

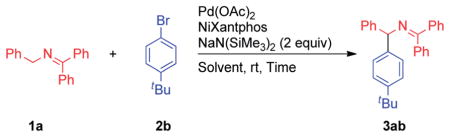
Abstract
Diarylmethylamines are of great interest due to their prevalence in pharmaceutical chemistry. As a result, new methods for their synthesis are in demand. Herein, we report a versatile protocol for the synthesis of diarylmethylamine derivatives involving palladium-catalyzed arylation of in situ generated 2-azaallyl anion intermediates. The 2-azaallyl anions are generated by reversible deprotonation of readily available aldimine and ketimine precursors. Importantly, the arylated aldimine and ketimine products do not undergo isomerization under the reaction conditions. Scale-up of the arylation and hydrolysis of the resulting products to furnish diarylmethylamines were also successfully performed.
Introduction
Molecules containing the diarylmethylamine scaffold are prevalent in biological and pharmaceutical sciences, with examples including Zyrtec,1 Levocetirizine,2 Meclozine,3 Solifenacin,4 BDF9148,5 SNC80,6 and ARM4347 (Fig. 1). Given the widespread utility of diarylmethylamines in medicinal chemistry, new syntheses of these pharmacophores have immediate potential applications.
Fig. 1.

Important diarylmethylamine containing molecules.
An appealing tactic for the synthesis of diarylmethylamines is functionalization of sp3-hybridized C–H bonds adjacent to nitrogen in benzylic amines.8 Retrosynthetically, such an approach represents an umpolung disconnection (Scheme 1).
Scheme 1.
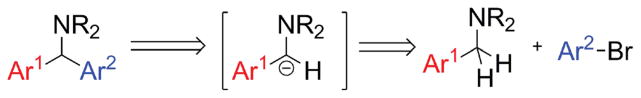
Umpolung retrosynthesis of diarylmethylamines.
Direct deprotonation of the benzylic C–H’s in benzyl amine derivatives under catalytic conditions is anticipated to be quite difficult. As a result, strategies to increase the acidity of the benzylic C–H’s have been explored. We recently employed arene activation by coordination of Cr(CO)3 in (η2-C6H5CH2NR2) Cr(CO)3 complexes, which were successfully coupled with aryl bromides, and enantioselectively with aryl triflates, under basic conditions.9 A different approach by Oshima and co-workers10 involved use of N-benzyl benzophenone ketimines and derivatives that were deprotonated with CsOH at 140 °C. In the presence of catalytic palladium and PCy3, cross-coupling of the intermediate 2-azaallyl anion with aryl chlorides was achieved in moderate yields (Scheme 2). Unfortunately, the harsh reaction conditions resulted in isomeric mixtures in every case, underminding the utility of this chemistry (Scheme 2).10 Oshima and coworkers later introduced a method to convert the isomeric mixtures to a single products, but this required additional steps.10a
Scheme 2.
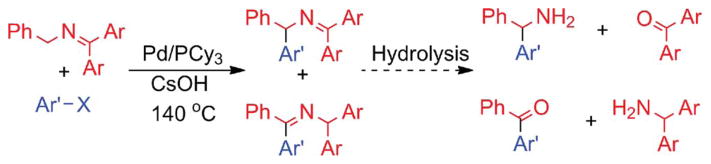
Arylation by Oshima and co-workers (X = Cl, Br).
Given the great impact of diarylmethylamines in medicinal chemistry, and the unrealized potential of this transformation, development of a viable arylation of benzylic ketimines is clearly of value. We hypothesized that two intertwined problems would need to be surmounted before the reaction in Scheme 2 could be rendered synthetically useful. First, a catalyst for the coupling that operates under mild conditions must be identified. Second, bases and conditions must be introduced that avoid the undesirable deprotonation and isomerization of the product that was observed by Oshima (Scheme 2).
Herein we report the first synthetically useful arylation of benzophenone imines. In the presence of a NIXANTPHOS-based catalyst we have reduced the reaction temperature by as much as 115 °C. Most importantly, under our conditions, no isomerization of the product is observed. We also demonstrate the first examples with aldimine substrates, ultimately providing a flexible route to diarylmethylamines via regioselective arylation of 1,1,3-triaryl-2-azaallyl anions.11
Results and discussion
To avoid simultaneous optimization of base, solvent, palladium source and ligand, we first focus only on the deprotonation step. We have found that trapping the resulting anion by benzylation allows us to determine which bases are capable of deprotonation of the substrates and serves as a starting point for optimization of the deprotonation step.12 The reaction between benzophenone imine 1a and benzyl chloride (Scheme 3) was examined in the presence of 12 bases [LiN(SiMe3)2, NaN(-SiMe3)2, KN(SiMe3)2, LiO–tBu, NaO–tBu, KO–tBu, NaH, LiOAc, KOAc, K3PO4, Cs2CO3, and KOPh, see ESI for details†]. Reactions were conducted in THF and CPME (cyclopentyl methyl ether) on microscale (10 μmol) at rt for 12 h. NaN(SiMe3)2, KN(SiMe3)2 and KO-tBu generated the benzylated product. On laboratory scale, the most promising result was obtained with NaN(SiMe3)2 in THF (95% assay yield).
Scheme 3.
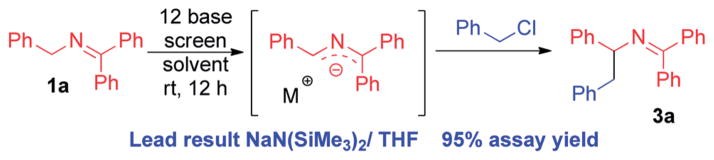
Use of benzylation to identify the base and solvent.
With the base/solvent combination outlined above, we conducted a room temperature screen of 24 electronically diverse, mono- and bidentate phosphine ligands with Pd(OAc)2 (see ESI for details†). The top 6 ligands, and their product : internal standard ratios, were: NIXANTPHOS13 (3.65), CataCXium A (2.96), PCy3 (1.22), dippf (1.10), SPhos (0.88) and XANTPHOS (0.77) (see Fig. 2 for select structures). It is particularly interesting that NIXANTPHOS outperformed PCy3 by a factor of three at rt and was also much more efficient than the structurally similar XANTPHOS.
Fig. 2.
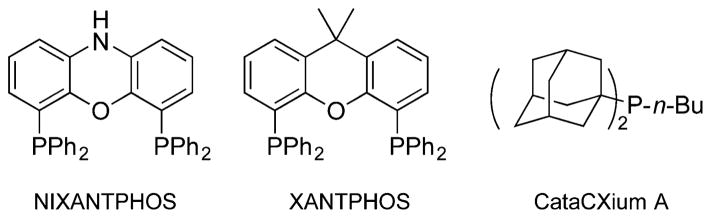
Structures of relevant phosphines used in screening reactions.
Based on the initial screening hits, we continued our optimization on laboratory scale with CPME and THF solvents, silylamide and alkoxide bases, NIXANTPHOS and Pd(OAc)2 (Table 1, entries 1–6). Consistent with HTE screening results, NaN(SiMe3)2 in THF and CPME were the most promising, with up to 80% assay yield. During the optimization, two byproducts were isolated and assigned as the isomerized aldimine 1a′ and the diastereomeric dimerization products 4a (Scheme 4). The assignment of aldimine 1a′ was confirmed by independent synthesis.14 Subjecting the benzophenone imine 1a and the isomeric aldimine 1a′ each to 1.5 equiv. NaN(SiMe3)2 in THF for 12 h at rt led to mixtures of 1a, 1a′ and 4a. In contrast, subjecting the arylation product 3ab to the deprotonation conditions did not lead to isomerization. Key to the outcome of the arylation reaction are the relative rates of the 2-azaallyl anion reacting with the starting imines to give the dimer vs. transmetalation to the palladium catalyst, as shown in Scheme 4.
Table 1.
| |||||
|---|---|---|---|---|---|
| Entry | Pd/L (%) | Solvent | Base | Time (h) | Yield (%) |
| 1 | 10/15 | CPME | NaN(SiMe3)2 | 1 h | 80 |
| 2 | 10/15 | CPME | KN(SiMe3)2 | 1 h | 41 |
| 3 | 10/15 | CPME | KO–tBu | 1 h | 77 |
| 4 | 10/15 | THF | NaN(SiMe3)2 | 1 h | 71 |
| 5 | 10/15 | THF | KN(SiMe3)2 | 1 h | 45 |
| 6 | 10/15 | THF | KO–tBu | 1 h | 64 |
| 7 | 10/15 | CPME | NaN(SiMe3)2 | 3 h | 99c |
| 8 | 10/15 | THF | NaN(SiMe3)2 | 3 h | 94c |
| 9 | 5/7.5 | CPME | NaN(SiMe3)2 | 3 h | 95c |
| 10 | 2.5/3.75 | CPME | NaN(SiMe3)2 | 3 h | 92c, (90)d |
| 11 | 1/1.5 | CPME | NaN(SiMe3)2 | 3 h | 78c |
Reactions conducted on a 0.1 mmol scale using 2 equiv. of 1a, 2 equiv. of NaN(SiMe3)2, and 1 equiv. of 2b at 0.1 M.
Yield determined by 1H NMR spectroscopy of the crude reaction mixture on a 0.1 mmol scale.
Base was added portionwise at 0.1 mL per 30 min.
Isolated yield after chromatographic purification.
Scheme 4.

Reaction pathway for arylation of the 1,1,3-triaryl-2-azaallyl anion.15
Attempts to eliminate the formation of the less reactive isomerized aldimine 1a′ or the dimerization product were unsuccessful. We hypothesized the impact of the byproducts on the arylation yield could be minimized by employing 2 equivalents of the benzophenone ketimine 1a in the coupling reaction. Furthermore, conversion to the desired arylation product, vs. the byproducts, could be improved by reducing the concentration of the azaallyl anion. We envisioned that this could be accomplished by slow addition of base to the reaction mixture. We found that portionwise addition of the base led to 99% assay yield of the arylation product in CPME (Table 1, entry 7) and 94% yield in THF (entry 8).
Further optimization of the catalyst loading indicated that the yield remained above 90% at 5 and 2.5 mol% Pd (entries 9 and 10), but dropped to 78% at 1 mol% (entry 11).
With the optimized reaction conditions in hand (Table 1, entry 10), the scope of aryl bromides in the arylation of benzophenone ketimine 1a was examined (Table 2). Bromobenzene and aryl bromides bearing electron donating alkyl substituents at para- and meta-positions furnished coupling products 3aa, 3ab, 3ac and 3ad in 83–90% yield (Table 2, entries 1–4). Aryl bromides possessing electron-donating 4-N,N-dimethylamino (2e) and 4-methoxy (2f) groups furnished coupling products (3ae, 3af) in 78 and 70% yield, respectively (5 mol% catalyst loading). Raising the palladium loading for these more challenging substrates to 10 mol% resulted in an increase in the yield to 93 and 90%, respectively (entries 5 and 6).
Table 2.
| |||
|---|---|---|---|
| Entry |
|
Pd(OAc)2 (mol%)/NiXANTPHOS (mol%) | Yield (%) |
| 1 |
|
2.5/3.75 | 90 (3aa) |
| 2 |

|
2.5/3.75 | 90 (3ab) |
| 3 |
|
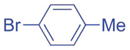
2.5/3.75 | 87 (3ac) |
| 4 |
|
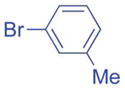
2.5/3.75 | 83 (3ad) |
| 5 |
|
10/15 | 93 (3ae) |
| 6 |
|
10/15 | 90 (3af) |
| 7 |
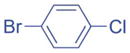
|
2.5/3.75 | 90 (3ag) |
| 8 |
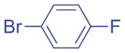
|
2.5/3.75 | 86 (3ah) |
| 9 |
|
2.5/3.75 | 75 (3ai) |
| 10 |
|
5/7.5 | 60 (3aj) |
| 11 |
|
10/15 | 64 (3ak) |
| 12 |
|
10/15 | 63 (3al) |
Reactions conducted on a 0.1 mmol scale using 2 equiv. of 1a, 2 equiv. of NaN(SiMe3)2, and 1 equiv. of aryl bromide at 0.1 M. Base was added portionwise at 0.1 mL per 30 min.
Isolated yield after chromatographic purification.
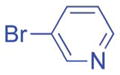
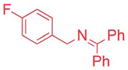
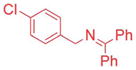
Aryl bromides with electron-withdrawing substituents were also suitable coupling partners. In particular, 4-chloro bromo-benzene (2g, entry 7) was transformed into the coupled product 3ag in 90% yield with the carbon-chloride bond remaining intact. 4-Fluoro- (2h) and 4-trifluoromethyl bromobenzene (2i) afforded the coupled products 3ah and 3ai in 86 and 75% yields, respectively (entries 8 and 9). Heterocyclic 3-bromopyridine 2j also coupled with 1a to give product in 60% yield at 5 mol% catalyst loading (entry 10). Aryl bromides bearing 4-cyano (2k) and 4-ethyl ester (2l) substituents provided arylation products in 64 and 63% yield with 10 mol% catalyst (entries 11 and 12). It is important to note that even with electron withdrawing substituents on the aryl substituted products, no isomerization was observed in Table 2.
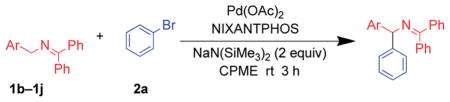
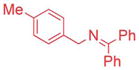
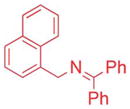
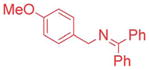
The scope of the N-benzyl group of the benzophenone ketimine coupling partner was determined (Table 3). Overall, ketimine substrates bearing electron-donating, electron-withdrawing and heterocyclic N-benzyl derived groups furnished the coupling products with bromobenzene in moderate to excellent yields with catalyst loadings of 2.5–10 mol%. Coupling of 4-methyl benzyl ketimine derivative furnished the product in 89% yield (Table 3, entry 1). The sterically more demanding 1-naphthyl substituted ketimine (1c) exhibited no reaction under the standard conditions. Optimization of this substrate led to 3ca in 71% yield at 80 °C after 12 h with 10 mol% palladium in THF and LiO–t-Bu (entry 2). The 4-methoxy benzyl amine derivative (1d) reacted sluggishly under our standard conditions, probably because of the increased pKa. Replacement of NaN(SiMe3)2 with a more reactive KN(SiMe3)2 resulted in 71% yield (entry 3).
Table 3.
| |||
|---|---|---|---|
| Entry | Ketimine | Pd(OAc)2/NIXANTPHOS (mol%) | Yield (product) |
| 1 |
|
5/7.5 | 89 (3ac) |
| 2 |
|
10/20 | 71c (3ca) |
| 3 |
|
5/7.5 | 71d (3af) |
| 4 |
|
2.5/3.75 | 83 (3ah) |
| 5 |
|
2.5/3.75 | 89 (3ag) |
| 6 |

|
2.5/3.75 | 91 (3ga) |
| 7 |
|
5/7.5 | 87 (3aj) |
| 8 |
|
10/20 | 90e (3ia) |
| 9 |

|
10/20 | 60f (3ja) |
Reactions conducted on a 0.1 mmol scale using 2 equiv. of imine, 2 equiv. of NaN(SiMe3)2, and 1 equiv. of bromobenzene at 0.1 M. Base was added portionwise at 0.1 mL per 30 min.
Isolated yield after chromatographic purification.
3 equiv. of LiO–tBu 80 °C in 3 mL THF for 12 h.
3 equiv of 1d and 3 equiv. of NaN(SiMe3)2.
2 equiv. LiN(SiMe3)2, 50 °C.
50 °C.
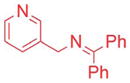
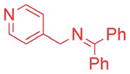
Ketimines containing halogens (1e, 1f, 1g) smoothly reacted with 2a (2.5 mol% Pd), generating the desired products in 83–91% yield (Table 3, entries 4–6). The 3-pyridyl derivative (1j) furnished the arylation product in 87% yield with 5 mol% catalyst loading (entry 7). With the 4-pyridyl substrate, use of LiN(SiMe3)2 at 50 °C was necessary, ultimately leading to 90% yield (entry 8). The 2-furyl-based ketimine underwent cross coupling with 10 mol% catalyst loading in 60% yield (entry 9). Along similar lines, Scheme 5 illustrates the coupling of two halogenated partners, followed by purification of the imine product and hydrolysis to give 3fh as the hydrochloride salt in 79% overall yield (Scheme 5).16 For this substrate, bearing two electron-withdrawing groups, and all the products in Tables 2 and 3, no product isomerization was observed under our reaction conditions.
Scheme 5.

Arylation of 1f with 2h.
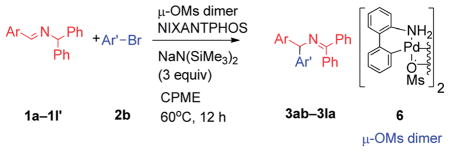
A wide variety of benzaldehyde derivatives are commercially available and inexpensive, inspiring us to explore the arylation of aldimines prepared from benzaldehyde and its analogues. Given the aldimine precursors 1′ and the isomeric ketimine 1 generate the same azaallyl anion, we examined coupling with aldimines 1′ under the standard conditions employed in Table 1 (entry 10), however, only 67% yield was obtained. In contrast, Buchwald’s 3rd-generation precatalyst in situ generated from 2.5 mol% of Buchwald’s μ-OMs Pd dimer with 5 mol% NI-XANTPHOS17 at 60 °C gave 94% yield (Table 4, entry 1). 4-N,N-Dimethylamino bromobenzene (2e) underwent coupling in 80% yield, providing product 3ae (entry 2). Aldimines containing heterocycles were also well tolerated. Both 3- and 4-pyridyl derived aldimines (1h′ and 1i′) furnished products in 77 and 91% yield with 10 and 5 mol% catalyst loading, respectively (entry 3 and 4). Notably, the 3-furyl-based substrate underwent cross-coupling at 5 mol% catalyst loading and furnished the product in 85% yield. Considering the price difference between 3-furylmethylamine ($750 per g) and 3-furancarboxaldehyde ($7.94 per g),18 the aldimine arylation proved to be much more cost effective.
Table 4.
| ||||
|---|---|---|---|---|
| Entry |

|
Ar’–Br | μ-OMs dimer/NIXANTHPOS (mol%) | Yield (%) |
| 1 |
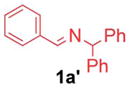 1a′ |
|
2.5/5 | 94 (3ab) |
| 2 |
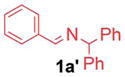 1a′ |
|
2.5/5 | 80 (3ae) |
| 3 |
 1i |
|
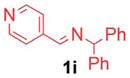
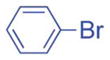
2.5/5 | 91 (3ia) |
| 4 |
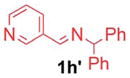 1h′ |
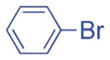
|
5/10 | 77 (3aj) |
| 5 |
 1l′ |
|
2.5/5 | 85c (3la) |
Reactions conducted on a 0.1 mmol scale using 2 equiv. of aldimine, 3 equiv. of NaN(SiMe3)2, and 1 equiv. of aryl bromide at 0.1 M. Base was added portionwise with speed 0.1 mL per 30 min.
Isolated yield after chromatographic purification.
THF, 3 h.
The success of the 3rd-generation Buchwald precatalyst in the aldimine arylation (Table 4) inspired us to exploit it with very difficult substrates. Coupling of ketimine 1a with N-(4-bromophenyl) acetamide, with a free N–H, was undertaken. As outlined in Scheme 6a, using μ-OMs dimer 6 enabled coupling of 1a with 2m to afford 75% yield of product 3am. No byproduct derived from Buchwald–Hartwig coupling19 was observed (1H NMR). In the case of the sterically hindered aryl bromide, 2-bromo toluene (2n), a synthetically useful 56% yield was obtained. It is noteworthy that precatalyst 7, in the presence of NIXANTPHOS, afforded bis-heterocyclic product 3ij in excellent yield (90%, Scheme 6c).
Scheme 6.

Coupling of aryl bromides with (a) a free N–H, (b) ortho-substitution, and (c) to give a bis-heterocyclic diarylmethylamine derivative.
To further increase the synthetic utility and efficiency of our arylation of 1,1,3-triaryl-2-azaallyl anions, we set out to develop a protocol for the in situ imine generation followed by cross-coupling. Subjecting the commercially available benzophenone imine Ph2C=NH to 1 equiv. of benzyl amine in THF at 50 °C for 12 h in a sealed microwave vial under nitrogen led to ketimine 1a (Table 5). Complete removal of the THF solvent under vacuum was followed by addition of 5 mol% Pd(OAc)2 and 7.5 mol% NIXANTPHOS. Next, aryl bromide was added followed by portionwise addition of NaN(SiMe3)2 in CPME (as outlined in entry 10 of Table 1). As illustrated in Table 5, aryl bromides bearing electron-withdrawing, neutral and electron-donating groups furnished the desired arylation products in 63–92% yield. In a similar fashion, a telescoped aldimine synthesis/cross-coupling procedure was conducted (see Experimental section). Aryl bromides bearing electron-withdrawing, neutral and electron-donating substituents furnished the desired arylation products in 73–93% yield (Table 6).
Table 5.
| |||
|---|---|---|---|
| Entry | Ar–Br | Pd(OAc)2 (mol%)/NIXANTPHOS (mol%) | Yield (%) |
| 1 |
|
5/7.5 | 90 (3ag) |
| 2 |
|
5/7.5 | 92 (3ab) |
| 3 |
|
5/7.5 | 63 (3ae) |
Reactions conducted on a 0.1 mmol scale using 3 equiv. of benzyl amine, 3 equiv. of benzophenone imine, 3 equiv. of NaN(SiMe3)2, and 1 equiv. of aryl bromide at 0.1 M. Base was added portionwise at 0.1 mL per 30 min.
Isolated yield after chromatographic purification.
Table 6.

| |||
|---|---|---|---|
| Entry | Ar–Br | μ-OMs dimer/NIXANTHPOS (mol%) | Yield (%) |
| 1 |
|
5/10 | 82 (3ah) |
| 2 |
|
5/10 | 93 (3ab) |
| 3 |
|
5/10 | 73 (3ae) |
Reactions conducted on a 0.1 mmol scale using 2 equiv. of benzaldehyde, 2 equiv. of diphenylmethanamine, 2 equiv. of NaN(SiMe3)2, and 1 equiv. of aryl bromide at 0.1 M. Base was added portionwise at 0.05 mL per 30 min.
Isolated yield after chromatographic purification.
For any method to be synthetically useful, it must be scalable. We scaled up the ketimine arylation using imine 1a with 1-bromo-4-tert-butylbenzene on a 3 mmol scale with catalyst loading of 2.5 mol%. The desired coupling product was isolated in 81% yield (0.98 g, Scheme 7A). We also scaled up the telescoped synthesis with 4-pyridyl imine 1i and cross-coupling with bromobenzene. Using the procedure outlined in Table 5, the reaction was conducted with 3 mmol 1i (Scheme 7B). The desired product 3ia was isolated in 74% yield (0.77 g).
Scheme 7.

Scale-up of the imine arylation.
Hydrolysis20 of several ketimine products afforded diaryl-methylamines 8–15 as crystalline hydrochloride salts in 92–97% yield (Table 7). Salts 9 and 10 were reported to have antispasmodic activity against furtrethonium.21 Other salts (8, 12, 13, and 14) are useful intermediates toward bioactive compounds.22 In each case, only a single diarylmethylamine product was isolated in high yield, indicating that isomerization of the arylated ketimines during their formation and hydrolysis did not take place. In contrast, hydrolysis of the products from the Oshima procedure in Scheme 2 would give a mixture of diarylmethylamines.
Table 7.
Hydrolysis of product ketimines
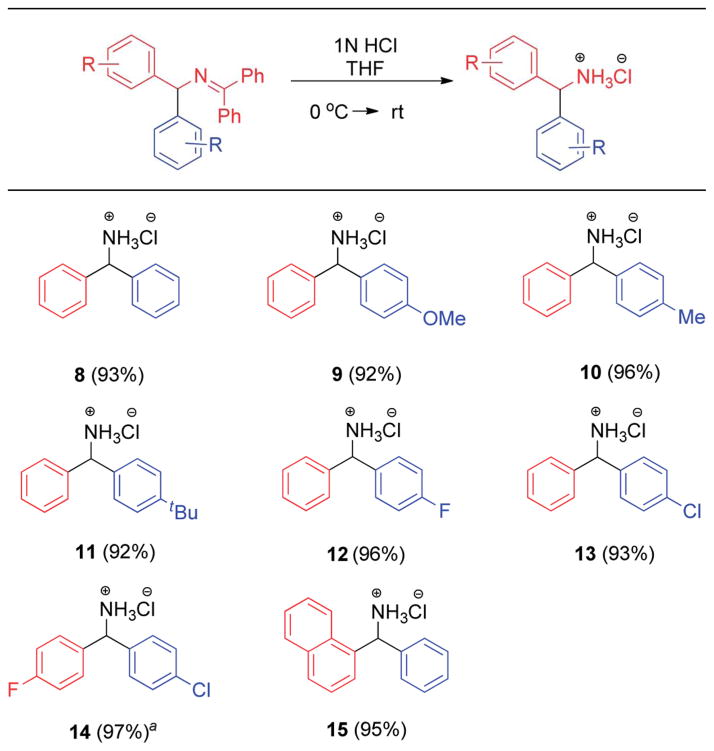
|
See ESI for the synthesis of corresponding imine precursor for hydrolysis.
Simple functionalization of our diarylmethylamine products afforded bioactive compounds in synthetically useful yields (Scheme 8). Acetylation of 12 was conducted to provide the amide 16. Acetamide 16 was reported to exhibit in vivo anti-hypoxic and anticonvulsant activity.22a N-Benzhydrylurea derivatives 17 was synthesized from 13 and urea. It was reported to have antihypoxic activity,23a in vivo effects on mouse liver microsome cytochrome P-450 content,23b and anticonvulsant activity.23c
Scheme 8.
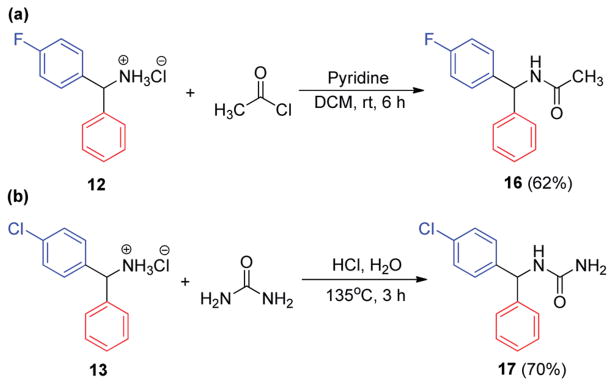
Functionalization of diarylmethylamines.
Conclusions
Diarylmethylamines have played a significant role in the betterment of human health and wellbeing. Herein, we have developed the first efficient approaches to the synthesis of this important class of compounds based on the arylation of 1,1,3-triaryl-2-azaallyl anions. The key to success is the advancement of a NIXANTPHOS-based catalyst that enables the arylation to be conducted under mild conditions and the identification of a hindered base [NaN(SiMe3)2], such that the product of the reaction is not deprotonated or isomerized, as observed previously. Finally, in contrast to other palladium catalyzed reactions of 1,1,3-triaryl-2-azaallyl anions,11 our catalyst enables excellent control over regiochemistry of the C–C bond forming reaction.24
Experimental section
General methods
All reactions were conducted under a nitrogen atmosphere with oven-dried glassware and standard Schlenk or vacuum line techniques. All solutions were handled under nitrogen and transferred via syringe. Anhydrous solvents, including CPME (cyclopentyl methyl ether), 1,4-dioxane, and 2-MeTHF were purchased from Sigma-Aldrich and directly used without further purification. Toluene and THF were dried through activated alumina columns. Unless otherwise stated, reagents were commercially available and used as purchased without further purification. Chemicals were purchased from Sigma-Aldrich, Acros, Alfa Aesar or Matrix Scientific, and solvents were purchased from Fisher Scientific. Progress of reactions was monitored by thin-layer chromatography using Whatman Par-tisil K6F 250 μm precoated 60 Å silica gel plates and visualized by short-wave ultraviolet light as well as by treatment with iodine or ceric ammonium molybdate (CAM) stain. Flash chromatography was performed with silica gel (230–400 mesh, Silicycle). 1H and 13C{1H} NMR spectra were obtained using a Brüker AM-500 Fourier-transform NMR spectrometer at 500 and 125 MHz, respectively. Chemical shifts were reported in units of parts per million (ppm) downfield from tetramethylsilane (TMS), and all coupling constants were reported in hertz. The infrared spectra were taken with KBr plates with a Perkin-Elmer Spectrum 100 Series spectrometer. High resolution mass spectrometry (HRMS) data were obtained on a Waters LC-TOF mass spectrometer (model LCT-XE Premier) using chemical ionization (CI) or electrospray ionization (ESI) in positive or negative mode, depending on the analyte. Melting points were determined on a Unimelt Thomas-Hoover melting point apparatus and were uncorrected. Deactivated silica gel was prepared by addition of 15 mL of Et3N to 1 L of silica gel. Note that in some cases, due to the large number of inequivalent aromatic carbons in the products, coincidental overlap of resonances prevented observation of all the expected resonances.
Procedure and characterization for the Pd catalyzed ketimine arylation
An oven-dried microwave vial equipped with a stir bar was charged with imine 1a (54.3 mg, 0.20 mmol) under a nitrogen atmosphere. A stock solution of Pd(OAc)2 (0.55 mg, 0.0025 mmol) and NIXANTPHOS (2.1 mg, 0.00375 mmol) under nitrogen in 0.5 mL dry CPME was taken up by syringe and added to the reaction vial. The vial was sealed, and 1-bromo-4-tert-butylbenzene (17.3 μL, 0.10 mmol) was added dropwise by syringe to this solution through the rubber septum. A solution of NaN(SiMe3)2 (36.7 mg, 0.20 mmol) in 0.5 mL CPME was added portionwise by syringe at 0.1 mL per 30 min at 24 °C. The reaction mixture was stirred for 3 h at 24 °C, opened to air, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with an additional 6 mL of ethyl acetate, and the combined solutions were concentrated in vacuo. The crude material was loaded onto a silica gel column via pipette and purified by flash chromatography (hexanes to diethyl ether–hexanes = 1 : 50).
Procedure and characterization for the Pd catalyzed aldimine arylation
An oven-dried microwave vial equipped with a stir bar was charged with aldimine 1a′ (54.3 mg, 0.20 mmol) under a nitrogen atmosphere. A stock solution of Buchwald’s 3rd generation pre-catalyst Pd dimer (1.8 mg, 0.0025 mmol) and NIXANTPHOS (2.8 mg, 0.0050 mmol) under nitrogen in 0.5 mL dry CPME was taken up by syringe and added to the reaction vial. The vial was sealed, and 1-bromo-4-tert-butylbenzene (17.3 μL, 0.10 mmol) was added dropwise by syringe to this solution through the rubber septum. A solution of NaN(SiMe3)2 (55.0 mg, 0.30 mmol) in 0.5 mL CPME was added portionwise by syringe at 0.1 mL per 30 min at 60 °C. The reaction mixture was stirred for 12 h at 60 °C, opened to air, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with an additional 6 mL of ethyl acetate, and the combined solutions were concentrated in vacuo. The crude material was loaded onto a silica gel column via pipette and purified by flash chromatography (hexanes to diethyl ether–hexanes = 1 : 50).
One-pot ketimine synthesis/Pd-catalyzed arylation
An oven-dried microwave vial equipped with a stir bar was charged with benzylamine (32.1 mg, 0.30 mmol) and benzophenone imine (54.4 mg, 0.30 mmol) under a nitrogen atmosphere. Next, 1 mL of dry THF was added under nitrogen via syringe and the vial was sealed. The reaction was then placed in an oil bath at 50 °C. After the reaction mixture was stirred for 12 h at 50 °C, the solvent was completely removed in vacuo and the vial was refilled with nitrogen. A stock solution of Pd(OAc)2 (0.55 mg, 0.0025 mmol) and NIXANTPHOS (2.1 mg, 0.00375 mmol) under nitrogen in 0.5 mL dry CPME was taken up by syringe and added to the same reaction vial through the rubber septum. 1-Bromo-4-tert-butylbenzene (17.3 μL, 0.10 mmol) was added dropwise. A solution of NaN(SiMe3)2 (55.0 mg, 0.30 mmol) in 0.5 mL CPME was added portionwise at 0.1 mL per 30 min at 24 °C. The reaction mixture was stirred for 3 h at 24 °C, opened to air, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with an additional 6 mL of ethyl acetate, and the combined solutions were concentrated in vacuo. The crude material was loaded onto a silica gel column via pipette and purified by flash chromatography (hexanes to diethyl ether–hexanes = 1 : 50).
One-pot aldimine synthesis/Pd-catalyzed arylation
An oven-dried microwave vial equipped with a stir bar was charged with benzaldehyde (21.2 mg, 0.20 mmol) and diphe-nylmethylamine (36.6 mg, 0.20 mmol) under a nitrogen atmosphere. Dry THF (1 mL) was then added under nitrogen via syringe and the vial was sealed. The reaction was then placed in an oil bath at 80 °C and stirred for 12 h. Next, the volatile materials were completely removed at rt and the remaining solid was dried under reduced pressure at 60 °C for 2 h. The vial was then backfilled with nitrogen and a stock solution of Buchwald’s 3rd generation pre-catalyst Pd dimer (3.7 mg, 0.005 mmol) and NIXANTPHOS (5.6 mg, 0.010 mmol) in 0.5 mL dry CPME was added by syringe through the rubber septum. Next, 1-bromo-4-tert-butylbenzene (17.3 μL, 0.10 mmol) was added dropwise by syringe through the rubber septum. A solution of NaN(SiMe3)2 (36.7 mg, 0.20 mmol) in 0.5 mL CPME was added portionwise at 0.05 mL per 30 min at 60 °C. The reaction mixture was stirred for 6 h at 60 °C, opened to air, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with an additional 6 mL of ethyl acetate, and the combined solutions were concentrated under reduced pressure. The crude material was loaded onto a silica gel column via pipette and purified by flash chromatography (hexanes to diethyl ether–hexanes = 1 : 50).
Supplementary Material
Acknowledgments
P. J. W. acknowledges the NIH (National Institute of General Medical Sciences NIGMS 104349) and the NSF (CHE-1152488). J. A. thanks Ministry of Education, Spain, for a Salvador de Madariaga mobility fellowship. B. Y. thanks the Scientific and Technical Research Council of Turkey for financial support. We thank Dr Simon Berritt (UPenn/Merck High-Throughput Experimentation Center) for assistance.
Footnotes
Electronic supplementary information (ESI) available: Data for new compounds, experimental procedures, 1H and 13C NMR spectra. See DOI: 10.1039/c3sc53526f
Notes and references
- 1.Li JJ. Contemporary Drug Synthesis. Wiley-Interscience; 2004. p. 221. [Google Scholar]
- 2.Grant JA, Riethuisen JM, Moulaert B, DeVos C. Ann Allergy, Asthma, Immunol. 2002;88:190. doi: 10.1016/S1081-1206(10)61995-3. [DOI] [PubMed] [Google Scholar]
- 3.Fuhrkop JH, Li G. Organic Synthesis. Concepts and Methods. Wiley-Interscience; 2003. p. 237. [Google Scholar]
- 4.Ko Y, Malone DC, Armstrong EP. Pharmacotherapy. 2006;26:1694. doi: 10.1592/phco.26.12.1694. [DOI] [PubMed] [Google Scholar]
- 5.Doggrell SA, Liang LC, Arch NS. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:126. doi: 10.1007/pl00005146. [DOI] [PubMed] [Google Scholar]
- 6.Plobeck N, Delorme D, Wei ZY, Yang H, Zhou F, Schwarz P, Gawell L, Gagnon H, Pelcman B, Schmidt R, Yue SY, Walpole C, Brown W, Zhou E, Labarre M, Payza K, St-Onge S, Kamassah A, Morin PE, Projean D, Ducharme J, Roberts E. J Med Chem. 2000;43:3878. doi: 10.1021/jm000228x. [DOI] [PubMed] [Google Scholar]
- 7.Wei ZY, Brown W, Takasaki B, Plobeck N, Delorme D, Zhou F, Yang H, Jones P, Gawell L, Gagnon H, Schmidt R, Yue SY, Walpole C, Payza K, St-Onge S, Labarre M, Godbout C, Jakob A, Butterworth J, Kamassah A, Morin PE, Projean D, Ducharme J, Roberts E. J Med Chem. 2000;43:3895. doi: 10.1021/jm000229p. [DOI] [PubMed] [Google Scholar]
- 8.Li Z, Li CJ. J Am Chem Soc. 2004;126:11810. doi: 10.1021/ja0460763.Li Z, Li CJ. Org Lett. 2004;6:4997. doi: 10.1021/ol047814v.Li Z, Li CJ. J Am Chem Soc. 2005;127:6968. doi: 10.1021/ja0516054.Li Z, Bohle DS, Li CJ. Proc Natl Acad Sci U S A. 2006;103:8928. doi: 10.1073/pnas.0601687103.Basle O, Li CJ. Org Lett. 2008;10:3661. doi: 10.1021/ol8012588.Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n.Muramatsu W, Nakano K, Li CJ. Org Lett. 2013;15:3650. doi: 10.1021/ol401534g.Dastbaravardeh N, Schnurch M, Mihovilovic MD. Org Lett. 2012;14:1930. doi: 10.1021/ol300627p.Dastbaravardeh N, Schnurch M, Mihovilovic MD. Org Lett. 2012;14:3792. doi: 10.1021/ol301680v.Pan S, Matsuo Y, Endo K, Shibata T. Tetrahedron. 2012;68:9009.For traditional synthetic approaches of diarylmethylamines. See: Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chem Soc Rev. 2006;35:454. doi: 10.1039/b600091f.Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069. doi: 10.1021/cr980414z.Petasis NA, Boral S. Tetrahedron Lett. 2001;42:539.Plobeck N, Powell D. Tetrahedron: Asymmetry. 2002;13:303.Kauffman MC, Walsh PJ. Science of Synthesis, Stereoselective Synthesis. Vol. 2. Thieme; Stuttgart: 2011. p. 449.Liu Y, Du H. J Am Chem Soc. 2013;135:6810. doi: 10.1021/ja4025808.Boone MP, Stephan DW. J Am Chem Soc. 2013;135:8508. doi: 10.1021/ja403912n.Das BG, Nallagonda R, Ghorai P. J Org Chem. 2012;77:5577. doi: 10.1021/jo300706b.For additional references related to functionalization adjacent to nitrogen. See: Zou LH, Mottweiler J, Priebbenow DL, Wang J, Stubenrauch JA, Bolm C. Chem–Eur J. 2013;19:3302. doi: 10.1002/chem.201204502.Zhang HJ, Bolm C. Org Lett. 2011;13:3900. doi: 10.1021/ol201431c.Kauffmann T, Berg H, Kiippelmann E, Kuhlmann D. Chem Ber. 1977;110:2659.Green JE, Bender DM, Jackson S, O’Donnell MJ, McCarthy JR. Org Lett. 2009;11:807. doi: 10.1021/ol802325h.Schmitt DC, Lee J, Dechert-Schmitt AM, Yamaguchi E, Krische MJ. Chem Commun. 2013;49:6096. doi: 10.1039/c3cc43463j.Cogan DA, Liu G, Ellman JA. Tetrahedron. 1999;55:8883.Xu HC, Chowdhury S, Ellman JA. Nat Protoc. 2013;8:2271. doi: 10.1038/nprot.2013.134.
- 9.(a) McGrew GI, Temaismithi J, Carroll PJ, Walsh PJ. Angew Chem, Int Ed. 2010;49:5541. doi: 10.1002/anie.201000957. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McGrew GI, Stanciu C, Zhang J, Carroll PJ, Dreher SD, Walsh PJ. Angew Chem, Int Ed. 2012;51:11510. doi: 10.1002/anie.201201874. [DOI] [PubMed] [Google Scholar]
- 10.(a) Niwa T, Yorimitsu H, Oshima K. Org Lett. 2008;10:4689. doi: 10.1021/ol802070d. [DOI] [PubMed] [Google Scholar]; (b) Niwa T, Suehiro T, Yorimitsu H, Oshima K. Tetrahedron. 2009;65:5125. [Google Scholar]
- 11.For additional references related to functionalization of 2-azaallyl anions. See: Yeagley AA, Chruma JJ. Org Lett. 2007;9:2879. doi: 10.1021/ol071080f.Fields WH, Khan AK, Sabat M, Chruma JJ. Org Lett. 2008;10:5131. doi: 10.1021/ol801986m.Yeagley AA, Lowder MA, Chruma JJ. Org Lett. 2009;11:4022. doi: 10.1021/ol901745x.Fields WH, Chruma JJ. Org Lett. 2010;12:316. doi: 10.1021/ol902651j.Li Z, Jiang YY, Yeagley AA, Bour JP, Liu L, Chruma JJ, Fu Y. Chem– Eur J. 2012;18:14527. doi: 10.1002/chem.201201425.Tunge JA, Burger EC. Eur J Org Chem. 2005:1715.Burger EC, Tunge JA. J Am Chem Soc. 2006;128:10002. doi: 10.1021/ja063115x.Grenning AJ, Tunge JA. J Am Chem Soc. 2011;133:14785. doi: 10.1021/ja205717f.Weaver JD, Recio A, III, Grenning AJ, Tunge JA. Chem Rev. 2011;111:1846. doi: 10.1021/cr1002744.
- 12.(a) Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J Am Chem Soc. 2012;134:13765. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]; (b) Bellomo A, Zhang J, Trongsiriwat N, Walsh PJ. Chem Sci. 2013;4:849. [Google Scholar]
- 13.For more references related to NIXANTPHOS. See: van der Veen LA, Keeven PH, Schoemaker GC, Reek JNH, Kamer PCJ, van Leeuwen PWNM, Lutz M, Spek AL. Organometallics. 2000;19:872.Sandee YAJ, Reek JNH, Kamer PCJ, van Leeuwen PWNM. J Am Chem Soc. 2001;123:8468. doi: 10.1021/ja010150p.
- 14.(a) Hatano M, Hattori Y, Furuya Y, Ishihara K. Org Lett. 2009;11:2321–2324. doi: 10.1021/ol900680f. [DOI] [PubMed] [Google Scholar]; (b) Cainelli G, Giacomini D, Trerè A, Boyl PP. J Org Chem. 1996;61:5134. [Google Scholar]
- 15.At this point we cannot rule out a mechanism in which the ketimine coordinates to palladium and is deprotonated on the metal. If this were the case, rearrangement from an N-bound azaallyl to a C-bound would be necessary for reductive elimination.
- 16.N-((4-Chlorophenyl)(4-fluorophenyl)methyl)acetamide, which could be synthesized easily from 3ha, is reported to have in vivo antihypoxic and anticonvulsant activity. See: Krasnov VA, Gorshkova VK, Bakibaev AA, Saratikov AS. Pharm Chem J. 1997;31:368.
- 17.(a) Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bruno NC, Buchwald SL. Org Lett. 2013;15:2876. doi: 10.1021/ol401208t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shu W, Buchwald SL. Chem Sci. 2011;2:2321. [Google Scholar]; (e) Biscoe MR, Fors BP, Buchwald SL. J Am Chem Soc. 2008;130:6686. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prices were found in Sigma-Aldrich website: http://www.sigmaaldrich.com/catalog/product/aldrich/278866?lang=en®ion=US, http://www.sigmaaldrich.com/catalog/product/aldrich/cds008929?lang=en®ion=US.
- 19.(a) Fors BP, Buchwald SL. J Am Chem Soc. 2010;132:15914. doi: 10.1021/ja108074t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vo GD, Hartwig JF. J Am Chem Soc. 2009;131:11049. doi: 10.1021/ja903049z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Maimone TJ, Buchwald SL. J Am Chem Soc. 2010;132:9990. doi: 10.1021/ja1044874. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Y, Deng L. J Am Chem Soc. 2012;134:14334. doi: 10.1021/ja306771n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) O’Donnell MJ, Polt RL. J Org Chem. 1982;47:2663. [Google Scholar]
- 21.Estiu GL, Cachau RE, Castro EA, Blanch LE. Bruno Farmaco. 1990;45:889. [Google Scholar]
- 22.(a) Krasnov VA, Gorshkova VK, Bakibaev AA, Saratikov AS. Pharm Chem J. 1997;31:368. [Google Scholar]; (b) Vernalis Res. Ltd. Azetidinecarboxamide derivatives and their use in the treatment of CB1 receptor mediated disorders. 2004GB01831. Eur Pat WO. 2004
- 23.(a) Bakibaev AA, Filimonov VD, Tignibidina LG, Gorshkova VK, Saratikov AS, Oleinik NB, Shtrykova VV. Pharm Chem J. 1993;27:254. [Google Scholar]; (b) Bakibaev AA, Akhmedzhanov RR, Novozheeva TP, Filimono VD, Tignibidina LG, Saratikov AS, Shtrykova VV. Pharm Chem J. 1993;27:395. [Google Scholar]; (c) Filimonov VD, Bakibaev AA, Pustovoitov AV, Tignibidina LG, Pechenkin AG, Gorshkova VK, Saratikov AS. Pharm Chem J. 1988;22:358. [Google Scholar]
- 24.While this manuscript was under review, Buchwald and Zhu published the enantioselective arylation of 9-aminofluorene-derived alkyl imines ( Zhu Y, Buchwald SL. J Am Chem Soc. 2014;136:4500. doi: 10.1021/ja501560x.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.