

Abstract
Animal behaviors are reinforced by subsequent rewards following within a narrow time window. Such reward signals are primarily coded by dopamine, which modulates the synaptic connections of medium spiny neurons in the striatum. The mechanisms of the narrow timing detection, however, remain unknown. Here, we optically stimulated dopaminergic and glutamatergic inputs separately and found that dopamine promoted spine enlargement only during a narrow time window (0.3 to 2 seconds) after the glutamatergic inputs. The temporal contingency was detected by rapid regulation of adenosine 3′,5′-cyclic monophosphate in thin distal dendrites, in which protein-kinase A was activated only within the time window because of a high phosphodiesterase activity. Thus, we describe a molecular basis of reinforcement plasticity at the level of single dendritic spines.
Animal behaviors are reinforced only when rewarded shortly after a motor or sensory event (1, 2). The neocortex, hippocampus, and amygdala process the sensorimotor signals and send glutamatergic synaptic output to the striatum (3), where connections can be modified by Hebbian learning mechanisms, such as spike-timing-dependent plasticity (STDP) (4). Animals learn to associate the sensorimotor signals with subsequent rewards through reinforcement of the neuronal circuits involving dopamine (5-7). Despite its importance, this narrow timing detection has never been demonstrated at the cellular level and might be ascribed to neural network properties (6, 8).
Dendritic spine morphology is correlated with spine function (9), and dendritic spines enlarge during long-term potentiation in the cortices (10-12). We examined the effects of dopamine on the structural plasticity in striatal medium spiny neurons (MSNs). Results show that dopamine affected spine structural plasticity in a narrow time window consistent with behavioral conditioning (5). Functional imaging revealed the molecular interrelationships between the reinforcement and Hebbian plasticity.
We investigated dopamine actions on glutamatergic synapses on MSNs using optogenetics and two-photon uncaging. For optogenetic stimulation of dopaminergic fibers, a Cre-dependent adeno-associated virus (AAV) vector expressing channelrhodopsin-2 (ChR2) was injected into the ventral tegmental area (VTA) of DAT-Cre mice expressing Cre specific to dopaminergic neurons (Fig. 1A and fig. S1). The direct pathway–constituting MSNs, which mainly express dopamine 1 receptors (D1Rs) (13), were labeled by an AAV vector with a specific promoter for D1R-MSNs (Fig. 1A and fig. S1). In acute coronal slices, including the nucleus accumbens (NAc) core, whole-cell recordings were obtained from the identified D1R-MSNs. Dendritic spines were visualized by means of two-photon microscopy (980 nm) detecting fluorescence of Alexa488 loaded through a recording pipette (Fig. 1B and fig. S1C). Stimulation of a single spine by means of two-photon uncaging (720 nm) of CDNI-Glu (Fig. 1B) induced two-photon excitatory postsynaptic potentials (2pEPSPs) with amplitudes similar to miniature EPSPs (mEPSPs) (Fig. 1C and fig. S1D). The amplitudes of 2pEPSCs positively correlated with spine volumes, as they did with pyramidal neurons (fig. S1E) (9). The STDP protocol of repetitive uncaging of glutamate paired with action potentials (APs) (Glu + AP) (Fig. 1D) (14) induced robust spine enlargement that was selective for the stimulated spine (Fig. 1, E and F) when dopamine (100 μM) was puff applied, whereas only a weak enlargement occurred in the absence of dopamine (Fig. 1G), which is consistent with previous findings (14, 15).
Fig. 1. A temporal profile of dopamine actions on spine enlargement.

(A) Injection of AAV vectors for ChR2 and the D1R-MSN marker (PPTA-mCherry) in 3-week-old DAT-Cre mice. (B) Selective stimulation of dopaminergic and glutamatergic inputs by means of blue laser field irradiation to ChR2 and two-photon uncaging of caged-glutamate at a single spine, respectively, in acute slices of NAc obtained from 5- to 7- week-old mice. (C) An amperometric measurement of dopamine (top) by carbon-fiber electrode and whole-cell recording of glutamate-induced current (bottom, 2pEPSP) in identified D1R-MSNs. (D) An STDP protocol with dopamine puff application. (E) Images of the dendritic spine (red arrowhead) that received STDP stimulation in the presence of dopamine (100 μM). (F and G) Time courses of spine enlargement in the presence [(F), 13 spines, 4 dendrites] and absence of dopamine [(G), 58 spines, 14 dendrites]. (H) Amplitudes of spine enlargements with or without dopamine. **P = 0.0041 by Mann-Whitney U test. (I) STDP with repetitive activation of dopaminergic fibers containing ChR2 (blue lines) at 30 Hz, 10 times (DAopto). (J) Images of the dendritic spine (arrowhead) that received STDP + DAopto with a delay of 1 s. (K to M) Time courses of spine enlargement induced by STDP + DAopto at 1 s [(K), 48 spines, 14 dendrites], −1 s [(L), 20 spines, 5 dendrites] and 5 s [(M), 28 spines, 7 dendrites] after STDP onset. (N) Timings of DAopto application. (O) Increases in spine volumes by STDP + DAopto plotted versus DAopto delay (fig. S2, A to C). Data are presented as mean ± SEM. P = 4.2 × 10−6 with Kruskal-Wallis and **P = 0.0018 (0.6 s) and 0.0027 (1 s) by Steel test in comparison with STDP in the absence of DAopto. Scale bars, 1 μm.
A single pulse of blue laser stimulation of ChR2-expressing dopaminergic fibers induced a transient increase in the tissue dopamine concentration (Fig. 1C). We tested the actions of a physiologically relevant phasic release of dopamine (30 Hz, 10 times) induced by the optogenetic stimulation of dopaminergic fibers (DAopto) and found that DAopto just after STDP stimulation (DAopto delay = 1 s) (Fig. 1I) induced robust spine enlargement (Fig. 1, J and K). Stimulation of dopaminergic fibers 1 s before (DAopto delay = −1 s) or 5 s after (DAopto delay = 5 s) STDP (Fig. 1, L and M) resulted in only a slight enhancement. The actions of DAopto were examined with various timings (Fig. 1N), revealing that DAopto timing was critical to enhance plasticity, with maximal effects for a delay of 0.6 s (Fig. 1O and fig. S2, A to C), and decayed in a few seconds, which is consistent with behavioral study results (5, 16). The spine enlargement was accompanied by an increase in the 2pEPSC (fig. S3). A similar DAopto timing was observed when we induced STDP through electrical stimulation of presynaptic fibers (fig. S4).
Pharmacological studies revealed that D1R-MSN structural plasticity was dependent on N-methyl-d-aspartate receptors (NMDARs), Ca2+/calmodulin–dependent protein kinase II (CaMKII), and protein synthesis (Fig. 2A and fig. S5A), suggesting that the molecular mechanisms for D1R-MSN plasticity are similar to those underlying structural plasticity in hippocampal pyramidal neurons (10-12). Plasticity also depended on D1R and protein kinase A (PKA), but not on D2R (Fig. 2B and fig. S5A). Spine enlargement was prevented by an inhibitory peptide blocking the interaction of dopamine- and adenosine 3′,5′-cyclic monophosphate (cAMP)–regulated phosphoprotein 32 kD (DARPP-32) with protein phosphatase 1 (PP-1) (17), but not its control peptide (Fig. 2, C and D). Moreover, spine enlargement was induced even in the absence of DAopto when PP-1 was inhibited by calyculin A (fig. S5, B and C). These results suggest that similar to hippocampal preparations (18), the phosphorylation of DARPP-32 by PKA would inhibit PP-1 and disinhibit CaMKII.
Fig. 2. Pharmacology of spine enlargement induced by STDP plus DAopto with a 0.6-s delay.

(A) Time courses of spine enlargement induced by STDP + DAopto with a 0.6-s delay in the absence (control, 24 spines, 7 dendrites) and presence of NMDAR antagonist (50 μM D-AP5, 22 spines, 6 dendrites), CaMKII inhibitor (3 μM KN62, 23 spines, 6 dendrites), or protein synthesis inhibitor (5 μM anisomycin, 25 spines, 6 dendrites). (B) Time courses of spine enlargement in the presence of D1R antagonist (3 μM SCH23390, 23 spines, 6 dendrites), D2R antagonist (10 μM sulpiride, 22 spines, 6 dendrites), or PKA inhibitor (10 μM PKI, in the pipette, 24 spines, 6 dendrites). (C) Time courses of spine enlargement in the presence of inhibitory (100 μM, in the pipette, 24 spines, 6 dendrites) or control peptide for DARPP-32 (100 μM, in the pipette, 24 spines, 6 dendrites). (D) Averaged volume changes in the absence and presence of the compounds. Data are presented as mean ± SEM. P = 3.4 × 10−6 with Kruskal-Wallis and *P = 0.023 (AP5), 0.023 (KN62), 0.037 (AIP) (fig. S5A), 0.023 (anisomycin), 0.035 (SCH23390), 0.023 (PKI), 0.037 (KT5720) (fig. S5A), and 0.023 (DARPP-32 inhibitory peptide) with Steel test.
To test whether changes in Ca2+ signaling account for DAopto timing (19), we imaged increases in cytosolic Ca2+ concentrations ([Ca2+]i) in spines using a low-affinity calcium indicator Fluo-4FF (KCa = 10 μM) to avoid saturation. The [Ca2+]i increases gradually built up and quickly waned after cessation of the STDP protocol (Fig. 3A). We found that DAopto did not affect Ca2+ transients (Fig. 3, B to D, and fig. S6, A to C), indicating that Ca2+ signaling modulation did not play a major role.
Fig. 3. DAopto effects on STDP stimulation-induced increases in [Ca2+]i and CaMKII activities.

(A and B) Increases in Fluo4-FF fluorescence, representing [Ca2+]i increases, within single spines in response to a train of STDP stimulation in the absence [(A), 20 spines, 15 dendrites] and presence of DAopto with a 0.6-s delay [(B), 15 spines, 8 dendrites]. Blue laser irradiation during DAopto is blanked by the blue bar. (C) STDP and DAopto protocols for Ca2+ and CaMKII imaging. Unlike plasticity induction (Fig. 1N), only one train was applied. (D) No effect of DAopto on the peak values of [Ca2+]i (fig. S6, A to C). P = 0.91 with Kruskal-Wallis test. (E and F) Ratiometric imaging with Camuiα-CR during STDP stimulation in the absence [(E), 33 spines, 14 dendrites] or presence of DAopto with a 0.6-s delay [(F), 42 spines, 14 dendrites]. Relative increases in the ratio are shown as pseudocolor coding in (E). (Bottom) Time courses of FRET ratios in the spines stimulated with glutamate uncaging or the neighboring spines. Scale bars, 1 μm. (G) Dependence of the peak Camuiα-CR FRET ratios on the DAopto delay (fig. S6, D to F). P = 1.3 × 10−5 with Kruskal-Wallis test and ***P = 8.4 × 10−5 with Steel test versus those without DAopto. (H) Normalized increases in Camuiα-CR ratios by STDP + DAopto with a 0.6-s delay in the stimulated (42 spines, 14 dendrites) and neighboring spines (42 spines, 14 dendrites), and in the presence of DARPP-32 inhibitory peptide in the stimulated spines (43 spines, 10 dendrites) (fig. S6G). Data are presented as mean ± SEM. P = 8.8 × 10−9 with Kruskal-Wallis test and *** P = 2.6 × 10−9 and 1.1 × 10−6 with Steel test.
We examined whether CaMKII activation was related to DAopto timing by using the Förster resonance energy transfer (FRET) indicator of CaMKII, Camuiα-CR (20-22), which was virally transfected into D1R-MSNs. CaMKII activation was weak in the absence of DAopto (Fig. 3E) but was greatly potentiated by DAopto (Fig. 3F), with timing similar to DAopto timing for spine enlargement (Fig. 3G and fig. S6, D to F). This enhancement was specific to the stimulated spine (Fig. 3H), abolished by the inhibitory peptide for DARPP-32 (Fig. 3H and fig. S6G), and mimicked by PP-1 inhibitors, calyculin A and tautomycetin (fig. S6, H and I). These results support the hypothesis that PKA/DARPP-32 disinhibits CaMKII via PP-1 (fig. S11).
We addressed whether the DAopto timing was formed at the level of PKA activation by using a FRET probe of PKA, AKAR2-CR (22), which was virally delivered to the D1R-MSNs. Unlike structural plasticity or Camuiα-CR activation, PKA activation in response to stimulation of a dendritic spine by STDP and DAopto was not restricted to the stimulated spine; neighboring spines also exhibited significant PKA activation (fig. S7, A to D). Even without glutamate uncaging, PKA activation was observed in the spine and dendritic shaft (Fig. 4A) in an AP-dependent manner (Fig. 4B), suggesting that PKA activation is a cell-wide phenomenon. When DAopto was applied at various times relative to APs, we obtained a timing (Fig. 4C and fig. S7, E to I) similar to DAopto timing on spine enlargement and CaMKII activation (Fig. 1O). The extent of spine enlargement positively correlated with that of PKA activation (Fig. 4D). APs themselves were not sufficient to activate PKA (fig. S8, C to E). The contingency between APs and DAopto might be detected by Ca2+/calmodulin–dependent adenylyl cyclase 1 (AC1), which is synergistically activated by Ca2+/calmodulin and Gs (23). Consistent with this, AC1 blocker (NB001) eliminated AKAR2 activation, as well as structural plasticity (fig. S9, A to D).
Fig. 4. AP effects on DAopto-induced PKA activation in proximal and distal dendrites.
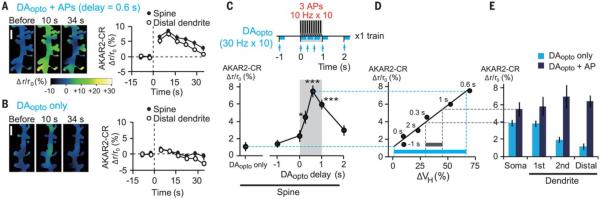
(A and B) Images and time courses of FRET ratios of AKAR2-CR in the spine and distal dendrites stimulated by APs + DAopto with a 0.6-s delay [(A), 153 spines, 25 dendrites] or DAopto only [(B), 158 spines, 26 dendrites]. The relative increases in the ratio were pseudocolor coded as shown in (A). Scale bar, 2 μm. (C) AKAR2-CR responses to APs + DAopto with various delays (fig. S7, E to I). P = 5.3 × 10−10 with Kruskal-Wallis test and *P = 0.012, ***P = 1.3 × 10−6 (0.6 s), and 4.0 × 10−4 (1 s) with Steel test versus DAopto only. (D) AKAR2-CR response (C) plotted against spine volume changes (Fig. 1O) for various DAopto timings. The Spearman’s correlation coefficient was 0.94, and P = 0.0048. The blue and gray bars indicate the dynamic ranges of volume changes predicted by the dynamic ranges of AKAR2 responses at dendritic spine (blue) and soma (gray). (E) AKAR2-CR responses at the soma and first, second, and distal dendrites in response to DAopto only and DAopto + APs with a 0.6-s delay (fig. S8, A and B).
In the soma and proximal (first branch) dendrites, however, DAopto alone was sufficient to activate PKA (Fig. 4E and fig S8, A and B) (24), and APs could only slightly enhance PKA. We predict that APs might have modulated spine enlargement to a small degree in these regions, if there had been spines (Fig. 4D). Thus, DAopto-induced PKA activation must be suppressed in distal dendrites in order to attain the large dynamic range for the timing detection. In fact, when phosphodiesterase 10A (PDE10A), the major phosphodiesterase in MSNs, was blocked by its inhibitor papaverine (25), PKA activations were similarly induced in distal dendrites as in the soma (fig. S9, E to G). Papaverine also disrupted the time window for structural plasticity (fig. S9, H and I). Why were PDE10A actions particularly potent in the distal dendrites? Subcellular differences in PDE10A expression might not account for this, considering that PDE10A is expressed at the plasma membrane and is uniformly distributed along the dendrites (26). Instead, we found a negative correlation between dendrite diameter and DAopto-induced PKA activity (fig. S10A, blue), which was lost when the phosphodiesterase was inhibited (fig. S10A, orange). Therefore, PDE10A might counteract the increases in cAMP more potently in the thin distal dendrites because of its high surface-to-volume ratios (fig. S10, B and C). Spines are only found in the distal thin dendrites of MSNs (fig. S10, D to E) (27), suggesting that spines are distributed to be efficiently modulated by dopamine timing in MSNs.
It has been enigmatic why dopamine reinforces preceding, but not subsequent, sensorimotor events. If dopamine always activates PKA, its effects should last long enough to reinforce the subsequent events over tens of seconds (Fig. 4A). However, dopamine did not activate PKA unless [Ca2+]i primed AC1 to outcompete the high phosphodiesterase activity in thin dendrites (fig. S11). Our data show that [Ca2+]i priming should occur strictly before dopamine delivery (0 s) (Figs. 1O, 3G, and 4C), which is reminiscent of serotonin’s action in the classical conditioning of the siphon withdrawal reflex in Aplysia (28), in which serotonin, carrying an aversive signal, was only effective when it was preceded by increases in [Ca2+]i for activation of calcium-dependent adenylyl cyclase (AC) and PKA in presynaptic terminals (29). The delay in [Ca2+]i priming of AC may compensate the time lag of monoaminergic signals after reward or punishment. Our data suggest that reinforcement plasticity occurs at the single spine level, even though PKA activation is a cell-wide phenomenon, in such a way that dopamine regulates the gain of NMDAR-dependent Hebbian plasticity via CaMKII activity (fig. S11). This interdependence between Hebbian and reinforcement plasticity has been implicitly assumed in the reinforcement learning theory, in which the Hebbian term is used for the credit assignment (6, 30), and the dopamine timing in our study corresponds to the eligibility trace that determines the time window for reward action (30). Thus, we have clarified a molecular and cellular basis of reinforcement plasticity at the level of single dendritic spines.
Supplementary Material
Acknowledgments
We thank K. Kaibuchi, M. Mishina, T. Tanabe, K. Doya, J. Wickens, T. Toyoizumi, and K. Kasai for helpful discussion; K. Deisseroth for the channelrhodopsin-2 plasmids; M. Z. Lin for the Camuiα-CR and AKAR2-CR plasmids; K. Iwamoto and S. Jinde for confocal microscopy; M. Ikeda for technical assistance; and M. Ogasawara and M. Tamura for laboratory facility. This work was supported by the Strategic Research Program for Brain Sciences (“Bioinformatics for Brain Sciences” to H.K. and S.I.) and Grants-in-Aid (2000009 and 26221011 to H.K.) from the Ministry of Education, Culture, Sports, Science, and Technology and by the National Institutes of Health GM53395, DA035612, and NS069720 (G.C.R.E.-D.). This work was also supported by the Human Frontier Science Program (H.K.). G.C.R.E.-D. has a patent in the United States for the synthesis of dinitroindolinyl-caged neurotransmitters.
Footnotes
Materials and methods-see supplementary materials.
References
- 1.Thorndike EL. Animal Intelligence. Macmillan; New York: 1911. [Google Scholar]
- 2.Pavlov IP. Conditioned Reflexes; An Investigation of the Physiological Activity of the Cerebral Cortex. Oxford Univ. Press; Humphrey Milford, London: 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roitman MF, Wheeler RA, Carelli RM. Nucleus accumbens neurons are innately tuned for rewarding and aversive taste stimuli, encode their predictors, and are linked to motor output. Neuron. 2005;45:587–597. doi: 10.1016/j.neuron.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 4.Dan Y, Poo MM. Spike timing-dependent plasticity: From synapse to perception. Physiol. Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- 5.Black J, Belluzzi JD, Stein L. Reinforcement delay of one second severely impairs acquisition of brain self-stimulation. Brain Res. 1985;359:113–119. doi: 10.1016/0006-8993(85)91418-0. [DOI] [PubMed] [Google Scholar]
- 6.Schultz W. Predictive reward signal of dopamine neurons. J. Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Smith-Roe SL, Kelley AE. Coincident activation of NMDA and dopamine D1 receptors within the nucleus accumbens core is required for appetitive instrumental learning. J. Neurosci. 2000;20:7737–7742. doi: 10.1523/JNEUROSCI.20-20-07737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakano T, Yoshimoto J, Doya K. A model-based prediction of the calcium responses in the striatal synaptic spines depending on the timing of cortical and dopaminergic inputs and post-synaptic spikes. Front. Comput. Neurosci. 2013;7:119. doi: 10.3389/fncom.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuzaki M, Ellis-Davies GCR, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr., Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 14.Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds JNJ, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature. 2001;413:67–70. doi: 10.1038/35092560. [DOI] [PubMed] [Google Scholar]
- 16.Rescorla RA. Behavioral studies of Pavlovian conditioning. Annu. Rev. Neurosci. 1988;11:329–352. doi: 10.1146/annurev.ne.11.030188.001553. [DOI] [PubMed] [Google Scholar]
- 17.Kwon YG, Huang HB, Desdouits F, Girault JA, Greengard P, Nairn AC. Characterization of the interaction between DARPP-32 and protein phosphatase 1 (PP-1): DARPP-32 peptides antagonize the interaction of PP-1 with binding proteins. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3536–3541. doi: 10.1073/pnas.94.8.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1943. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 19.Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci. STKE. 2006;2006:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- 20.Takao K, Okamoto K, Nakagawa T, Neve RL, Nagai T, Miyawaki A, Hashikawa T, Kobayashi S, Hayashi Y. Visualization of synaptic Ca2+/calmodulin-dependent protein kinase II activity in living neurons. J. Neurosci. 2005;25:3107–3112. doi: 10.1523/JNEUROSCI.0085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SJR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, McKeown MR, Wiedenmann J, Davidson MW, Schnitzer MJ, Tsien RY, Lin MZ. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods. 2012;9:1005–1012. doi: 10.1038/nmeth.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson GD, Storm DR. Why calcium-stimulated adenylyl cyclases? Physiology (Bethesda) 2004;19:271–276. doi: 10.1152/physiol.00010.2004. [DOI] [PubMed] [Google Scholar]
- 24.Castro LRV, Brito M, Guiot E, Polito M, Korn CW, Hervé D, Girault JA, Paupardin-Tritsch D, Vincent P. Striatal neurones have a specific ability to respond to phasic dopamine release. J. Physiol. 2013;591:3197–3214. doi: 10.1113/jphysiol.2013.252197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishi A, Kuroiwa M, Miller DB, O’Callaghan JP, Bateup HS, Shuto T, Sotogaku N, Fukuda T, Heintz N, Greengard P, Snyder GL. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J. Neurosci. 2008;28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charych EI, Jiang LX, Lo F, Sullivan K, Brandon NJ. Interplay of palmitoylation and phosphorylation in the trafficking and localization of phosphodiesterase 10A: Implications for the treatment of schizophrenia. J. Neurosci. 2010;30:9027–9037. doi: 10.1523/JNEUROSCI.1635-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson CJ, Groves PM. Fine structure and synaptic connections of the common spiny neuron of the rat neostriatum: A study employing intracellular inject of horseradish peroxidase. J. Comp. Neurol. 1980;194:599–615. doi: 10.1002/cne.901940308. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins RD, Abrams TW, Carew TJ, Kandel ER. A cellular mechanism of classical conditioning in Aplysia: Activity-dependent amplification of presynaptic facilitation. Science. 1983;219:400–405. doi: 10.1126/science.6294833. [DOI] [PubMed] [Google Scholar]
- 29.Yovell Y, Abrams TW. Temporal asymmetry in activation of Aplysia adenylyl cyclase by calcium and transmitter may explain temporal requirements of conditioning. Proc. Natl. Acad. Sci. U.S.A. 1992;89:6526–6530. doi: 10.1073/pnas.89.14.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutton RS, Barto AG. Reinforcemnet Learning. MIT Press; Cambridge, MA: 1998. [Google Scholar]
- 31.Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron. 2010;66:896–907. doi: 10.1016/j.neuron.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Xu H, Wu LJ, Kim SS, Chen T, Koga K, Descalzi G, Gong B, Vadakkan KI, Zhang X, Kaang BK, Zhuo M. Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci. Transl. Med. 2011;3:65ra3. doi: 10.1126/scitranslmed.3001269. [DOI] [PubMed] [Google Scholar]
- 34.Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science. 2013;341:1394–1399. doi: 10.1126/science.1239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis-Davies GCR, Matsuzaki M, Paukert M, Kasai H, Bergles DE. 4-Carboxymethoxy-5,7-dinitroindolinyl-Glu: An improved caged glutamate for expeditious ultraviolet and two-photon photolysis in brain slices. J. Neurosci. 2007;27:6601–6604. doi: 10.1523/JNEUROSCI.1519-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda Y, Marzo A, Otani S. The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J. Neurosci. 2006;26:4803–4810. doi: 10.1523/JNEUROSCI.5312-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, Koos T. Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J. Neurosci. 2010;30:7105–7110. doi: 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–266. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu NL, Harnett MT, Williams SR, Huber D, O’Connor DH, Svoboda K, Magee JC. Nonlinear dendritic integration of sensory and motor input during an active sensing task. Nature. 2012;492:247–251. doi: 10.1038/nature11601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.