

Abstract
Combination chemotherapy is a common practice in clinical management of malignancy. Synergistic therapeutic outcome is only achieved when tumor cells are exposed to cells in an optimal ratio. However, due to diverse physicochemical properties of drugs, no free drug cocktails or nanomaterials are capable of co-loading and co-delivering drugs at an optimal ratio. Herein, we develop a novel nano-platform with precise ratiometric co-loading and co-delivery of two hydrophilic drugs for synergistic anti-tumor effects. Based on previous work, we utilize a solvent displacement method to ratiometrically load dioleoyl phosphatidic acid (DOPA)-gemcitabine monophosphate and DOPA coated cisplatin-precipitate nanocores into the same PLGA NP. These cores are designed to have similar hydrophobic surface properties. GMP and cisplatin are engineered into PLGA NP at an optimal synergistic ratio (5:1, mol:mol) with over 70% encapsulation efficiency and were ratiometrically taken up by tumor cells in vitro and in vivo. These PLGA NP exhibit synergistic anti-cancer effects in a stroma-rich bladder tumor model. A single injection of dual drugs in PLGA NP can significantly inhibit tumor growth. This nanomaterial-system solves problems related to ratiometric co-loading and co-delivery of different hydrophilic moieties and provides possibilities for co-loading hydrophilic drugs with hydrophobic drugs for combination therapy.
Keywords: combination therapy, cisplatin, gemcitabine, ratiometric loading, ratiometric delivery
1. Introduction
Combination therapy is particularly effective in the treatment of HIV/AIDs and cancer. It provides a general means to maximize therapeutic efficacy, overcome treatment resistance, and diminish adverse effects.[1] Optimized doses and molar ratios of combined drugs are critical to promote synergistic rather than antagonistic effects.[2] However, differential pharmacokinetics and distribution of individual drugs within the conventionally administered “cocktail” lead to deviation from the optimized ratio during systemic delivery. This fact makes predicting improved in vivo therapeutic outcomes from in vitro synergistic effects a real clinical challenge.[3] Nanomaterial-based delivery is one approach to unifying dual-drug pharmacokinetics.[2c] However, it is rather difficult to load drugs with drastically different physical chemistry into the designed nano-carriers, which is why only a few nanoparticulate formulations [4] were able to reach the goal. Although attempts have been made, precise loading and ratiometric delivery of drugs with diverse solubility, steric configuration and other physicochemical properties still remains a challenge.[5] Moreover, combining individual therapeutic blocks together without interference their own functionalities adds to the complexity of compact nanostructures for combination drug delivery.[6]
Cisplatin is considered the gold standard in several first-line combination therapies.[7] A nanoparticulate approach used to enhance the ratio-dependent synergistic cisplatin-related combination therapy is rarely reported due to the difficulties in loading cisplatin along with other types of drugs into a single NP and the possible chemical interference with other groups of drugs such as nucleic acids.[8] Limited solubility of inorganic cisplatin in both water and oil significantly hinders the development of NP with high drug loading and encapsulation efficacy.[9] Gemcitabine monophosphate (GMP), an organic hydrophilic drug, was chosen for combination therapy with cisplatin. It is well known that Gemcitabine is widely used as a first line therapy in combination with cisplatin for the treatment of bladder cancer. However, Gemcitabine relies on nucleoside transporters [10] to enter into cells where it is subsequently phosphorylated by deoxycytidine kinase to form active intermediates for DNA synthesis interference. GMP is one of the active intermediates of Gemcitabine.[11] Since the addition of the first phosphate group in GMP formation is the rate-limiting step, we anticipate GMP to be an efficient therapeutic drug candidate with great commercial value that can exhibit a synergistic effect in combination with cisplatin.[12] Due to the significant difference in physicochemical properties, co-encapsulation of cisplatin and GMP is difficult. Therefore, NP that can ratiometrically co-encapsulate and co-deliver native cisplatin and GMP while not compromising the drug activity, are highly desired.
Previously in our lab, we developed dioleoyl phosphatidic acid (DOPA) coated calcium phosphate cores with the capability of loading hydrophilic phosphorylated drugs (such as GMP core)[13], small interfering RNA (siRNA)[14], DNA[15] and peptides[16]; as well as DOPA coated cisplatin cores (CP core), where cisplatin serves as both nanocarrier and anti-cancer drug.[9, 17] The surface and size similarities between these two categories of cores provide us a methodology to unify a wide range of drugs or biomolecules with drastically disparate solubility and polarity into a standardized hydrophobic physicochemical property. Unifying physicochemical characteristics of dual drugs is the prerequisite for ratio-controlled loading and delivery.
Based on this rationale, we report a novel strategy to achieve both precise ratiometric loading and delivery of cisplatin with GMP. Cisplatin and GMP were originally formulated into DOPA coated CP cores and DOPA coated GMP cores. As shown in Figure 1A, PLGA NP are used to incorporate these two separate hydrophobic cores. Since CP cores and GMP cores have similar surface properties, we therefore hypothesize that these two drugs can be ratiometrically encapsulated into the same PLGA NP. To demonstrate our hypothesis, CP cores and GMP cores were co-loaded into single PLGA NP using the solvent displacement method (Figure 1A). Ratiometric loading of GMP and cisplatin were first examined. With the confirmation of ratiometric loading property of PLGA NP, we further proposed that this dual-drug containing NP could be ratiometrically delivered to the site of malignancy at the optimal ratio (Figure 1B). This hypothesis was tested in vitro via release kinetics study and cellular uptake study, and in vivo via tumor accumulation analysis. We then hypothesized that co-delivery of both drugs at an optimized ratio would result in synergistic anticancer efficacy. A stroma-rich human bladder cancer xenograft model was used to evaluate the anti-tumor efficacy of dual-drug containing NP at optimized ratio.[18] Synergistic anti-cancer effect was further determined via protein based mechanistic analysis. To our knowledge, this is the first time that cisplatin has been reported to be co-encapsulated with another hydrophilic drug in the same NP with precise ratiometric control.
Figure 1.

Fabrication of PLGA-PEG-Anisamide NP (PLGA NP) containing CP cores and GMP cores via a single step solvent displacement method (A). Cisplatin and GMP, which are ratiometrically encapsulated in PLGA NP, are ratiometrically delivered into the tumor and exhibit strong synergistic anti-tumor efficacy (B).
2. Results and Discussions
2.1. Preparation and Characterization of Single Drug Loaded PLGA NP
GMP cores and CP cores were prepared as previously mentioned [9, 13] and characterized as 8-12 nm in diameter as determined by transmission electron microscopy (TEM) (Figure S1). Encapsulation efficiency (EE) of GMP in the GMP core was 60.6 ± 4.3% (n=5) as measured by absorbance of GMP at 273 nm. CP cores were also prepared with an EE of 40.4 ± 1.4% (n=5) as measured by inductively coupled plasma mass spectrometry (ICP-MS). Both GMP cores and CP cores could be well dispersed into organic solvent, such as tetrahydrofuran (THF). The above results indicated that hydrophilic GMP and cisplatin have been successfully loaded into hydrophobic cores respectively and these cores were ready to be further incorporated into PLGA NP.
High and comparable encapsulation efficiency of each component is a prerequisite for controlled loading of several modalities in the same nanoparticle. Therefore, single drug loaded PLGA NP were initially investigated and characterized. PLGA NP were originally conjugated with polyethylene glycol (PEG) to prolong systemic circulation time and then self-assembled with PLGA and DOPA coated cores into PLGA NP via single step solvent displacement (Figure 1). Briefly, polymer and drug containing cores were dissolved in THF, a water-miscible solvent, and poured drop wise into water. NP was formed instantaneously during this rapid solvent diffusion process. Anisamide, an agonist of the sigma receptor, was also introduced into PLGA NP as a ligand to enhance internalization in epithelium-derived cancer cells, which overexpress the sigma-receptor (Figure S2).[19] Results in Figure S3 indicate that both GMP and cisplatin in DOPA coated core structures can be encapsulated into PLGA NP separately with high EE (70.6 ± 2.5% and 74.0 ± 10.1%, respectively, n=5) at drug loading (DL) of up to approximately 5 wt%. This is the first time that GMP and cisplatin have been engineered into PLGA NP using solvent displacement method. This method proved much more efficient than loading free gemcitabine and cisplatin into PLGA NP via the double emulsion method, whose maximum loading is only around 1 wt%.[20] Notably, free cisplatin and GMP are quite polar and cannot be loaded into PLGA NP using the solvent displacement method; and thus, DOPA coated cores not only provide an approach to load different types of drugs, especially hydrophilic drugs, into PLGA NP using solvent displacement, but also facilitate hydrophilic drugs to be loaded into PLGA NP with higher DL and EE. More importantly, the EE for single free drugs in PLGA NP using this novel preparation method is quite comparable to each other, suggesting the possibility of loading different drug moieties simultaneously into the same NP at similar EE but different dual-drug ratios,which is one indispensable parameter for ratiometric loading.
2.2. Precise Ratiometric Control over Dual-Drug Loading in Combo NP
The success of loading GMP cores and CP cores into PLGA NP provides us with the possibility to encapsulate two different drug-containing cores into a single NP in a ratiometric manner. To confirm that CP cores and GMP cores can be ratiometrically co-loaded into PLGA NP (Combo NP), several further studies were investigated. Firstly, total feed loading of GMP and cisplatin in Combo NP was fixed at 6 wt% while the feed molar ratio between GMP and cisplatin was altered from 0.5:1 to 5:1 (Figure 2A). Results indicated that the measured molar ratio between the two drugs in Combo NP was almost the same as the feed molar ratio (0.52 vs 0.5; 0.97 vs 1; 3.3 vs 3, 5.3 vs 5) and the EE of both drugs, which all remained above 70% with subtle fluctuation, was almost identical as well. Next, the feed molar ratio of GMP to cisplatin was set at 5 (Figure 2B). It was found that the measured molar ratio of GMP to cisplatin in Combo NP was approximately 5 when the total loading of the two drugs was below 6 wt%. Additionally, greater than 80% EE was achieved. In both experiments, particle size measured by dynamic light scattering (DLS) was under 120 nm and polydispersity of the dual drug particles was around 0.2 (Figure S4). Thus, these results demonstrated that ratiometric loading of distinct types of drugs in DOPA coated cores could be achieved over a wide dual drug ratio range and loading efficiency.
Figure 2.

Dual-drug ratiometric loading in Combo NP. EE and DL of GMP and cisplatin in Combo NP while the total loading of drugs was fixed at 6 wt% (A); EE and DL of GMP and cisplatin in Combo NP while the feed molar ratio of GMP to cisplatin was fixed at 5:1 (B); TEM image of 5.5 wt% total drug loading of Combo NP with molar ratio of GMP and cisplatin of 5.3:1 (C). EDS spectra of Combo NP (D). Both platinum from CP cores and fluorine from GMP cores were observed in a single NP indicating actual loading of dual drugs in single NP. XPS spectrum of Combo NP (E). Molar ratio of GMP and cisplatin was also quantified using atomic ratio of fluorine and platinum. Spectrum of Pt 4F and spectrum of F 1S, from which, area of peaks are integrated for atom quantification.
2.3. Characterization of Dual-Drug Loaded Combo NP using TEM and XPS
To demonstrate that GMP cores and CP cores are homogenously distributed in each Combo NP, we further characterized the Combo NP with total drug feeding ratio of 6 wt% and feed GMP/cisplatin ratio of 5, whose determined loading was 5.5 ± 0.8 wt% (n=5) and molar ratio between GMP and cisplatin was 5.3. TEM revealed Combo NP as spherical and mono-dispersed with a diameter of approximately 90-120 nm (Figure 2C), which is consistent with the value measured by DLS (average 120 nm) (Figure S5). In addition, large quantities of well-dispersed cores were clearly clustered in each NP, further confirming the hypothesis of a nanocapsule-like structure with high and efficient drug loading (Figure 2C). Notably, each NP contained a similar amount of cores. However, TEM result alone cannot show the homogeneous distribution of cores in NP. Therefore, we further characterized the Combo NP using high resolution TEM with energy dispersive spectroscopy (EDS) analysis and x-ray photoelectron spectroscopy (XPS). Chemical element analysis using EDS indicated that both fluorine (characteristic element of GMP) and platinum (characteristic element of cisplatin) were present in single NP (Figure 2D). Over 20 particles were analyzed to determine the average molar ratio of GMP and cisplatin inside each NP. The ratio of fluorine to platinum, representing the ratio of GMP to cisplatin, was approximately 4.9 ± 1.9, which is comparable to the feed ratio of 5 and the determined ratio in bulk solution of 5.3. This result demonstrated that the two distinct cores were present in single NP and their ratio was precisely controlled. To avoid disturbance of neighboring oxygen on fluorine quantification, XPS was carried out to further confirm the ratiometric distribution of the two drugs. Combo NP was dissolved in THF, and a 5 nm layer of particle lysates were analyzed by XPS. The spectrum in Figure 2E indicates that fluorine could be separated well from oxygen, and the calculated molar ratio of GMP to cisplatin was approximately 5.6, similar to the results determined using other techniques. Therefore, quantifications from the single particle nano-layer of particle lysate as well as the bulk solution strongly suggest the fact that the dual-drug combination has been successfully, homogenously loaded into single Combo NP with relatively precise ratiometric control.
2.4. In Vitro Ratiometric Control over Dual-drug Cellular Uptake
In vitro synergy studies of free cisplatin and GMP (Combo free) using Chou-Talalay method[21] indicated that Combo free exhibited the strongest synergy at a GMP/cisplatin ratio of 5 in human urinary bladder carcinoma UMUC3 cell line.[18] We incorporated 3H-labeled CMP (cytidine monophosphate) into single GMP cores in PLGA NP (GMP NP) as a marker to detect the concentration of GMP. In vitro cellular uptake (Figure S6) of free GMP and free cisplatin indicated that UMUC3 cells exhibited an equivalent uptake of GMP and cisplatin, suggesting that the feed ratio and the actual intracellular ratio of the drug combination was almost identical in the in vitro assay (Figure 3A). However, the uptake of drugs in the tumor cells in vivo will be much different due to differing PK profiles and the complicated tumor microenvironment. In order to maintain the ratio of drugs in vivo and utilize the strongest synergy of Combo free at a GMP/cisplatin ratio of 5, PLGA NP with a total drug loading of 5.5 ± 0.8 wt% (n=5) and molar ratio between GMP and cisplatin of 5.3 were further investigated in the following studies. Single drug PLGA NP with a feed ratio of 6 wt% was used for comparison (Table S1). Notably, the size of CP cores in single PLGA NP (cisplatin NP) was smaller (approximately 60 nm) than that of GMP NP and Combo NP. This is not surprising considering that CP cores are denser than GMP cores which are mainly composed of calcium phosphate.
Figure 3.
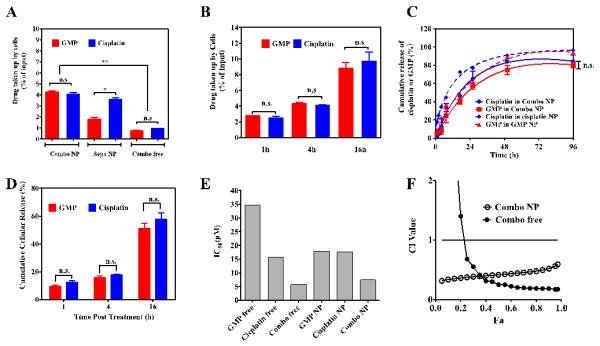
Ratiometric cellular uptake and release of dual drugs from Combo NP. Uptake of cisplatin and GMP in Combo NP, Sepa NP, and free drugs at 37 °C for 4 h in UMUC3 cells (A). Accumulative uptake of Combo NP loaded with cisplatin and GMP in UMUC3 Cells (B). In vitro release kinetics of cisplatin and GMP from Combo NP and single NP in PBS at 37 °C (C) and intracellular release of cisplatin and GMP from Combo NP (D). IC50 of free GMP, cisplatin, and Combo free at molar ratio 5.3:1, as well as single drug NP and Combo NP at molar ratio 5.3:1 (E). X-axis indicated the total concentration of dual drugs or single drug formulations. The corresponding CI vs Fa plots of Combo NP and Combo free were shown (F). DL of cisplatin and GMP in Combo NP is 0.8 wt% and 4.6 wt% respectively, while DL of cisplatin and GMP in single NP is 4.4 wt% and 4.2 wt% respectively. n.s.: no significant difference; * P<0.05; ** P<0.01.
Ratiometric cellular uptake of both GMP and cisplatin by UMUC3 cells is a prerequisite to evaluating synergistic effects. Cellular uptake of GMP and cisplatin in separate NP was compared with that of the dual-drug combination in Combo NP (Figure 3A). Results indicated that Combo NP ratiometrically transported drugs into cells, which is consistent with the results from Combo free, while a mixture of separate NP (Sepa NP) cannot maintain the predetermined ratio of drugs because smaller cisplatin NP deliver their cargo into cells more efficiently than the larger GMP NP. This ratiometric uptake of Combo NP was also observed over a longer incubation of NP with cells (Figure 3B).
2.5. In Vitro Ratiometric Control over Dual-drug Release from PLGA NP
After verifying that Combo NP can ratiometrically transport the drugs into cells, we then studied the extracellular and intracellular release of Combo NP. The in vitro release kinetics of cisplatin and GMP from Combo NP, cisplatin NP and GMP NP were first investigated via dialysis in PBS (pH=7.4) at 37 °C for 96 h. The amount of platinum released from NP was measured by ICP-MS, while 3H-labeled CMP served as a marker for the measurement of GMP. It is notable that only negligible burst release was observed when the drugs inside DOPA-coated cores were encapsulated in PLGA NP (Figure 3C), although burst release phenomenon is well known and commonly observed for hydrophilic drugs in PLGA nanoparticulate formulation.[20, 22] For example, cisplatin incorporated PLGA15K-PEG5000 NP have shown a burst release in the initial 4 h with a release fraction of approximately 50% and gemcitabine encapsulated PLGA NP have shown 60% liberated drug in the initial 6 h[20a, 23]. This suggests that the DOPA layer prevents burst release of GMP and cisplatin from PLGA NP. Release kinetics of these two drugs in combination was further analyzed by grouped t-tests, which showed that there was no significant difference between these two drugs (p=0.78). This observation suggests that dual drugs in Combo NP followed a ratiometric release profile. The subtle difference in release rate may be due to the different composition of CP cores and GMP cores, yet the difference can be neglected when compared to the release rate of drugs from PLGA NP, which is a key rate-limiting step of the procedure. This indicates that release of cisplatin and GMP can be controlled at a similar rate and in a ratiometric manner when co-encapsulated into single PLGA NP. In order to further mimic the acidic endosome microenvironment[24], a release kinetics study was also carried out in pH 5.6 PBS for 96h. There were subtle changes in the release kinetics of the drugs and the ratio-controlled release of the dual drugs in Combo NP was still well-maintained (Figure S7).
Intracellular release of drugs from Combo NP was then studied. UMUC3 Cells were first incubated with Combo NP for 1, 4, or 16 h and subsequently washed. At each time points, cells were lysed with RIPA buffer, followed by separation of NP and free drugs via centrifugation at 16,000 g for 20 min. We found this method can extract more than 98% of NP and free drugs from cells with little destruction of NP. Results in Figure 3D indicated that a controlled and ratiometric release of cisplatin and GMP were also observed in the UMUC3 at the cellular level.
2.6. In Vitro Synergistic Effect of Combo NP
The in vitro cytotoxicity of free drugs and drug-loaded PLGA NP were evaluated by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Results showed that although subtle differences between the half-maximal inhibitory concentration (IC50) of free cisplatin and cisplatin NP existed, GMP NP resulted in a much lower IC50 of 17.8 μM compared with GMP free drug (IC50 of 34.8 μM), indicating that targeted NP delivery can maintain or enhance the cytotoxicity in vitro (Figure 3E). In addition, data revealed blank PLGA NP containing CaP core with negligible toxicity (data not shown). To validate the in vitro synergistic effect of Combo NP with dual-drug molar ratio of 5.3:1 (GMP:cisplatin), the combination index (CI) was further determined using the isobologram equation of Chou–Talalay.[21] As shown in Figure 3F, Combo NP displayed an overall CI value < 1 when Fa value was in the validated range of 0.2 to 0.8, indicating the pronounced and clear synergy of PLGA combo therapy in vitro.
2.7. In Vivo Anti-cancer Efficacy of Combo NP on Stroma-riched Bladder Xenograft Tumor Model
As previously mentioned, one of the most fundamental principles behind this formulation is to controllably deliver dual drugs into the tumor with an optimized ratio so as to achieve an enhanced anti-tumor efficacy in vivo. Therefore, different treatments were evaluated in an aggressive stroma-rich bladder cancer model, which was established by subcutaneously co-inoculating UMUC3 cells along with fibroblast NIH 3T3 cells in matrigel. Tumors were allowed to develop until their volume reached 100~150 mm3. Tumor bearing mice were then treated with a total of 3 injections at a dose of 12 mg/kg GMP and 1.9 mg/kg cisplatin in Combo NP. Cisplatin and GMP prepared in separate PLGA NP (Sepa NP) were administrated simultaneously in a mixture for comparison. Previous study in our lab has shown that blank PLGA NP have no tumor inhibition effect.[25] As shown in Figure 4A, free drugs showed little inhibitory effect at the same dose and dose schedule, possibly due to low tumor accumulation; while single drugs in PLGA NP demonstrated an enhanced therapeutic efficacy compared with free drugs. This is due to the EPR effect and receptor mediated endocytosis mentioned earlier. Dual drugs in Combo NP inhibited the growth of UMUC3 tumors most significantly without reducing the body weight (Figure 4A and Figure S8), indicating the enhanced anti-cancer effect and the safety of cisplatin and GMP in combination compared to single drugs. However, when the dual drugs were dosed together in a mixture (i.e., Sepa NP), tumor inhibition seemed to be compromised and the tumor weight on the last day of measurement was significantly higher than that of the Combo NP (Figure 4A). To further confirm the potent anti-cancer efficacy of Combo NP in the aggressive UMUC3 tumor model, a single injection of high dose Combo NP was administered and compared with low dose at regular dosing intervals. Results indicated that GMP and cisplatin in single high dose Combo NP showed potent efficacy, which is comparable to the effect of low dose at regular dosing intervals. Thus, only single injection could inhibit tumor growth in the aggressive stroma-rich tumor model (Figure 4C).
Figure 4.
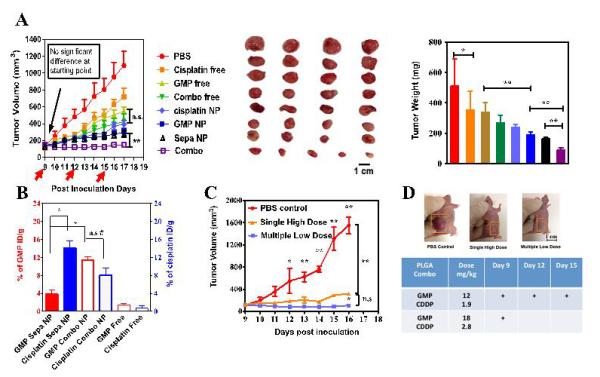
Tumor inhibition effects of free drugs, Combo free, cisplatin NP, GMP NP, Sepa NP and Combo NP on a stroma-rich UMUC3 bladder cancer xenograft model (A). Red arrows in panel A indicate time of injection. The tumors were treated with three IV injections at a dose of 1.9 mg/kg cisplatin and 12 mg/kg GMP in all the treatment groups. Tumor accumulation of cisplatin and GMP was calculated 10 h post injection of Combo NP, Sepa NP and Combo free at the injection dose of 1.9 mg/kg cisplatin and 12 mg/kg GMP into nude mice bearing stroma-rich bladder cancer xenograft tumors (B). Anti-tumor effects of multiple low dosing schedule and single high dosing schedule were compared (C). N=5; * P<0.05; ** P<0.01; #P >0.2; ## P>0.5; n.s: non-significant difference. ID/g: injected dose per gram tissue (tumor)
2.8. In Vivo Ratiometric Control over Dual-drug Tumor Accumulation in Xenograft Tumor Model
We postulated that Combo NP were more efficient in inhibiting growth of the tumor than Sepa NP due to the fact that Combo NP may deliver cisplatin and GMP into the tumor at the predetermined optimized synergistic ratio and dose. Tumor accumulation data indicated ratiometric accumulation of GMP and cisplatin from Combo NP over 10 h post injection (Figure 4B). However, higher uptake of cisplatin NP and lower uptake of GMP NP was observed after dosing with Sepa NP. On one hand, smaller particle size (around 60 nm) can account for higher tumor accumulation of cisplatin in single PLGA NP, while on the other hand, compared with 5.5 wt% loading of dual drugs in single PLGA NP, the same dose of 4.4 wt% cisplatin and 4.2 wt% GMP in separate PLGA NP doubles the amount of injected anisamide modified PLGA NP, which can result in saturation of sigma receptors and subsequently reduce the accumulation of GMP in tumors. This observation suggests advantages in controlling the ratio of drugs in DOPA coated cores in single PLGA NP over a mixture of separate NPs, which have variant physicochemical properties and distinct pharmacokinetics. Variations in the loaded ratio and actual amount of drug taken up by tumor tissues can directly affect the anti-tumor efficacy induced by synergy. In addition, nanoparticles also increased the tumor accumulation of free drugs from 2% ID/g to more than 10% ID/g due to the EPR effect and enhanced internalization into tumor cells through a receptor mediated pathway.
2.9. Combo NP Triggered Significant Tumor Cell Apoptosis and Inhibited Tumor Cell Proliferation Effectively In Vivo in Stroma-rich UMUC3 Xenografts
Enhanced antitumor efficacy of Combo NP was confirmed via analysis of apoptosis and proliferation. Tumor tissues after treatment were further sectioned for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay and PCNA (proliferating cell nuclear antigen) immunohistochemistry (Figure 5A and Figure 5B). Results indicated that Combo NP induced apoptosis in 28.8% of cells in UMUC3 xenograft tumors. Dual drugs in Sepa NP caused more cell apoptosis compared with cisplatin NP and GMP NP treatment, but were still significantly less efficient in inducing apoptosis than Combo NP. Free drugs induced few apoptotic cells in vivo, probably because the majority of the free drugs were metabolized and cleared before they accumulated in the tumor. In addition, the inhibition of tumor cell proliferation was investigated using PCNA assay. PCNA is expressed in the cell nuclei during DNA synthesis and can be used as a marker for cell proliferation. PCNA results were consistent with those of TUNEL assay. Combo NP showed minimal amounts of PCNA positive cells. These data further illustrated that combined drugs in a single NP inhibited the growth of the tumor through enhanced induction of apoptosis and reduced cell proliferation.
Figure 5.

Apoptosis (A) and proliferation (B) of tumor cells in vivo after administration of different treatments. Expression of XPA and ERCC-1, common in nucleotide excision repair (NER) systems, after three dosage systemic treatments (C). The formation of Pt-DNA adduct (green) in tumor cells detected by anti-Pt-DNA adduct antibody after systemic treatment (D). Bar chart in D is a quantitative analysis of % of Pt-DNA adduct in tissue sections. Five randomly selected microscopic fields were quantitatively analyzed on Image J. * P<0.05; ** P<0.01.
2.10. Mechanism of Synergistic Effect of the Dual-Drug Combo NP
In order to validate the observed enhanced antitumor effect of Combo NP is a synergistic effect imposed by GMP and cisplatin in the NP, subsequent studies were designed accordingly from a mechanistic basis. It is reported that gemcitabine potentiates the accumulation of cisplatin damage by suppressing the expression of key proteins involved in nucleotide excision repair (NER) and mismatch repair (MMR), leading to a decreased repair of Pt-DNA adducts, and thereby suppressed repair of cisplatin-induced DNA lesions.[12b, 26] Therefore, intensified inhibition of DNA repair and Pt-DNA adduct removal are two signs of synergistic interaction. The effect of combination therapy on ERCC1 and XPA[12b], two major proteins with key roles in NER was first examined by western blotting and showed that down-regulation of ERCC1 and XPA was induced by GMP free drug and enhanced by GMP NP treatment (Figure 5C). Combo NP almost completely depleted the expression of ERCC1 and XPA and was more efficient than Sepa NP. To study the effect of down-regulation of ERCC1 and XPA on Pt-DNA repair, Pt-DNA adducts were stained with FITC-labeled anti Pt-DNA adduct antibody. As shown in Figure 5D, a significant increase in the amount of Pt-DNA adducts was observed when tumors were treated with Combo NP, compared with that of Sepa NP.
The level of cleaved PARP and Caspase-3 were observed in order to further investigate the relationship of the suppressed DNA repair proteins and apoptosis. During the execution phase of apoptosis, intact PARP is mainly cleaved by caspase-3 or caspase-7 to a larger fragment and a smaller fragment. Therefore, PARP cleavage serves as a reliable marker of apoptosis.[13, 27] Figure S9 indicated that cleaved PARP was significantly elevated after treatment with Combo NP, which is consistent with the results of the DNA repair proteins and Pt-DNA adduct formation. Caspase-3 was also elevated after Combo NP treatment. Conclusively, Combo NP exhibited greater efficacy in inhibiting DNA repair and suppressing the removal of Pt-DNA adducts, leading to intensified apoptosis compared to dual drugs in separate NP in vivo. These results further verify that Combo NP acted in a synergistic fashion rather than only additive fashion to induce the enhanced anti-cancer effect in the stroma-rich bladder cancer xenograft model.
2. 11. Evaluation of Systemic Toxicity of Combo NP
Another important issue involved with combination therapy is the dual-drug distribution and ratio in major organs, as well as, the association of synergistic effects with toxicity in these organs. Quantitative biodistribution analyses of GMP and cisplatin in Combo NP indicated that the ratio of dual drugs remained constant in almost all organs (Figure S10). Similar to other nano-platforms, the major particle uptake organs were the liver (approximately 20% ID/g tissue) and the spleen (approximately 40% ID/g tissue) 10 h post injection. However, free drugs were eliminated rapidly from the body leaving the kidney as the major accumulation organ, which also explains the common nephrotoxicity of free cisplatin. Due to different particle size, cisplatin and GMP in separate nanoparticles presented very different distribution behaviors in vivo. Notably, cisplatin NP showed significantly higher accumulation in spleen, which might be a potential factor for inducing spleen toxicity.
Since the major side effect of GMP is myelosuppression and cisplatin can also induce an accumulated decrease in hematopoietic cell counts, a blood routine test was performed on healthy nude mice with three dosages of the 8 treatment groups. Both free GMP and cisplatin significantly reduced the levels of red blood cells (RBC), platelets (PLT) and white blood cells (WBC) compared to untreated control (Figure S12). Combination of these free drugs slightly potentiates the toxicity. Although there was an inevitable amount of accumulation of NP in the liver and kidney, blood biochemistry tests showed that NP coating can slightly alleviate the chemo-drug induced myelosuppression. There is no noticeable aggravation of blood toxicity in Combo NP. WBC, RBC, hematocrit (HCT), hemoglobin (HGB) of Combo NP were all close to the value of the untreated control (Figure S12).
Other hematological parameters showed that no detectable damage was caused; aspartate aminotransferase (AST), alanine aminotransferase (ALT) and blood urea nitrogen (BUN) analyses were all within the normal range (Table S2). No noticeable histological changes were seen in H&E-stained tissue sections of the liver, kidney and spleen (Figure S11). These studies demonstrated that Combo NP, with the most significant synergistic therapeutic efficacy, have elevated tumor uptake and low spleen accumulation, and as well exhibited no significant toxicity to major organs and tissues. Therefore, ratiometric synergistic combination therapy with non-overlapping toxicity is a promising strategy in overcoming drug resistance while enhancing anti-cancer effect.
3. Conclusions
Developing NP to simultaneously encapsulate drugs with different physicochemical properties with precise ratiometric loading and delivery is extremely important in the combination chemotherapy of malignant diseases. In the present study, we have successfully developed single nanocapsule-like PLGA particles with payloads of GMP cores and cisplatin cores. These dual-drug loaded NP exhibited precise ratiometric control over drug loading, cellular uptake, in vitro release and in vivo tumor accumulation. Furthermore, this single NP with well-controlled optimal dual-drug ratio exhibited a more significant antitumor efficacy compared with dual drugs in a mixture of separate NPs. Overall, our studies provide a solution to the problems of formulating cisplatin and other groups of hydrophilic drugs for ratiometric combination therapy and have therefore distinguished this single nanoparticulate delivery platform as an efficient and relatively safe candidate in the treatment of human bladder cancer.
This nanomaterial-system with spatially separated modalities prevents functional interference between individual molecules. Also, this system provides a possible well controlled platform for co-delivery chemotherapy with other hydrophobic ligand coated inorganic NP (e.g. ion oxide NP, gold NP, quantum dots and upconversion NP) for photothermal and theranostic purposes.
4. Experimental Section
Chemicals and Materials
Gemcitabine monophosphate disodium salt (GMP, purity ≥97%) was generously provided by Qualiber, Inc. (Chapel Hill, NC). Cisplatin was purchased from Sigma-Aldrich (Dorset, UK). Dioleoyl phosphatidic acid (DOPA) was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). mPEG3000-NH2.HCl and tBOC-PEG3500-NH2.TFA were purchased from JenKem Technology USA Inc. (Allen, TX). Acid terminated PLGA was ordered from DURECT Corporation (Cupertino, CA). N,N-diisopropylethylamine (DIPEA), 1-ethyl-3-(3-(dimethylamino)-propyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), dichloromethane, triton™ X-100, Igepal® CO-520, p-Anisic acid, silver nitrate and cyclohexane were purchased from Sigma-Aldrich (St Louis, MO) without further purification.
Cell Culture
The mouse embryonic fibroblast cell line (NIH 3T3) was obtained from UNC Tissue Culture Facility. The human bladder transitional cell line (UMUC3) was generously provided by Dr. William Kim (University of North Carolina at Chapel Hill, NC). These two cell lines were cultured in Dulbecco’s Modified Eagle’s Media (DMEM) (Invitrogen, Carlsbad, CA), supplemented with streptomycin (100 Ug/mL) (Invitrogen), penicillin (100 U/mL), and 10% Bovine calf serum (Hyclone, Logan, Utah) or 10% fetal bovine serum (Sigma, St. Louis, MO) respectively. Cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Experimental Animals
Female athymic nude mice used in all the studies weighed between 28-22 g and were 6-8 weeks of age. They were provided by the University of North Carolina animal facility. Animals were cared for in the Center for Experimental Animals (an AAALAC accredited experimental animal facility) at the University of North Carolina. Experimental animal handling procedures were performed following the protocols conformed to the Guide for the Care and Use of Laboratory Animals and approved by the University of North Carolina Institutional Animal Care and Use Committee.
Synthesis of PLGA-PEG-MBA and PLGA-mPEG
Briefly, for the synthesis of PLGA-PEG-MBA, tBoc-PEG3000-NH2.HCl (1 eq), anisic acid (8 eq) and DIPEA (4 eq) were dissolved in DCM and added with DIC (8 eq) to react for 26 h to obtain MBA-PEG-Boc. After purification and structure confirmation by NMR, Boc protecting group was removed using a TFA/DCM (1:2, v/v) mixture to achieve MBA-PEG-NH2.TFA. Afterwards, MBA-PEG-NH2.TFA was conjugated to PLGA (15 kDa, 0.1 mmol) in the presence of DIPEA and DIC for 26 h and purified. PLGA-PEG-MBA structure was confirmed by NMR. In the synthesis of PLGA-mPEG, mPEG-NH2.TFA (3000, 0.126 mmol), PLGA (15 kDa, 0.1 mmol) and DIPEA (0.5 mmol) were dissolved in 6 ml DCM and reacted with DIC (1.0 mmol) for 24 h.
Preparation of CP cores
CP cores were prepared as previously mentioned with a little adjustment.[9, 17] First, 300 μL of 200 mM cis-[Pt(NH3)2(H2O)2](NO3)2 was dispersed in a mixed solution of cyclohexane/triton-X100/hexanol (75:15:10, V:V:V) and cyclohexane/Igepal CO-520 (71:29, V:V) to form a well-dispersed reversed micro-emulsion. Another reversed micro-emulsion containing KCl was prepared by adding 300 μL of 800 mM KCl aqueous solution to a separate oil phase. Then, 500 μL of DOPA (20 mM) was added to the cisplatin precursor phase and the mixture was stirred. Twenty minutes later, the two emulsions were mixed and reacted for another 20 min. Forty mL of ethanol was then added to break the micro-emulsion and the mixture was centrifuged at 10,000 g for at least 15 min. The pellets were washed with ethanol 2 more times to ensure the complete removal of the surfactants and cyclohexane, and then re-dispersed in 2.0 mL of chloroform for storage.
Preparation of GMP cores
GMP cores were synthesized according to our previous work with a little adjustment.[27] Briefly, 100 μL of 60 mM GMP was mixed with 500 μL of 25 mM Na2HPO4 and then dispersed in 20 mL of oil phase containing Igepal CO-520/cyclohexane (29:71, V:V). The other emulsion was prepared by adding 600 μL of 2.5 M CaCl2 into a separate oil phase. Six-hundred mL of 20 mM DOPA was added to the phosphate phase before mixing of the two separate emulsions. Another 400 μL of 20mM DOPA was added to the combined emulsion 5 min after mixture. The emulsion was stirred for another 20 min and then 40 mL of ethanol was added. Next, the mixture was centrifuged at 10,000 g for 15 min to remove the surfactants and cyclohexane. After being washed with ethanol 2–3 times, the pellets were re-dispersed in 2.0 ml of chloroform for storage.
Preparation of PLGA/PLGA-PEG/PLGA-PEG-MBA (4:4:2) NP (PLGA NP) Loaded with cores
Drug encapsulated cores were loaded into PLGA NP using a single step solvent dispersion method as previously described with little adjustment.[25] Briefly, 2 mg of polymers and the cores were dissolved in 200 Ul of THF and added dropwise into 2 ml of water with continuous stirring at room temperature. Then, the NP suspension was stirred uncovered for 6 h at room temperature in order to remove the residual THF. The resulting NP were further purified by ultrafiltration (3000 × g, 15 min, Amicon Ultra, Ultracel membrane with 50,000 NMWL, Millipore, Billerica, MA). The obtained PLGA NP were then re-suspended, washed twice with water, centrifuged at 14,000 rpm for 20 min to further remove free lipids and micelles. And then re-suspended again and centrifuged at 800 rpm to remove nanocore aggregations.
Characterization of PLGA NP
DL and EE of cisplatin were measured using Inductively Coupled Plasma-Mass Spectroscopy (ICP-MS, NexIONTM 300, Perkin Elmer Inc); LE and EE of GMP were both measured by Ultraviolet–Visible Spectroscopy (UV, DU®800, Beckman Coulter) and 3H labeled cytidine 5’ monophosphate (CMP) [5-3H] disodium salt (Moravek Bio Inc, 1 mCi/mL) incorporation using a Liquid Scintillation Analyzer (TRI-CARB 2900 TR, Packard Bioscience Co). The size distribution of particles was determined using a Malvern ZetaSizer Nano series (Westborough, MA). TEM images of NP were obtained using a JEOL 100CX II TEM (JEOL, Japan). For NP imaging, the NP were negatively stained with 2% uranyl acetate. The composition of PLGA combo NP was studied using Electron Dispersive Spectroscopy (EDS) (Oxford instruments, INCA PentaFET -x3) and X-ray photoelectron spectroscopy (XPS) (Kratos Axis Ultra DLD X-ray Photoelectron Spectrometer).
Cellular Uptake Study in UMUC3 Cell Lines
UMUC3 cells were seeded into a 12-well plate (1.5 × 105 cells/well) containing 1 ml of media. Twenty-four hours later, 1 ml of the free drug combination, targeted PLGA Combo NP, targeted PLGA Sepa NP, 20%-targeted PLGA Combo NP or non targeted PLGA Combo at a concentration of 20 μM GMP and 3.8 μM cisplatin were incubated with cells in a serum-free medium. Four hours later, cells were treated with RIPA buffer (Sigma-Aldrich). The concentration of cisplatin was measured using ICP-MS and GMP was measured as 3H-CMP using a scintillation counter as previously mentioned.
In Vitro Release and Intracellular Release of cisplatin and GMP from PLGA NP
The dialysis technique was employed to study the in vitro release of GMP and cisplatin from PLGA in phosphate buffered saline (PBS) (pH 7.4 or pH 5.6) at 37 °C. Five hundred μL of PLGA NP loaded with 100 μg/mL of GMP and cisplatin separately or co-loaded with 100 μg/mL of GMP and cisplatin at a ratio of 5.33:1 were added into the dialysis tube with a molecular weight cut off of 3000 Da and dialyzed against 15 mL of PBS (pH 7.4 or pH 5.6) in a thermo-controlled shaker with a stirring speed of 200 rpm at 37 °C for 96 h. In the preparation of GMP cores, a trace amount of radioactive 3H-CMP was mixed with GMP to serve as a marker for the entrapped GMP. At each predetermined time point, 400 μL samples were taken and replaced with fresh media. Platinum and GMP concentrations were then determined by ICP-MS and scintillation analyzer respectively at specified times. All experiments were performed in triplicate and the data were reported as mean ± SD of the three individual experiments. Measuring of intracellular release of free drugs from the nanoparticles was carried out according to a previous protocol.[9] Briefly, a 12-well plate of UMUC3 cells was prepared as mentioned in the uptake study and incubated with 20 μM of GMP and 3.8 μM of cisplatin encapsulated into PLGA Combo NP. After 1, 4, and 16 hours, the cells were treated with 50 μL of RIPA buffer (Sigma-Aldrich) at 4°C for 10 min and the cell lysate was centrifuged at 14,000 rpm for 20 min at 4°C to separate nanoparticle and cell lysate from free drugs. Free drugs and nanoparticles were measured using ICP-MS and 3H-labeled scintillation. All experiments were performed in four replicates and the data reported as mean ± SD.
In vitro Cell Viability on UMUC3 Cells and Analysis of Synergistic Effects of Drug Combinations
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay was conducted to detect in vitro viability of free GMP, cisplatin and their combinations as well as PLGA GMP NP, PLGA cisplatin NP and PLGA Combo NP. In Brief, cells were seeded in 96-well plates with a density of 3,000 cells per well 24 h prior to drug treatment. On the second day, cells were treated with free drugs or the drug combination at a series of dilutions with various molar ratios. Forty-eight h post treatment, 20 μL of MTT (5 mg/mL) reagent was added for another 4 h at 37 °C. The medium was then discarded and the formed formazan salt was dissolved in 150 μL of DMSO. The absorbance in each well was read at the wavelength of 570 nm using a multidetection microplate reader (Plate CHAMELEON™ V-Hidex).Each concentration was tested in five wells and data presented as mean ± SD. The mean drug concentration required for 50% growth inhibition (IC50) was calculated using CompuSyn software (Version 1.0, Combo-Syn Inc., U.S.) with the median effect equation: Fa=[1+(IC50/D)m]−1, where, m is the Hill slope, D is drug concentration and Fa is the fraction of affected cells.
Combination Index (CI) Analysis of free drug combination based on the Chou and Talalay method[21] was conducted using CompuSyn software. Briefly, for each level of Fa, the CI values of cisplatin and GMP combinations were calculated according to the following equation: CI=(D)1/(Dx)1+(D)2/(Dx)2, where (Dx)1 and (Dx)2 are the concentrations of the drugs alone resulting in Fa×100% growth inhibition, while (D)1 and (D)2 are the concentrations of each drug in the combination leading to Fa×100% growth inhibition. CI values of the drug combinations were drawn as a function of Fa. CI values more than 1 or less than 1 indicate antagonism or synergism of drug combinations, respectively. Notably, CI values between Fa 0.2 to 0.8 are considered validate.[28]
Tumor Accumulation of GMP and Cisplatin in Stroma-rich Xenograft Bladder Tumor Model
A stroma-rich subcutaneous xenograft bladder tumor model was established previously in our lab.[18] Briefly, UMUC3 (5×106) and NIH 3T3 cells (2×106) in 100 μL of PBS were subcutaneously co-injected into the right flank of mice along with Matrigel (BD Biosciences, CA) at a ratio of 3:1 (v/v). When the tumor reached 100-150 mm2 in size, animals were randomly divided into three groups (n=8) and intravenously injected with free GMP and cisplatin (Combo Free), PLGA Combo NP and PLGA Sepa NP at a dose of 12 mg/kg GMP and 1.9 mg/kg cisplatin respectively. Trace fraction of 3H-CMP was added to the GMP related groups for the measurement of tumor accumulation of GMP. Four mice from each group were sacrificed at each predestined time point and tumor tissues were collected. Tumor uptake of GMP and cisplatin was expressed as the percentage of the injected dose per gram tumor. For measurement of GMP, 10-20 mg of tumor tissue was immediately mixed with 10× NCS® II Tissue Solubilizer (Amersham Biosciences, Inc) and digested at 60°C overnight. Three hundred μL of hydrogen peroxide (30% in water, Fisher) was then added to the samples and vortexed to bleach the color. The sample was then mixed with 4 mL of scintillation cocktail (Fisher Inc). The radioactivity of 3H in the tumor samples was counted using a liquid scintillation analyzer (TRI-CARB 2900 TR, Packard Bioscience Co.). For the measurement of cisplatin, 25-35 mg of tumor tissue was digested with 400 μL 60% nitric acid (Acros Organic) at 70°C overnight and the amount of platinum was measured by ICP-MS.
Biodistribution of Dual Drug in Major Organs
Mice were administered a single dose of Combo Free, PLGA Sepa NP and PLGA Combo NP respectively at a dose of 1.9 mg/kg cisplatin and 12 mg/kg GMP. Each group contained four mice, which were sacrificed 10 h following injection. Tissue samples were digested as previously mentioned in the tumor accumulation study. Cisplatin was quantified via ICP-MS and GMP via scintillation counter.
Anti-tumor Efficacy in Stroma-rich Xenografts
When the inoculated tumor reached 100-150 mm2 in size, mice were randomized into eight groups (n=5) as follows: Untreated Control (PBS), free GMP (GMP free), free cisplatin (Cisplatin Free), combination of free GMP and cisplatin (Combo free), PLGA GMP NP, PLGA cisplatin NP, GMP and cisplatin PLGA NP mixtures (PLGA Sepa NP) as well as PLGA Combo NP. IV injections were performed every three days for a total of three injections with a GMP dose of 12 mg/kg and a cisplatin dose of 1.9 mg/kg. Tumor volume was measured every day. Body weight was also recorded. Mice were sacrificed two days after the last injection and tumor tissues were collected for further study.
TUNEL Assay
All the immunofluorescence detections mentioned in this manuscript on UMUC tumor bearing mice were prepared using paraffin embedded sections (prepared by the UNC Tissue Procurement Core). Slides were deparaffinized through xylene and a graded alcohol series and prefixed with 4% formaldehyde. Apoptosis was then detected by TdT-dependent dUTP-biotin nick end labeling (TUNEL) assay using an apoptosis detection kit (Promega, Madison, WI) following the manufacturer’s instructions. Slides were then coverslipped using VECTASHIELD with DAPI (Vector laboratories, Burlingame, CA). All staining was evaluated and digital images were acquired by an Eclipse Ti-U inverted microscope (Nikon Corp., Tokyo, Japan) at 20× magnification. Five randomly selected microscopic fields were quantitatively analyzed using Image J (National Institutes of Health).
PCNA Assay
Proliferation of tumor cells after the aforementioned treatments was detected by antibody against proliferating cell nuclear antigen (PCNA) (1:200 dilution, Santa Cruz).[13] The paraffin embedded tissue sections were deparaffinized, antigen recovered, blocked and probed with PCNA antibody overnight at 4°C, and then detected using a mouse-specific HRP/DAB detection IHC kit as recommended by the manufacturer (Abcam, Cambridge, MA). Cell nuclei were counter-stained with hematoxylin. The percentage of proliferative cells was calculated by dividing the number of PCNA positive cells (shown as brown dots) by the number of total cells (blue nuclei stained by hematoxylin). Five representative microscopic fields were randomly selected in each treatment group for quantification.
Platinum Adduct staining
The platinum-DNA adducts were detected using anti-cisplatin modified DNA antibodies [CP9/19] (Abcam, Cambridge, MA).[9] The tumor sections were deparaffinized, antigen recovered, blocked with 1% BSA/PBS for 1h at room temperature, incubated with a 1:250 dilution of anti-cisplatin modified DNA antibody [CP9/19] at 4°C overnight, and then incubated with FITC-labeled goat anti-rat IgG antibody (1:200,Santa Cruz, CA). The sections were also counter-stained with VECTASHIELD mounting media with DAPI (Vector laboratories, Burlingame, CA). The tumor sections were observed and quantified using a Nikon light microscope (Nikon Corp., Tokyo, Japan).
Western-blot Analysis
Two days after three daily IV injections, UMUC tumor bearing mice were sacrificed and tumor tissues were collected and lysed using radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich). The concentration of total protein in the tumor lysate was quantified using bicinchoninic acid (BCA) protein assay reagent following the manufacturer’s instruction (Invitrogen). After dilution with 4× sample buffer containing reducing agent and heating at 95 °C for 5 min, forty μg of protein per lane was separated by 4-12% SDS-PAGE electrophoresis (Invitrogen). The proteins were then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The membranes were blocked with 5% skim milk for 1 h and incubated overnight at 4°C with mouse monoclonal poly(ADP-ribose) polymerase-1 (PARP-1) antibodies, mouse monoclonal ERCC1, mouse monoclonal XPA (12F5). GAPDH antibody (1:4000 dilution; Santa Cruz biotechnology, Inc.) was used as the internal loading control. The membranes were washed three times and then incubated with a secondary antibody (1:4000 dilution; Santa Cruz biotechnology, Inc.) at room temperature for 1 h. Goat anti-mouse secondary antibody was used for PARP, XPA and ERCC-1 primary antibody. Goat anti-rabbit secondary antibody was used for GAPDH primary antibody. Finally, the membranes were washed four times and detected using the Pierce ECL Western Blotting Substrate according to the manufacturer’s instructions (Thermo Fisher Scientific).
Serum biochemical value analysis and hematology assay
After three injections, blood was collected and centrifuged at 4000 rpm for 5 min to obtain the serum. Blood urea nitrogen (BUN), creatinine, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels, were assayed as indicators of renal and hepatic function. Whole blood was collected from healthy nude mice after three repeated treatments. Red blood cells (RBC), white blood cells (WBC), platelets (PLT), hemoglobin (HGB) and hematocrits (HCT) were counted for the detection of myelosuppression. Organs (heart, liver, spleen, lung, and kidney) were fixed and sectioned for H&E staining as to evaluate the organ-specific toxicity.
Statistical Analysis
Quantitative results were expressed as mean ± SD. The analysis of variance was completed using student's t-test and one-way analysis of variance (ANOVA). A p value of p < 0.05 was considered statistically significant.
Supplementary Material
Table 1.
Characteristic features of the optimized single drug PLGA NP and dual Drug PLGA Combo NP
| Optimal | CDDP PLGA NP |
GMP PLGA NP |
GMP & CDDP PLGA Combo NP |
|---|---|---|---|
| DL (wt%) | 4.4 ± 0.6 | 4.2 ± 0.3 | 5.5 ± 0.8 |
| EE (%) | 74.0 ± 10.0 | 69.5 ± 1.6 | 86.6 ± 1.9 & 92.4 ± 1.6 |
(n=3)
Table 2.
Effect of Different Treatments on serum ALT, AST, BUN and creatinine levels
| Treatment | BUN mg/dL | Creatinine mg/dL | AST U/L | ALT U/L |
|---|---|---|---|---|
| PBS | 19 ± 1 | 0.2 | 228 ± 13 | 60 ± 14 |
| Cisplatin free | 25 ± 1 | 0.2 | 216 ± 15 | 59 ± 1 |
| GMP free | 24 ± 2 | 0.2 | 122 ± 20 | 47 ± 3 |
| Combo free | 22 ± 5 | 0.2 | 116 ± 18 | 60 ± 4 |
| Cisplatin NP | 28 ± 3 | 0.2 | 245 ± 22 | 58 ± 11 |
| GMP NP | 21 ± 1 | 0.2 | 86 ± 6 | 42 ± 2 |
| Sepa NP | 29 ± 2 | 0.3 | 238 ± 10 | 55 ± 8 |
| Combo NP | 18 ± 3 | 0.2 | 122 ± 12 | 52 ± 12 |
| Normal Range | 12 - 33 | 0.2 - 0.9 | 54 - 298 | 17 - 132 |
(n=5)
Acknowledgements
This work was supported by NIH grants CA151652 and CA149363. We thank Steven Glenn Plonk and Andrew Mackenzie Blair for their assistance in manuscript preparation. We thank Dr. Yuan Zhang for providing method of preparing GMP cores. We also thank Nazar Filonov and Nanomedicines Characterization Core Facility at UNC for platinum quantification using ICP-MS.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Reference
- [1].a) Greco F, Vicent MJ. Adv. Drug Deliv. Rev. 2009;61:1203. doi: 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]; b) Broxterman HJ, Georgopapadakou NH. Drug Resist. Updat. 2005;8:183. doi: 10.1016/j.drup.2005.07.002. [DOI] [PubMed] [Google Scholar]
- [2].a) Mayer LD, Janoff AS. Mol. Interv. 2007;7:216. doi: 10.1124/mi.7.4.8. [DOI] [PubMed] [Google Scholar]; b) Mayer LD, Harasym TO, Tardi PG, Harasym NL, Shew CR, Johnstone SA, Ramsay EC, Bally MB, Janoff AS. Mol. Cancer Ther. 2006;5:1854. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]; c) Tardi PG, Dos Santos N, Harasym TO, Johnstone SA, Zisman N, Tsang AW, Bermudes DG, Mayer LD. Mol. Cancer Ther. 2009;8:2266. doi: 10.1158/1535-7163.MCT-09-0243. [DOI] [PubMed] [Google Scholar]
- [3].Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Proc. Natl. Acad. Sci. USA. 2010;107:17939. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Licona Limon P, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM. Nat. Mater. 2012;11:895. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat. Mater. 2011;10:389. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang C, Li Z, Cao D, Zhao YL, Gaines JW, Bozdemir OA, Ambrogio MW, Frasconi M, Botros YY, Zink JI, Stoddart JF. Angew Chem. Int. Ed. Engl. 2012;51:5460. doi: 10.1002/anie.201107960. [DOI] [PubMed] [Google Scholar]; d) Wang H, Wu Y, Zhao R, Nie G. Adv. Mater. 2013;25:1616. doi: 10.1002/adma.201204750. [DOI] [PubMed] [Google Scholar]
- [5].a) Aryal S, Hu CM, Zhang L. Mol. Pharm. 2011;8:1401. doi: 10.1021/mp200243k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hu CM, Zhang L. Biochem. Pharmacol. 2012;83:1104. doi: 10.1016/j.bcp.2012.01.008. [DOI] [PubMed] [Google Scholar]
- [6].a) Hu SH, Gao X. J. Am. Chem. Soc. 2010;132:7234. doi: 10.1021/ja102489q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen XW, Sneed KB, Pan SY, Cao C, Kanwar JR, Chew H, Zhou SF. Current drug metabolism. 2012;13:640. doi: 10.2174/1389200211209050640. [DOI] [PubMed] [Google Scholar]
- [7].Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J. N. Engl. J. Med. 2010;362:1273. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- [8].a) Lee SM, O'Halloran TV, Nguyen ST. Journal of the American Chemical Society. 2010;132:17130. doi: 10.1021/ja107333g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18638. doi: 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guo S, Wang Y, Miao L, Xu Z, Lin CM, Zhang Y, Huang L. ACS Nano. 2013;7:9896. doi: 10.1021/nn403606m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Cancer research. 1998;58:4349. [PubMed] [Google Scholar]
- [11].Plunkett W, Huang P, Gandhi V. Anti-cancer drugs. 1995;6(Suppl 6):7. doi: 10.1097/00001813-199512006-00002. [DOI] [PubMed] [Google Scholar]
- [12].a) Moufarij MA, Phillips DR, Cullinane C. Mol. Pharmacol. 2003;63:862. doi: 10.1124/mol.63.4.862. [DOI] [PubMed] [Google Scholar]; b) Besancon OG, Tytgat GA, Meinsma R, Leen R, Hoebink J, Kalayda GV, Jaehde U, Caron HN, van Kuilenburg AB. Cancer Lett. 2012;319:23. doi: 10.1016/j.canlet.2011.12.016. [DOI] [PubMed] [Google Scholar]
- [13].Zhang Y, Kim WY, Huang L. Biomaterials. 2013;34:3447. doi: 10.1016/j.biomaterials.2013.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li J, Yang Y, Huang L. J. Control. Release. 2012;158:108. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hu Y, Haynes MT, Wang Y, Liu F, Huang L. ACS Nano. 2013;7:5376. doi: 10.1021/nn4012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu Z, Ramishetti S, Tseng YC, Guo S, Wang Y, Huang L. J. Control. Release. 2013;172:259. doi: 10.1016/j.jconrel.2013.08.021. [DOI] [PubMed] [Google Scholar]
- [17].Guo S, Miao L, Wang Y, Huang L. J. Control. Release. 2014;174:137. doi: 10.1016/j.jconrel.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang J, Miao L, Guo S, Zhang Y, Zhang L, Satterlee A, Kim WY, Huang L. J. Control. Release. doi: 10.1016/j.jconrel.2014.03.016. DOI 10.1016/j.jconrel.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Vivero-Escoto JL, Taylor-Pashow KM, Huxford RC, Della Rocca J, Okoruwa C, An H, Lin W, Lin W. Small. 2011;7:3519. doi: 10.1002/smll.201100521. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nakagawa O, Ming X, Huang L, Juliano RL. J. Am. Chem. Soc. 2010;132:8848. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. J. Control. Release. 2002;79:123. doi: 10.1016/s0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]; b) Aggarwal S, Yadav S, Gupta S. J. Biomed. Nanotechnol. 2011;7:137. doi: 10.1166/jbn.2011.1238. [DOI] [PubMed] [Google Scholar]
- [21].Chou TC, Talalay P. Adv. Enzyme Regul. 1984;22:27. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- [22].Pantazis P, Dimas K, Wyche JH, Anant S, Houchen CW, Panyam J, Ramanujam RP. Methods Mol. Biol. 2012;906:311. doi: 10.1007/978-1-61779-953-2_25. [DOI] [PubMed] [Google Scholar]
- [23].Martin-Banderas L, Saez-Fernandez E, Holgado MA, Duran-Lobato MM, Prados JC, Melguizo C, Arias JL. Int. J. Pharm. 2013;443:103. doi: 10.1016/j.ijpharm.2012.12.048. [DOI] [PubMed] [Google Scholar]
- [24].Tong Z, Luo W, Wang Y, Yang F, Han Y, Li H, Luo H, Duan B, Xu T, Maoying Q, Tan H, Wang J, Zhao H, Liu F, Wan Y. PloS one. 2010;5:e10234. doi: 10.1371/journal.pone.0010234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guo S, Lin CM, Xu Z, Miao L, Wang Y, Huang L. ACS Nano. doi: 10.1021/nn5010815. DOI: 10.1021/nn5010815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) CJA van Moorsel HP, Veerman G, Bergman AM, Kuiper CM, Vermorken JB, van der Vijgh WJF. Br. J. Cancer. 1999;80:10. doi: 10.1038/sj.bjc.6690452. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liedert B, Pluim D, Schellens J, Thomale J. Nucleic Acids Res. 2006;34:e47. doi: 10.1093/nar/gkl051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Y, Peng L, Mumper RJ, Huang L. Biomaterials. 2013;34:8459. doi: 10.1016/j.biomaterials.2013.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, Kabanov AV. Mol. Pharm. 2012;9:2302. doi: 10.1021/mp300159u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.