50 is defined as the concentration needed to reduce the growth of treated cells to half that of untreated cells. Briefly, cells were seeded in quadruplicates on 96-well plates, exposed for 48 hours to proteasome inhibitors in 7 dilutions ranging from 10 nM to 1000 nM (for BSc2118) and from 1 nM to 100 nM (for bortezomib). The cytostatic/cytotoxic effects of both BSc2118 and bortezomib on treated cells were compared to that of control cells. The mean viability for the whole cell panel was calculated in two ways, thereafter. First, average viability for the entire panel was calculated for each concentration point followed by calculation of the average GI50 value. Second, GI50 was averaged for each cell line individually. Both methods of calculation resulted in similar results.
Isolation and Activity Measurement of 20S Proteasomes
20S proteasomes were isolated from both red blood cells of healthy volunteers and from murine organs [31]. Lysates from murine organs after injection of inhibitors were obtained by homogenization in 100 mM HEPES, pH 7.4, 2 mM MgCl2, 0.1% NP-40, 5 mM dithiothreitol and completed by Ultra-Turrax T8 (IKA-Werke). Chymotrypsin-like activity of the 20S proteasome was measured with a fluorogenic substrate (Suc-LLVY-AMC, Bachem, Germany). Briefly, 100 ng of purified proteasomes were exposed to proteasome inhibitors (0-1000 nM) and incubated with 50 μM of fluorogenic substrate for up to 60 min. Lysates normalized to protein content were directly incubated with 50 μM of fluorogenic substrate. The fluorescence was measured with POLARstar reader (BMG Labtech, Germany). The excitation and emission wavelengths were 390 nm and 460 nm, respectively. All experiments were performed in quadruplicates and repeated at least three times. Differences between groups were calculated by a Student’s t test. A P value of < 0.05 was considered to be statistically significant.
The Influence of Microsomal Enzymes on Inhibitor Stability
For analysis of inhibitor stability in the presence of microsomal enzymes, BSc2118 and MG132 (at 0.1 to 5 μM, respectively) were incubated with mouse (Balb/c) microsomes (GIBCO) for up to 24 hours according to manufacturer instructions. The proteasome activity (20S isolated from mouse muscles) was measured in the presence of inhibitors with/without microsomal fraction as described in the section above. The results are displayed as relative 20S activity in the presence of inhibitors incubated with microsomal fraction. Inhibitors incubated with PBS were defined as 100%. The data are displayed as decrease of inhibitory activity in the presence of microsomal enzymes. Differences between groups were calculated by Student’s t test. A P value of < 0.05 was considered to be statistically significant.
Immunoblot Analysis
The procedure has been described previously [27]. The polyubiquitinated proteins were detected with a polyclonal rabbit anti-ubiquitin antibody (Dako, Germany), and GAPDH was detected using a monoclonal anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase) antibody (Santa Cruz, Germany). A peroxidase-coupled anti-rabbit or anti-mouse antibody was used as secondary antibody (Jackson Immunoresearch, UK).
Incubation with BSc2118-FL and Confocal Microscopy
HeLa cells and C26 cells were directly incubated with BSc2118-FL, washed with PBS, fixed with 70% ethanol and probed with either rabbit anti-proteasome antibody (anti α4 subunit, Inst. for Biochemistry, Charité, Berlin, Germany) or with a mouse anti-ubiquitin conjugated antibody FK1 (Biomol, Germany). After washing, they were probed with secondary antibodies coupled with a fluorescent dye such as FITC or Alexa-fluor 540 (Jackson Immunoresearch, UK). All antibody solutions were made up in PBS containing 5% bovine serum albumin. Actin filaments were stained by incubation for 1 hour with Phalloidin coupled to Atto647N (Sigma Aldrich, Germany). Cell nuclei were visualized by staining with DAPI (Sigma Aldrich, Germany). Specimens were embedded in Vecta shield medium (Reactolab SA, Switzerland). The sections were examined with a Leica confocal laser scanning microscope (Leica Microsystems, Germany) equipped with a krypton-argon laser. Sequential scans at a series of optical planes were performed with a 63 × oil immersion objective lens through specimens.
Experimental Design for In Vivo Studies
Female C57BL/6 and BALB/c mice, 8 to 12 weeks of age, were obtained from the Animal House of the Polish Academy of Sciences, Medical Research Center (Warsaw, Poland). All In Vivo experiments were performed according to EU guidelines for the care and use of laboratory animals and approved by local authorities. The inhibitor BSc2118 was administered intraperitoneally (i.p.) in 50 μl DMSO or intratumorally (i.t.) in 20 μl DMSO. As controls, mice were treated with appropriate volumes of DMSO. Bortezomib was given to mice at 1 mg/kg i.p. in 50 μl PBS, for which mice treated with 50 μl PBS i.p. served as controls.
For studies on biodistribution and kinetics of inhibitors, BALB/c mice received one single dose of inhibitor at 5 and 10 mg/kg. After 1 hour or 24 hours post injection, mice were sacrificed; tissue samples were collected and divided into halves. For direct observations of BSc2118-FL tissue samples were embedded in OCT and immediately frozen at − 70°C. For biochemical analysis, tissue samples were frozen at − 70°C and kept until preparation. An initial study was carried out in melanoma bearing C57BL/6 mice to determine the potency of BSc2118 to inhibit the proteasome activity In Vivo either within red blood cells or within tumor tissue after i.t. or i.p. injection of the inhibitor.
Melanoma Anti-Tumor Therapy in Mice by BSc2118
Female C57BL/6 mice of 8 to 9 weeks of age were inoculated into the footpad on day 0 with 1 × 106 B16F10 cells in 20 μl of PBS. Tumor cell viability measured by trypan blue exclusion was above 98%. Beginning from day 7 after inoculation, melanoma bearing mice were injected with 5, 10, or 15 mg per kg body weight of BSc2118 in a volume of 20 μl i.t. for 7 consecutive days. For i.p. injection protocols, BSc2118 was given at dose of 15, 30, or 60 mg per kg body weight for seven consecutive days. Bortezomib was given at 1 mg/kg i.p. 7 times every second day. Each group contained 7 animals. Appropriate volumes of the solvents were given as control. During the experiments melanoma bearing mice were observed daily for survival and adverse effects. Tumor size of melanoma-bearing mice was measured every 2 days. Tumor volume was determined according to the formula: tumor volume = (shorter diameter2 × longer diameter)/2. Differences in tumor volume were analyzed for significance by the Student’s t test. A P value of < 0.05 was considered to be statistically significant. Log-rank test was used to analyze survival.
In Vivo Angiogenesis Assay
Mice were anesthetized and received sterile abdominal injections of 250 μl of Matrigel (Becton Dickinson, Germany) subcutaneously containing 50 nM βFGF (Sigma Aldrich, Germany). Thereafter, mice were i.p. treated with BSc2118 at 30 mg/kg for 7 consecutive days. At day 8, vascularization of the Matrigel was quantified by intravenous injecting of 0.1 ml (0.25 mg/ml stock solution) of FITC-dextran (125,000 molecular weight, Sigma Aldrich, Germany) into mice, which allowed blood vessels within plugs to be visualized. Animals were sacrificed 20 minutes after injection, when Matrigel plugs were removed and digested in Dispase reagent (Becton Dickinson, Germany). The fluorescence of the solution obtained was measured using a fluorimeter (POLARstar, BMG Labtech, Germany) with an excitation at 480 nm and an emission wavelength at 520 nm. Differences between groups were calculated by Student’s t test. A P value of < 0.05 was considered to be statistically significant.
In Vivo Metastasis Assay
Experimental lung metastases were performed as described by Feleszko et al. [32]. Briefly, experimental lung metastases were induced by injection of 2 × 105 of B16F10 cells/100 μl PBS into the tail vein of anesthetized female C57BL/6 mice. Mice (5 to 7 per group) were i.p. injected for 7 consecutive days with either DMSO or BSc2118 (15 mg/kg body weight/day). The animals were then sacrificed on day 21 after inoculation of tumor cells. An average number of metastases were calculated for every mouse by two independent observers blinded to the experimental groups. Differences between experimental groups were analyzed using the Mann–Whitney U test. A P value of < 0.05 was considered to be statistically significant.
Results
BSc2118 is Cytotoxic for a Panel of Solid Tumor Cell Lines
An In Vitro cytotoxicity screening was performed to characterize the anti-tumor potential of BSc2118. For this purpose, a panel of solid tumor cell lines, most of them originating from malignant melanoma, was incubated either with BSc2118 or with bortezomib as a reference inhibitor (Figure 1). The average GI50 value was estimated for each cell line and across the entire tumor cell panel. We found an average GI50 value of 76 nM for BSc2118, whereas the average GI50 value for bortezomib across 22 cell lines was 6.3 nM. For comparison, on NCI cell panel the GI50 value for bortezomib was given at 7 nM [30]. Figure 1 shows average viability for seven concentrations as percentages regarding controls for both BSc2118 and bortezomib.
Figure 1.
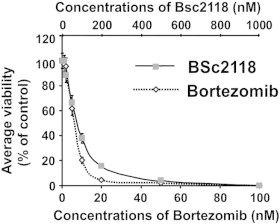
BSc2118 affects viability of 22 tumor cell lines. Graphs were prepared by averaging viability of 22 cell lines from seven concentration points of inhibitors. Results are displayed as percentage of controls. The discrepancy in the concentrations of inhibitors is a result of different Ki-values for both drugs.
Characterization of Inhibitory Properties of BSc2118 and BSc2118-FL
The proteasome inhibitor BSc2118 was described previously by Braun et al. [28]. To better track the biological properties of this inhibitor in living organisms, we synthesized a dye-coupled version of this molecule (Figure 2A). The Bodipy FL-BSc2118 (thereafter named as BSc2118-FL) inhibited proteasome activity similarly to non-fluorescent BSc2118 in HeLa cells (Figure 2B), suggesting that this chemical modification does not change the inhibitory properties of the compound. A 24-hour incubation of HeLa cells with 1 μg/ml of BSc2118-FL resulted in formation of aggregates that co-localized with both ubiquitin and the proteasome (Figure 2C). Furthermore, we found an accumulation of polyubiquitinylated proteins after 24 hours of incubation of C26 cells with BSc2118 as indicated by Western blotting (Figure 2D). Like the non-fluorescent compound, BSc2118-FL induces apoptosis in C26 colon cancer cells as exemplarily shown in (Figure 2E).
Figure 2.
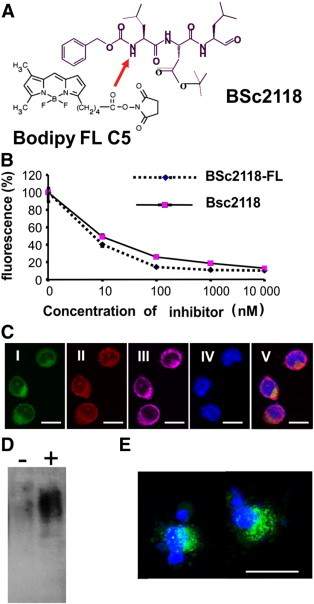
Characteristics of BSc2118-FL. (A) Chemical structure of BSc2118 and BSc2118-FL. The arrow indicates the position of covalent modification by binding of the fluorescent group (Bodipy FL C5). (B) Comparison of 20S inhibition profiles of parental BSc2118 and its modified version BSc2118-FL within HeLa cells. The error bars (SD), best visible at 0 and 10 nM, at the other points are very small. (C). Confocal microscopy of C26 colon cancer cells incubated for 24 hours with BSc2118-FL (1 μg/μl). (I) Direct fluorescence signal of BSc2118-FL (green), (II) antibody-mediated staining against the 20S proteasome (red), (III) antibody-mediated staining against ubiquitin (magenta). BSc2118-FL co-localizes with intracellular 20S proteasome and ubiquitin. Nuclei were stained with DAPI (IV). Merged photo is shown in (V). (D). Accumulation of polyubiquitin conjugates in soluble fraction obtained from C26 colon cancer cells after 24 hours of exposure to 130 nM FL-BSc2118 (+) compared to cell fractions from non-treated cells (−) using Western blotting. (E) BSc2118-FL (green, 500 nM for 24 h) induces apoptosis in C26 colon cancer cells as shown by morphological analysis. Nuclear staining with DAPI is shown in blue. Scale bar: 15 μm.
Analysis of inhibition of proteasomes derived from different murine tissues revealed that BSc2118 is sufficient to inhibit 20S activity in a concentration dependent manner in all organs analyzed (Supplementary Figure 1).
BSc2118 and BSc2118-FL Inhibit 20S Activity In Vivo
We next analyzed the inhibition of 20S proteasomal activity induced by BSc2118 as compared to bortezomib In Vivo. For this purpose, mice were i.p. injected with either BSc2118 or bortezomib at different concentrations followed by sacrifice of mice after 1 hour or 24 hours post-injection. Lung, heart, spleen, liver, kidney, skeletal muscle, brain and blood were collected for each time point. A dose of at least 10 mg/kg of BSc2118 was sufficient to inhibit 20S activity in mice organs (Figure 3). One hour after injection of BSc2118 (30 mg/kg), the proteasome activity was reduced to 15% or less in all organs with the exception of the brain and the kidney (Figure 3). Nevertheless, at 24 hours post-injection 20S activities recovered from 60% up to 100% as compared to controls (Figure 3). Similar inhibition patterns were shown for bortezomib (Figure 3) with a reduction of 80% to 90% at 1 hour following its inoculation in most organs. In the brain, however, only a 10% reduction of 20S proteasome activity at 1 hour after treatment was observed, whereas 24 hours after treatment 20S activity was found to be inhibited about 20%. In contrast to other tissues, no recovery of proteasome inhibition 24 hours post-injection was detected within the brain. In line with this, BSc2118-FL at a dose of 5 mg/kg effectively inhibited the 20S activity in mice after i.p. administration (Supplementary Figure 2). In the selected organs, there was a strong 20S inhibition after 1 hour, whereas after 24 hours proteasomal activity was almost completely restored.
Figure 3.
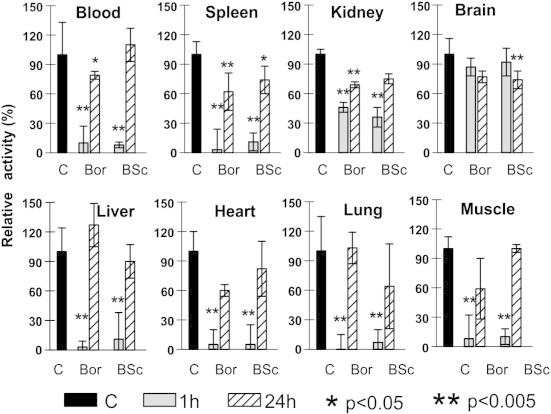
Inhibition of proteasome activities in different tissues after intraperitoneal administration of inhibitors in mice. Application of both BSc2118 (BSc) and bortezomib (Bor) yield similar inhibitory patterns of the 20S proteasome activity, which is given as relative activity of controls (abbreviated as “C”). All controls have received respectively: DMSO or 0.9% NaCl at both time points and did not show any significant differences, thus for better visibility one bar for averaged controls is shown. Mice were sacrificed either after 1 hour (grey boxes) or after 24 hours (striated boxes) post-injection, respectively. Significantly different from controls with *P < 0.05 and **P < 0.005.
None of the BSc2118-treated animals (n = 7) demonstrated any signs of toxicity that could be identified by observation. No weight loss, diarrhea, hair loss, or neurological symptoms (like tremor, ataxia, paralysis) were observed in BSc2118-treated mice, even if mice were treated daily with a 60 mg/kg dose for 7 consecutive days. In mice treated with bortezomib the drug was administered at 1 mg/kg every second day. Animals that were given higher daily doses of bortezomib died on day three after start of treatment (data not shown). Concluding, BSc2118 inhibits the proteasome activity In Vivo to a similar extend as bortezomib (Figure 3), but is better tolerated and less toxic in spite of daily administration.
Analysis of Stability of the BSc2118 Against Microsomal Proteases
Some proteasome inhibitors like MG132 become instable in presence of microsomal fractions due to various mechanisms involving binding or modification. To analyze the resistance of BSc2118 against microsomal proteins, measurement of 20S inhibitory activity in the presence/absence of liver microsomes was performed. As shown in Figure 4, BSc2118 is stable for up to 4 hours of incubation with microsomes. After 8 hours of incubation with liver microsomes, there is an observed 30% loss of inhibitory activity. Thus, BSc2118 turned out to be more stable than MG132, which lost its activity at 4 hours in a dose and time dependent manner, i.e., 50% of activity loss at 4 hours and 70% of activity loss at 8 hours, After 24 hours of treatment, the activity loss for both MG132 and BSc2118 was comparative.
Figure 4.
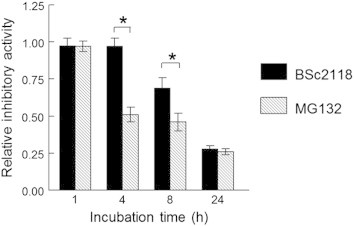
Analysis of stability of BSc2118 in microsomal fraction. Relative stability of indicated inhibitors (at 500 nM each) in the presence of liver microsomal fraction is shown. BSc2118 is more stable in liver microsomal fraction in comparison to MG132. *Significant difference between BSc2118 and MG132 in the indicated time point (*P < 0.05).
Analysis of Biodistribution Using BSc2118-FL
To track the biodistribution of BSc2118, the Bodipy-labeled BSc2118 (BSc2118-FL) was directly analyzed in tissue sections detecting spontaneous fluorescence by means of confocal microscopy. In order to achieve sufficient direct fluorescence signal, dosages of BSc2118-FL had to be at least 10 mg/kg body weight. Although dosages of 5 mg/kg body weight significantly inhibited 20S activities and induced accumulation of polyubiquitinated proteins, spontaneous fluorescence signal was too weak for direct observations (data not shown). The fluorescence within erythrocytes mounted after 1 hour or 24 hours post i.p. injection (10 mg/kg) is depicted in (Figure 5A). Erythrocytes (0 h) from vehicle injected mice served as control to visualize the autofluorescence signal of hemoglobin. The inhibitor-specific fluorescence was most pronounced in animals at 1 hour after inhibitor injection. After 24 hours the fluorescence intensity was approximating the baseline due to inhibitor fluorescence instability or to the low reversibility of Bodipy-BSc2118. In organs, bright specific BSc2118-FL fluorescence was observed exemplarily in intestine (I), heart (II) and in proximal tubules of kidney (III) (Figure 5B). Remarkably, no fluorescence was observed in intact brain (not shown), confirming the 20S activity data of brain lysates included in Figure 3.
Figure 5.
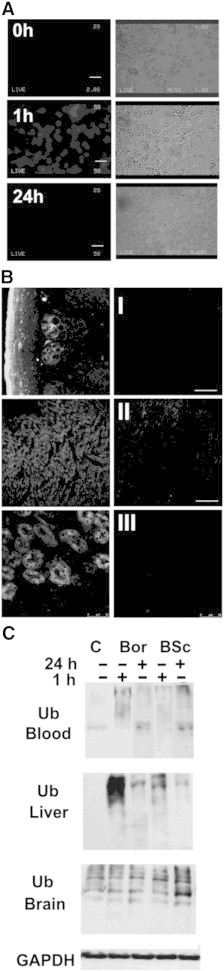
Detection of fluorescence intensities of BSc2118-FL after In Vivo administration. (A) Left panel: direct observations of fluorescence within red blood cells isolated either from untreated mice (0 h) or after 1 hour or 24 hours post-administration of BSc2118-FL (10 mg/kg) in mice. Right panel: phase contrast pictures of erythrocytes from left panel. Scale bars: 15 μm. (B) Observations of BSc2118-FL direct fluorescence by means of confocal microscopy within the small intestine (I), heart (II) and kidney (III), which were isolated from mice injected with BSc2118-FL at 10 mg/kg (left panel) or treated with vehicle (right panel). Internal organs were collected after 1 hour and 24 hours post-inhibitor injection. Scale bars: 50 μm. (C) Western blotting for detection of polyubiquitin conjugates in lysates from organs after i.p. injection of BSc2118 or Bortezomib. Time points given refer to time of injection of inhibitors. Controls (C) were injected with vehicle and here one representative control is shown. GAPDH (here exemplary shown in the liver) served as loading control.
To further demonstrate the In Vivo effects of proteasome inhibition, we examined tissue lysates for polyubiquitin conjugates using Western blotting (Figure 5C). In blood and liver there was an accumulation of polyubiquitin conjugates observed that correlated with proteasome inhibition displayed in Figure 3. In the brain, however, a constitutive high amount of polyubiquitin conjugates was detected that did not increase upon administration of inhibitors. A moderate accumulation of polyubiquitin was only found in brain homogenates after 24 hours of BSc2118 treatment.
BSc2118 Exerts Local Anti-Tumor Effects in a Mouse Melanoma Model
Proteasome inhibitors are approved for use in the treatment of multiple myeloma and mantle cell lymphoma. Moreover, a number of clinical studies investigated the anti-tumor effects of bortezomib in patients with solid tumors. Therefore, we decided to compare potential anti-tumor effects of BSc2118 to bortezomib in the B16F10 melanoma model in mice (Figure 6). First, we compared the efficiency of BSc2118 to inhibit the 20S activity directly within tumor tissues after i.t. or i.p. administration. The inhibition in tumor tissues was compared to the inhibition in red blood cells. After i.p. administration of BSc2118 (30 mg/kg),the activity of 20S proteasomes was reduced by up to 65% within tumors when measured at 1 hour post-injection. However, the 20S proteasome activity recovered to control levels within 24 hours (Figure 6). BSc2118 when given i.t. (10 mg/kg) almost completely inhibited 20S proteasome activity at 1 hour after administration. Activities also remained inhibited by 90% during the following 24 hours. BSc2118 given i.p. reduced the 20S activity in blood cells of mice bearing melanoma (Figure 6) similarly to healthy ones (Figure 3), i.e. there was an initial reduction of 20S activity in the 1-hour group that increased in the 24-hour group. After i.t. injection of BSc2118, the initial inhibition of the 20S proteasome in the 1 hour groups was 40% and it dropped towards 50% in the 24-hour groups. Taken together with proteasome inhibition within erythrocytes after i.t. injection of BSc2118, there is an evidence that at least half of the initial proteasome activity is still inhibited within the tumor after 24 hours post-injection and that the inhibitor is slowly released from the place of injection. The activity data correlated with accumulation of polyubiquitin conjugates within tumors that are shown in Figure 6E.
Figure 6.
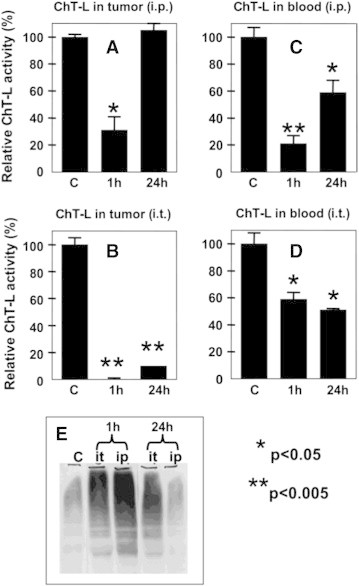
Comparison of proteasome inhibition after intraperitoneal and after intratumoral injection of BSc2118 in a mouse B16F10 tumor model. ChT-L activity was determined either in melanoma homogenates (A-B) or in red blood cells (C-D) after intraperitoneal (i.p.; A and C) or intratumoral (i.t.; B and D) injection of BSc2118. Each diagram displays bars as follows: activity in vehicle treated controls (C), activity 1 hour post-injection and 24 hours post-injection of BSc2118. Controls were defined as 100%. (E) Western blot analysis of polyubiquitynated proteins from tumor lysates displayed in (A-B). Accumulation of polyubiquitinylated proteins correlates with proteasome inhibition profile displayed in A and B. Significantly different from controls with *P < 0.05 and **P < 0.005.
Tumor size or survival of treated mice after i.p. administration with either BSc2118 or bortezomib was, however, not affected by the inhibitors (data not shown). On the contrary, when BSc2118 was administered i.t. at a 5mg/kg dose, a complete tumor regression was observed, which led to prolonged animal survival for up to two months (Figure 7, A–B). Unexpectedly, when BSc2118 was injected i.t. at 10 or 15 mg/kg doses, a significant local toxicity (edema and ischemia) was observed in 30% of animals that precluded further analysis of tumor growth (Figure 7, A–B).
Figure 7.
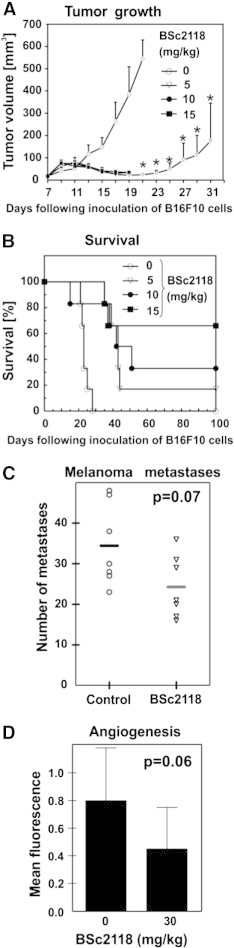
Anti-tumor effects of BSc2118. (A) BSc2118 induced significant melanoma tumor growth retardation in mice treated i.t. with BSc2118 at 5 mg/kg for up to 40 days of treatment. BSc2118 at 10 mg/kg and 15 mg/kg induced foot loose in 3 mice at day 19. (B) Kaplan-Meier curves showing survival comparison between control and drug-treated mice. Log-rank test showed significant difference in survival time among all treated groups compared to controls at P < 0.0005. (C) In Vivo melanoma lung metastases assay displaying a trend towards reduced numbers of lung metastasis due to treatment with BSc2118 (30 mg/kg). Horizontal lines show means from 7 displayed points. (D) Indirect In Vivo assessment of angioneogenesis within matrigels followed by staining of blood vessels with dextran FITC. Mean fluorescence of matrigels are displayed on y-axis and corresponds to the number of blood vessels. There was a trend towards a reduction of fluorescence intensity detectable in BSc2118-treated mice.
To better characterize potential mechanisms of anti-tumor action of BSc2118, a metastasis assay as well as a matrigel angiogenesis assay were performed. For these assays, BSc2118 had to be i.p. administered for technical reasons, yielding no optimal inhibition of the 20S proteasome within the 24-hour animal groups. Nevertheless, animals treated with BSc2118 at 30 mg/kg revealed a tendency to reduce the number of metastases as compared to controls (Figure 7C). In spite of a tendency of BSc2118 (30 mg/kg) to reduce angiogenesis, significant results were lacking (Figure 7D; P = 0.06). Taken together, BSc2118 exerts local antitumor activity in a mouse melanoma model.
Discussion
Novel proteasome inhibitors are intensively developed and studied in order to find more specific and safer inhibitors with a broad spectrum of therapeutic applications [15], [33], [34], [35]. In this context, we studied for the first time the biodistribution of the novel proteasome inhibitor BSc2118 In Vivo followed by an analysis of its therapeutic potential and therapeutic safety in the context of malignant melanoma.
For inhibitor tracking in living organisms, the fluorescent variant of BSc2118, BSc2118-FL, was synthesized. BSc2118-FL was cell-permeable, targets the proteasome specifically, co-localizes with the proteasome and had a similar inhibition profile in comparison to its non-fluorescent variant. The bright fluorescence signal facilitated rapid and sensitive detection of proteasomes by fluorescence-based microscopy in living cells and in tissues. Because the proteasome inhibitor BSc2118 had a low toxicity, even the use of higher concentrations that allows monitoring of inhibitor biodistribution, was well tolerated in experimental models.
The biodistribution and inhibition profile of proteasomes inhibited by BSc2118 in a mouse model was compared to bortezomib and was similar in equivalent concentrations. BSc2118 was given daily at maximal doses of 60 mg/kg body weight for 7 days, which was well tolerated by mice with no signs of toxicity. Using this application schedule, no lethality was observed. Moreover as it was shown in a different publication, BSc2118 up to 60 mg/kg daily dose did not affect peripheral blood morphology in C57BL/6 mouse [36]. In contrast, bortezomib had to be given with at least a one-day break, whereas daily injection of 1 mg/kg body weight was lethal in most animals. As such, BSc2118 might serve as a potential, low toxic and well tolerated novel drug [30].
Therefore, we analyzed the potential for BSc2118 usage in different application forms to be considered for proteasome inhibition. These typically include anti-tumor effects based on cell cycle arrest and on inducing apoptosis [34], [35]. Although Bortezomib was developed and approved for therapy of multiple myeloma and mantle cell lymphoma only, therapeutic potential for other tumors was investigated within the last years as well [37]. However, bortezomib was not effective in treatment of solid tumors until recently [38]. BSc2118 tested on a panel of 22 tumor cell lines derived from solid tumors exerted cytotoxic and cytostatic effects with a GI50 of 76 nM, whereas for bortezomib 6.3 nM was calculated. Due to different Ki-values for both inhibitors, previously data has shown that concentration ratios giving similar 20S inhibition patterns for BSc2118 and bortezomib is 10:1 [27]. Thus, compilation of equally potent concentrations of both BSc2118 and bortezomib revealed that these inhibitors comparatively inhibit growth of the 22 tumor cell lines analyzed.
BSc2118 and BSc2118-FL induce both accumulation of polyubiquitin conjugates and apoptosis in a broad spectrum of cells, as has been exemplarily shown in C26 colon cancer cells. Efficiency of inhibitors in organisms is highly dependent on bioavailability, stability and reversibility of the compounds. BSc2118 is partially instable in liver microsomal fraction. Whereas Bortezomib is irreversible, binding of BSc2118 is reversible [36]. Proteasome inhibition induces compensatory De Novo synthesis of proteasomes [39]. Whereas reversible inhibition affects more proteasomes in cells positively correlating with exposition time (binding-dissociation-rebinding), more stable inhibition rather acts like a pulse inhibition. This means that cells which are able to compensate proteasome inhibition via De Novo synthesis do survive, but cells that are incapable of doing so suffer from UPR stress and accumulation of oxidized proteins [40]. In this context, the majority of tumor cells are more sensible to proteasome inhibition than their parental cells [27].
In order to study possible therapeutic potentials of BSc2118, we studied BSc2118-mediated effects in a mouse model of malignant melanoma. BSc2118 in experimental melanoma therapy revealed some unexpected findings. First of all, neither BSc2118 nor bortezomib injected i.p. had any effects on tumor growth or survival of B16F10 tumor bearing mice (data not shown). It is known that tumor tissue has its own milieu and drugs working well In Vitro might not be effective In Vivo due to the existence of the tumor matrix [41]. Therefore, the inhibitor was injected directly into the tumor. Comparison of proteasome inhibition profiles after both i.p. and i.t. injection of BSc2118 revealed that BSc2118 completely inhibited proteasome activity after i.t. injection, which lasted for at least 24 h. This result prompted us to check the effects of BSc2118 on tumor growth when injected i.t. We obtained tumor growth retardation and complete remission with a survival for up to two months in 38.5% of mice receiving BSc2118 from all experimental groups. However, BSc2118 at 10 and 15 mg/kg induced local toxicity, suggesting that local levels of proteasome inhibition within the tissue should not exceed 80%. On the contrary, increased proteasome inhibition might be toxic as has been demonstrated for bortezomib in primates [42]. In humans the inhibition of 20S activity with bortezomib does not exceed 70% [43].
To better characterize the mechanisms of action of BSc2118 in a tumor model we studied BSc2118-mediated De Novo angiogenesis and metastasis in a model of malignant melanoma. Noteworthy, our experimental model needed i.p. injections of BSc2118 that might not induce satisfactory level of proteasome inhibition and had no effects on tumor growth. However, in this application design there was a tendency at the border of significance to reduce the number of metastases and De Novo arising blood vessels. It seems that BSc2118 retards tumor growth by means of 20S inhibition within tumor cells and additionally might reduce both metastasis and angiogenesis.
In conclusion, we characterized a novel proteasome inhibitor that has a similar proteasome inhibition spectrum compared to bortezomib In Vitro and In Vivo, but has under the conditions tested less signs of toxicity. We hypothesize that BSc2118 is a therapeutic alternative to bortezomib in therapy of solid tumors, for which further studies will be needed.
The following are the supplementary data related to this article.
Inhibition of tissue-derived proteasomes In Vitro. 20S proteasomes were isolated from indicated mouse organs and incubated with indicated concentrations of BSc2118. A final concentration of 50 nM of BSc2118 is sufficient to reduce 40% to 50% of initial 20S activity. *Significantly different from controls (*P < 0.05 and **P < 0.005).
Inhibition profile of BSc2118-FL in selected mouse organs. Proteasomal activity was measured in selected organs 1 hour or 24 hours after i.p. injection of BSc2118-FL in mice. Groups injected with solvent for BSc2118 (C) served as controls, here one representative bar for averaged controls is shown. Data are given as relative fluorescence units. *Significantly different from controls, p< 0.05. **Significantly different from controls, P < 0.005.
Acknowledgments
This work was in part supported by the Polish Ministry of Science and higher Education (grant number: N405 007 31/0544 to IMB).
We thank Dr. Anna Ratajska from the Department of Pathological Anatomy, Medical University of Warsaw, Poland, for assistance in preliminary experiments on BSc2118-FL stability in mice.
References
- 1.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 2.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 3.Rechsteiner M, Hoffman L, Dubiel W. The multicatalytic and 26 S proteases. J Biol Chem. 1993;268:6065–6068. [PubMed] [Google Scholar]
- 4.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seufert W, Jentsch S. In vivo function of the proteasome in the ubiquitin pathway. EMBO J. 1992;11:3077–3080. doi: 10.1002/j.1460-2075.1992.tb05379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner RC, Assinder SJ, Christie G, Mason GG, Markwell R, Wadsworth H, McLaughlin M, King R, Chabot-Fletcher MC, Breton JJ. Characterization of peptidyl boronic acid inhibitors of mammalian 20 S and 26 S proteasomes and their inhibition of proteasomes in cultured cells. Biochem J. 2000;346(Pt 2):447–454. [PMC free article] [PubMed] [Google Scholar]
- 7.Wojcik C. Proteasomes in apoptosis: villains or guardians? Cell Mol Life Sci. 1999;56:908–917. doi: 10.1007/s000180050483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. 2009;15:781–787. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- 9.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 10.Dahlmann B, Ruppert T, Kuehn L, Merforth S, Kloetzel PM. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J Mol Biol. 2000;303:643–653. doi: 10.1006/jmbi.2000.4185. [DOI] [PubMed] [Google Scholar]
- 11.Marfella R, Di Filippo C, Portoghese M, Siniscalchi M, Martis S, Ferraraccio F, Guastafierro S, Nicoletti G, Barbieri M, Coppola A. The ubiquitin-proteasome system contributes to the inflammatory injury in ischemic diabetic myocardium: the role of glycemic control. Cardiovasc Pathol. 2009;18:332–345. doi: 10.1016/j.carpath.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Tie L, Xu Y, Lin YH, Yao XH, Wu HL, Li YH, Shen ZF, Yu HM, Li XJ. Down-regulation of brain-pancreas relative protein in diabetic rats and by high glucose in PC12 cells: prevention by calpain inhibitors. J Pharmacol Sci. 2008;106:28–37. doi: 10.1254/jphs.fp0071092. [DOI] [PubMed] [Google Scholar]
- 13.Petroski MD. The ubiquitin system, disease, and drug discovery. BMC Biochem. 2008;9(Suppl. 1):S7. doi: 10.1186/1471-2091-9-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Discovery and development of the anticancer agent salinosporamide A (NPI-0052) Bioorg Med Chem. 2009;17:2175–2180. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15:243–249. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999;5:2638–2645. [PubMed] [Google Scholar]
- 18.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 19.Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov Today. 2003;8:307–315. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- 20.O'Connor OA, Czuczman MS. Novel approaches for the treatment of NHL: Proteasome inhibition and immune modulation. Leuk Lymphoma. 2008;49(Suppl. 1):59–66. doi: 10.1080/10428190802365033. [DOI] [PubMed] [Google Scholar]
- 21.Goy A, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009;20:520–525. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Einsele H. Bortezomib. Recent Results Cancer Res. 2010;184:173–187. doi: 10.1007/978-3-642-01222-8_12. [DOI] [PubMed] [Google Scholar]
- 23.Gilardini A, Marmiroli P, Cavaletti G. Proteasome inhibition: a promising strategy for treating cancer, but what about neurotoxicity? Curr Med Chem. 2008;15:3025–3035. doi: 10.2174/092986708786848622. [DOI] [PubMed] [Google Scholar]
- 24.Meregalli C, Canta A, Carozzi VA, Chiorazzi A, Oggioni N, Gilardini A, Ceresa C, Avezza F, Crippa L, Marmiroli P. Bortezomib-induced painful neuropathy in rats: a behavioral, neurophysiological and pathological study in rats. Eur J Pain. 2010;14:343–350. doi: 10.1016/j.ejpain.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Argyriou AA, Iconomou G, Kalofonos HP. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood. 2008;112:1593–1599. doi: 10.1182/blood-2008-04-149385. [DOI] [PubMed] [Google Scholar]
- 26.Nowis D, Maczewski M, Mackiewicz U, Kujawa M, Ratajska A, Wieckowski MR, Wilczynski GM, Malinowska M, Bil J, Salwa P. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol. 2010;176:2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mlynarczuk-Bialy I, Roeckmann H, Kuckelkorn U, Schmidt B, Umbreen S, Golab J, Ludwig A, Montag C, Wiebusch L, Hagemeier C. Combined effect of proteasome and calpain inhibition on cisplatin-resistant human melanoma cells. Cancer Res. 2006;66:7598–7605. doi: 10.1158/0008-5472.CAN-05-2614. [DOI] [PubMed] [Google Scholar]
- 28.Braun HA, Umbreen S, Groll M, Kuckelkorn U, Mlynarczuk I, Wigand ME, Drung I, Kloetzel PM, Schmidt B. Tripeptide mimetics inhibit the 20 S proteasome by covalent bonding to the active threonines. J Biol Chem. 2005;280:28394–28401. doi: 10.1074/jbc.M502453200. [DOI] [PubMed] [Google Scholar]
- 29.Kern MA, Helmbach H, Artuc M, Karmann D, Jurgovsky K, Schadendorf D. Human melanoma cell lines selected in vitro displaying various levels of drug resistance against cisplatin, fotemustine, vindesine or etoposide: modulation of proto-oncogene expression. Anticancer Res. 1997;17:4359–4370. [PubMed] [Google Scholar]
- 30.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- 31.Kuckelkorn U, Ruppert T, Strehl B, Jungblut PR, Zimny-Arndt U, Lamer S, Prinz I, Drung I, Kloetzel PM, Kaufmann SH. Link between organ-specific antigen processing by 20S proteasomes and CD8(+) T cell-mediated autoimmunity. J Exp Med. 2002;195:983–990. doi: 10.1084/jem.20011199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feleszko W, Mlynarczuk I, Olszewska D, Jalili A, Grzela T, Lasek W, Hoser G, Korczak-Kowalska G, Jakobisiak M. Lovastatin potentiates antitumor activity of doxorubicin in murine melanoma via an apoptosis-dependent mechanism. Int J Cancer. 2002;100:111–118. doi: 10.1002/ijc.10440. [DOI] [PubMed] [Google Scholar]
- 33.Ling YH, Liebes L, Jiang JD, Holland JF, Elliott PJ, Adams J, Muggia FM, Perez-Soler R. Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase arrest and apoptosis in human non-small cell lung cancer cell lines. Clin Cancer Res. 2003;9:1145–1154. [PubMed] [Google Scholar]
- 34.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–3076. [PubMed] [Google Scholar]
- 35.Lu G, Punj V, Chaudhary PM. Proteasome inhibitor Bortezomib induces cell cycle arrest and apoptosis in cell lines derived from Ewing's sarcoma family of tumors and synergizes with TRAIL. Cancer Biol Ther. 2008;7:603–608. doi: 10.4161/cbt.7.4.5564. [DOI] [PubMed] [Google Scholar]
- 36.Doeppner TR, Mlynarczuk-Bialy I, Kuckelkorn U, Kaltwasser B, Herz J, Hasan MR, Hermann DM, Bahr M. The novel proteasome inhibitor BSc2118 protects against cerebral ischaemia through HIF1A accumulation and enhanced angioneurogenesis. Brain. 2012;135:3282–3297. doi: 10.1093/brain/aws269. [DOI] [PubMed] [Google Scholar]
- 37.Jones RJ, Chen Q, Voorhees PM, Young KH, Bruey-Sedano N, Yang D, Orlowski RZ. Inhibition of the p53 E3 ligase HDM-2 induces apoptosis and DNA damage–independent p53 phosphorylation in mantle cell lymphoma. Clin Cancer Res. 2008;14:5416–5425. doi: 10.1158/1078-0432.CCR-08-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Zonder JA, Dou QP. Clinical development of novel proteasome inhibitors for cancer treatment. Expert Opin Investig Drugs. 2009;18:957–971. doi: 10.1517/13543780903002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Kruger E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J Biol Chem. 2003;278:21517–21525. doi: 10.1074/jbc.M301032200. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki E, Demo S, Deu E, Keats J, Arastu-Kapur S, Bergsagel PL, Bennett MK, Kirk CJ. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS One. 2011;6:e27996. doi: 10.1371/journal.pone.0027996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabelloni P, Da Pozzo E, Bendinelli S, Costa B, Nuti E, Casalini F, Orlandini E, Da Settimo F, Rossello A, Martini C. Inhibition of metalloproteinases derived from tumours: new insights in the treatment of human glioblastoma. Neuroscience. 2010;168:514–522. doi: 10.1016/j.neuroscience.2010.03.064. [DOI] [PubMed] [Google Scholar]
- 42.Adams J. Development of the proteasome inhibitor PS-341. Oncologist. 2002;7:9–16. doi: 10.1634/theoncologist.7-1-9. [DOI] [PubMed] [Google Scholar]
- 43.Hamilton AL, Eder JP, Pavlick AC, Clark JW, Liebes L, Garcia-Carbonero R, Chachoua A, Ryan DP, Soma V, Farrell K. Proteasome inhibition with bortezomib (PS-341): a phase I study with pharmacodynamic end points using a day 1 and day 4 schedule in a 14-day cycle. J Clin Oncol. 2005;23:6107–6116. doi: 10.1200/JCO.2005.01.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inhibition of tissue-derived proteasomes In Vitro. 20S proteasomes were isolated from indicated mouse organs and incubated with indicated concentrations of BSc2118. A final concentration of 50 nM of BSc2118 is sufficient to reduce 40% to 50% of initial 20S activity. *Significantly different from controls (*P < 0.05 and **P < 0.005).
Inhibition profile of BSc2118-FL in selected mouse organs. Proteasomal activity was measured in selected organs 1 hour or 24 hours after i.p. injection of BSc2118-FL in mice. Groups injected with solvent for BSc2118 (C) served as controls, here one representative bar for averaged controls is shown. Data are given as relative fluorescence units. *Significantly different from controls, p< 0.05. **Significantly different from controls, P < 0.005.