

Summary
Background
Glycosylation plays an important role in protein function. The importance of glycosylation for tissue factor (TF) function is unclear.
Objective
The aim of the present study is to investigate the importance of TF glycosylation in transport to the cell surface and its coagulant and signaling functions.
Methods
Endothelial cells and peripheral blood mononuclear cells (PBMC) were treated with tunicamycin to inhibit N-linked glycosylation. Site-specific mutagenesis of one or more potential N-linked glycosylation sites in TF was used to generate TF mutants lacking glycans. TF expression at the cell surface was determined in binding assays using 125I-FVIIa or 125I-TF mAb and confocal microscopy. TF coagulant activity was measured by factor (F) Xa generation assay, and TF signaling function was assessed by measuring cleavage of protease activated receptor 2 (PAR2) and activation of p44/42 MAPK.
Results
Tunicamycin treatment reduced TF activity at the endothelial cell surface; however, this reduction was found to be the result of decreased TF protein production in tunicamycin-treated cells. Tunicamycin treatment had no significant effect on TF activity or antigen levels in PBMC. No significant differences were observed in TF protein expression and procoagulant activity among cells transfected to express either wild-type TF or TF mutants. A fully non-glycosylated TF is shown to bind FVIIa and interact with FX with the same efficiency as that of wild-type TF. Non-glycosylated TF is also capable of supporting FVIIa cleavage of PAR2 and PAR2-dependent p44/42 MAPK activation.
Conclusions
Glycosylation is not essential for TF transport and coagulant or signaling functions.
Keywords: factor VIIa, factor X, glycosylation, tissue factor
Introduction
Tissue factor (TF) is a transmembrane glycoprotein that initiates the extrinsic coagulation cascade by serving as a cofactor for plasma coagulation factor (F)VII/FVIIa. Recent studies have shown that TF-FVIIa may also induce cell signaling via activation of protease activated receptors (PAR) and thus modulate a number of non-hemostatic functions [1–3]. It has been reported earlier that TF undergoes a series of post-translational modifications such as N-linked glycosylation, phosphorylation, palmitoylation and S-nitrosylation (see [4]). At present, it is not entirely clear whether and how these post-translational modifications modulate TF biological functions. It is well recognized that glycosylation plays an important role in cell surface receptor functions such as intracellular trafficking, ligand binding, aiding in protein maturation by protecting the protein from degradation, and signal transduction [5]. The amino acid sequence analysis of TF indicates that there are four potential N-linked glycosylation sites; three of them are in the extracellular domain at Asn11, Asn124 and Asn137 and one in the cytoplasmic domain at Asn261. Studies of Paborsky et al. [6] revealed the presence of carbohydrates at all three glycosylation sites of the extracellular domain.
Experimental data regarding the importance of glycosylation for TF function vary. Pitlick et al. [7] demonstrated that concanavalin A, a member of the lectin family of carbohydrate binding proteins that preferentially bind to glucosyl and mannosyl residues [8], reversibly inhibited TF procoagulant activity. Tunicamycin, the inhibitor of N-linked glycosyl reaction, was found to inhibit surface TF procoagulant activity in LPS-stimulated murine macrophages [9]. In other studies, tunicamycin treatment was found to inhibit the transport of TF to the cell surface, which in turn decreased TF procoagulant activity [10]. In contrast to the above observations that suggest TF glycosylation could play an important role in its function, Paborsky et al. [6,11] reported that glycosylation is not required for TF procoagulant activity The observation that a series of glycosylation site mutants of soluble rTF expressed in yeast exhibit a similar procoagulant activity as of rTF produced in E.coli and Chinese hamster ovary (CHO) cells also suggests that glycosylation does not influence TF procoagulant activity [12].
Recently, the comparison of TF activities when TF was derived from different sources (rTF1-243 produced in E. coli, rTF1-263 produced in insect sf9 cells, and natural placental TF) showed that placental TF was more catalytically active than other forms of TF. Deglycosylation of placental TF led to a significant decrease in the catalytic constant [13]. Mass spectrophotometric analysis revealed that rTF1-243 had no carbohydrates attached to the backbone of the protein as expected, and placental TF was more heavily modified than rTF1-263. Although all three potential glycosylation sites in the extracellular domain of both rTF1-263 and placental TF have carbohydrate attachments, the extent of glycosylation and carbohydrate composition was different between the two proteins, as well as between each glycosylated site within the protein [13]. Taken together, these data indicate that the heterogeneity in the carbohydrate composition, as well as the presence of carbohydrates, would significantly influence TF procoagulant activity. From this study, however, it is not clear why the deglycosylated rTF1-263 expressed in Sf9 insect cells did not show altered TF activity, as one would expect if the glycosylation is important for TF coagulant function [13].
Interestingly, studies designed to examine the influence of cysteine disulfide bond formation in the extracellular domain on TF structure and function suggested that mutation of Cys186 and/or Cys209 residues resulted in impaired or aberrant glycosylation of TF and decreased TF procoagulant activity [14,15]. Mutation of Cys186 residue to Ser resulted in creation of a new carbohydrate signal. In addition to a decrease in TF procoagulant activity, the synthesis of the mutant proteins was also severely impaired [15]. Although there was no direct evidence in these studies that impaired glycosylation is responsible for either decreased TF procoagulant activity or the transport of TF to the plasma membrane, the data are consistent with the hypothesis that glycosylation modulates TF coagulant activity and its transport. We are not aware of any studies that have investigated the role of glycosylation in TF signaling function.
In the present study, we have used a specific and direct approach to explore the importance of TF glycosylation in modulating its cell surface expression and functions. We have used site-specific mutagenesis to generate TF deficient in glycosylation to specifically delineate the role of carbohydrates at the individual glycosylation sites in modulating TF procoagulant and signaling functions. Using these mutants, we demonstrate clearly that glycosylation does not influence FVIIa binding to TF or the interaction of FX with the TF-FVIIa complex. Non-glycosylated mutants of TF are as catalytically efficient as that of wild-type TF. Our data also demonstrate that glycosylation has no effect on either TF synthesis or its transport to the plasma membrane. Furthermore, our data also suggest that glycosylation does not have an effect on TF signaling potential.
Materials and methods
Primary human umbilical vein endothelial cells (HUVEC) were obtained from Lonza Walkersville, Inc (Walkersville, MD, USA) and Chinese hamster ovary cells were from American Type Culture Collection (Manassas, VA, USA). The cells were cultured as described earlier [15]. Peripheral blood mononuclear cells (PBMC) from human blood were isolated by gradient centrifugation using Ficoll-Paque Plus (1.077 g mL−1) (GE Healthcare, Uppsala, Sweden). To prevent N-linked glycosylation of TF protein during translation, HUVEC and PMBC were treated with varying concentrations of tunicamycin during their stimulation with TNF-α + IL-1β and LPS, respectively. The QuikChange II XL® Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) was used to generate single, double or triple point mutations at sites Asn11, Asn124, Asn137 and Asn261, in each case changing the Asn to Ala. A plasmid pacAd5 CMVK-NpA Shuttle vector (Cell Biolabs, San Diego, CA, USA) containing the TF cDNA fused to green fluorescent protein (GFP) at the C-terminus was used to construct the mutants. Wild-type TF-GFP and the triple mutant TF-GFP were also prepared as adenoviral vectors. CHO cells were transfected transiently with wild-type TF-GFP or TF mutant-GFP using Fugene HD. HUVEC were transduced with adenoviral vectors encoding wild-type TF-GFP and the triple mutant TF-GFP (20 MOI per cell). TF procoagulant activity was measured in a FX activation assay as described earlier [16]. TF antigen was measured in an ELISA as described earlier [17]. FVIIa and TF mAb TF9 9C3 were labeled with 125I and radioligand binding assays were performed as described previously [15]. Immunofluorescence staining of TF and confocal microscopy were performed as described earlier [18]. TF-FVIIa cleavage of PAR2 was determined using AP-PAR2 reporter constructs [19]. Activation of p44/42 MAPK was analyzed on immunoblot analysis using phospho-specific and total p44/42 MAPK antibodies (Cell Signaling Technology, Boston, MA, USA). TR3 mRNA levels were measured by real-time qPCR analysis. A full description of Materials and Methods is provided as Data S1.
Results
TF expression in stimulated endothelial cells and PBMC treated with tunicamycin
To determine whether glycosylation of TF modulates its cell surface expression or procoagulant activity, HUVEC were stimulated with TNFα + IL1β for 6 h and simultaneously treated with varying concentrations of tunicamycin, an inhibitor of the N-linked glycosyl reaction. As shown in Fig. 1(A), treatment of HUVEC with increasing concentrations of tunicamycin progressively reduced the amount of TF protein that migrates at 47 kDa, representing the fully glycosylated form of TF. Non-glycosylated TF protein in tunicamycin-treated cells migrated near 35 kDa. However, the intensity of this band was very low and faintly detectable. As a control, the same samples were analyzed for ICAM-1, a cell surface glycoprotein that is absent on unperturbed endothelial cells but induced by cytokines in a similar fashion to that of TF. Immunoblot analysis of ICAM-1 showed clearly that tunicamycin treatment, in a dose-dependent manner, inhibited glycosylation of ICAM-1, with 1 μg mL−1 tunicamycin completely inhibiting glycosylation of ICAM-1. In contrast to TF, a decrease in intensity of the glycosylated ICAM-1 band in tunicamycin-treated cells correlated well with an increase in non-glycosylated ICAM-1. These data raise the possibility that tunicamycin treatment either selectively inhibited the synthesis and/or surface expression of TF protein, or TF antibodies fail to effectively recognize the non-glycosylated TF protein in Western blot analysis. Next, we investigated the effect of tunicamycin (1 μg mL−1) on the expression of TF antigen and its activity at the cell surface. As shown in Fig. 1(B), cell surface TF activity was reduced by about 80% in HUVEC treated with tunicamycin. Similarly, tunicamycin treatment also reduced the expression of TF protein at the cell surface by about 80% or more (Fig. 1C). These data indicate that reduced TF activity observed in cells treated with tunicamycin is the result of reduced TF protein expression at the cell surface rather than reduced TF procoagulant activity of non-glycosylated TF. Measurement of total TF antigen levels and activity in cell lysates revealed that tunicamycin treatment also inhibited total TF protein levels and subsequently the activity by about 60% and 80%, respectively (Fig. 1D,E). These data suggest that the impaired TF protein expression at the cell surface in tunicamycin-treated cells primarily stems from reduction in total TF protein synthesis rather than impaired transport of TF protein from intracellular sources to the cell surface membrane. In additional studies, we investigated whether tunicamycin inhibited TF protein synthesis via a pre- or post-transcriptional mechanism. Analysis of TF mRNA levels with quantitative RT-PCR showed that TNFα + IL1β induction increased TF mRNA levels by about 150-fold over the control unstimulated HUVEC and tunicamycin (1 μg mL−1) treatment had no effect on cytokine-induced TF mRNA (TF mRNA levels as fold increase over the uninduced HUVEC: TNFα + IL1β, 158 ± 27; tunicamycin + TNFα + IL1β, 169 ± 35, n = 3, mean ± SEM). These data indicate that tunicamycin inhibited TF protein expression via a post-transcriptional mechanism.
Fig. 1.
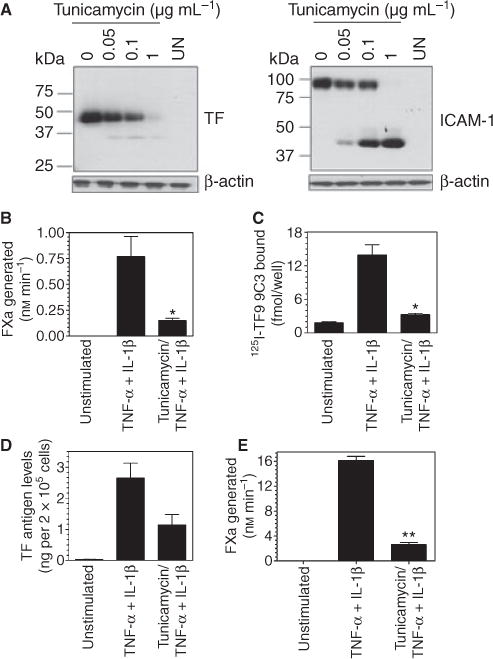
Tunicamycin treatment inhibits tissue factor (TF) protein expression and activity in stimulated endothelial cells. (A) Monolayers of human umbilical vein endothelial cells (HUVEC) were stimulated with TNF-α + IL1-β (20 ng mL−1 each) in the presence or absence of varying concentrations of tunicamycin (A) or 1 μg mL−1 (B–D) for 6 h. At the end of stimulation, (A) cells were lysed in non-reducing SDS-PAGE buffer and the samples were subjected to immunoblot analysis using polyclonal antibodies against TF or ICAM-1; (B) cell surface TF activity was determined by adding factor (F) VIIa (10 nM) and FX (175 nM) and measuring FXa generation; (C) cell surface TF antigen levels were determined in a binding assay using 125I-TF9 9C3 mAb (10 nM); (D) total TF antigen in cell lysates was determined in ELISA with TF polyclonal antibody; (E) TF activity in cell lysates was determined by adding FVIIa (10 nM) and FX (175 nM) and measuring FXa generation. *Denotes significant difference from TNF-α + IL1-β alone treated cells (P ≤ 0.02).
Next, we examined the effect of tunicamycin on TF expression in PBMC. As shown in Fig. 2(A), as expected, no detectable TF protein was expressed in unperturbed PBMC and LPS stimulation markedly increased TF protein expression in PBMC. LPS-stimulated PBMC were more resistant to tunicamycin treatment as compared with HUVEC and complete deglycosylation of TF required a five times higher concentration of tunicamycin; that is, 5 μg mL−1. Analysis of intact cells showed LPS markedly increased TF activity while pretreatment of PBMC with tunicamycin had no statistically significant effect on TF activity induced by LPS (Fig. 2B). A similar trend was noted in TF activity measured in cell lysates (data not shown). Although TF protein levels appeared to decrease in LPS-stimulated PBMC upon treatment with tunicamycin on western blots (Fig. 2A), measurement of TF antigen levels quantitatively by ELISA revealed no significant differences in TF protein levels in PBMC treated with tunicamycin or control vehicle and stimulated with LPS (Fig. 2C). These data suggest that TF lacking carbohydrate is not recognized well by western blot analysis (see the following section for more on this). Because inhibition of N-glycosylation by tunicamycin treatment affected the cell surface expression of TF activity and protein differently in HUVEC and PBMC and seeing that it is difficult to draw a firm conclusion on whether TF glycosylation plays a role in regulating TF procoagulant activity from these data alone, we examined next the role of glycosylation on TF activity by site-specific mutagenesis in which one or more of the Asn residues in potential glycosylation sites were mutated to Ala.
Fig. 2.
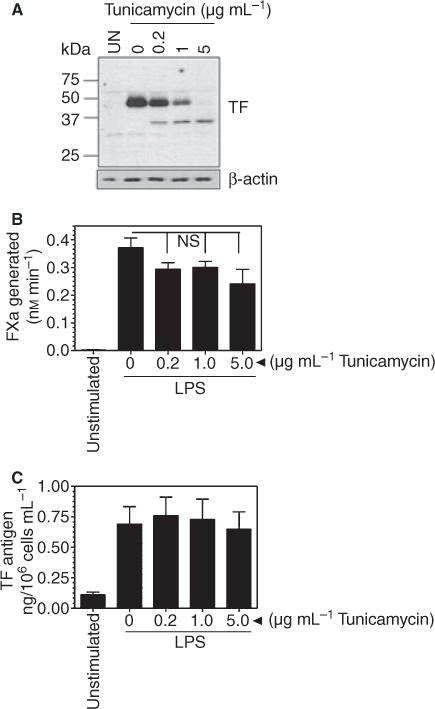
Effect of tunicamycin on tissue factor (TF) activity and antigen in peripheral blood mononuclear cells (PBMC). PBMC (2 × 106 cells mL−1) in serum-rich RPMI 1640 medium were stimulated with 100 ng mL−1 LPS for 6 h in the presence or absence of varying concentrations of tunicamycin. Where tunicamycin was added, cells were preincubated with it for 1 h. (A) Immunoblot analysis using TF polyclonal antibodies; (B) TF activity on intact cells as determined in factor X activation assay; (C) TF antigen levels determined in ELISA. Data are mean ± SEM (n = 4). ns, not statistically significant.
Tissue factor mutants lacking the potential glycosylation sites are expressed normally and exhibit no defect in tissue factor procoagulant activity
To determine the functional importance of N-glycosylation, Asn residues at one, or various combinations, of the four potential glycosylation sites were mutated to Ala, and plasmid constructs encoding wild-type TF-GFP or one of these TF-GFP mutants were used for transfection in CHO cells. Only potential glycosylation sites in the extracellular domain of TF were used in double- or triple-mutation combinations. Analysis of total TF antigen in the transfected cells by immunoblot analysis with TF polyclonal antibodies demonstrated an apparent increase in mobility of the glycosylation-deficient mutants as compared with the wild-type TF-GFP protein (Fig. 3A). In view of the fact that these constructs had GFP tagged to the cytoplasmic tail of TF, wild-type TF fusion protein migrated at approximately 72 kDa, whereas glycosylation-deficient mutants migrated at 69 (single mutant, TFN11A, TFN124A, TFN137A), 65 (double mutant, TFN11A/N124A, TFN11A/N137A and TFN124A/N137A mutant) or 62 kDa (triple mutant, TFN11A/N124A/N137A). The intensity of the TF band recognized by TF antibodies on immunoblot analysis was significantly lower in cell lysates harvested from cells transfected with double mutants, and markedly lower in cells transfected with the triple mutant, thereby giving the impression that lack of carbohydrates at more than one N-linked glycosylation site impairs TF protein expression. However, when cells transfected with wild-type TF-GFP or TF-GFP mutants were examined for GFP fluorescence by fluorescence microscopy, all transfectants showed similar levels of TF expression (data not shown). Similarity in levels of TF expression among cells transfected to express wild-type TF-GFP and TF-GFP mutants was confirmed by immunoblot analysis using GFP antibodies that recognize the TF fusion protein (Fig. 3B). Both wild-type TF-GFP and mutant TF-GFP were in monomeric form as we observed a single band corresponding to the molecular weight of monomeric fusion protein.
Fig. 3.
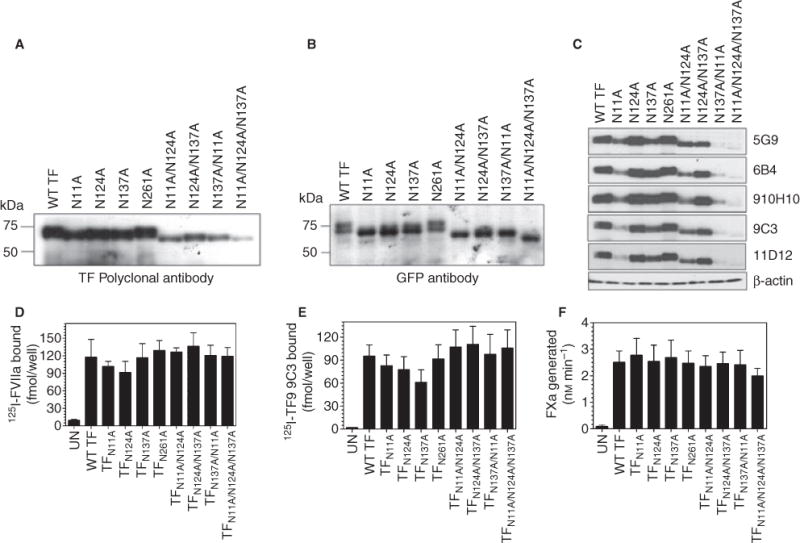
Glycosylation does not play a role in tissue factor (TF) cell surface expression or procoagulant activity. CHO cells were transfected with wild-type TF-GFP, single, double or triple glycosylation defective TF-GFP mutants. After 48 h of transfection, cells were lysed in non-reducing SDS-PAGE and an equal amount of protein was subjected to SDS-PAGE followed by immunoblot analysis with TF polyclonal antibodies (A), GFP peptide antibodies (B), or different TF monoclonal antibodies (C). Cell surface expression of TF was determined in radioligand binding assays using 125I-FVIIa (10 nM) (D) or 125I-TF9 9C3 mAb (10 nM) (E). (F) TF procoagulant activity at the cell surface was measured in a factor (F) Xa generation assay after adding FVIIa (10 nM) and FX (175 nM). Data are mean ± SEM (n = 4–6).
Carbohydrate moieties on a protein are known to be highly antigenic and thus it may be possible that our TF polyclonal antibodies raised using native (presumably fully glycosylated) TF purified from human brain extract [20] may have reduced reactivity towards non-glycosylated TF protein. This could be a possible explanation for the observed differences in band intensities of wild-type and mutant TF fusion protein when probed with anti-TF antibodies and GFP antibodies. In additional studies, we evaluated TF protein expression levels using multiple TF mAb (TF9 10H10, TF8 5G9, TF9 6B4, TF9 9C3 and TF811 D12) that may bind to TF at different epitopes [21]. Interestingly, all of the antibodies barely detected TF from cells transfected with the triple mutant (Fig. 3C). The mutation at Asn11 resulted in a marked reduction in detection of TF protein by immunoblotting when this mutation was present alone or in combination with other Asn mutants (TFN11A/N124A and TFN11A/N137A). The inability of TF antibodies to detect the non-glycosylated TF mutant appears to be limited to immunoblot analysis as we were able to measure similar trends in TF antigen levels in cell lysates of cells transfected with wild-type TF-GFP or glycosylation-deficient mutant TF-GFP in ELISA using TF polyclonal antibodies (data not shown). Next, we evaluated the expression of TF antigen at the cell surface in cells transfected with wild-type TF-GFP or glycosylation-deficient TF-GFP mutants in TF-specific binding assays using 125I-FVIIa or 125I-TF mAb (TF9 9C3). As shown in Fig. 3(D,E), there were no significant differences in 125I-FVIIa or 125I-TF mAb binding to cells expressing wild-type-GFP or mutant TF-GFP. Measurement of TF procoagulant activity in a FX activation assay showed no significant differences between wild-type and TF mutants, including the triple mutant that is completely devoid of glycosylation (Fig. 3F). Overall, these data indicate that glycosylation does not affect TF synthesis, transport to the cell surface, or its coagulant activity.
As the above studies showed that mutation at different potential glycosylation sites, at one, two or all three of the extracellular sites, resulted in similar behavior of TF, further studies were employed to examine the role of TF glycosylation in TF procoagulant activity and signaling function when expressed in endothelial cells. For these studies, we used the triple mutant (TFN11A/N124A/N137A) as this TF mutant would be completely devoid of glycosylation. In order to have a better control of expression levels and be able to perform these studies in HUVEC, we generated an adenovirus expression system encoding wild-type TF-GFP or TFN11A/N124A/N137A-GFP (both contain GFP at the C-terminus). To determine if N-linked glycosylation plays a role in subcellular localization and distribution of TF, HUVEC transduced with adenovirus encoding wild-type TF-GFP or TFN11A/N124A/N137A-GFP were analyzed for TF distribution by confocal microscopy. Analysis of TF expression by examining the fluorescence of tagged-GFP revealed that both wild-type and the triple mutant exhibited a similar pattern of TF distribution, that is, predominantly localized at the cell surface with a substantial amount present in the perinuclear compartment, and a small fraction in endosome-like structures. Immunostaining of non-permeabilized cells with TF polyclonal antibodies also showed comparable expression of wild-type TF and the triple mutant TF at the cell surface. TF antibody staining completely overlapped with the GFP fluorescence. These data further support the observation that N-linked glycosylation of TF is not necessary for transport of TF to the cell surface (Fig. 4).
Fig. 4.
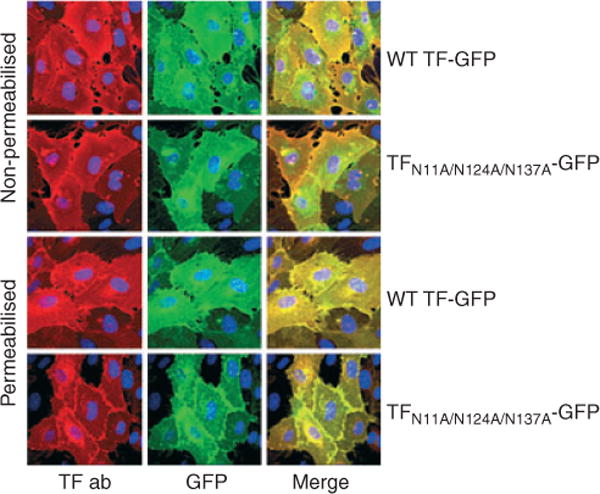
No impairment in cellular expression of glycosylation-deficient tissue factor (TF). Human umbilical vein endothelial cells (HUVEC) monolayers cultured on glass coverslips were transduced with 20 MOI per cell of wild-type or TFN11A/N124A/N137A adenovirus tagged with GFP. After 48 h of transduction, the monolayers were fixed, and one set permeabilised with 0.025% Triton-X-100 and stained with TF polyclonal antibodies (5 μg mL−1) and DAPI (nuclear staining). Fluorescence was analyzed by confocal microscopy. Upper two rows, non-permeabilised cells; bottom two rows, permeabilised cells. Left panel, TF expression analyzed by immunofluorescence staining; middle panel, TF expression by GFP fluorescence; right panel, merged image of GFP and TF immunofluorescence.
Next, we evaluated the procoagulant function of the triple mutant in comparison with wild-type TF-GFP in a FX activation assay in HUVEC. As observed with CHO cells transfected with wild-type and TFN11A/N124A/N137A plasmid constructs, mutation of all three putative glycosylation sites in the extracellular domain did not alter the procoagulant activity of TF in endothelial cells. Given that the glycosylation sites are located in the region of FVIIa interaction, it is conceivable that carbohydrates attached to TF may contribute to FVIIa interaction with TF. However, if this effect is not substantial, measuring FX activation at 10 nM FVIIa may mask the subtle effect of TF glycosylation on its interaction with FVIIa. Nevertheless, analysis of FVIIa interaction with TF using varying concentrations of FVIIa in a FX activation assay showed similar Kd values for FVIIa binding to coagulant-active wild-type TF-GFP and the triple mutant (Fig. 5A; Kd values as follows – wild-type TF-GFP, 0.18 ± 0.028 nM and TFN11A/N124A/N137A-GFP, 0.17 ± 0.039 nM). Recent studies have shown that deglycosylation of natural placental TF decreases the affinity of FX for both the TF-FVIIa complex (albeit minimally) and Kcat (significantly), thus contributing to a 6-fold difference in TF activity between glycosylated and deglycosylated TF [13]. Our comparative analysis of wild-type TF-GFP and the glycosylation-deficient mutant expressed at the endothelial cell surface for their ability to activate varying concentrations of FX showed no significant differences in the Vmax or the Km of FX activation (Fig. 5B: values for Vmax for wild-type TF-GFP, 7.70 ± 0.61 nM min−1, and TFN11A/ N124A/N137A-GFP, 7.21 ± 0.81 nM min−1; values for Km for wild-type TF-GFP, 0.65 ± 0.16 μM, and TFN11A/N124A/N137A-GFP, 1.0 ± 0.30 μM; a small difference in Km values between them is not statistically significant). In order to minimize error as the result of differences in the expression levels between wild-type and mutant TF, cell surface 125I-TF9 9C3 mAB binding was performed and data were normalized with protein amounts. Finally, to evaluate FVIIa binding to cryptic TF, which generally has lower affinity for FVIIa compared with coagulant-active TF, we analyzed 125I-FVIIa binding to HUVEC expressing wild-type TF-GFP or the triple mutant (because most of the TF on cell surfaces is cryptic [22,23], the binding parameters observed with 125I-FVIIa reflect its binding to cryptic TF). The binding data showed no significant differences between the mutant and wild-type TF-GFP in their affinity and binding capacity (Fig. 5C; Kd values as follows – wild-type TF-GFP, 15.5 ± 3.57 nM, and TFN11A/N124A/N137A-GFP, 13.4 ± 4.00 nM; Bmax values as follows – wild-type TF-GFP, 63 ± 4.7 fmol 10−5 cells, and TFN11A/N124A/N137A-GFP, 52 ± 4.8 fmol 10−5 cells).
Fig. 5.
Non-glycosylated tissue factor (TF) interacts with factor (F) VIIa and FX with a similar efficiency to that of wild-type TF. Human umbilical vein endothelial cells (HUVEC) monolayers were transduced with 20 MOI per cell of wild-type TF-GFP or TFN11A/N124A/N137A-GFP adenovirus. After 48 h of transduction, TF activity at the cell surface was measured in a FX activation assay using varying concentrations of FVIIa (0.01–10 nM) and a fixed concentration of FX (175 nM) (A), or varying concentrations of FX (0.025–3 μM) and a fixed concentration of FVIIa (10 nM) (B). Specific binding of 125I-FVIIa to the surface TF was measured in a radioligand binding assay (C). (●) Wild-type TF-GFP; (○) TFN11A/N124A/N137A-GFP. Data are mean ± SEM (n = 3).
Glycosylation of tissue factor is not essential for its de-encryption
To investigate the potential effect of glycosylation on TF de-encryption, HUVEC expressing either the wild-type TF-GFP or mutant TFN11A/N124A/N137A-GFP were exposed to either the oxidizing agent HgCl2 or ionomycin, and TF coagulant activity at the cell surface was measured by FX activation assay. To allow for complete de-encryption of TF, the cells were lysed by two freeze-thaw cycles before measuring TF activity. HgCl2 and ionomycin treatments increased TF activity at the cell surface to a similar extent; that is, approximately 3-fold in cells expressing either wild-type TF-GFP or the triple mutant (Fig. 6). Measurement of TF activity in cell lysates also showed no significant differences in TF activity between the two cell lysates. These results suggest that both the wild-type TF-GFP and mutant TF-GFP were activated to a similar extent, excluding the possible role of glycosylation in TF encryption/de-encryption.
Fig. 6.
Glycosylation is not a determinant of tissue factor (TF) de-encryption. Human umbilical vein endothelial cells (HUVEC) monolayers transduced with wild-type TF-GFP or TFN11A/N124A/N137A-GFP adenovirus were treated either with a control vehicle, HgCl2 (100 μM) or ionomycin (10 μM) for 5 min. Cells were washed once before the cell surface TF activity was determined in a factor (F) X activation assay using FVIIa (10 nM) and FX (175 nM). In parallel, cell lysates were prepared by two cycles of freeze-thaw, and TF activity in cell lysates, after an appropriate dilution, was measured as described above. Data are mean ± SEM (n = 3).
Tissue factor glycosylation does not affect TF-FVIIa-mediated PAR2-dependent cell signaling
To investigate whether TF glycosylation plays a role in modulating TF-FVIIa-mediated signaling, we examined TF-FVIIa cleavage of PAR2 and PAR2-dependent p44/42 MAPK activation. CHO cells were co-transduced with wild-type TF-GFP or TFN11A/N124A/N137A-GFP and AP-PAR2. The transduced cells were exposed to FVIIa and TF-FVIIa cleavage of PAR2 was determined by measuring the alkaline phosphatase activity released into the medium. As shown in Fig. 7(A), both wild-type TF-GFP and TFN11A/N124A/N137A-GFP activated PAR2 to a similar extent; that is, approximately 15–20% of that observed with trypsin. Consistent with the observation that FVIIa bound to wild-type TF-GFP or the triple mutant cleaves PAR2 with a similar efficiency, there was no observable difference in FVIIa-induced, time-dependent, activation of p44/42 MAPK in cells expressing wild-type TF-GFP or TFN11A/N124A/N137A-GFP (Fig. 7B). Consistent with the data obtained in heterologus CHO cells, HUVEC transduced with either wild-type TF-GFP or TFN11A/N124A/N137A-GFP supported FVIIa cleavage of PAR2 to a similar extent (Fig. 7C). We also analyzed FVIIa-induced p44/42 MAPK activation in HUVEC transduced with wild-type TF-GFP or TFN11A/N124A/N137A-GFP in the presence of EPCR blocking mAb (to block EPCR-dependent FVIIa-induced p44/42 MAPK activation [24]). The data showed that endothelial cells expressing either wild-type TF-GFP or TFN11A/N124A/N137A-GFP supported FVIIa activation of p44/42 MAPK to a similar extent (approximately 2.5-fold increase over the control; data not shown). In additional studies, we examined TF-FVIIa-PAR2 signaling in endothelial cells by measuring FVIIa induction of TR3 in HUVEC transduced to express PAR2 and either wild-type TF-GFP or TFN11A/N124A/N137A-GFP. As shown in Fig. 7(D), FVIIa induction of TR3 is strictly dependent on TF and no significant differences were found in TR3 levels between the cells expressing wild-type TF or the glycosylation-deficient TF mutant (Fig. 7D).
Fig. 7.
Factor (F) VIIa cleavage of PAR2 and activation of p44/42 MAPK in cells expressing wild-type or non-glycosylated tissue factor (TF). (A and B) CHO cells were co-transduced with 100 MOI per cell of wild-type TF-GFP and TFN11A/N124A/N137A-GFP adenovirus with 25 MOI per cell of AP-PAR2 adenovirus or with AP-PAR2 alone for 48 h. (A) The transduced cells were incubated at 37°C with a control vehicle, FVIIa (10 nM) or trypsin (5 nM), and at the end of 1 h incubation, soluble alkaline phosphatase activity released in the medium was measured. The values obtained in control vehicle treatment were subtracted from FVIIa and trypsin treatments and the value obtained in trypsin treatment was designated 100% of PAR2 cleavage. (B) Cells deprived of serum overnight were treated with FVIIa (10 nM) for varying times (0–30 min) or trypsin (5 nM) for 5 min and the cell lysates were subjected to SDS-PAGE and immunoblotted with phospho p44/42 or total MAPK antibodies. (C) Human umbilical vein endothelial cells (HUVEC) were infected with 20 MOI per cell of wild-type TF-GFP or TFN11A/N124A/N137A-GFP adenovirus and 10 MOI per cell of AP-PAR2 adenovirus. Cells were treated with FVIIa and trypsin and cleavage was determined as described for panel A. (D) HUVEC were transduced with PAR2 (10 MOI per cell) and either wild-type TF-GFP or TFN11A/N124A/N137A-GFP (20 MOI per cell) adenovirus for 48 h. The cells were serum starved for 2 h in EBM-2 SFM and then treated with FVIIa (10 nM) for 90 min. Total RNA was isolated and subjected to real-time qPCR analysis in triplicates. Fold-increase in TR3 mRNA levels was measured relative to TR3 mRNA levels in cells expressing PAR2 (no TF) and treated with control vehicle. ns, no statistically significant difference.
In additional studies, we also examined whether TF glycosylation plays a role in cell adhesion by plating CHO cells expressing wild-type TF-GFP or TFN11A/N124A/N137A-GFP on different extracellular matrix proteins (collagen, laminin and fibronectin) or BSA-coated wells. The results showed no differences between wild-type TF-GFP and TFN11A/N124A/N137A-GFP in their attachment to extracellular matrix proteins, their morphology or spreading pattern (data not shown). We also attempted to investigate the role of carbohydrate attachment in TF in its interaction with β1-integrin. Co-immunoprecipitation studies revealed that although TF constitutively expressed in cancer cells (MDA-MB-231) could associate with the β1-integrin as reported earlier [25], this interaction is not detectable in HUVEC transduced with wild-type TF-GFP or TFN11A/N124A/N137A-GFP either in the presence or absence of FVIIa.
Discussion
Glycosylation is the most common and complex form of post-translational modification of proteins and usually has an effect on many properties of proteins, including their stability, transport and function [5,26]. Evidence for the presence of carbohydrates in TF was first provided by Pitlick [7], who demonstrated reversible inhibition of TF procoagulant activity by concanavalin A, a lectin that binds to carbohydrates. Using TF apoprotein isolated from different species and tissues, binding studies of these isolates to concanavalin A suggested that glycosylation of TF from various species is different, and human TF was heterogeneous with respect to its carbohydrate moiety [27]. The primary structure of the TF protein revealed that TF contains three potential glycosylation sites in the extracellular domain and one in the cytoplasmic domain [28,29]. At present, the influence of glycosylation on TF protein function is unclear. Previously, limited and indirect studies that examined the potential role of TF glycosylation in its procoagulant activity yielded contradicting results [6,9]. Recent comparative analysis of the carbohydrate composition of full-length recombinant TF (expressed in Sf9 insect cells) and natural placental TF did reveal glycosylation at Asn 11, 124 and 137 in both rTF and natural TF, but the composition of carbohydrates at each site varied between the rTF and natural human TF [13]. Analysis of TF activity before and after enzymatic removal of oligosaccharides revealed that glycosylation does alter TF activity [13]. Thus, it is somewhat surprising that in the present study, which was directly aimed at investigating the contribution of glycosylation of specific Asn by site-specific mutagenesis, we found that glycosylation plays no role in modulating either TF coagulant or signaling activity.
Although both rTF expressed in insect cells and placental TF are glycosylated at all three extracellular sites, carbohydrate composition between them varies. rTF was found to contain high levels of mannose, hybrid and fucosylated glycans, whereas placental TF contained highly fucosylated and sialylated sugars [13]. It is interesting to note that while glycosylated rTF had much lower coagulant activity than placental TF, deglycosylation of rTF altered TF procoagulant activity only minimally, whereas deglycosylation of placental TF caused a pronounced reduction in the kcat of FX activation [13]. These data suggest that the presence of specific carbohydrates and not just any carbohydrate per se may be important for TF function. Although we have no information regarding the composition of carbohydrates on rTF expressed on cell surfaces in the present study, it is likely that the carbohydrate composition of rTF expressed in our cell system would be comparable to placental human TF as we have employed eukaryotic expression systems, including human endothelial cells. However, one cannot rule out the possibility that glycosylation of TF in placental tissue may vary from that which occurs in endothelial cells. Such a possible variation may explain differences in findings on the importance of carbohydrates for TF coagulant function between the current study and an earlier study by Krudysz-Amblo et al. [13]. Another difference between the current study and the earlier one is that we have evaluated TF activity in its native phospholipid environment whereas Krudysz-Amblo et al. [13] performed their studies using relipidated TF after purification.
Tunicamycin, an inhibitor that specifically blocks the N-linked glycosylation reaction, is used widely to probe the importance of glycosylation in protein function. Tunicamycin treatment was shown to completely inhibit the production of TF procoagulant activity in macrophages in response to LPS [9]. Tunicamycin treatment was also found to inhibit cell surface TF activity significantly but not completely in HL-60 cells stimulated with PMA [10]. However, there was no information in those studies on whether reduced TF activity in tunicamycin-treated cells stems from reduced activity of non-glycosylated TF or diminished TF antigen present at the cell surface. In our studies we found that PBMC and HUVEC showed different sensitivities to tunicamycin in terms of inhibition of N-linked glycosylation, TF procoagulant activity and cell surface antigen expression. Both TF activity and protein synthesis were markedly decreased in HUVEC treated with tunicamycin. Our data show that diminished TF proco-agulant activity at the endothelial cell surface associated with tunicamycin treatment is primarily the result of decreased TF antigen present at the cell surface. In the case of PBMC, although a higher concentration of tunicamycin was needed to deglycosylate TF, this did not significantly inhibit either TF activity or protein synthesis. It is unlikely in endothelial cells, as suggested earlier [10], that decreased TF antigen at the cell surface represents impaired transport of non-glycosylated TF from intracellular compartments to the plasma membrane as we also found significant reduction in TF activity and antigen in total cell lysates from tunicamycin-treated HUVEC. The observation that TFN11A/N124A/N137A is expressed at the cell surface to the same extent as that of wild-type TF further confirms that glycosylation plays no role in the transport of TF in mammalian cells. Here it may be pertinent to note that TF expression levels were found to be sensitive to the mutations in glycosylation sites when soluble TF was expressed in yeast expression system [12].
An earlier TF mutagenesis study involving Cys186 and Cys209 showed a decrease in activity of the mutant protein [14]. Our recent study demonstrated a marked defect in the expression of these mutant proteins [15]. The mutation appeared to alter the glycosylation pattern of TF as the mutant proteins migrated at a higher (in the case of Cys186, conversion of Cys to Ser at this position creates a new carbohydrate signal) or lower molecular weight (in the case of Cys209 and the double mutant) on SDS-PAGE [14,15]. This has raised the possibility that defects in glycosylation of the mutant could be a contributing factor in the decreased procoagulant activity [13]. However, the observation in the present study that TF mutants with varying degrees of deglycosylation were expressed normally in the same cell model systems as employed in the earlier studies rules out such a possibility.
We are not aware of any reported studies in the literature addressing the importance of carbohydrates in the encryption/decryption of TF or TF-FVIIa activation of PAR2-mediated cell signaling. As far as we know, the present study is the first report to show that glycosylation is not required for TF decryption. The observation that TFN11A/N124A/N137A-FVIIa complexes can cleave PAR2 and activate PAR2-mediated cell signaling indicates that carbohydrate moieties of TF are not important for TF-FVIIa recognition and binding of PAR2.
A puzzling auxiliary observation of the present study is that all TF monoclonal and polyclonal antibodies tested failed to effectively recognize the non-glycosylated TF in immunoblot analyses, while they were fully capable of binding to the mutant TF under native conditions. The epitopes for most of the mAb fall into two groups that react with non-overlapping epitopes (locus 1, Thr106-Lys165; locus 2, Thr40-Val83) [30]. Both 5G9 and 10H10 recognize locus 1, whereas 6B4 and other mAb recognize locus 2. The crystal structure of the TF.5G9 complex revealed that 5G9 recognition involves 18 TF antigen residues [31] and none of them were in the N-linked glycosylation sites. In theory, one expects all mAb would at least bind to TFN11A as this mutation was completely outside of both loci 1 and 2. However, the antibodies showed maximum impairment in binding to this particular variant on western blot. It is possible that the removal of N-glycans changes the conformation of the epitope, creating perhaps a faster off-rate for the mAb, which would be more significant in the context of the longer time frame of western blot rather than the shorter time frame of cell binding studies. However, performing the western blot for a shorter duration did not alter the results. It is possible that TF lacking carbohydrates unfolds differently on SDS-PAGE compared with wild-type TF and therefore loses the majority of the epitopes recognized by the antibodies. Although at present it is difficult to explain this particular finding, it suggests that one should exercise caution in analyzing expression levels of TF mutant proteins solely based on immunoblot analyses.
Overall, our present data suggest that N-linked glycosylation of TF is not important for its transport or coagulant and signaling functions. It remains an open question whether differences in glycosylation among various cell types and subsequent, possible alterations in the carbohydrate(s) attached to TF affect TF transport and function differently in different cell types. This requires further and extensive systematic studies.
Supplementary Material
Data S1. Materials and methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Acknowledgments
The study was supported by grants HL58869 (LVMR) and HL65500 (UP). The authors are thankful to P. Sen for his assistance in performing p44/42 MAPK activation in HUVEC, W. Ruf, Scripps Research Institute, La Jolla, CA, USA, for providing TF mAb and C. Esmon, Oklahoma Medical Research Foundation, for EPCR blocking antibodies. We appreciate the help received from C. Clark in editing the manuscript.
Footnotes
Addendum
H. Kothari performed all the experiments described in the manuscript and analyzed the data. U.R. Pendurthi designed the generation of TF mutants used in the study. All authors contributed to the research design and preparation of the manuscript.
Disclosure of Conflicts of Interest
The authors state they have no conflicts of interest.
Additional Supporting Information may be found in the online version of this article:
References
- 1.Rao LVM, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol. 2005;25:47–56. doi: 10.1161/01.ATV.0000151624.45775.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belting M, Ahamed J, Ruf W. Signaling of the tissue factor coagulation pathway in angiogenesis and cancer. Arterioscler Thromb Vasc Biol. 2005;25:1545–50. doi: 10.1161/01.ATV.0000171155.05809.bf. [DOI] [PubMed] [Google Scholar]
- 3.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24:1015–22. doi: 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- 4.Egorina EM, Sovershaev MA, Osterud B. Regulation of tissue factor procoagulant activity by post-translational modifications. Thromb Res. 2008;122:831–7. doi: 10.1016/j.thromres.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–67. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Paborsky LR, Harris RJ. Post-translational modifications of recombinant human tissue factor. Thromb Res. 1990;60:367–76. doi: 10.1016/0049-3848(90)90219-3. [DOI] [PubMed] [Google Scholar]
- 7.Pitlick FA. Concanavalin A inhibits tissue factor coagulant activity. J Clin Invest. 1975;55:175–9. doi: 10.1172/JCI107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstein IJ, Hollerman CE, Smith EE. Protein-carbohydrate interaction. II. Inhibition studies on the interaction of concanavalin A with polysaccharides. Biochemistry. 1965;4:876–83. doi: 10.1021/bi00881a013. [DOI] [PubMed] [Google Scholar]
- 9.Shands JW., Jr Macrophage factor X activator formation: metabolic requirements for synthesis of components. Blood. 1985;65:169–75. [PubMed] [Google Scholar]
- 10.Bona R, Lee E, Rickles F. Tissue factor apoprotein: intracellular transport and expression in shed membrane vesicles. Thromb Res. 1987;48:487–500. doi: 10.1016/0049-3848(87)90405-1. [DOI] [PubMed] [Google Scholar]
- 11.Paborsky LR, Tate KM, Harris RJ, Yansura DG, Band L, McCray G, Gorman CM, O’Brien DP, Chang JY, Swartz JR, Fung VP, Thomas JN, Vehar GA. Purification of recombinant human tissue factor. Biochemistry. 1989;28:8072–7. doi: 10.1021/bi00446a016. [DOI] [PubMed] [Google Scholar]
- 12.Stone MJ, Ruf W, Miles DJ, Edgington TS, Wright PE. Recombinant soluble human tissue factor secreted by Saccharomyces cerevisiae and refolded from Escherichia coli inclusion bodies: glycosylation of mutants, activity and physical characterization. Biochem J. 1995;310:605–14. doi: 10.1042/bj3100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krudysz-Amblo J, Jennings ME, Mann KG, Butenas S. Carbohydrates and activity of natural and recombinant tissue factor. J Biol Chem. 2010;285:3371–82. doi: 10.1074/jbc.M109.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehemtulla A, Ruf W, Edgington TS. The integrity of the cysteine 186-cysteine 209 bond of the second disulfide loop of tissue factor is required for binding of factor VII. J Biol Chem. 1991;266:10294–9. [PubMed] [Google Scholar]
- 15.Kothari H, Nayak RC, Rao LV, Pendurthi UR. Cystine186-cystine 209 disulfide bond is not essential for the procoagulant activity of tissue factor or for its de-encryption. Blood. 2010;115:4273–83. doi: 10.1182/blood-2009-09-241356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pendurthi UR, Ghosh S, Mandal SK, Rao LV. Tissue factor activation: is disulfide bond switching a regulatory mechanism? Blood. 2007;110:3900–8. doi: 10.1182/blood-2007-07-101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almus FE, Rao LVM, Pendurthi UR, Quattrochi L, Rapaport SI. Mechanism for diminished tissue factor expression by endothelial cells cultured with heparin binding growth factor-1 and heparin. Blood. 1991;77:1256–62. [PubMed] [Google Scholar]
- 18.Mandal SK, Pendurthi UR, Rao LVM. Cellular localization and trafficking of tissue factor. Blood. 2006;107:4746–53. doi: 10.1182/blood-2005-11-4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849–57. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao LVM. Characterization of anti-tissue factor antibody and its use in immunoaffinity purification of human tissue factor. Thromb Res. 1988;51:373–84. doi: 10.1016/0049-3848(88)90373-8. [DOI] [PubMed] [Google Scholar]
- 21.Morrissey JH, Fair DS, Edgington TS. Monoclonal antibody analysis of purified and cell-associated tissue factor. Thromb Res. 1988;52:247–61. doi: 10.1016/0049-3848(88)90084-9. [DOI] [PubMed] [Google Scholar]
- 22.Rao LVM, Pendurthi UR. Tissue factor on cells. Blood Coagul Fibrinolysis. 1998;9:S27–35. [PubMed] [Google Scholar]
- 23.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–61. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- 24.Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood. 2011;117:3199–208. doi: 10.1182/blood-2010-09-310706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111:190–9. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart GW. Glycosylation. Curr Opin Cell Biol. 1992;4:1017–23. doi: 10.1016/0955-0674(92)90134-x. [DOI] [PubMed] [Google Scholar]
- 27.van den Besselaar AMHP, Bertina RM. Interaction of thromboplastin apoprotein of different tissues with concanavalin A- evidence for heterogeneous glycosylation of the human apoprotein. Thromb Haemost. 1984;52:192–5. [PubMed] [Google Scholar]
- 28.Spicer EK, Horton R, Bloem L, Back R, Williams KR, Guha A, Kraus J, Lin TC, Nemerson Y, Konigsberg WH. Isolation of cDNA clones for human tissue factor: primary structure of the protein and cDNA. Proc Natl Acad Sci U S A. 1987;84:5148–52. doi: 10.1073/pnas.84.15.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scarpati EM, Wen D, Broze GJ, Jr, Miletich JP, Flandermeyer RR, Siegel NR, Sadler JE. Human tissue factor: cDNA sequence and chromosome localization of the gene. Biochemistry. 1987;26:5234–8. doi: 10.1021/bi00391a004. [DOI] [PubMed] [Google Scholar]
- 30.Ruf W, Rehemtulla A, Edgington TS. Antibody mapping of tissue factor implicates two different exon-encoded regions in function. Biochem J. 1991;278(Pt 3):729–33. doi: 10.1042/bj2780729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang M, Syed R, Stura EA, Stone MJ, Stefanko RS, Ruf W, Edgington TS, Wilson IA. The mechanism of an inhibitory antibody on TF-initiated blood coagulation revealed by the crystal structures of human tissue factor, Fab 5G9 and TF.G9 complex. J Mol Biol. 1998;275:873–94. doi: 10.1006/jmbi.1997.1512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.