

Abstract
Rationale: Progress has been made in understanding how the cystic fibrosis (CF) basic defect produces lung infection susceptibility. However, it remains unclear why CF exclusively leads to chronic infections that are noninvasive and highly resistant to eradication. Although biofilm formation has been suggested as a mechanism, recent work raises questions about the role of biofilms in CF.
Objectives: To learn how airway conditions attributed to CF transmembrane regulator dysfunction could lead to chronic infection, and to determine if biofilm-inhibiting genetic adaptations that are common in CF isolates affect the capacity of Pseudomonas aeruginosa to develop chronic infection phenotypes.
Methods: We studied P. aeruginosa isolates grown in agar and mucus gels containing sputum from patients with CF and measured their susceptibility to killing by antibiotics and host defenses. We also measured the invasive virulence of P. aeruginosa grown in sputum gels using airway epithelial cells and a murine infection model.
Measurements and Main Results: We found that conditions likely to result from increased mucus density, hyperinflammation, and defective bacterial killing could all cause P. aeruginosa to grow in bacterial aggregates. Aggregated growth markedly increased the resistance of bacteria to killing by host defenses and antibiotics, and reduced their invasiveness. In addition, we found that biofilm-inhibiting mutations do not impede aggregate formation in gel growth environments.
Conclusions: Our findings suggest that conditions associated with several CF pathogenesis hypotheses could cause the noninvasive and resistant infection phenotype, independently of the bacterial functions needed for biofilm formation.
Keywords: cystic fibrosis, chronic infection, Pseudomonas aeruginosa, biofilm
At a Glance Commentary
Scientific Knowledge on the Subject
Pseudomonas aeruginosa infections in cystic fibrosis remain localized to the airway surface, and are highly resistant to eradication even at their onset. This is surprising because in patients who have defective lung host defenses, P. aeruginosa can be highly invasive, and the bacteria are antibiotic-sensitive until later in disease. The bacterial mechanisms and host conditions producing the chronic and eradication-resistant infection phenotype remain poorly understood.
What This Study Adds to the Field
We show that conditions associated with several cystic fibrosis pathogenesis hypotheses cause P. aeruginosa to grow in cell aggregates, and that aggregate formation does not require functions needed for biofilm development. Aggregates are highly resistant to antibiotics and host defenses, and exhibit reduced invasive virulence. These findings could explain how cystic fibrosis airway conditions promote the noninvasive and resistant infection phenotype, and shed light on the growth mode of bacteria in cystic fibrosis sputum.
Cystic fibrosis (CF) leads to life-long airway infections and eventual respiratory failure (1). Many patients are infected with Pseudomonas aeruginosa, and most deaths are attributed to this organism (2). Among the many poorly understood aspects of CF infections, two characteristics standout in terms of clinical importance.
First, CF P. aeruginosa infections are almost always chronic and noninvasive (1, 2), and it is unclear how a defect compromising host defenses would exclusively produce this infection phenotype. Other genetic immunodeficiency diseases cause a wider spectrum of illness ranging from recurrent acute infections, to systemic disease that disseminates, and chronic infections (3, 4). The noninvasive phenotype of CF infections is also surprising given the virulence potential of P. aeruginosa, and the enormous burden of organisms present in CF lungs (1, 2).
Second, chronic CF infections are extremely resistant to eradication by host defenses and antibiotics (1, 2). This resistance is not caused by insufficient inflammatory responses or inadequate antibiotic levels, because inflammation is intense (5, 6) and high concentrations of drugs reach the airways (7). Genetic resistance mechanisms of infecting bacteria are also not responsible, because treatment fails early on, when the bacteria are antibiotic-sensitive (8, 9).
Thus, the noninvasive and eradication-resistant nature of CF infections is not explained by the intrinsic properties of P. aeruginosa or other CF pathogens, or solely by the existence of an innate immune defect. This fact has led to the hypothesis that conditions produced by the basic CF defect somehow induce these infection phenotypes. However, how airway conditions could do this remains unclear.
Work by several laboratories (including ours) suggested that biofilm formation could be responsible (10–12). Biofilms are surface-attached, matrix-encased bacterial structures. However, recent work raises questions about the role of biofilms, as typically defined, in CF infections. For example, bacteria in CF lungs appear as aggregates suspended in airway sputum, and the morphology of these structures differs markedly from laboratory biofilms (10–16). Perhaps more importantly, conditions in CF airways consistently select against bacterial functions deemed essential for biofilm formation during in vivo bacterial evolution (17–21). In fact, P. aeruginosa isolates from chronically infected patients are often impaired in forming biofilms (22–25). Finally, the link between biofilms and reduced invasive virulence is limited. Indeed, some studies indicate that biofilm growth can actually increase the capacity of bacteria to disseminate in vivo (27, 28).
Here we used a system involving gels as growth substrates for P. aeruginosa to address two key questions about chronic CF infections. First, we investigated how airway conditions attributed to CF transmembrane regulator (CFTR) dysfunction could produce the phenotypes characteristic of CF infections: resistance to eradication and reduced invasiveness. Second, we examined whether biofilm-inhibiting genetic adaptations, which occur commonly in CF, affect the capacity of bacteria to develop these phenotypes when bacteria are grown in sputum and agar gels. Some of the results of these studies have been previously reported in the form of an abstract (29).
Methods
Bacterial Strains, Chemicals, and Growth Conditions
P. aeruginosa strains were obtained from the University of Washington’s transposon library (30), and ΔflgK mutant was complemented as described (31). Clinical isolates and sputum samples were obtained after informed consent. Bacteria were grown in Mueller-Hinton broth unless indicated. Sputum supernatants were generated by 1:1 H2O dilution, followed by sonication. Heat treatment used 100°C for 10 minutes. Agar gels were made using 50–100 bacteria per milliliter, 100 μM sodium nitrate, Mueller-Hinton broth, and molten noble agar; and incubated at 37°C for 24 hours before microscopy and antimicrobial treatment. Mucus gels were generated using 2.5% or 8% porcine gastric mucus as described (32). Where indicated 10% sputum supernatants, serine protease inhibitor, neutrophil elastase (NE) inhibitor, or NE were added. Additional information is in the online supplement.
Antimicrobial Treatments, Biofilm and Motility Assays, and Elastase Measurements
Gels were treated with 10 μg/ml tobramycin, 1 mg/ml ceftazidime, or 2.5 and 5 μg/ml ciprofloxacin for 3 hours; hydrogen peroxide (100 mM) or hypochlorous acid (2 mM) for 2 hours; or neutrophils at a 1:1 polymorphonuclear leukocyte/bacterial ratio for 2 hours. Biofilm, swimming motility assays, and elastase activity measurements are described (33–35). Additional information is in the online supplement.
Epithelial Cell Culture and Animal Infections
Bronchial epithelial cell culture and transepithelial resistance measurements were performed as described (36, 37). Western blots were performed with anti-Occludin, anti–junctional adhesion molecule-A, and anti–glyceraldehyde phosphate dehydrogenase. All animal experiments were approved by the University of Washington’s Animal Care Committee. Additional information is in the online supplement.
Results
As a first step in exploring the link between the CF basic defect and the noninvasive and eradication-resistant infection phenotypes, we examined how conditions associated with major CF pathogenesis hypotheses could affect the growth mode of P. aeruginosa.
High-Density Gels Promote the Formation of Bacterial Aggregates
One group of CF hypotheses implicates increased density of airway mucus. Postulated mechanisms include impaired bicarbonate secretion, which could alter mucus gel properties (38); increased sodium channel (ENaC) activity (39, 40), which could produce sodium and water hyperabsorption; and abnormalities in mucus carbohydrate or sulfate composition (41, 42).
We began studying the effect of gel density using gels of varying agar concentrations. We chose this approach because the hypotheses invoking increased mucus density involve different mechanisms, and our initial objective was to study the general effects of gel density on bacterial phenotypes. In low-agar concentrations, bacteria grew in a dispersed manner with cells distributed throughout the gel (Figures 1A and 1B). At agar concentrations of 0.6% or higher, the bacteria grew as multicellular aggregates, separated from one another (Figures 1A and 1B; see Figure E1A in the online supplement). Differential growth yields did not account for this phenotype, because bacterial density was similar in agar gels ranging from 0% to 1% (see Figure E2). Consistent with previous work (32), increased mucus gel density also promoted aggregate formation (Figure 1C).
Figure 1.

High-density gels promote the growth of Pseudomonas aeruginosa in cell aggregates. (A) Photographs of agar gels in which P. aeruginosa had been grown for 24 hours. In low-density agar (0.2%), cells grow in a dispersed manner producing a haze that darkens the gel. Bacterial aggregates (which appear as dark colored spheres) are apparent in high-density (0.8%) agar. Low- and high-density gels contain similar numbers of bacteria (see Figure E2). Scale bar represents 1 mm. (B) Confocal microscopy images showing the dispersed growth mode of SYTO 9–stained P. aeruginosa in low-density agar gels (0.2%), and an aggregate that formed in high-density (0.8%) agar gels (aggregates appear as dark colored spheres in the photograph of Figure 1A). Scale bars represent 40 μm. (C) Fluorescent microscopy images of green fluorescent protein–labeled P. aeruginosa grown for 24 hours in low- (2.5%) and high-density (8%) mucus gels. In low-density mucus, bacteria grow as dispersed cells, and in high-density mucus the bacteria grow as aggregates. Scale bars represent 100 μm. (D) Photographs of agar gels in which a P. aeruginosa swimming motility mutant (ΔflgK) had been grown for 24 hours. The motility mutant grows as cell aggregates in low- and high-density agar gels. Scale bar represents 1 mm. (E) Confocal microscopy images showing the aggregated growth mode of SYTO 9–stained ΔflgK P. aeruginosa in low- and high-density agar gels. Scale bars represent 40 μm. (F) Fluorescent microscopy images of GFP-labeled ΔflgK P. aeruginosa grown for 24 hours in low- (2.5%) and high-density (8%) mucus gels. ΔflgK P. aeruginosa grow as aggregates in low- and high-density mucus. Scale bars represent 100 μm. Images in A–F are representative of five experiments.
Restricted Bacterial Motility Mediates Aggregate Formation
We performed additional studies to understand the mechanism of aggregate formation. A simple explanation was that high gel density restricts motility and forces bacteria into localized groups. If true, inactivating motility genes should promote aggregate formation in low-density gels. As expected, a P. aeruginosa motility mutant (ΔflgK) formed aggregates in low-density agar (Figures 1D and 1E; see Figure E1B) and mucus gels (Figure 1F), and genetic complementation reversed this phenotype (see Figure E3). Thus, high gel density likely promotes aggregate formation by limiting bacterial motility.
CF Sputum Promotes Aggregate Formation in Low-Density Gels
Although hypotheses involving increased mucus density are compelling, other work implicates defects that do not involve physical changes in mucus. For example, inactivation of CFTR has been reported to impair bacterial killing by epithelial or phagocytic defenses (43–48), and cause hyperactive inflammation (49–51).
We explored the possibility that CF-related conditions other than increased gel density could promote aggregate formation by adding soluble components of CF sputum into agar gels, where the density could be controlled. Gels containing 10% sputum supernatants supported similar growth yields as the laboratory medium we used, and was used for additional studies (see Figure E4). Whereas P. aeruginosa grew in a dispersed manner in low-density agar and mucus gels (0.2% agar and 2.5% mucus) containing laboratory medium (Figures 1A–1C), the addition of sputum supernatants produced aggregated bacterial growth (Figure 2A). Supernatants from all five patients with CF tested produced the same effect.
Figure 2.

Neutrophil elastase in cystic fibrosis (CF) sputum promotes aggregated bacterial growth in low-density gels. (A) CF sputum supernatants (10%) caused aggregated growth of wild-type Pseudomonas aeruginosa in low-density agar (left) and mucus (right) gels (top panels), but heat-treated supernatants did not (bottom panels). Images of low-density (0.2%) agar gels (left) are photographs, scale bar represents 1 mm. Images of mucus gels are fluorescence micrographs of green fluorescent protein–labeled bacteria, scale bar represents 100 μm. Images are representative of five experiments. (B) P. aeruginosa swimming motility is inhibited by 10% CF sputum supernatant but not by heat-treated (HT) CF supernatants. The swimming distance was measured after overnight incubation at 37°C. Data are the mean of three experiments, error bars represent SEM. *P ≤ 0.05. (C) Neutrophil elastase activity measurements in CF sputum, HT CF sputum, and sputum treated with a specific neutrophil elastase inhibitor (NEI) at 100 μM. Data are the means of four measurements of sputum from one subject, and representative of samples from five other subjects. Error bars represent SEM. *P ≤ 0.05 compared with untreated sputum. (D) Neutrophil elastase in CF sputum mediates aggregate formation. Purified neutrophil elastase (1 μM) restored the aggregate-promoting effect of HT sputum (left panel). The elastase inhibitors NEI (100 μM; middle panel) and PMSF (100 μM; right panel) inhibit the aggregate-promoting effect of CF sputum. Scale bar represents 1 mm. Images are representative of five experiments. (E) CF sputum supernatants produce flagellar degradation. Equal volumes of lysates from P. aeruginosa cultures were not treated (no treatment), or exposed to CF sputum supernatants (CF sputum) or HT CF sputum supernatants, and Western blots using P. aeruginosa anti-flagellan antibodies were performed. Purified flagella (from the same P. aeruginosa culture) was used as a control. Results are representative of three experiments. PMSF = phenylmethylsulfonyl fluoride.
Neutrophil Elastase in CF Sputum Promotes Aggregate Formation
Two facts led us to hypothesize that neutrophil elastase (NE) in CF sputum was responsible for promoting aggregate formation. First, high levels of NE are among the first abnormalities detected in the airways of infants with CF (5, 52–54). This is important because conditions present early on could induce bacterial phenotypes that determine whether acute or chronic infection develops. Second, NE may reduce bacterial motility by several mechanisms (35, 55–57), and the studies above implicate impaired motility in aggregate formation. We tested this hypothesis in several ways.
First, we incubated P. aeruginosa in 10% sputum supernatants, and found markedly reduced swimming motility (Figure 2B), consistent with previous findings (35, 55–57). Second, we heat-treated supernatants to disrupt NE activity (Figure 2C), and found that the aggregate-promoting and motility-blocking effect was lost (Figure 2A). Third, we added purified NE to heat-treated supernatants, and found that the aggregate-promoting effect of heat-treated supernatants was restored (Figure 2D). Fourth, we treated supernatants with two elastase inhibitors. Both reduced elastase activity (Figure 2C; see Figure E5), and prevented aggregate formation (Figure 2D).
Previous work indicating that elastase can cleave flagella (58) led us to investigate whether the sputum supernatants we used had this effect. Ten percent CF sputum supernatants rapidly degraded P. aeruginosa flagella, whereas heat-treated supernatants did not (Figure 2E). Together, these experiments suggest that inflammation caused either directly by the CF defect, or secondarily by impaired bacterial killing, could mediate aggregate formation by elevating airway elastase levels, even in low-density gels.
Aggregates Are Antibiotic-Tolerant
CF infections resist treatment, even when bacteria are antibiotic-sensitive (1, 59). To determine if aggregated growth produces antibiotic tolerance, we compared the susceptibility of dispersed bacteria with aggregates generated in several ways. Aggregates produced by growing wild-type P. aeruginosa in high-density gels (Figure 3A), the motility mutant in low-density gels (Figure 3B), and wild-type P. aeruginosa in low-density gels containing sputum supernatants (Figure 3C) showed 100- to 1,000-fold less killing by tobramycin as compared with dispersed cells. Similar antibiotic tolerance has been observed with P. aeruginosa aggregates that form in stationary phase cultures (60). Although aggregates were also tolerant of ceftazidime (Figure 3A) they were relatively sensitive to ciprofloxacin (see Figure E6).
Figure 3.

Aggregate formation decreases antibiotic sensitivity of Pseudomonas aeruginosa. (A) P. aeruginosa aggregates that form in 0.8% agar (black bars) are approximately 100-fold less sensitive to killing by tobramycin and ceftazidime than dispersed cells grown in 0.2% agar gels (gray bars). Data are the mean of three experiments, error bars represent SEM. *P ≤ 0.05 compared with 0.2% agar gels. (B) P. aeruginosa aggregates that form in 0.2% agar because of inactivation of motility genes (ΔflgK mutant, black bar) are approximately 1,000-fold less sensitive to killing by tobramycin than wild-type P. aeruginosa that grow as dispersed cells in 0.2% agar gels (gray bar). Complementation with flgK prevented aggregate formation (see Figure E3) and antibiotic tolerance (gray bar). Data are the mean of three experiments; error bars represent SEM. *P ≤ 0.05 compared with dispersed growth (gray bars). (C) P. aeruginosa aggregates that form in 0.2% agar containing cystic fibrosis (CF) sputum supernatants (black bar) are less sensitive to tobramycin killing than dispersed cells that grow in 0.2% agar in the absence of sputum supernatants, or dispersed cells that grow in 0.2% agar containing sputum supernatants and the elastase inhibitor PMSF at 100 μM (gray bars). Data are the means of three experiments; error bars represent SEM. *P ≤ 0.05 compared with dispersed growth (gray bars). PMSF = phenylmethylsulfonyl fluoride.
Control experiments in liquid cultures showed that neither CF sputum supernatants nor the inactivation of motility genes increased antibiotic tolerance by themselves (see Figure E7). We also considered the possibility that the tolerance of aggregates in 0.8% agar was caused by limited drug diffusion or drug binding. To explore this, we used a high inoculum of P. aeruginosa to produce dispersed growth in both 0.2% and 0.8% agar gels, and found similar tobramycin susceptibility (see Figure E8). Furthermore, the fact that aggregates that formed in both low- (Figures 3B and 3C) and high-density gels (Figure 3A) were antibiotic-tolerant implicates aggregate formation, rather than gel-density in the tolerance phenotype.
Biofilm-Defective Mutants Form Antibiotic Tolerant Aggregates
Whereas motility is required for biofilm formation in several systems (33, 61, 62), our experiments indicate that motility loss enhances aggregate formation. This prompted us to investigate whether other functions, deemed “essential” for biofilm formation, are dispensable for aggregate formation and killing resistance.
We studied P. aeruginosa mutants inactivated in quorum sensing (lasRrhlR−) (63), a two-component regulator (gacA) that regulates biofilm formation (64), pili biosynthesis (pilA−) (33), secretion of the iron chelator pyoverdine (pvdA−) (65), and production of all known exopolysaccharides (pelA/pslBCD/algD) (66, 67). Whereas the mutants were compromised for biofilm formation (see Figure E9), they all formed aggregates in high-density gels (Figure 4A) and low-density gels containing CF sputum supernatants (see Figure E10), and aggregated growth increased their antibiotic tolerance (Figure 4A).
Figure 4.

Gene inactivation mutants that are biofilm-deficient form tobramycin-tolerant aggregates in gels. Images show the growth phenotype of Pseudomonas aeruginosa mutants in which genes important for biofilm formation have been inactivated (A), and of the stringent response mutant (B) in low- (0.2%) and high-density (0.8%) agar gels. All mutants formed aggregates in high-density gels. Scale bar represents 1 mm. Graphs show sensitivity of the dispersed cells that grow in 0.2% agar (gray bars) and aggregates that grow in 0.8% agar (black bars) to tobramycin killing. Data represent mean log reduction in bacterial counts from three experiments, error bars represent SEM. *P ≤ 0.06; **P ≤ 0.005.
We also studied a mutant in the P. aeruginosa starvation-sensing system known as the stringent response, which affects the antibiotic tolerance of biofilms rather than biofilm formation per se (68). Inactivation of the stringent response prevented aggregate-mediated tolerance without blocking aggregate formation (Figure 4B). Thus, although inhibiting biofilm formation functions may not prevent bacteria from growing in aggregates, targeting mechanisms that lead to biofilm-mediated antimicrobial tolerance may have therapeutic potential.
Chronic Infection Isolates Form Drug-Tolerant Aggregates
During infection, selective pressures in lungs of patients with CF select for isolates that are biofilm-deficient (22–26). Given that aggregates may more closely resemble bacteria in CF secretions than laboratory biofilms (10–15), we hypothesize that aggregate-forming capacity may be preserved in evolved CF clinical isolates.
To test this, we studied isogenic pairs of CF isolates obtained at early and late stages of infection (between 5 and 20 yr apart) from five different patients (18). Consistent with previous work (18), four of five of the late isolates were defective in biofilm formation as compared with the early isolate (see Figure E11A). However, the biofilm-deficient late-stage isolate from each pair readily formed aggregates when grown in 0.8% agar (see Figure E12). Furthermore, all but one late isolate formed aggregates in 0.2% agar, even in the absence of CF sputum (see Figure E12). The isolates’ ability to form aggregates in low-density gels suggested that they may be motility deficient, and swimming assays confirmed this (see Table E1).
We also studied motile clinical isolates. Whereas these isolates also varied in the ability to form biofilms (see Figure E11B), all grew as dispersed cells in 0.2% agar, and formed aggregates if gel density was increased or if CF sputum was added (Figure 5).
Figure 5.

Clinical Pseudomonas aeruginosa isolates form tobramycin-tolerant aggregates in gels. Motile clinical P. aeruginosa isolates grew as dispersed cells in low-density agar (0.2%), and as aggregates in high-density agar (0.8%) or in low-density agar containing sputum supernatants (0.2% agar + cystic fibrosis [CF] sputum). Scale bar represents 1 mm. Images are representative of five experiments.
As seen with the laboratory strains, antibiotic tolerance tracked with aggregate formation. Isolates that formed aggregates in 0.2% agar (CF1–4L) developed substantial increases in tobramycin tolerance when grown in 0.2% agar (Figure 6). In contrast, motile isolates (CF5L, CF6-CF8) required 0.8% agar for both aggregate formation and increased tobramycin tolerance (Figure 6).
Figure 6.

Aggregates formed by clinical isolates are tobramycin-tolerant. Eight clinical isolates from chronically infected patients with cystic fibrosis (CF) were grown in 0, 0.2, and 0.8% agar. The growth phenotype observed in each condition is indicated below each bar. Liquid = bacteria were grown in shaken liquid culture; Disp. and Agg. = dispersed and aggregated growth in agar gels. Graphs show sensitivity of isolates grown in different conditions to tobramycin killing. Isolates CF1L–CF5L were nonmotile, and with the exception of CF5L, growth in 0.2% agar was sufficient to produce aggregate formation and major decreases in antibiotic sensitivity. Isolates CF6–CF8 were motile and for these 0.8% agar was required to produce aggregate formation and major decreases in antibiotic sensitivity. Data represent mean log reduction in bacterial counts from three experiments, error bars represent SEM. *P ≤ 0.05 compared with 0% agar control; **P ≤ 0.05 compared with 0.2% agar.
Notably, isolates CF3L, CF4L, and CF8 were approximately 10- to 100-fold more resistant to tobramycin killing in liquid cultures than other isolates, likely because of resistance-producing mutations (Figure 6). Aggregate formation produced an additional 100- to 1,000-fold reduction in susceptibility (Figure 6). Thus, biofilm-deficient clinical isolates readily form aggregates, and aggregate formation increases the antibiotic tolerance of genetically sensitive and resistant bacteria.
Antibiotic Treatment Selects for Loss of Motility in Gels
Our finding that loss of motility promotes formation of drug-tolerant aggregates led us to hypothesize that antibiotic treatment could serve as a selective pressure for motility loss. Notably, CF isolates frequently acquire mutations in motility genes (18, 20, 21).
To test this, we mixed isogenic motile and nonmotile P. aeruginosa strains (1:1) in low-density gels, and exposed them to tobramycin. The motile strain was advantaged in the absence of tobramycin (Figure 7A), possibly because motility enhanced access to nutrients and oxygen.
Figure 7.

Motility loss increases bacterial fitness during tobramycin treatment in gels. Motile and nonmotile bacteria with similar tobramycin susceptibility (see Figure E7B) were inoculated at a 1:1 ratio in 0.2% agar gels, grown for 24 hours, exposed to tobramycin (or were untreated), and then viable bacteria were enumerated. (A) Shows the relative abundance of nonmotile Pseudomonas aeruginosa when wild-type P. aeruginosa PA01 and its isogenic motility mutant (ΔflgK) were competed. (B) Shows the relative abundance of nonmotile P. aeruginosa when clonally related motile and nonmotile clinical isolates that infected an individual patient with cystic fibrosis were competed. Data are mean relative abundance measurements from three experiments, error bars represent SEM. *P ≤ 0.05.
In contrast, nonmotile P. aeruginosa (which formed aggregates) were more fit during tobramycin treatment (Figure 7A). Competing clonally related motile and nonmotile isolates that coexisted in the sputum of a patient with CF produced a similar result (Figure 7B). These findings suggest that antibiotics could select for adaptations like motility loss, which enhance survival in vivo, but are unrelated to conventional resistance mechanisms. These data could also explain why a valuable pathogenesis function like motility is consistently mutated during CF infections.
Sputum-generated Aggregates Resist Killing by Innate Immune Defenses
Although aggregate-mediated antibiotic tolerance could thwart treatment, chronic infection initiation is likely more dependent on the ability of bacteria to resist host defenses. We tested dispersed and aggregated P. aeruginosa produced by growth in gels containing untreated and heat-treated sputum supernatants, and by using the wild-type and motility-deficient P. aeruginosa, and found that aggregates were more resistant to killing by hypochlorous acid and human neutrophils (Figure 8), and H2O2 (see Figure E13).
Figure 8.
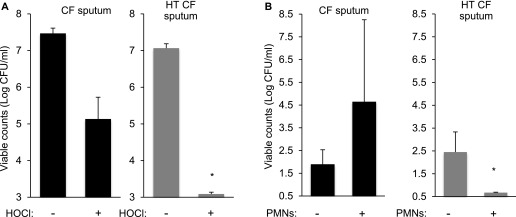
Sputum-induced aggregates resist killing by host defense oxidants and neutrophils. Growth of bacteria in gels containing cystic fibrosis (CF) sputum supernatants but not heat-treated (HT) supernatants induced aggregate formation (Figure 2A) and tolerance of HOCl (A) and resistance to polymorphonuclear leukocyte (PMN) killing (B). Data are mean bacteria counts with and without treatment from three experiments, error bars represent SEM. *P ≤ 0.05 for the log reduction produced by HOCl and PMNs after growth in CF sputum as compared with HT sputum.
Sputum-mediated Aggregate Formation Attenuates Epithelial Injury and Inflammation
CF infections are noninvasive even at their onset, despite the high burden of virulent organisms, compromised lung defenses, and periodic use of immunosuppressives like corticosteroids (1, 52). These facts suggest that conditions in the lungs of patients with CF somehow produce an immediate (and sustained) moderation of P. aeruginosa’s invasive functions.
We investigated whether aggregate formation reduced the invasiveness of infections in two ways. First, we infected air–liquid interface airway epithelial cultures with P. aeruginosa in the presence of untreated and heat-treated sputum supernatants. CF sputum supernatants (but not heat-treated supernatants) promoted bacterial aggregate formation on epithelial cells (see Figure E14), and markedly attenuated epithelial injury (Figure 9A), tight junction protein degradation (Figure 9B), and IL-8 and tumor necrosis factor-α mRNA induction (Figure 9C).
Figure 9.

Growth in cystic fibrosis (CF) sputum attenuates the invasive virulence of Pseudomonas aeruginosa. (A) CF sputum reduces epithelial injury produced by P. aeruginosa. Transepithelial resistance (TER) measured after airway epithelial cells were inoculated with 0.2% agar gels containing P. aeruginosa with CF sputum supernatants (solid circles), P. aeruginosa alone (solid squares), or P. aeruginosa and heat-treated (HT) CF sputum (open squares). HT CF sputum supernatants without bacteria (labeled “no bacteria” and indicated with open circles) had no effect. Data are representative of three independent experiments. Each data point is the mean of measurements from three epithelial culture inserts; error bars represent STD. (B) Western blot analysis of epithelial cell lysates for occludin and junctional adhesion molecule-A (JAM-A) following infection of epithelia with 0.2% agar gels containing no bacteria, HT CF sputum alone, P. aeruginosa and CF sputum (PA + sputum), P. aeruginosa and HT CF sputum (PA + HT CF sputum), and P. aeruginosa alone (PA alone). Glyceraldehyde phosphate dehydrogenase (GAPDH) expression is a control for protein loading; results are representative of three independent experiments. (C) CF sputum reduces epithelial inflammation induced by P. aeruginosa. Quantitative reverse-transcriptase polymerase chain reaction measurements of tumor necrosis factor (TNF)-α and IL-8 transcript levels from epithelial cells infected with P. aeruginosa in 0.2% agar gels containing either CF sputum supernatants or HT CF sputum. Data are the mean of three experiments, and fold induction is relative to uninfected epithelia normalized to a control gene transcript (gapdh). Error bars represent STD. *P ≤ 0.03. (D) Survival of mice with skin wounds infected with P. aeruginosa incubated in CF sputum (open circles) or heat inactivated CF sputum (solid circles). Data represent survival of six mice in each group, and similar results were seen in two additional experiments.
Second, we investigated whether sputum supernatants could reduce invasive virulence in vivo. We used a murine wound-infection model (69) because untreated or heat-treated sputum supernatants could be applied topically along with the bacteria. As shown in Figure 9D, infections initiated with P. aeruginosa incubated in CF sputum supernatants produced less mortality as compared with those incubated in heat-treated supernatants. Thus, soluble sputum components can mediate the noninvasive phenotype characteristic of CF lung infections.
Discussion
The CF defect has been postulated to derange many defense functions; however, it is unclear why CF infections are exclusively chronic in nature and eradication-resistant even when infecting bacteria are antibiotic-sensitive. Our work suggests two general mechanisms by which CF-associated conditions could lead to aggregated bacterial growth, and the hallmark noninvasive and resistant infection phenotype.
Our experiments and previous work (15, 32) indicate that increased density of airway secretions could lead to aggregate formation by constraining bacterial motility. This mechanism is of particular interest given recent work showing that a direct consequence of CFTR dysfunction, impaired bicarbonate secretion, may increase airway mucus density (38).
However, increased gel density is not required, because NE in CF sputum promotes aggregate formation in low-density gels by cleaving flagella and inhibiting bacterial motility (35, 55, 57, 58). Importantly, elevated elastase is one of the earliest and most reproducible abnormalities seen in infants with CF, it can predate chronic infection (5, 52–54), and it strongly predicts future lung disease (70).
Defective CFTR could elevate NE levels by several mechanisms: CF may increase inflammatory responses (49–51), impair epithelial or phagocytic host defenses (43–48), or disrupt lung mucus clearance (39, 40). Any of these mechanisms could lead to neutrophil influx, and high levels of elastase. If the CF defect simultaneously increased neutrophil influx and the density of secretions, the combined aggregate-promoting effect may be magnified.
Although the agar and mucus gel models reproduce the aggregated morphology and some infection phenotypes seen in CF, the models have limitations. The most significant is that the airway surface environment is poorly understood, particularly at infection onset. Thus, conditions are difficult to replicate. It will be important to modify the model as the understanding of the lung environment in CF and healthy children improves.
It also would be interesting to study pristine environmental isolates, which cause initial infection. We replicated key findings with isolates from early stages of CF infection and with the P. aeruginosa strain PAO1, which is often used as a “wild-type” strain. However, it is possible that environmental strains behave differently. It is also not yet clear whether in vivo aggregates are of comparable sizes as those we studied (16), or are composed of genetically homogenous bacterial populations. Because of these and other factors, we do not know how closely the infection phenotypes of the aggregates we studied resemble those in CF.
If our in vitro and in vivo findings with aggregates reflect bacterial functioning in humans, aggregate formation could explain aspects of chronic infection pathogenesis, treatment resistance, and have implications for therapeutics development in CF and other diseases. For example, epidemiologic evidence indicates that chronic CF P. aeruginosa airway infections are often preceded by transient infections with other pathogens (1, 52). Our data raise the possibility that these antecedent infections could induce NE secretion, thereby generating conditions that promote P. aeruginosa aggregate formation.
It has also been suggested that CF P. aeruginosa infections begin in the sinuses (71, 72), and sinus inflammation could promote aggregate formation. If this were true, aspirated P. aeruginosa could already have the noninvasive and resistant phenotype on arrival in the lung. After treatment begins, aggregate formation could contribute to the failure of antibiotic therapy, and explain why minimum inhibitory concentration testing does not predict treatment efficacy (73).
Inflammation and a gel-like environment may also present in such settings as wounds; non-CF sinusitis and bronchiectasis; and perhaps during biliary, gastrointestinal, and renal tract pathology. Thus, the mechanisms of aggregate formation identified here could contribute to chronic infection pathogenesis in other disease states.
The resistance phenotype of aggregates could also shape the evolutionary adaptations of infecting bacteria. Competition experiments in low-density gels showed that nonmotile strains (which grew as aggregates) had an antibiotic survival advantage over motile strains (which grew dispersed in the gels). These findings raise the possibility that adaptations like motility loss, which arise at high frequency in CF, could be selected for because they enhance aggregate growth.
Finally, our data may have implications in the search for new therapeutic targets against chronic infections. Much interest has been directed toward targeting functions needed for the biofilm formation, such as surface adherence, motility, and signaling systems. This focus is logical because interfering with these functions is effective in in vitro systems. However, the fact that many CF chronic infection isolates are already biofilm-deficient (22–26), and that mutants impaired in biofilm formation readily produce aggregates in gel conditions raise questions about targeting biofilm-formation functions in CF. Our experiments suggest that aggregated growth may in some ways be a default growth mode in sputum gels if NE is present, gel densities are high, or if infecting bacteria are nonmotile. If this is true, the formation of aggregates may not require a developmental program (like biofilms may) that is amenable to targeting with therapeutics (16).
An alternative is to target the physiologic functions that mediate important phenotypes of bacterial aggregates. For example, aggregated growth may produce gradients of oxygen, nutrients, and metabolic products and induce bacterial stress responses, as do biofilms (74). These characteristics may also be shared by aggregates in other infections, such as chronic wounds and medical device infections, even though pathogenesis mechanisms differ. The idea that physiologic functions could be valuable targets in a variety of growth conditions is supported by our finding that inactivation of starvation response systems sensitized laboratory biofilms (68) and gel-grown aggregates to killing. It may be fruitful to identify and target other conserved functions needed for aggregated growth or aggregate-mediated drug tolerance.
Acknowledgments
Acknowledgment
The authors thank E. Bauerle, M. Parsek, C. Manoil, and J. Mougous for helpful discussions.
Footnotes
Supported by grants to P.K.S. from National Institutes of Health (R01HL110879 and R01AI101307), the Cystic Fibrosis Foundation, and the Burroughs Wellcome Fund. B.J.S. was supported by a National Institutes of Health training grant and the Cystic Fibrosis Foundation LeRoy Matthews Physician Scientist award. NIH awards R01HL103965 and P30DK089507 supported the acquisition of clinical material and bacterial strains.
Author Contributions: Conception and design, B.J.S., J.F.M., S.H., B.B., D.N., H.R., O.B., M.G., G.H.G., and P.K.S. Analysis and interpretation, B.J.S., J.F.M., S.H., and P.K.S. Drafting the manuscript for important intellectual content, B.J.S., J.F.M., S.H., and P.K.S.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201312-2142OC on January 27, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 2.Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballow M, Notarangelo L, Grimbacher B, Cunningham-Rundles C, Stein M, Helbert M, Gathmann B, Kindle G, Knight AK, Ochs HD, et al. Immunodeficiencies. Clin Exp Immunol. 2009;158:14–22. doi: 10.1111/j.1365-2249.2009.04023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orange JS. Congenital immunodeficiencies and sepsis. Pediatr Crit Care Med. 2005;6:S99–S107. doi: 10.1097/01.PCC.0000164488.19810.DB. [DOI] [PubMed] [Google Scholar]
- 5.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 6.Wagener JS, Kahn TZ, Copenhaver SC, Accurso FJ. Early inflammation and the development of pulmonary disease in cystic fibrosis. Pediatr Pulmonol Suppl. 1997;16:267–268. doi: 10.1002/ppul.19502308138. [DOI] [PubMed] [Google Scholar]
- 7.Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev KM, Borowitz D, Bowman CM, Marshall BC, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study group. N Engl J Med. 1999;340:23–30. doi: 10.1056/NEJM199901073400104. [DOI] [PubMed] [Google Scholar]
- 8.Treggiari MM, Retsch-Bogart G, Mayer-Hamblett N, Khan U, Kulich M, Kronmal R, Williams J, Hiatt P, Gibson RL, Spencer T, et al. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch Pediatr Adolesc Med. 2011;165:847–856. doi: 10.1001/archpediatrics.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tramper-Stranders GA, van der Ent CK, Molin S, Yang L, Hansen SK, Rau MH, Ciofu O, Johansen HK, Wolfs TF. Initial Pseudomonas aeruginosa infection in patients with cystic fibrosis: characteristics of eradicated and persistent isolates. Clin Microbiol Infect. 2012;18:567–574. doi: 10.1111/j.1469-0691.2011.03627.x. [DOI] [PubMed] [Google Scholar]
- 10.Lam J, Chan R, Lam K, Costerton JW. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect Immun. 1980;28:546–556. doi: 10.1128/iai.28.2.546-556.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoiby N, Krogh Johansen H, Moser C, Song Z, Ciofu O, Kharazmi A. Pseudomonas aeruginosa and the in vitro and in vivo biofilm mode of growth. Microbes Infect. 2001;3:23–35. doi: 10.1016/s1286-4579(00)01349-6. [DOI] [PubMed] [Google Scholar]
- 12.Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- 13.Bjarnsholt T, Jensen PO, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Hoiby N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol. 2009;44:547–558. doi: 10.1002/ppul.21011. [DOI] [PubMed] [Google Scholar]
- 14.Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109:317–325. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sriramulu DD, Lunsdorf H, Lam JS, Romling U. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol. 2005;54:667–676. doi: 10.1099/jmm.0.45969-0. [DOI] [PubMed] [Google Scholar]
- 16.Bjarnsholt T, Alhede M, Alhede M, Eickhardt-Sorensen SR, Moser C, Kuhl M, Jensen PO, Hoiby N. The in vivo biofilm. Trends Microbiol. 2013;21:466–474. doi: 10.1016/j.tim.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Wilder CN, Allada G, Schuster M. Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect Immun. 2009;77:5631–5639. doi: 10.1128/IAI.00755-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci USA. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hancock RE, Mutharia LM, Chan L, Darveau RP, Speert DP, Pier GB. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: a class of serum-sensitive, nontypable strains deficient in lipopolysaccharide o side chains. Infect Immun. 1983;42:170–177. doi: 10.1128/iai.42.1.170-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luzar MA, Thomassen MJ, Montie TC. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect Immun. 1985;50:577–582. doi: 10.1128/iai.50.2.577-582.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. 1994;62:596–605. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee B, Haagensen JA, Ciofu O, Andersen JB, Hoiby N, Molin S. Heterogeneity of biofilms formed by nonmucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. J Clin Microbiol. 2005;43:5247–5255. doi: 10.1128/JCM.43.10.5247-5255.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deligianni E, Pattison S, Berrar D, Ternan NG, Haylock RW, Moore JE, Elborn SJ, Dooley JS. Pseudomonas aeruginosa cystic fibrosis isolates of similar RAPD genotype exhibit diversity in biofilm forming ability in vitro. BMC Microbiol. 2010;10:38. doi: 10.1186/1471-2180-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rau MH, Hansen SK, Johansen HK, Thomsen LE, Workman CT, Nielsen KF, Jelsbak L, Hoiby N, Yang L, Molin S. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol. 2010;12:1643–1658. doi: 10.1111/j.1462-2920.2010.02211.x. [DOI] [PubMed] [Google Scholar]
- 25.Head NE, Yu H. Cross-sectional analysis of clinical and environmental isolates of Pseudomonas aeruginosa: biofilm formation, virulence, and genome diversity. Infect Immun. 2004;72:133–144. doi: 10.1128/IAI.72.1.133-144.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaber JA, Carty NL, McDonald NA, Graham ED, Cheluvappa R, Griswold JA, Hamood AN. Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J Med Microbiol. 2004;53:841–853. doi: 10.1099/jmm.0.45617-0. [DOI] [PubMed] [Google Scholar]
- 27.Ciornei CD, Novikov A, Beloin C, Fitting C, Caroff M, Ghigo JM, Cavaillon JM, Adib-Conquy M. Biofilm-forming Pseudomonas aeruginosa bacteria undergo lipopolysaccharide structural modifications and induce enhanced inflammatory cytokine response in human monocytes. Innate Immun. 2010;16:288–301. doi: 10.1177/1753425909341807. [DOI] [PubMed] [Google Scholar]
- 28.Gursoy UK, Pollanen M, Kononen E, Uitto VJ. Biofilm formation enhances the oxygen tolerance and invasiveness of Fusobacterium nucleatum in an oral mucosa culture model. J Periodontol. 2010;81:1084–1091. doi: 10.1902/jop.2010.090664. [DOI] [PubMed] [Google Scholar]
- 29.Staudinger B, Muller JM, Halldórsson S, Angermeyer A, Boles BR, Baldursson O, Rosen H, Singh PK. CF sputum enhances the formation of Pseudomonas aeruginosa aggregates and promotes tolerance to antibiotics and host defenses. Pediatr Pulmonol. 2010;45:1–76. [Google Scholar]
- 30.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range flp-frt recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 32.Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, Taylor RM, II, Superfine R, Rubinstein M, Iglewski BH, et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA. 2006;103:18131–18136. doi: 10.1073/pnas.0606428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 34.Drake D, Montie TC. Flagella, motility and invasive virulence of Pseudomonas aeruginosa. J Gen Microbiol. 1988;134:43–52. doi: 10.1099/00221287-134-1-43. [DOI] [PubMed] [Google Scholar]
- 35.Sonawane A, Jyot J, During R, Ramphal R. Neutrophil elastase, an innate immunity effector molecule, represses flagellin transcription in Pseudomonas aeruginosa. Infect Immun. 2006;74:6682–6689. doi: 10.1128/IAI.00922-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halldorsson S, Asgrimsson V, Axelsson I, Gudmundsson GH, Steinarsdottir M, Baldursson O, Gudjonsson T. Differentiation potential of a basal epithelial cell line established from human bronchial explant. In Vitro Cell Dev Biol Anim. 2007;43:283–289. doi: 10.1007/s11626-007-9050-4. [DOI] [PubMed] [Google Scholar]
- 37.Halldorsson S, Gudjonsson T, Gottfredsson M, Singh PK, Gudmundsson GH, Baldursson O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol. 2010;42:62–68. doi: 10.1165/rcmb.2008-0357OC. [DOI] [PubMed] [Google Scholar]
- 38.Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372:415–417. doi: 10.1016/S0140-6736(08)61162-9. [DOI] [PubMed] [Google Scholar]
- 39.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 40.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 41.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, al-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature. 1991;352:70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Doranz B, Yankaskas JR, Engelhardt JF. Genotypic analysis of respiratory mucous sulfation defects in cystic fibrosis. J Clin Invest. 1995;96:2997–3004. doi: 10.1172/JCI118372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pier GB, Grout M, Zaidi TS. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci USA. 1997;94:12088–12093. doi: 10.1073/pnas.94.22.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol. 2008;83:1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol. 2006;8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 46.Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialogm1 which is increased on the surface of cystic fibrosis epithelial cells. J Clin Invest. 1993;92:1875–1880. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith JJ, Travis S, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 48.Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol. 1995;13:257–261. doi: 10.1165/ajrcmb.13.3.7544594. [DOI] [PubMed] [Google Scholar]
- 50.Weber AJ, Soong G, Bryan R, Saba S, Prince A. Activation of NF-kappaB in airway epithelial cells is dependent on CFTR trafficking and Cl–channel function. Am J Physiol Lung Cell Mol Physiol. 2001;281:L71–L78. doi: 10.1152/ajplung.2001.281.1.L71. [DOI] [PubMed] [Google Scholar]
- 51.Eidelman O, Srivastava M, Zhang J, Leighton X, Murtie J, Jozwik C, Jacobson K, Weinstein DL, Metcalf EL, Pollard HB. Control of the proinflammatory state in cystic fibrosis lung epithelial cells by genes from the TNF-alpha/NFkappaB pathway. Mol Med. 2001;7:523–534. [PMC free article] [PubMed] [Google Scholar]
- 52.Accurso FJ. Early pulmonary disease in cystic fibrosis. Curr Opin Pulm Med. 1997;3:400–403. doi: 10.1097/00063198-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Pillarisetti N, Williamson E, Linnane B, Skoric B, Robertson CF, Robinson P, Massie J, Hall GL, Sly P, Stick S, et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am J Respir Crit Care Med. 2011;184:75–81. doi: 10.1164/rccm.201011-1892OC. [DOI] [PubMed] [Google Scholar]
- 54.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, Olinsky A, Phelan EM, Robertson CF, Phelan PD. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156:1197–1204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- 55.Jyot J, Sonawane A, Wu W, Ramphal R. Genetic mechanisms involved in the repression of flagellar assembly by Pseudomonas aeruginosa in human mucus. Mol Microbiol. 2007;63:1026–1038. doi: 10.1111/j.1365-2958.2006.05573.x. [DOI] [PubMed] [Google Scholar]
- 56.Palmer KL, Mashburn LM, Singh PK, Whiteley M. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J Bacteriol. 2005;187:5267–5277. doi: 10.1128/JB.187.15.5267-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolfgang MC, Jyot J, Goodman AL, Ramphal R, Lory S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients 10. 1073/pnas.0307553101. Proc Natl Acad Sci USA. 2004;101:6664–6668. doi: 10.1073/pnas.0307553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez-Boado YS, Espinola M, Bahr S, Belaaouaj A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J Immunol. 2004;172:509–515. doi: 10.4049/jimmunol.172.1.509. [DOI] [PubMed] [Google Scholar]
- 59.Flume PA, O'Sullivan BP, Robinson KA, Goss CH, Mogayzel PJ, Jr, Willey-Courand DB, Bujan J, Finder J, Lester M, Quittell L, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957–969. doi: 10.1164/rccm.200705-664OC. [DOI] [PubMed] [Google Scholar]
- 60.Alhede M, Kragh KN, Qvortrup K, Allesen-Holm M, van Gennip M, Christensen LD, Jensen PO, Nielsen AK, Parsek M, Wozniak D, et al. Phenotypes of non-attached pseudomonas aeruginosa aggregates resemble surface attached biofilm. PloS ONE. 2011;6:e27943. doi: 10.1371/journal.pone.0027943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DG. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol. 2002;184:1140–1154. doi: 10.1128/jb.184.4.1140-1154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramsey MM, Whiteley M. Pseudomonas aeruginosa attachment and biofilm development in dynamic environments. Mol Microbiol. 2004;53:1075–1087. doi: 10.1111/j.1365-2958.2004.04181.x. [DOI] [PubMed] [Google Scholar]
- 63.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 64.Parkins MD, Ceri H, Storey DG. Pseudomonas aeruginosa gaca, a factor in multihost virulence, is also essential for biofilm formation. Mol Microbiol. 2001;40:1215–1226. doi: 10.1046/j.1365-2958.2001.02469.x. [DOI] [PubMed] [Google Scholar]
- 65.Banin E, Vasil ML, Greenberg EP. Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci USA. 2005;102:11076–11081. doi: 10.1073/pnas.0504266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jackson KD, Starkey M, Kremer S, Parsek MR, Wozniak DJ. Identification of PSL, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J Bacteriol. 2004;186:4466–4475. doi: 10.1128/JB.186.14.4466-4475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colvin KM, Gordon VD, Murakami K, Borlee BR, Wozniak DJ, Wong GC, Parsek MR. The PEL polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011;7:e1001264. doi: 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 2011;334:982–986. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao G, Hochwalt PC, Usui ML, Underwood RA, Singh PK, James GA, Stewart PS, Fleckman P, Olerud JE. Delayed wound healing in diabetic (db/db) mice with Pseudomonas aeruginosa biofilm challenge: a model for the study of chronic wounds. Wound Repair Regen. 2010;18:467–477. doi: 10.1111/j.1524-475X.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sagel SD, Sontag MK, Accurso FJ. Relationship between antimicrobial proteins and airway inflammation and infection in cystic fibrosis. Pediatr Pulmonol. 2009;44:402–409. doi: 10.1002/ppul.21028. [DOI] [PubMed] [Google Scholar]
- 71.Taylor RF, Morgan DW, Nicholson PS, Mackay IS, Hodson ME, Pitt TL. Extrapulmonary sites of Pseudomonas aeruginosa in adults with cystic fibrosis. Thorax. 1992;47:426–428. doi: 10.1136/thx.47.6.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hansen SK, Rau MH, Johansen HK, Ciofu O, Jelsbak L, Yang L, Folkesson A, Jarmer HO, Aanaes K, von Buchwald C, et al. Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J. 2012;6:31–45. doi: 10.1038/ismej.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith AL, Fiel SB, Mayer-Hamblett N, Ramsey B, Burns JL. Susceptibility testing of Pseudomonas aeruginosa isolates and clinical response to parenteral antibiotic administration: lack of association in cystic fibrosis. Chest. 2003;123:1495–1502. doi: 10.1378/chest.123.5.1495. [DOI] [PubMed] [Google Scholar]
- 74.Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]