

Abstract
Introduction
The tertiary structure of normal podocytes prevents protein from leaking into urine. Patients with lupus nephritis (LN) develop proteinuria, and kidney biopsies from these patients display a number of podocyte abnormalities including retraction of podocyte processes. Autoantibodies have been shown to deposit in the kidneys of patients and mice with LN and are believed to play a key role in causing renal inflammation and dysfunction. The objective of this research was to study the effects of IgG antibodies from patients with LN on cultured human podocytes.
Methods
We exposed a human podocyte cell line to heat-inactivated (HI) plasma and purified polyclonal IgG from the following groups of subjects; patients with LN, patients with lupus without nephritis, patients with rheumatoid arthritis and healthy controls. We measured expression and intracellular distribution of podocyte-specific proteins and global phosphorylation of tyrosine. We then used mass spectrometry to identify the major protein targets of this phosphorylation.
Results
HI LN plasma did not alter expression or cellular distribution of podocyte-specific proteins but caused a significant reduction in podocyte protein tyrosine phosphorylation compared with plasma from healthy controls (p=0.0008). This result was replicated using purified IgG but was not seen with plasma from rheumatoid arthritis or non-renal lupus patients. The dominant tyrosine phosphorylated protein in podocytes was 55 kDa in size and was identified as tubulin.
Conclusions
Since tubulin is an important component of podocyte major processes, these results suggest that autoantibodies from LN patients may exert an important pathogenic effect by dephosphorylation of this protein in podocytes.
Keywords: nephritis, podocyte, phosphorylation
Key messages.
Lupus nephritis is characterised by proteinuria and dysfunction of renal podocytes.
This paper shows a specific effect of IgG from patients with lupus nephritis, but not non-renal lupus, rheumatoid arthritis or healthy controls in reducing tyrosine phosphorylation of podocyte proteins.
Tubulin, a key structural protein in podocytes is a major target for this dephosphorylation.
Introduction
The management of patients with lupus nephritis (LN) continues to be dependent on use of corticosteroids and broad spectrum immunosuppressants. Introduction of agents such as rituximab and belimumab, which target specific components of the immune system, may be an improvement. A recent prospective observational study suggested that rituximab may be useful in treating patients with LN without using corticosteroids.1 However, randomised controlled clinical trials have not yet proved the efficacy of either belimumab or rituximab in LN.2–4 An alternative approach would be to study end organ effects by probing the effect of sera from patients with LN on glomerular cells in order to identify novel therapeutic targets. This study was designed to test the hypothesis that pathogenic autoantibodies from patients with systemic lupus erythematosus (SLE) can have a direct effect on the glomerular epithelial cell, the podocyte.
Podocytes are highly specialised cells with interdigitating, tubulin-based, primary and secondary extensions (also termed major processes), and smaller, actin-based, tertiary or foot processes, which are tethered to the glomerular basement membrane by integrin molecules. Maintaining the tertiary structure of the podocyte is critical for preventing the leak of protein into urine. Certain podocyte proteins, notably nephrin, podocin and CD2-associated protein (CD2AP), are critical to this function since lack of any of these proteins in genetic disorders and/or knockout mice causes severe nephrotic syndrome.5–7 A fourth protein, α-actinin 4, is also expressed in podocytes and mutations in this protein are associated with focal segmental glomerulosclerosis.8 Antibodies to α-actinin have been suggested to play a pathogenic role in LN based on both murine and clinical studies,9–11 though more recent evidence from clinical studies12 and electron microscopy of renal biopsies from human and murine LN did not support this hypothesis.13 14
Patients with LN almost always present with proteinuria and have podocyte abnormalities on biopsy.15 Furthermore, the degree of podocyte pathology, as measured by foot process effacement, correlates with proteinuria.16 More recently, Perysinaki et al17 showed a correlation between increased foot process effacement, reduced glomerular expression of nephrin and podocin and progressive worsening of histological nephritis and proteinuria in both NZB/W F1 mice and patients with LN. The presence of immune deposits in a subepithelial distribution (ie, around the podocyte), as seen in membranous LN, is associated with more severe proteinuria. It is not known, however, whether podocytes are targeted directly by autoantibodies.18 There is evidence that exposure to plasma from children with nephrotic syndrome can cause relocation and altered expression of nephrin, podocin and CD2AP in cultured human podocytes.19 This study only included one patient (aged 14) with LN. There is no similar evidence using samples from adult patients or purified IgG from either children or adults with LN.
In the context of SLE, it is the immunoglobulin fraction of plasma that is of particular interest, as generation of autoantibodies is considered to be critical in disease pathogenesis. There are observational data from human studies (reviewed in20) and experimental data from murine models21 22 to support the concept that these antibodies can be directly pathogenic. Here, we describe a series of experiments investigating the effects of plasma and purified IgG on cultured human podocytes. We compared effects of plasma samples from patients with LN, patients with non-renal lupus, patients with rheumatoid arthritis and healthy control subjects. In particular, we looked for effects on expression, distribution and tyrosine phosphorylation of key podocyte proteins.
Patients and methods
Patients and controls
This study received approval from the Thames Valley Multi-Centre Research Ethics Committee. Patients and age/sex matched healthy controls were recruited from the University College Hospital and St Thomas’ Hospital, London, after receiving informed written consent. Blood was collected in heparinised tubes spun at 4°C, 500 g for 10 min and then plasma was stored at −20°C. Demographic information about all the patients with LN, and the healthy and disease controls, is summarised in table 1. More detailed information about the patients with LN is given in table 2.
Table 1.
Demographics of patients and controls used in podocyte experiments
| Group | Mean age* (range) | Sex | Ethnicity |
|---|---|---|---|
| Lupus nephritis (n=17) | 32 (18–56) | 16F 1M |
7A, 3B, 7W |
| Non-renal lupus (n=5) | 40 (30–64) | 5F | 1A, 4W |
| Rheumatoid arthritis (n=5) | 62 (40–78) | 3F 2M |
5W |
| Normal controls (n=30) | 39 (24–64) | 24F 6M |
4A, 5B, 21W |
*Age in years.
A, Asian; B, Black; F, female; M, male; W, White.
Table 2.
Baseline details of LN patients
| Patient ID | Sex | Age | Ethnicity | Biopsy result | Urine PCR (mg/mmol) | Serum albumin (g/L) | Treatment at time of biopsy* (mg) |
|---|---|---|---|---|---|---|---|
| LN1 | F | 49 | Asian | III | 58 | 38 | P20, H400 |
| LN2 | F | 34 | White | IV,V | 457 | 30 | P20, MP |
| LN3 | F | 20 | Black | V | 123 | 28 | P30, H400 |
| LN4 | F | 31 | White | IV | 533 | 21 | P10, H400, M1500 |
| LN5 | F | 30 | White | V | 569 | 23 | P10, H400 |
| LN6 | F | 35 | Black | III | 202 | 30 | P5, C |
| LN7 | F | 29 | Asian | III | 96 | 20 | P15, A100 |
| LN8 | F | 33 | Asian | III/V | 208 | 36 | P12.5, H200 |
| LN9 | F | 27 | Asian | III | 34 | 28 | P60, C |
| LN10 | F | 18 | Asian | IV | 66 | 44 | P10, M2000 |
| LN11 | M | 19 | Asian | IV | 1017 | 17 | P20, M1000, C |
| LN12 | F | 43 | White | II | 312 | 34 | H400 |
| LN13 | F | 48 | White | II | 127 | 33 | H400, MP |
| LN14 | F | 34 | White | V | 366 | 34 | P20, A100 |
| LN15 | F | 56 | White | III/V | 88 | 43 | P5, A100 |
| LN16 | F | 28 | Black | IV/V | 297 | 37 | P20 |
| LN17 | F | 21 | Asian | IV | 65 | 43 | P15, A 150 |
*In a number of cases, the treatment was changed as a result of the biopsy (eg, the addition of immunosuppressant drugs to high-dose corticosteroids) but those drugs are not included in the table as they would have no effect on samples taken before the drug was started, that is, at the time of the biopsy.
(M)P, (methyl)prednisolone; A, azathioprine; C, cyclophosphamide; H, hydroxychloroquine; LN, lupus nephritis; M, mycophenolate mofetil; PCR, protein creatinine ratio.
IgG purification
IgG was purified from plasma samples using HiTrap Protein G Columns (GE Healthcare). Aliquots of IgG were tested for LPS using the E-Toxate kit (Sigma), and samples that tested positive were treated with Detoxi-Gel Endotoxin Removing columns (Pierce). The concentration of IgG was confirmed in an IgG ELISA as described previously.23
Plasma and IgG exposure experiments
The conditionally immortalised human podocytes were cultured as described previously.24 Fully differentiated podocytes were then exposed to medium containing heat-inactivated (HI) human plasma, or purified IgG, for a prespecified time—5 min to 48 h. Thereafter, cells were trypsinised, washed in ice cold PBS and lysed in ice cold lysis buffer. Lysates were stored at −80°C.
Immunofluorescence microscopy
Podocytes were cultured on sterilised coverslips in medium containing plasma from LN patients or from healthy controls. After 48 h exposure of the podocytes to plasma, the effect on the localisation of α-actinin and CD2AP was determined by immunofluorescent staining. Cells were fixed with paraformaldehyde and permeabilised with triton-X 100. Non-specific binding was blocked with 2%FCS/2%BSA/0.1%Tween/PBS containing 10% normal rabbit serum (sigma), and then cells were incubated with the primary antibodies (rabbit-anti-α-actinin-4 (ImmunoGlobe), rabbit-anti-CD2AP (Santa Cruz Biotechnology)). After washing, goat antirabbit IgG-FITC conjugate (Sigma) was added and the cover slips were mounted on glass slides using Mowiol mountant. The slides were viewed on a Biorad confocal microscope and images captured using LaserSharp 2000 and analysed using confocal assistant V.4.02 software.
Western blot analysis
Changes in protein expression and phosphorylation were assessed by western blotting. Equal amounts of protein (30 μg per well) were separated on SDS-polyacrylamide gels. Proteins were electrophoretically transferred onto methanol activated polyvinylidene difluoride (PVDF) membranes (Millipore). Following blocking, membranes were probed with antiphosphotyrosine (Upstate), antipodocin (Santa Cruz), antinephrin (kind gift of Dr Lawrence Holzman), anti-CD2AP (Santa Cruz), actin (Sigma) and anti-GAPDH (Chemicon) antibodies. Visualisation of the protein bands was achieved using horseradish peroxidase (HRP)-conjugated secondary antibodies and chemiluminescence. Expression of podocin, nephrin, CD2AP and tyrosine phosphorylated proteins was quantified by normalising the strength of the relevant protein band to that of the housekeeping protein (actin or GAPDH) measured on the same gel using densitometry.
Immunoprecipitation
Tyrosine phosphorylated podocyte proteins were isolated using an antiphosphotyrosine antibody bound to protein G beads (Amersham, UK). Cell lysates were clarified by centrifugation, then added to the washed beads, along with protease and phosphatase inhibitors, and left to bind overnight.
Following a brief spin, the supernatant was removed and the beads washed in ice-cold lysis buffer. The pellet was resuspended in Laemmli sample buffer containing 2-mercaptoethanol. Bound proteins were released by boiling and run on an SDS-polyacrylamide gel. Protein bands were visualised by silver staining and were carried out using a Silver Stain Plus Kit (Bio-Rad, UK), as per the manufacturer's instructions.
Mass spectrometry
Relevant protein bands were cut out of the polyacrylamide gel and partially dehydrated using acetonitrile (ACN). The ACN was discarded and the gel pieces were dehydrated completely by centrifugal evaporation. Samples were reduced with dithiothreitol and alkylated with iodoacetamide. In-gel trypsinisation was followed by release of peptides using trifluoroacetic acid. Peptides were separated out on a 25 cm C18 Aqua HPLC column prior to mass spectrometry. Continuum LC-MS data were processed and searched using ProteinLynx GlobalServer versions 2.2.5 and 2.3 (Waters, UK). Protein identifications were obtained by searching the human entries of the UniProt/Swiss-Prot (V.53.5) database, and the presence/position of tyrosine phosphorylation sites investigated using the Phosphosite database (http://www.phosphosite.org).
Statistics
Data were analysed using non-parametric statistical tests using GraphPad Prism.
Results
Position, expression and phosphorylation of podocyte proteins following exposure to plasma from LN patients or healthy controls
Plasma obtained from six patients with LN and six healthy donors was heat inactivated to remove viable cytokines and complement but retain immunoglobulins. The six patients whose samples were used in this part of the study were LN4, LN8, LN9, LN10, LN11 and LN17. They were chosen because they were among the earliest samples available in this study and not on the basis of particular clinical characteristics. The cellular distributions of α-actinin-4 and CD2AP, both proteins important for podocyte function, were examined by immunofluorescence following 48 h culture with HI plasma from patients and controls (figure 1A,B,D and E show representative immunofluorescence images). The characteristic peripheral pattern of staining for CD2AP and α-actinin-419 was assessed; however, quantitative analysis revealed no significant difference between podocytes exposed to plasma from LN patients compared with healthy donors (figure 1C,F).
Figure 1.

Intracellular position of podocyte proteins after exposure to healthy or lupus nephritis plasma. Human podocytes were incubated for 48 h with heat-inactivated plasma from six lupus nephritis (LN) patients and six healthy donors. Cells were then fixed and stained for α-actinin and CD2AP (CD2-associated protein) using immunofluorescence staining (see ‘Methods”). Cells were examined by confocal microscopy, by two independent assessors and the number of cells with membrane staining was noted. Representative images showing α-actinin staining after culture with LN (A) and healthy (B) plasma. The percentage of cells with membrane staining was compared between the two groups using a t test (C). Representative images showing CD2AP staining after culture with LN (D) and healthy (E) plasma. The percentage of cells with membrane staining was compared between the two groups using a t test (F). No significant differences in distribution of α-actinin or CD2AP were found.
Next, we assessed the effect of exposing podocytes to healthy and LN plasma over a time course (5 min, 30 min, 1 h, 4 h, 14 h, 24 h and 48 h). No differences in the total expression of α-actinin-4, CD2AP or podocin at any of the time points assayed were observed by western blotting (data not shown).
However, a significant reduction in global protein tyrosine phosphorylation was observed in podocytes exposed to plasma from LN patients compared with the effect of plasma from healthy controls after 24 h culture (p=0.0008) (figure 2). By 48 h, differences between the groups were no longer seen (figure 2).
Figure 2.

Tyrosine phosphorylation of proteins in podocyte lysates following exposure to healthy or lupus nephritis plasma. Human podocytes were incubated with heat-inactivated (HI) plasma from five lupus nephritis (LN) patients and five healthy donors. Cells were lysed following 24 (i) or 48 (ii) h exposure, and protein tyrosine phosphorylation assessed by western blot. This demonstrated globally reduced tyrosine phosphorylation at 24 h in the lupus nephritis plasma exposed-cells, which had returned to normal levels by 48 h. Representative western blots (A) showing tyrosine phosphorylation of podocyte proteins following exposure to plasma from LN patients or from normal controls (NCs). Actin was used to check equal loading of protein. Results were quantified using densitometry and the median and IQR from 5 NCs and 5 patients are shown (B). Data from the two groups were compared using the Mann–Whitney test for significance.
This observation was specific to LN patients since similar changes were not observed when podocytes were exposed to plasma from patients with rheumatoid arthritis (a disease where podocyte pathology is not observed) or from patients with SLE, but with no history of renal involvement (figure 3A–C). In addition, this effect appeared to be specific to podocytes since no changes in protein tyrosine phosphorylation were observed when cells from an irrelevant epithelial line (HeLa cells) were cultured in medium containing plasma from LN patients (data not shown).
Figure 3.

Comparison of the effects of growing podocytes in plasma from patients with lupus nephritis (LN), rheumatoid arthritis or non-renal lupus. Representative antiphosphotyrosine blots from experiments comparing the effect of growing podocytes for 24 h in normal control (NC) plasma versus plasma from patients with LN (A), rheumatoid arthritis (RA, B) or non-renal systemic lupus erythematosus (NR-SLE, C). Cells were lysed and protein tyrosine phosphorylation assessed by western blot. The graphs display densitometry, with the median and IQR for each group. Medians were compared using the Mann–Whitney test. As already shown, LN plasma leads to a reduction in protein tyrosine phosphorylation, whereas the effect of plasma from patients with RA or NR-SLE is no different to NC. Equal loading was ensured by blotting for either actin or GAPDH.
Thus, the results indicated that only plasma from LN patients contained a factor that influenced tyrosine phosphorylation of podocyte proteins.
Tyrosine phosphorylation of podocyte proteins following exposure to purified IgG
In order to prove that antibodies, rather than any other plasma component, were responsible for influencing protein tyrosine phosphorylation, IgG was purified from LN and healthy control plasma samples. Podocytes were grown in medium containing purified IgG at two concentrations: 100 μg/mL, a concentration typically used in experiments with monoclonal antibodies,25 and 1.5 mg/mL to mirror the amount of IgG present in the plasma exposure experiments. Exposing podocytes to purified IgG from patients with LN at the higher (1.5 mg/mL) (figure 4B), but not at the lower (100 µg/mL) concentration (figure 4A), led to a relative decrease in tyrosine phosphorylation compared with IgG from healthy donors, thus recapitulating the initial plasma exposure experiments (p=0.001).
Figure 4.

The effect of purified IgG from healthy controls or patients with lupus nephritis on tyrosine phosphorylation in podocytes at two different IgG concentrations. Podocytes were grown in medium containing purified IgG from patients with LN, or normal controls. Lysates were blotted with antiphosphotyrosine antibody and equal loading of protein confirmed with GAPDH. The groups were compared using the Mann–Whitney test for significance. (A) Medium containing 100 μg/mL IgG, with representative western blots. No difference was seen between normal control and lupus nephritis IgG-exposed cells. (B) Medium containing 1.5 mg/mL IgG, with representative western blots. Exposure to this concentration of IgG from patients with lupus nephritis led to a relative reduction in protein tyrosine phosphorylation compared with healthy control IgG.
Identification of proteins with variable tyrosine phosphorylation
Examination of the western blots shown in figure 2A revealed dominant tyrosine phosphorylated protein bands at approximately 55 and 80 kDa. In order to understand the functional implications of the observed dephosphorylation, the tyrosine phosphorylated proteins were isolated by immunoprecipitation using an antiphosphotyrosine antibody before separation on polyacrylamide gels, and the protein bands visualised by silver staining (figure 5). The proteins of interest, and a number of control bands, were excised from the polyacrylamide gel and subjected to in-gel proteolytic digestion before analysis and identification using electrospray QTOF mass spectrometry.
Figure 5.
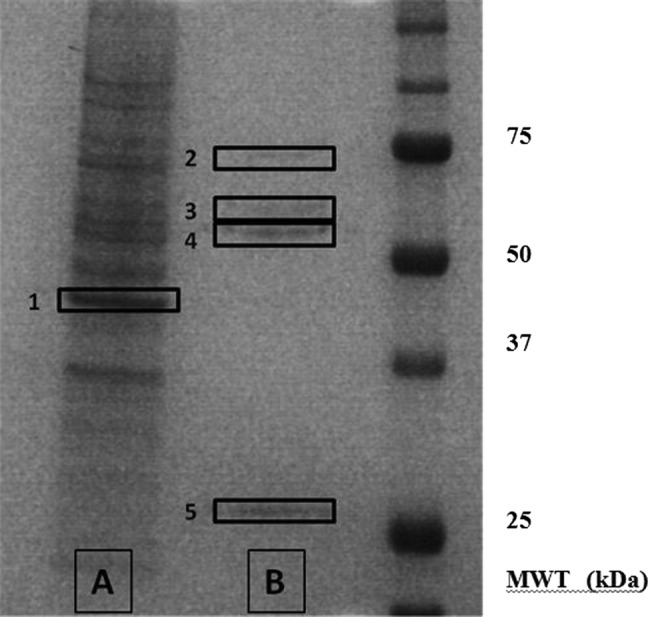
Silver-stained gel showing the major tyrosine-phosphorylated proteins in podocytes. In order to identify the key tyrosine phosphorylated proteins, podocyte lysates were subjected to immunoprecipitation with mouse antiphosphotyrosine antibody. The products of the immunoprecipitation were run on polyacrylamide gels and visualised by silver staining. The unbound proteins are shown in lane A, and the bound (ie, tyrosine phosphorylated) proteins in lane B. Proteins bands 1–5 were excised from the polyacrylamide gel and subjected to in-gel proteolytic digestion before their analysis and identification using mass spectrometry.
A number of tyrosine phosphorylated proteins were obtained from the bands. However, apart from the expected contaminants—keratin, trypsin and IgG—the results revealed two specific proteins: tubulin in the 55 kDa band and glucose-regulated protein 78 (GRP78) in the 80 kDa band (see table 3). The results were confirmed since the proteins were isolated in the replicate sample, but not in any of the control bands, and were of the correct molecular weight.
Table 3.
Summary of proteins identified for each sample
| Band number (see gel in figure 5) | Approximate molecular weight (kDa) | Isolated proteins (species) |
|---|---|---|
| 1 | 40 | Actin |
| 2 | 80 | GRP78 (multiple) |
| 3 | 60 | IgG2β (mouse) |
| 4 | 50 | IgG2β (mouse), tubulin α (multiple), tubulin β (multiple) |
| 5 | 25 | IgGκ (mouse) |
Analysis of the spectra obtained from the mass spectrometry sequence data for tubulin added further support to the validity of the finding, in that there was a low signal-to-noise ratio, and that the isolated peptide had an identical amino acid sequence when read in either direction.
All proteins were sequenced using electrospray QTOF-MS, except trypsin and keratin, if number of matches ≥3.
Discussion
These experiments were designed to investigate possible mechanisms by which IgG from patients with LN could have a direct effect on podocytes. Our key finding is that growing podocytes in medium containing plasma or purified IgG from patients with active LN for 24 h leads to reduced tyrosine phosphorylation of podocyte proteins, and the effect of purified IgG is dose dependent. This effect is seen only with samples from patients with active LN and not with IgG from non-nephritic lupus. These same LN samples had no similar effect on an irrelevant cell line. Importantly, these changes in phosphorylation were reversible, demonstrating that the cells were not simply killed, and normal levels of phosphorylation were noted by 48 h. In particular, tubulin, a protein known to play a key role in maintaining the structure of podocytes, was identified among the proteins that were dephosphorylated. Ideally, these results could be further confirmed by carrying out western blots with specific antibodies to tyrosine phosphorylated forms of tubulin. However, no such antibodies are available.
There are a number of limitations to our experiments. First, we exposed podocytes to IgG for a limited period only as the culture was not replenished with IgG following addition of the culture medium at time 0. In vivo, autoantibodies would be present continuously as they have been shown to deposit in chromatin-rich areas near podocytes by electron microscopy.13 14 Thus, rather than the transient effect on tyrosine phosphorylation that we have shown in vitro, we propose that there would be a longer lasting effect in vivo, which could contribute to the pathogenesis of LN. This might be investigated, for example, by using immunofluorescence to look for reduced tyrosine phosphorylation of podocyte proteins in renal biopsies from human or murine LN. Although we showed no effect on podocyte tyrosine phosphorylation exerted by serum from patients with rheumatoid arthritis or non-renal lupus, we did not have access to samples from patients with other forms of autoimmune nephritis. It would be interesting to compare the effects of serum and IgG from those patients with the effects of LN IgG and also to see whether the effect in LN is specific to particular IgG isotypes and/or varies with the histological class of LN or with degree of proteinuria. Finally, for pragmatic reasons (the amount of serum and IgG that we could obtain from individual donors under the conditions of our ethics approval), the normal control (NC) samples used were not the same in all parts of the experiment and there is thus a variation in the results obtained from NC in different figures of the results section. Although an imperfect compromise, use of densitometry and normalisation to actin or GAPDH does reduce the effect of this variation and shows statistically significant differences between LN and NC samples.
Changes in phosphorylation of tubulin alter podocyte morphology. Polymerisation of tubulin is important in the formation of thick tubular structures that support the major processes of podocytes.26 These tubulin-containing major processes connect the podocyte cell body with the actin network of the foot processes. Inhibition of microtubule function using vinblastine causes collapse and thinning of podocyte major processes, but leaves the actin-based foot processes unaffected.27 Xing et al28 reported that exposure to dexamethasone promotes maturation and process formation of cultured human podocytes in vitro and that this is associated with increased expression of tubulin and nephrin but not podocin or CD2AP. Although these authors did not look at tyrosine phosphorylation of tubulin or other podocyte proteins, there is evidence that this process is important in podocyte function. In the normal rat kidney, tyrosine phosphorylated proteins are concentrated in the glomerulus, particularly the podocyte foot processes.29 Furthermore, treatment of cultured mouse podocytes with protamine sulfate or puromycin aminonucleoside caused retraction of podocyte cell processes, increase in cytoplasmic staining of tyrosine phosphorylated proteins and a reduction of tyrosine phosphorylation at focal contacts.30
Tyrosine phosphorylation of tubulin by the tyrosine kinase Fes promotes the formation of microtubule-like polymers.31 This may have implications for cell differentiation as shown in a study of myelocytic leukemic HL-60 cells.32 Induction of differentiation using tetradecanoyl phorbol acetate led to increased tyrosine phosphorylation of tubulin α and β, stretching of microtubules and the development of cell projections. In contrast to the evidence that increased tyrosine phosphorylation promotes microtubule formation, there is less evidence relating to reduced phosphorylation. Kobayashi et al33 used a conditionally immortalised mouse cell line to demonstrate that microtubule elongation and major podocyte process formation was inhibited following suppression of serine/threonine phosphatase activity by treatment with okadaic acid. By contrast, cells treated with vanadate, which inhibits tyrosine phosphorylation, did develop processes, although with fewer cell contacts, and at higher concentrations, vanadate caused cell detachment. The finding that the serine/threonine phosphatase PP2A is absent in adult kidney34 might suggest a greater role for tyrosine phosphorylation in the mature podocyte.
We also detected tyrosine phosphorylation of GRP78 in podocytes. GRP78, also known as binding immunoglobulin protein (BiP), is involved in endoplasmic reticulum stress, which involves attenuation of new protein synthesis and degradation of misfolded proteins.35 36 GRPs play a key role in the latter process by binding to defective proteins and marking them out for degradation. As terminally differentiated cells, podocytes require a well-developed adaptive response to stress and there is evidence linking GRP78 to podocyte dysfunction. Inagi et al36 studied megsin transgenic rats, in which overexpression and accumulation of megsin in podocytes leads to cell damage, proteinuria and renal impairment. By immunohistochemistry they demonstrated marked upregulation of GRP78 in glomeruli of these rats. GRP78 has also been shown to be upregulated in renal biopsies from patients with focal segmental glomerulosclerosis and membranoproliferative glomerulonephritis.37 Mice carrying a mutation in BiP developed tubular interstitial injury, which is accelerated by protein overload.38 However, there is no information in mice, humans or rats regarding any effect of tyrosine phosphorylation on the function of GRP78.
We did not see any effect on total expression of CD2AP, podocin or α-actinin-4 in podocytes exposed to IgG or plasma from patients with LN despite testing this at a wide range of time points. There is conflicting evidence on overall expression of podocyte proteins in LN in the literature. Koop et al39 found that expression of nephrin and podocin was reduced at the protein level but increased at the mRNA level in renal tissue from seven patients with LN compared with 13 healthy kidneys. Perysinaki et al17 found that expression was dependent on the histological type of nephritis. In NZB/W F1 mice, nephrin and podocin expression were both reduced at both protein and mRNA level in proliferative nephritis but only nephrin was reduced in mesangial disease. In human renal biopsies, nephrin expression was normal in type II LN but reduced in type IV or V.17 Thus, the expression of these proteins may be affected by the structural milieu within the inflamed kidney, which would not be reproduced in our experiments on cultured cells.
Coward et al19 showed that the characteristic peripheral distribution of CD2AP in the same cultured human podocyte line that we used was disrupted by addition of nephrotic syndrome plasma (but not healthy control plasma) and postulated that this disruption was important in altering podocyte function. In contrast, we found that LN plasma did not cause translocation of podocyte slit diaphragm proteins away from the cell membrane. The patients studied by Coward et al19 were all children with nephrotic syndrome severe enough to require plasma exchange and only one had SLE. Our patients were all adults and had much less severe proteinuria. None had plasma exchange. Our results do not exclude the possibility that plasma samples from adults with severe LN might produce the same effects on protein distribution seen by Coward et al, but suggest that at earlier and less severe stages of LN a different mechanism operates.
We propose a mechanism in which IgG from patients binds directly to podocytes, leading to changes in phosphorylation of key structural proteins (especially tubulin), which alters morphology and function of the cells. IgG from patients with SLE have previously been shown to bind more strongly to human mesangial cells40 and pleural mesothelial cells41 than IgG from healthy controls. Affinity purified anti-DNA antibodies have been shown to bind to surface annexin II on a human mesangial cell line, following which they become internalised, causing activation of cell kinases and increased production of the pro-inflammatory cytokine IL-6.42
Conclusion
In summary, these results suggest that pathogenic IgG in patients with LN interact directly with podocytes to cause changes in intracellular tyrosine phosphorylation of proteins such as tubulin. Further investigation of this mechanism could be important in the identification of novel treatments that do not rely upon immunosuppression.
Footnotes
Contributors: JJM and AR conceived the idea of the project and wrote the final paper. JJM and LM carried out the laboratory experiments on human podocytes. AR, DI and DDC recruited patients and obtained clinical details. EJ took part in the cell culture and immunological experiments. KM took part in the mass spectrometry experiments. LN, MS and PM developed the human podocyte line and method for assessing staining of podocyte specific proteins using immunofluorescence. All authors reviewed and agreed on the final manuscript.
Funding: This work was funded by Arthritis Research UK Research Grants 16555 and 17045 and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. The funders had no role in the acquisition or analysis of the data or writing the manuscript.
Competing interests: None.
Ethics approval: Thames Valley Multi-Centre Research Ethics Committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Condon MB, Ashby D, Pepper RJ, et al. Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann Rheum Dis 2013;72:1280–6. [DOI] [PubMed] [Google Scholar]
- 2.Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010;62:222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Navarra SV, Guzman RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011;377:721–31. [DOI] [PubMed] [Google Scholar]
- 4.Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum 2012;64:1215–26. [DOI] [PubMed] [Google Scholar]
- 5.Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1998;1:575–82. [DOI] [PubMed] [Google Scholar]
- 6.Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000;24:349–54. [DOI] [PubMed] [Google Scholar]
- 7.Shih NY, Li J, Karpitskii V, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 1999;286:312–15. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000;24:251–6. [DOI] [PubMed] [Google Scholar]
- 9.Mostoslavsky G, Fischel R, Yachimovich N, et al. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur J Immunol 2001;31:1221–7. [DOI] [PubMed] [Google Scholar]
- 10.Deocharan B, Qing X, Lichauco J, et al. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J Immunol 2002;168:3072–8. [DOI] [PubMed] [Google Scholar]
- 11.Mason LJ, Ravirajan CT, Rahman A, et al. Is alpha-actinin a target for pathogenic anti-DNA antibodies in lupus nephritis? Arthritis Rheum 2004;50:866–70. [DOI] [PubMed] [Google Scholar]
- 12.Manson JJ, Ma A, Rogers P, et al. Relationship between anti-dsDNA, anti-nucleosome and anti-alpha-actinin antibodies and markers of renal disease in patients with lupus nephritis: a prospective longitudinal study. Arthritis Res Ther 2009;11:R154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalaaji M, Mortensen E, Jorgensen L, et al. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am J Pathol 2006;168:1779–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalaaji M, Sturfelt G, Mjelle JE, et al. Critical comparative analyses of anti-alpha-actinin and glomerulus-bound antibodies in human and murine lupus nephritis. Arthritis Rheum 2006;54:914–26. [DOI] [PubMed] [Google Scholar]
- 15.Ng WL, Chan KW, Ma L. A scanning electron microscope study of isolated glomeruli in glomerulonephritis. Pathology 1983;15:139–46. [DOI] [PubMed] [Google Scholar]
- 16.Kraft SW, Schwartz MM, Korbet SM, et al. Glomerular podocytopathy in patients with systemic lupus erythematosus. J Am Soc Nephrol 2005;16:175–9. [DOI] [PubMed] [Google Scholar]
- 17.Perysinaki GS, Moysiadis DK, Bertsias G, et al. Podocyte main slit diaphragm proteins, nephrin and podocin, are affected at early stages of lupus nephritis and correlate with disease histology. Lupus 2011;20:781–91. [DOI] [PubMed] [Google Scholar]
- 18.Trivedi S, Zeier M, Reiser J. Role of podocytes in lupus nephritis. Nephrol Dial Transplant 2009;24:3607–12. [DOI] [PubMed] [Google Scholar]
- 19.Coward RJ, Foster RR, Patton D, et al. Nephrotic plasma alters slit diaphragm-dependent signaling and translocates nephrin, Podocin, and CD2 associated protein in cultured human podocytes. J Am Soc Nephrol 2005;16:629–37. [DOI] [PubMed] [Google Scholar]
- 20.Manson JJ, Isenberg DA. The origin and pathogenic consequences of anti-dsDNA antibodies in systemic lupus erythematosus. Expert Rev Clin Immunol 2006;2:377–85. [DOI] [PubMed] [Google Scholar]
- 21.Ehrenstein MR, Katz DR, Griffiths MH, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int 1995;48:705–11. [DOI] [PubMed] [Google Scholar]
- 22.Ravirajan CT, Rahman MA, Papadaki L, et al. Genetic, structural and functional properties of an IgG DNA-binding monoclonal antibody from a lupus patient with nephritis. Eur J Immunol 1998;28:339–50. [DOI] [PubMed] [Google Scholar]
- 23.Giles IP, Haley J, Nagl S, et al. Relative importance of different human aPL derived heavy and light chains in the binding of aPL to cardiolipin. Mol Immunol 2003;40:49–60. [DOI] [PubMed] [Google Scholar]
- 24.Saleem MA, O'Hare MJ, Reiser J, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 2002;13:630–8. [DOI] [PubMed] [Google Scholar]
- 25.Raschi E, Testoni C, Bosisio D, et al. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood 2003;101:3495–500. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi N, Gao SY, Chen J, et al. Process formation of the renal glomerular podocyte: is there common molecular machinery for processes of podocytes and neurons? Anat Sci Int 2004;79: 1–10. [DOI] [PubMed] [Google Scholar]
- 27.Andrews PM. The effect of vinblastine-induced microtubule loss on kidney podocyte morphology. Am J Anat 1977;150:53–61. [DOI] [PubMed] [Google Scholar]
- 28.Xing CY, Saleem MA, Coward RJ, et al. Direct effects of dexamethasone on human podocytes. Kidney Int 2006;70:1038–45. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Yoshida Y, Nameta M, et al. Glomerular proteins related to slit diaphragm and matrix adhesion in the foot processes are highly tyrosine phosphorylated in the normal rat kidney. Nephrol Dial Transplant 2010;25:1785–95. [DOI] [PubMed] [Google Scholar]
- 30.Reiser J, Pixley FJ, Hug A, et al. Regulation of mouse podocyte process dynamics by protein tyrosine phosphatases rapid communication. Kidney Int 2000;57:2035–42. [DOI] [PubMed] [Google Scholar]
- 31.Laurent CE, Delfino FJ, Cheng HY, et al. The human c-Fes tyrosine kinase binds tubulin and microtubules through separate domains and promotes microtubule assembly. Mol Cell Biol 2004;24: 9351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katagiri K, Katagiri T, Kajiyama K, et al. Tyrosine-phosphorylation of tubulin during monocytic differentiation of HL-60 cells. J Immunol 1993;150:585–93. [PubMed] [Google Scholar]
- 33.Kobayashi N, Reiser J, Schwarz K, et al. Process formation of podocytes: morphogenetic activity of microtubules and regulation by protein serine/threonine phosphatase PP2A. Histochem Cell Biol 2001;115:255–66. [DOI] [PubMed] [Google Scholar]
- 34.Everett AD, Xue C, Stoops T. Developmental expression of protein phosphatase 2A in the kidney. J Am Soc Nephrol 1999;10:1737–45. [DOI] [PubMed] [Google Scholar]
- 35.Cybulsky AV. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int 2010;77:187–93. [DOI] [PubMed] [Google Scholar]
- 36.Inagi R, Nangaku M, Onogi H, et al. Involvement of endoplasmic reticulum (ER) stress in podocyte injury induced by excessive protein accumulation. Kidney Int 2005;68:2639–50. [DOI] [PubMed] [Google Scholar]
- 37.Markan S, Kohli HS, Joshi K, et al. Up regulation of the GRP-78 and GADD-153 and down regulation of Bcl-2 proteins in primary glomerular diseases: a possible involvement of the ER stress pathway in glomerulonephritis. Mol Cell Biochem 2009;324:131–8. [DOI] [PubMed] [Google Scholar]
- 38.Kimura K, Jin H, Ogawa M, et al. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem Biophys Res Commun 2008;366:1048–53. [DOI] [PubMed] [Google Scholar]
- 39.Koop K, Eikmans M, Baelde HJ, et al. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol 2003;14:2063–71. [DOI] [PubMed] [Google Scholar]
- 40.Chan TM, Leung JK, Ho SK, et al. Mesangial cell-binding anti-DNA antibodies in patients with systemic lupus erythematosus. J Am Soc Nephrol 2002;13:1219–29. [DOI] [PubMed] [Google Scholar]
- 41.Guo H, Leung JC, Chan LY, et al. The pathogenetic role of immunoglobulin G from patients with systemic lupus erythematosus in the development of lupus pleuritis. Rheumatology (Oxford) 2004;43:286–93. [DOI] [PubMed] [Google Scholar]
- 42.Yung S, Cheung KF, Zhang Q, et al. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J Am Soc Nephrol 2010;21:1912–27. [DOI] [PMC free article] [PubMed] [Google Scholar]