

Abstract
Rationale: Ivacaftor is a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator recently approved for patients with CF age 6 and older with the G551D mutation.
Objectives: To evaluate ivacaftor in a postapproval setting and determine mechanism of action and response of clinically relevant markers.
Methods: We conducted a longitudinal cohort study in 2012–2013 in G551D CF patients age 6 and older with no prior exposure to ivacaftor. Study assessments were performed at baseline, 1, 3, and 6 months after ivacaftor initiation. Substudies evaluated mucociliary clearance, β-adrenergic sweat secretion rate, gastrointestinal pH, and sputum inflammation and microbiology
Measurements and Main Results: A total of 151 of 153 subjects were prescribed ivacaftor and 88% completed the study through 6 months. FEV1 % predicted improved from baseline to 6 months (mean absolute change, 6.7%; P < 0.001). Similarly, body mass index improved from baseline to 6 months (mean change, 0.8 kg/m2; P < 0.001). Sweat chloride decreased from baseline to 6 months (mean change, −53.8 mmol/L; 95% confidence interval, −57.7 to −49.9; P < 0.001), reflecting augmented CFTR function. There was significant improvement in hospitalization rate (P < 0.001) and Pseudomonas aeruginosa burden (P < 0.01). Significant improvements in mucociliary clearance (P < 0.001), gastrointestinal pH (P = 0.001), and microbiome were also observed, providing clinical mechanisms underlying the therapeutic benefit of ivacaftor.
Conclusions: Significant clinical and physiologic improvements were observed on initiation of ivacaftor in a broad patient population, including reduced infection with P. aeruginosa. Biomarker studies substantially improve the understanding of the mechanistic consequences of CFTR modulation on pulmonary and gastrointestinal physiology.
Keywords: cystic fibrosis, CFTR modulator, ivacaftor, Pseudomonas aeruginosa, pH
At a Glance Commentary
Scientific Knowledge on the Subject
Two randomized controlled trials have reported the efficacy of ivacaftor in G551D-mediated cystic fibrosis (CF), but no studies have reported the effectiveness following approval or established physiologic implications of modulating CF transmembrane conductance regulator (CFTR) in humans.
What This Study Adds to the Field
This study establishes the effectiveness of ivacaftor in patients with CF carrying a copy of the G551D CFTR mutation and involves a generalized population nearly 40% larger than the phase three program. Ivacaftor confers robust clinical benefit, including an unexpected improvement in the growth of Pseudomonas aeruginosa and other microbiologic changes. Improved mucociliary clearance and intestinal bicarbonate secretion provide novel insights into the clinical mechanism of CFTR modulators, and may explain the marked benefit in lung function and gastrointestinal health conferred by ivacaftor.
Cystic fibrosis (CF) is caused by mutations in CF transmembrane conductance regulator (CFTR), which functions as an anion channel expressed predominantly on epithelial tissues (1). In the respiratory tract, absent or dysfunctional CFTR results in mucus obstruction of the small airways and early infection with pathogenic bacteria, leading to bronchiectasis and ultimately respiratory failure (2). Gastrointestinal (GI) obstruction, dysbacteriosis, and inflammation contribute to disease burden (3). Salt loss may contribute to symptoms early in life and is the traditional diagnostic test for CF (4). Despite several supportive therapies, the median age of death is still 27 years (5).
There are more than 2,000 disease-causing mutations in CFTR, although a minority of mutations account for approximately 90% of CFTR alleles (6). The relatively common G551D-CFTR mutation, which accounts for approximately 4% of CFTR alleles, results in defective channel gating and little or no anion transport. Recently, the CFTR potentiator ivacaftor was approved for the treatment of patients with CF age 6 and older with the G551D-CFTR mutation. Phase 3 studies demonstrated marked improvement in lung function, body weight, and quality of life, in addition to sweat chloride, a measure of CFTR activity (7, 8). However, questions remain regarding the mechanistic basis for improved outcomes with ivacaftor treatment. For example, whether augmented CFTR activity would alter mucociliary transport, intestinal pH profiles, the nature of chronic infection or pulmonary inflammation, or sweat gland secretion are untested questions, and could reveal important insights into the long-term effects of CFTR modulators. Furthermore, the effectiveness of ivacaftor in a larger population of individuals with CF has not yet been assessed.
To address these questions, we conducted a multicenter clinical study entitled the “G551D Observational Study” among patients with CF with G551D-CFTR mutation, and captured clinical status before and after initiation of ivacaftor at the time of its approval by the US Food and Drug Administration. The study also included biospecimen collection and four concurrent substudies to evaluate mechanism of action and test new markers of disease activity. Some of the results of these studies have been previously reported in the form of abstracts (9–13).
Methods
We conducted a longitudinal cohort study involving 28 centers within the Cystic Fibrosis Therapeutics Development Network to capture clinical measures and biospecimens in patients with CF age 6 and older with at least one G551D mutation and no prior exposure to ivacaftor (http://clinicaltrials.gov/ct2/show/NCT01521338). Study assessments conducted in all enrolled subjects (i.e., the “core” study) included spirometry; body weight; sweat chloride; and the patient-reported outcome instruments Cystic Fibrosis Questionnaire-Revised (CFQ-R) (14), Cystic Fibrosis Respiratory Symptom Diary (15), and Sino-Nasal Outcome Test-20 (16). Samples collected and subsequently stored at the Cystic Fibrosis Foundation Therapeutics Biorepository for future research included plasma, serum, buffy coat, urine, and expectorated sputum. Study assessments were conducted at one or two baseline visits, and then 1, 3, and 6 months after initiation of ivacaftor. Participants who were never prescribed ivacaftor during the study period had a visit at baseline and 6 months postenrollment. Linked participant data from the Cystic Fibrosis Foundation National Patient Registry (CFFNPR) augmented the study data by providing microbiologic data, hospitalization history, and follow-up. Spirometry was performed according to American Thoracic Society standards (17) and percent predicted calculated using reference equations (18, 19). Participants or their guardians provided written informed consent, and the study was approved by site institutional review boards.
The GOAL study also included four substudies with additional assessments, including the evaluation of mucociliary clearance (MCC), GI pH profiles, measures of sputum inflammation and microbiology, and of β-adrenergic sweat rate.
MCC
Mucociliary and cough clearance was measured in a subset using γ scintigraphy at baseline and following 1 and 3 months of ivacaftor therapy at four participating sites. All sites received on-site training and followed a standard operating procedure that closely mirrored published methods and is described in the online supplement (20).
GI pH
GI pH was measured in a subset of adult participants at baseline and 1 month after initiation of ivacaftor at three sites as previously described (21) and in the online supplement.
Sputum Inflammation and Microbiome
Induced sputum was collected at baseline and 6 months after initiation of ivacaftor from participants older than 10 years of age. Sputum induction was performed on all substudy participants according to a standard operating procedure (22) and sputum specimens were processed using a standard operating procedure (23). Free neutrophil elastase activity, α1-antitrypsin, secretory leukoprotease inhibitor, IL-1β, IL-6, and IL-8 were measured in sputum supernatants, as described in the online supplement. For microbiome analysis sequence counts from bacterial genera known to contain traditional CF pathogens Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus, Stenotrophomonas, Achromobacter, and Burkholderia were combined to represent total CF pathogen relative abundance (24). Total and P. aeruginosa–specific bacterial quantitative polymerase chain reaction assays were also performed, as detailed in the online supplement.
β-Adrenergic Sweat Secretion Rate
The β-adrenergic sweat secretion rate was measured in a subset of participants at baseline, 1, and 3 months after initiation of ivacaftor as previously described (25). See the online supplement for additional details.
Statistical Analyses
Paired mean change and SD from baseline (the last visit before ivacaftor initiation) in continuous outcomes were calculated along with 95% confidence intervals (CI) and tested by paired t test, and reported in Results unless otherwise noted. Difference in proportions were compared cross-sectionally with Fisher exact test and paired with Wilcoxon sign test. Hospitalization rates (adjusted for participant follow-up) were compared with Wilcoxon sign test; reported P values are two-sided. Area under the curve was calculated for time series (pH and lung clearance) and compared with the paired t test. Pearson correlation coefficient (r) was used to explore relationship between change in clinical measures and change in sweat chloride. Analyses were performed using SAS (version 9.2, Cary, NC) and R (version 2.15, Vienna, Austria).
Results
A total of 153 participants were enrolled over a 4.5-month enrollment period (February to June 2012). A total of 151 were prescribed ivacaftor and 133 (88%) completed the study through the 6-month follow-up visit (Figure 1). Participant demographics and baseline clinical characteristics are shown in Tables 1 and 2. Age (mean [SD]) was 21.1 (11.4) years and 46.4% were female. All participants had one copy of the G551D mutation, and 72.2% were compound heterozygous with F508del on the other allele.
Figure 1.

Diagram of participants screened, eligible, enrolled in core and substudies, follow-up, and withdrawals.
Table 1.
Demographics and Baseline Characteristics
| Taking Ivacaftor (n = 151) | Not Taking Ivacaftor (n = 2) | All Participants (n = 153) | |
|---|---|---|---|
| Female, n (%) | 70 (46) | 0 (0) | 70 (46) |
| Age, mean (SD) | 21.1 (11.4) | 19.7 (7.8) | 21.1 (11.3) |
| Age category, n (%) | |||
| 6–11 | 38 (25) | 0 (0) | 38 (25) |
| 12–17 | 32 (21) | 1 (50) | 33 (22) |
| 18–29 | 51 (34) | 1 (50) | 52 (34) |
| 30+ | 30 (20) | 0 (0) | 30 (20) |
| Race, n (%) | |||
| White | 145 (96) | 2 (100) | 147 (96) |
| Hispanic | 2 (1) | 0 (0) | 2 (1) |
| Black/African American | 3 (2) | 0 (0) | 3 (2) |
| Other/unknown | 1 (1) | 0 (0) | 1 (1) |
| Genotype class of non-G551D allele, n (%) | |||
| Class I | 10 (7) | 1 (50) | 11 (7) |
| Class II | 114 (75) | 1 (50) | 115 (75) |
| Class III | 2 (1) | 0 (0) | 2 (1) |
| Class IV | 4 (3) | 0 (0) | 4 (3) |
| Class V | 8 (5) | 0 (0) | 8 (5) |
| Unknown | 13 (9) | 0 (0) | 13 (8) |
| Activity of non-G551D CFTR mutation*, n (%) | |||
| Not active | 132 (87) | 2 (100) | 134 (88) |
| Partially active | 7 (5) | 0 (0) | 7 (5) |
| Unknown | 12 (8) | 0 (0) | 12 (8) |
| Body mass index, mean (SD) | 21.3 (4.5) | 24.5 (6.5) | 21.3 (4.5) |
| FEV1, % predicted†, mean (SD) | 82.6 (25.6) | 66.0 (52.8) | 82.4 (25.9) |
| FVC, % predicted, mean (SD) | 101.4 (24.2) | 70.6 (44.3) | 101.0 (24.6) |
| Pseudomonas aeruginosa culture positive‡, n (%) | 75 (52) | 1 (50) | 76 (52) |
Definition of abbreviation: CFTR = cystic fibrosis transmembrane conductance regulator.
CFTR activity determined CFTR2 mutation database.
% predicted spirometry is calculated using the Wang equations (Peds Pulm, 1994) for females younger than 16 years and males younger than 18 years of age and the Hankinson equations (AJRCCM 1999) for others.
Any positive culture within 6 months before starting ivacaftor (n = 143 with culture results in Cystic Fibrosis Foundation National Patient Registry) or before enrollment in those who did not initiate ivacaftor (n = 2 with culture results).
Table 2.
Clinical Characteristics and Change from Baseline at Each Time Point
| Estimate |
Absolute Change from Baseline |
|||||
|---|---|---|---|---|---|---|
| N | Mean (SD) | N | Mean | 95% CI | P Value | |
| Weight, kg | ||||||
| Baseline | 151 | 55.2 (19.7) | — | — | — | — |
| 1 mo | 144 | 55.6 (19.4) | 144 | 1.2 | (0.9 to 1.4) | <0.001 |
| 3 mo | 140 | 55.9 (19.5) | 140 | 1.7 | (1.3 to 2.1) | <0.001 |
| 6 mo | 133 | 56.0 (18.8) | 133 | 2.5 | (1.9 to 3.1) | <0.001 |
| BMI, kg/m2 | ||||||
| Baseline | 151 | 21.3 (4.5) | — | — | — | — |
| 1 mo | 144 | 21.4 (4.3) | 144 | 0.4 | (0.3 to 0.5) | <0.001 |
| 3 mo | 140 | 21.6 (4.4) | 140 | 0.6 | (0.4 to 0.7) | <0.001 |
| 6 mo | 133 | 21.7 (4.4) | 133 | 0.8 | (0.6 to 1.0) | <0.001 |
| FEV1, L | ||||||
| Baseline | 151 | 2.5 (1.0) | — | — | — | — |
| 1 mo | 144 | 2.7 (1.1) | 144 | 0.2 | (0.2 to 0.3) | <0.001 |
| 3 mo | 138 | 2.6 (1.1) | 138 | 0.2 | (0.2 to 0.2) | <0.001 |
| 6 mo | 131 | 2.7 (1.0) | 131 | 0.3 | (0.2 to 0.3) | <0.001 |
| FEV1, % predicted | ||||||
| Baseline | 151 | 82.6 (25.6) | — | — | — | — |
| 1 mo | 144 | 89.4 (25.6) | 144 | 6.7 | (5.2 to 8.3) | <0.001 |
| 3 mo | 138 | 88.7 (25.8) | 138 | 5.4 | (4.0 to 6.7) | <0.001 |
| 6 mo | 131 | 90.1 (25.0) | 131 | 6.7 | (4.9 to 8.5) | <0.001 |
| FVC, L | ||||||
| Baseline | 151 | 3.3 (1.2) | — | — | — | — |
| 1 mo | 144 | 3.5 (1.3) | 144 | 0.2 | (0.2 to 0.3) | <0.001 |
| 3 mo | 138 | 3.4 (1.3) | 138 | 0.2 | (0.1 to 0.2) | <0.001 |
| 6 mo | 131 | 3.5 (1.3) | 131 | 0.3 | (0.2 to 0.3) | <0.001 |
| FVC, % predicted | ||||||
| Baseline | 151 | 101.4 (24.2) | — | — | — | — |
| 1 mo | 144 | 106.9 (24.9) | 144 | 5.7 | (4.2 to 7.2) | <0.001 |
| 3 mo | 138 | 106.2 (24.8) | 138 | 4.4 | (3.0 to 5.8) | <0.001 |
| 6 mo | 131 | 108.3 (26.4) | 131 | 6.2 | (4.3 to 8.1) | <0.001 |
| Sweat chloride, mEq/L | ||||||
| Baseline | 147 | 102.9 (13.8) | — | — | — | — |
| 1 mo | 135 | 55.2 (23.6) | 132 | −47.3 | (−50.9 to −43.7) | <0.001 |
| 3 mo | 128 | 50.8 (23.3) | 127 | −52.2 | (−56.0 to −48.3) | <0.001 |
| 6 mo | 127 | 49.0 (23.1) | 125 | −53.8 | (−57.7 to −49.9) | <0.001 |
| CFQR Respiratory Domain, points | ||||||
| Baseline | 151 | 70.7 (19.9) | — | — | — | — |
| 1 mo | 142 | 80.9 (13.6) | 142 | 9.7 | (7.1 to 12.4) | <0.001 |
| 3 mo | 139 | 82.2 (16.5) | 139 | 10.9 | (8.1 to 13.7) | <0.001 |
| 6 mo | 133 | 78.9 (17.0) | 133 | 7.4 | (4.1 to 10.7) | <0.001 |
| CFRSD Severity Score, points | ||||||
| Baseline | 146 | 27.7 (14.9) | — | — | — | — |
| 1 mo | 142 | 20.4 (13.4) | 138 | −7.0 | (−9.3 to −4.8) | <0.001 |
| 3 mo | 140 | 19.2 (14.7) | 135 | −8.0 | (−10.3 to −5.7) | <0.001 |
| 6 mo | 131 | 18.6 (15.0) | 128 | −9.0 | (−11.6 to −6.4) | <0.001 |
| SNOT-20, points | ||||||
| Baseline | 151 | 0.9 (0.8) | — | — | — | — |
| 1 mo | 142 | 0.7 (0.7) | 142 | −0.3 | (−0.4 to −0.2) | <0.001 |
| 3 mo | 140 | 0.6 (0.7) | 140 | −0.3 | (−0.4 to −0.2) | <0.001 |
| 6 mo | 133 | 0.7 (0.7) | 133 | −0.2 | (−0.3 to −0.1) | <0.001 |
Definition of abbreviations: BMI = body mass index; CFQR = Cystic Fibrosis Questionnaire-Revised; CFRSD = Cystic Fibrosis Respiratory Symptom Diary; CI = confidence interval; SNOT-20 = Sino-Nasal Outcome Test-20.
Lung function improved from baseline percent of predicted FEV1% of 82.6 (25.6) to 90.1 (25.0) at 6 months (mean change, 6.7; 95% CI, 4.9–8.5; P < 0.001) (Figure 2A and Table 2); FEV1% improvement was detectable as soon as the 1-month follow-up visit (mean change, 6.7; 95% CI, 5.2–8.3; P < 0.001). A similar increase was seen in percent of predicted FVC (mean change at 6 months, 6.2; 95% CI, 4.3–8.1; P < 0.001). Body weight improved from baseline to 6 months (mean change, 2.5 kg; 95% CI, 1.9–3.1; P < 0.001); body mass index (BMI) also increased 0.8 kg/m2 (95% CI, 0.6–1.0; P < 0.001) by 6 months (Figure 2B). After initiation of ivacaftor, decline in sweat chloride from baseline of 102.9 (13.8) mmol/L was detected by 1 month (55.2 [23.6] mmol/L) and was maintained at 6 months (mean change, −53.8; 95% CI, −57.7 to −49.9; P < 0.001) (Figure 2C). There was significant improvement in all measures of quality of life, including the respiratory domain of the CFQ-R (7.4; 95% CI, 4.1–10.7; P < 0.001) (Figure 2D), Cystic Fibrosis Respiratory Symptom severity score (−9.0; 95% CI, −11.6 to −6.4; P < 0.001), and the Sino-Nasal Outcome Test-20 (−0.24; 95% CI, −0.35 to −0.13; P < 0.001) at the end of 6 months (Table 2; see Figure E1 in the online supplement). Baseline and changes in clinical outcome at 6 months subdivided by age group are shown in Table E1. Changes in outcome measures were similar across age groups with the exception of the youngest age group (age 6–11), which had a smaller change in FEV1% and CFQ-R, likely caused by less severe disease at baseline.
Figure 2.
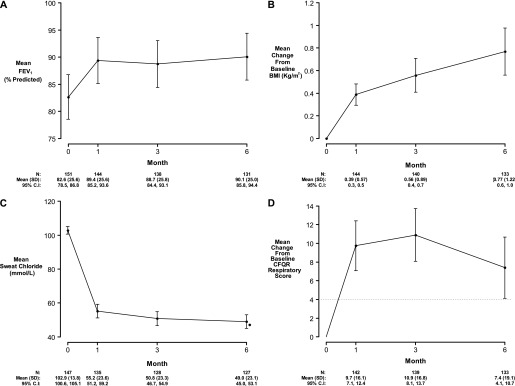
Clinical measures at baseline, 1, 3, and 6 months postivacaftor. (A) FEV1 percent predicted. Change from baseline in (B) body mass index (BMI) and (C) sweat chloride. (D) Change from baseline in Cystic Fibrosis Questionnaire-Revised (CFQR) Respiratory Score. Means and 95% confidence intervals. Horizontal hashed line in D at 4 represents minimal clinically important difference (41).
Hospitalization and P. aeruginosa history from the year before ivacaftor initiation and up to 12 months following commencement (mean [SD] follow-up, 7.5 [3.0] months) were acquired from the CFFNPR, which records data from spontaneously expectorated and oropharyngeal cultures. In the 6 months before starting ivacaftor 41 of 150 (27.3%) had been hospitalized (one participant did not have data during that interval) and 75 of 143 (52.4%) were P. aeruginosa positive on at least one culture. The percent of participants who were hospitalized during the 6 months following ivacaftor declined by 19.1% (95% CI, 10.8–27.5; P < 0.001) compared with the 6 months before ivacaftor; the total number of hospitalizations was also reduced by 16.3% (95% CI, 8.1–24.4; P < 0.001) compared with the same 6-month period 1 year earlier (Figure 3A). Adjusting for differential follow-up, there were on average (SD) 0.70 (1.2) hospitalizations per participant year before ivacaftor, and 0.30 (1.2) postivacaftor, for a paired rate reduction of 0.35 hospitalizations per participant year (95% CI, 0.19–0.52; P < 0.001). There were significant reductions in the percent of participants with at least one documented isolation of P. aeruginosa from a respiratory culture 6 months before initiation of ivacaftor compared with 6 months afterward (18.8% fewer; 95% CI, 7.1–30.6; P = 0.003) (Figure 3B). As a control and to account for possible seasonality, we also evaluated the 6-month intervals starting from 1 year before initiation; a similar reduction in P. aeruginosa isolation was observed following ivacaftor treatment (21.5%; 95% CI, 9.5–33.6; P < 0.001). A sensitivity analysis accounting for missing data confirmed a statistically significant reduction in P. aeruginosa growth (P < 0.05).
Figure 3.

Clinical measures obtained through Cystic Fibrosis Foundation National Patient Registry in 6-month intervals preivacaftor and postivacaftor. (A) Participants with at least one hospitalization and (B) Pseudomonas aeruginosa positivity among those with a recorded respiratory culture in 6-month intervals. Means and 95% confidence intervals; *P < 0.001.
MCC
Twenty-three subjects participated in the MCC substudy at four sites. Twenty-two subjects completed baseline MCC assessments, and 21 completed all assessments: mean (SD) age, 25.2 (7.7) years; 36.4% were female. This cohort had similar improvements in FEV1, BMI, patient-reported outcomes, and sweat chloride as the core study. Particle clearance from the whole right lung was markedly increased from baseline at both 1 and 3 months postivacaftor, with no difference between the post-treatment time points (Figure 4A). Average clearance through 60 minutes at 1 month post-treatment was more than twice the baseline value (P < 0.001), reflecting substantially improved MCC (Figure 4B).
Figure 4.

Substudy mucociliary clearance (A) mean particle retention curves from the whole right lung at baseline and 1 month following ivacaftor treatment. (B) Average (± SD) whole lung clearance for 60 minutes after inhaling Tc99m-SC aerosol. *P < 0.001 versus baseline.
GI pH
Eleven participants enrolled in this substudy at four sites; complete data before and after ivacaftor administration were available in seven participants: mean (SD) age, 32.2 (8.7) years; four were female. Improvements in FEV1 and sweat chloride were similar to the core study although weight gain was not observed. Intestinal pH profiles before and after ivacaftor outline a significant improvement in early ability to neutralize gastric acid (Figure 5). Average clearance 0–30 minutes after gastric emptying was significantly higher after ivacaftor (mean difference, 2,632 pH seconds; P = 0.001), translating to an average pH increase of 1.46 (95% CI, 0.86–2.06).
Figure 5.

Substudy small intestinal pH profile. Mean pH readings every minute at baseline (gray) and 1 month postivacaftor (black). GE = gastric emptying.
Sputum Inflammation and Microbiome
Paired induced sputum specimens were obtained preivacaftor and postivacaftor in 14 substudy participants at five sites; analysis of the sputum inflammation and microbiome was limited to these samples. The mean (SD) age was 27 (14) years, six were females, and baseline mean (SD) FEV1 was 84 (23) % predicted. This cohort experienced significant improvements in FEV1 and reductions in sweat chloride that resembled the core study (P < 0.001 for both). There were no significant changes in any sputum markers of inflammation, including neutrophil elastase activity (mean change [SD], −0.1 [0.37] log10 μg/ml; P = 0.29) (see Figure E2). Although sputum bacterial diversity did not change significantly with treatment (Shannon Diversity: mean change [SD], 0.13 [0.52]; P = 0.34), the combined relative abundance of traditional CF bacterial pathogens (listed in the Methods) trended down with treatment (mean change [SD], −13.9 [30.7]; P = 0.11) (see Figure E3A). Prevotella relative abundance significantly increased with treatment (mean change [SD], 8.8 [11.2]; P = 0.01) (see Figure E3B). By quantitative polymerase chain reaction, neither total bacterial load (mean change [SD], −0.18 (0.60) log10 gene copies/ml; P = 0.28) nor P. aeruginosa load (mean change [SD], −0.76 [2.5] log10 gene copies/ml; P = 0.27) changed significantly following ivacaftor administration.
β-Adrenergic Sweat Secretion Rate
Fifteen participants (mean [SD] age, 23.4 [9.2] years; 10 female) completed the substudy and data from 54 of 57 test recordings were suitable for masked analysis. This cohort demonstrated similar improvement in FEV1 and sweat chloride (49.1 mEq/L; 95% CI, 40.4–57.7 reduction; P < 0.001) in 1 month as the core study. At baseline the β-adrenergic sweat secretion rate was absent in all CF participants with an absolute rate of 0.4 (95% CI, −0.3 to 1.0). There was no detectable increase in β-adrenergic sweat secretion following initiation of ivacaftor at 1 or 3 months (mean change, 0.05 g/m2/h, 95% CI, −0.8 to 0.9, P = 0.91; and 0.2 g/m2/h, 95% CI, −0.7 to 1.1, P = 0.66, respectively) (see Figure E4). At 1 month and 3 months the absolute β-adrenergic sweat secretion rate was 0.3 (95% CI, −0.3 to 1.0) and 0.2 (95% CI, −0.5 to 0.9), respectively.
Discussion
Ivacaftor is the first CFTR modulator clinically approved for the treatment of the CF basic defect. Its initial use provided a unique opportunity to explore mechanism of action and evaluate the effectiveness of ivacaftor on a larger and more diverse population than previously studied. Furthermore, the efficacy demonstrated in phase 2 and 3 testing (7, 8, 26) has engendered considerable momentum toward developing an approach to the most common CFTR mutations and highlighted the need to understand the benefits and limitations of modulating CFTR (27). Our findings, in a sample nearly 40% larger than those receiving ivacaftor in the phase 3 program, indicate that ivacaftor confers substantial benefit to individuals with the G551D-CFTR mutation when used outside of a research setting. We observed significant clinical benefit on all outcomes tested, including spirometry, BMI, and several independent markers of quality of life. The effect was somewhat smaller than that observed in randomized phase 3 testing (7, 8), with the biggest discrepancies occurring in the 6–11 years age group: the phase 3 trial reports FEV1% improvement larger than observed here, and respiratory symptoms were similarly discrepant (see Table E1). In contrast, improved weight gain and BMI were consistently observed across all age groups in the two settings. The reduced pulmonary improvement observed in the age 6–11 cohort was likely caused by the higher FEV1 at baseline in our study (baseline FEV1% was 104% in the GOAL study compared with 85% in the phase 3 cohort) (8); high baseline spirometry is known to diminish measureable treatment effect because of a ceiling effect (28). Taken together, the results indicate that the effect of ivacaftor in G551D patients is robust, even outside the scope of a select patient population.
Remarkably, the effect of ivacaftor on sweat chloride, the principal biomarker of CFTR activity used in phase 3 development, matched the effect in phase 3 testing, even without controlling for time of collection in relation to drug administration; these data suggest sweat chloride is a highly robust biomarker for CFTR potentiator therapy, even when used in the routine clinical setting. Nevertheless, like that recently reported by Durmowicz and coworkers (29), changes in sweat chloride did not correlate with clinical response on an individual level (see Figure E5); further studies are needed to better understand the relationship between measures of CFTR activity and individual treatment response.
It has been suggested that efficient modulation of CFTR function should result in alterations in the natural history of the disease (1). CFTR dysfunction alters the endobronchial environment; 80% of patients with CF eventually have airway colonization with P. aeurginosa (3). The finding that recovery of P. aeruginosa significantly decreased with ivacaftor treatment provides the first empiric evidence that CFTR modulation alters CF microbiology in a beneficial way (Figure 3); reduced infection with pathogenic bacteria, such as P. aeruginosa, would be expected to substantially improve long-term prognosis (30–33). A trend toward reduced CF pathogens, particularly those with high relative abundance, was also observed in the microbiome analysis of a small subset of individuals, supporting observations in the core cohort. Additionally, an increased abundance of Prevotella, a bacteria associated with higher lung function in CF (24), was observed. It is possible that increased Prevotella may reflect increased microbial diversity, which is associated with improved lung function in CF (34); alternatively increased contamination from mouth flora is possible, but seems unlikely given all patients underwent induced sputum collection for this analysis. Given the rapidity of P. aeruginosa reduction, these findings suggest CFTR plays an important role in innate host defense. Additional research is needed to determine whether reduced colonization of P. aeruginosa is associated with increased microbial diversity and sustained long-term, and the clinical parameters that predict its successful elimination. If confirmed, there may be opportunities to eradicate P. aeruginosa in individuals treated with a CFTR modulator. Of note, despite altered colonization of P. aeruginosa and improved airway obstruction, biomarkers of airway inflammation did not meaningfully change with addition of ivacaftor. Although this study was not powered to detect significant changes in sputum markers of inflammation, significant improvements in airway inflammation likely require longer durations of treatment.
Defective MCC has long been postulated to be a fundamental defect underlying CF airway disease, although in vivo comparisons with healthy subjects have yielded inconsistent results, likely because of altered deposition of inhaled radiolabeled particles in CF caused by heterogeneous airway obstruction (35). Our findings indicate a large improvement in MCC following administration of ivacaftor that was sustained following more prolonged (3 mo) administration. The magnitude of improvement compares favorably with other therapies thought to augment MCC, including hypertonic saline (36). These results indicate that CFTR has a central role in regulating MCC, and its activation can have a large beneficial effect on this innate defense pathway. We speculate that the change in MCC caused mucus plugging to improve relatively quickly, resulting in a rapid improvement in spirometry that did not increase over time; this could explain the kinetics of FEV1 improvement observed with CFTR potentiator therapy shown here (Figure 2) and in prior studies (7, 8). The results also suggest that oral agents may be particularly well suited toward targeting the small airways, the site of earliest mucus obstruction in CF, which may not be reached as readily with inhaled therapeutics.
The effect of ivacaftor on body weight and BMI began shortly after initiation of treatment and was robust, both within the present study and the prior phase 3 testing (7, 8), suggesting that CFTR may have a direct effect on GI absorption. Although CFTR is well known for its activity as a chloride transporter in the airways, its role as a bicarbonate transporter is of renewed interest (37, 38), and is particularly important in the regulation of pancreatic and GI function (39). Our results indicated that ivacaftor had pronounced effects on duodenal alkalization that was sustained throughout the distal intestine, approaching levels seen in normal individuals (21). These findings suggest that normalization of GI pH via CFTR potentiation may be a key feature that improves food and nutrient assimilation, and suggest that additional studies to evaluate changes in pancreatic function, macronutrient and micronutrient absorption, and intestinal mucosal health are needed.
A key goal of the present study was to evaluate mechanistic biomarkers sensitive to CFTR modulation. Recently, sweat rate measured by evaporative water loss was shown to reflect CFTR activity in secretory glands (25). We did not observe an improvement in β-adrenergic sweat rate despite improved sweat chloride in the same individuals. This may be a problem with lower-end sensitivity of the assay, or increased CFTR-mediated fluid absorption in the duct overriding a small increase in secretion in the coil, or insufficient CFTR activity to improve glandular secretion. Further studies in individuals with greater levels of CFTR function are needed to understand these distinctions.
Although this study was observational in nature, we believe the conclusions are significant and reliable. Execution of the core study clearly benefited from the relatively rapid uptake of ivacaftor among eligible individuals (enrollment exceeded expectations and concluded within 4.5 mo). Furthermore, historical data provided by the CFFNPR provided an accurate comparison from which to base conclusions. The study also benefited from experienced research sites and a motivated patient population (40). Nevertheless, further studies are warranted to follow up on a number of novel observations because the study did not include a placebo control. Although analyses of biomarkers were standardized and quantitative, subjective assessments, such as patient-reported outcomes, may be biased by treatment benefit. Similarly, longer periods of observation may further the understanding of this novel treatment strategy. Nevertheless, the results of this study confirm the robust effect of stimulating CFTR among individuals with CF, provide seminal information regarding the mechanism underlying clinical benefit of respiratory and GI disease, and indicate the surprising potential to drastically alter the natural history of the disease by altering colonization of P. aeruginosa, suggesting a promising future for individuals with G551D-mediated CF.
Acknowledgments
Acknowledgment
This study was conducted with the participation of the study site principal investigators and coordinators listed in the online supplement. The study was executed with the assistance of the TDN Coordinating Center, which included the contributions of Nicole Rogers, Rachael Buckingham, Rose Mitchell, and Lynette Brown; in addition to Heather Hathorne (University of Alabama at Birmingham). The authors acknowledge assistance from the CFF National Patient Registry Team, including Dr. Bruce Marshall and Alex Elbert, and support for study planning and execution provided by Dr. Preston W. Campbell III.
The GOAL Study included four substudies, and the authors acknowledge the following key personnel on the GOAL Investigative Team:
The Mucociliary Clearance Substudy was led by Dr. Scott Donaldson and included the following site investigators: Drs. Timothy E. Corcoran, Landon W. Locke, Michael M. Myerburg, and Joseph M. Pilewski (University of Pittsburgh); Drs. Beth Laube, Pam Zeitlin, and Michael Boyle (Johns Hopkins University); and Drs. Pradeep Bhambhvani, Brian Flanagan, and Steven M. Rowe (University of Alabama at Birmingham). Drs. William Bennett and Kirby Zeman (University of North Carolina at Chapel Hill) provided a leadership role and conducted education, training, coordination, and centralized image analysis.
The Sweat Rate Substudy was led by Dr. Tanja Gonska and included the following site investigators: Dr. Peter Durie (University of Toronto); Drs. Paul Quinton, A. K. M. Shamsuddin, Douglas J. Conrad, and Mark Pian (University of California, San Diego); Drs. Jeffrey Wine and Carlos Milla (Stanford University); Dr. Frank Accurso (University of Colorado); and Dr. Steven M. Rowe (University of Alabama at Birmingham). Dr. Erik Frisbee also provided technical assistance in designing the study. The study was possible because of the support of the CF Foundation Sweat Rate Consortium organized by Drs. Elizabeth Joseloff and Diane Wetmore (Cystic Fibrosis Foundation).
The Sputum Substudy was led by Dr. Scott Sagel and included the following site investigators: Dr. Jennifer L. Taylor-Cousar (National Jewish Health), Dr. Christopher M. Oermann (Texas Children's Hospital, Baylor College of Medicine), Dr. Joanne L. Billings (University of Minnesota), and Dr. Ronald C. Rubenstein (Children's Hospital of Philadelphia). Drs. J. Kirk Harris and Edith T. Zemanick (Children's Hospital Colorado) performed and helped to interpret the sputum microbiome analyses. Peggy Emmett (Director of the Pediatric Clinical Translational Research Core Laboratory at Children's Hospital Colorado) oversaw the measurements of sputum inflammatory mediators. Dr. Brandie D. Wagner (Colorado School of Public Health) performed the biostatistical analyses.
The pH Pill Substudy was led by Drs. Drucy Borowitz and Daniel Gelfond and included the following site investigators: Dr. Ahmet Z. Uluer (Children's Hospital Boston); Dr. Leonard Sicilian (Massachusetts General Hospital); Dr. Carla A. Fredrick (Women and Children's Hospital of Buffalo); Dr. John P. Clancy (Cincinnati Children's Hospital Medical Center); and Dr. Michael W. Konstan (Rainbow Babies and Children's Hospital).
Footnotes
Supported by the Cystic Fibrosis Foundation Therapeutics (GOAL11K1) and the National Institutes of Health (DK072482, DK089507, UL1TR000165, UL1TR000423, UL1TR001111, and UL1TR001082).
Author Contributions: All authors contributed to the study design, data collection, and data interpretation for this manuscript. GOAL Site Investigators performed data collection. S.L.H., T.G., S.H.D., D.B., D.G., S.D.S., and U.K. performed analyses. S.M.R., S.L.H., T.G., S.H.D., D.B., D.G., and S.D.S. drafted the manuscript. N.M.-H., J.M.V.D., E.J., and B.W.R. also contributed significantly to the manuscript.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201404-0703OC on June 13, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- 3.Gelfond D, Borowitz D.Gastrointestinal complications of cystic fibrosis Clin Gastroenterol Hepatol 201311333–342.quiz e330–331 [DOI] [PubMed] [Google Scholar]
- 4.Collie JT, Massie RJ, Jones OA, Legrys VA, Greaves RF. Sixty-five years since the New York heat wave: advances in sweat testing for cystic fibrosis. Pediatr Pulmonol. 2014;49:106–117. doi: 10.1002/ppul.22945. [DOI] [PubMed] [Google Scholar]
- 5.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation National Patient Registry annual data report 2012. Bethesda, MD: Cystic Fibrosis Foundation; 2013
- 6.Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45:1160–1167. doi: 10.1038/ng.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, et al. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, Mainz JG, Rodriguez S, Li H, Yen K, et al. VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219–1225. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donaldson SH, Zeman K, Laube B, Corcoran T, Locke LW, Pilewski J, Hanes J, Schuster B, Kanzawa M, Rowe SM, et al. Effect of ivacaftor on mucociliary clearance and mucus rheology in patients with a g551d CFTR mutation [abstract] Pediatr Pulmonol Supp. 2013;48:279–280. [Google Scholar]
- 10.Gelfond D, Borowitz D, Frederick CA, Uluer A, Sicilian L, Konstan MW, Rowe SM. Impact of ivacaftor therapy on the intestinal pH profile in CF subjects with g551d mutation. Pediatr Pulmonol. 2013;48:405. [Google Scholar]
- 11.Gonska T, Shamsuddin A, Conrad D, Wine J, Milla C, Accurso FJ, Dupuis A, Avolio J, Hamblett NM, Heltshe SL, et al. Effect of ivacaftor on b-adrenergic sweat secretion by evaporimetry in g551d patients [abstract] Pediatr Pulmonol Supp. 2013;48:279. [Google Scholar]
- 12.Rowe SM, Heltshe SL, Gonska T, Donaldson SD, Borowitz D, Gelfond D, Sagel SD, Khan U, Hamblett NM, VanDalfsen J, et al. Results of the g551d observational study: the effect of ivacaftor in g551d patients following FDA approval [abstract] Pediatr Pulmonol Supp. 2013;48:278. [Google Scholar]
- 13.Sagel SD, Harris JK, Wagner BD, Zemanick ET, Emmett P, Taylor-Cousar JL, Oermann CM, Billings JL, Rubenstein RC, Rowe SM. Effects of ivacaftor on airway microbiome and inflammation in g551d patients [abstract] Pediatr Pulmonol Supp. 2013;48:285. [Google Scholar]
- 14.Quittner AL, Buu A, Messer MA, Modi AC, Watrous M. Development and validation of The Cystic Fibrosis Questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest. 2005;128:2347–2354. doi: 10.1378/chest.128.4.2347. [DOI] [PubMed] [Google Scholar]
- 15.Goss CH, Edwards TC, Ramsey BW, Aitken ML, Patrick DL. Patient-reported respiratory symptoms in cystic fibrosis. J Cyst Fibros. 2009;8:245–252. doi: 10.1016/j.jcf.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Piccirillo JF, Merritt MG, Jr, Richards ML. Psychometric and clinimetric validity of the 20-item Sino-Nasal Outcome Test (SNOT-20) Otolaryngol Head Neck Surg. 2002;126:41–47. doi: 10.1067/mhn.2002.121022. [DOI] [PubMed] [Google Scholar]
- 17.American Thoracic Society. Standardization of spirometry, 1994 update. American Thoracic Society. Am J Respir Crit Care Med. 1995;152:1107–1136. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 18.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–187. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG., Jr Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol. 1993;15:75–88. doi: 10.1002/ppul.1950150204. [DOI] [PubMed] [Google Scholar]
- 20.Bennett WD, Laube BL, Corcoran T, Zeman K, Sharpless G, Thomas K, Wu J, Mogayzel PJ, Jr., Pilewski J, Donaldson S. Multisite comparison of mucociliary and cough clearance measures using standardized methods. J Aerosol Med Pulm Drug Delivery. 2013;26:157–164. doi: 10.1089/jamp.2011.0909. [DOI] [PubMed] [Google Scholar]
- 21.Gelfond D, Ma C, Semler J, Borowitz D. Intestinal pH and gastrointestinal transit profiles in cystic fibrosis patients measured by wireless motility capsule. Dig Dis Sci. 2013;58:2275–2281. doi: 10.1007/s10620-012-2209-1. [DOI] [PubMed] [Google Scholar]
- 22.Ordoñez CL, Henig NR, Mayer-Hamblett N, Accurso FJ, Burns JL, Chmiel JF, Daines CL, Gibson RL, McNamara S, Retsch-Bogart GZ, et al. Inflammatory and microbiologic markers in induced sputum after intravenous antibiotics in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:1471–1475. doi: 10.1164/rccm.200306-731OC. [DOI] [PubMed] [Google Scholar]
- 23.Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med. 2012;186:857–865. doi: 10.1164/rccm.201203-0507OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zemanick ET, Harris JK, Wagner BD, Robertson CE, Sagel SD, Stevens MJ, Accurso FJ, Laguna TA. Inflammation and airway microbiota during cystic fibrosis pulmonary exacerbations. PLoS ONE. 2013;8:e62917. doi: 10.1371/journal.pone.0062917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinton P, Molyneux L, Ip W, Dupuis A, Avolio J, Tullis E, Conrad D, Shamsuddin AK, Durie P, Gonska T. β-adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med. 2012;186:732–739. doi: 10.1164/rccm.201205-0922OC. [DOI] [PubMed] [Google Scholar]
- 26.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clancy JP, Jain M. Personalized medicine in cystic fibrosis: dawning of a new era. Am J Respir Crit Care Med. 2012;186:593–597. doi: 10.1164/rccm.201204-0785PP. [DOI] [PubMed] [Google Scholar]
- 28.Montgomery AB, Abuan T, Yeager MA. Regulatory aspects of phase 3 endpoints for new inhaled antibiotics for cystic fibrosis patients with chronic pseudomonas aeruginosa infections. J Aerosol Med Pulm Drug Delivery. 2012;25:198–203. doi: 10.1089/jamp.2011.0911. [DOI] [PubMed] [Google Scholar]
- 29.Durmowicz AG, Witzmann KA, Rosebraugh CJ, Chowdhury BA. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest. 2013;143:14–18. doi: 10.1378/chest.12-1430. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, Collins J, Rock MJ, Splaingard ML. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 2005;293:581–588. doi: 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- 31.Mayer-Hamblett N, Kronmal RA, Gibson RL, Rosenfeld M, Retsch-Bogart G, Treggiari MM, Burns JL, Khan U, Ramsey BW EPIC Investigators. Initial Pseudomonas aeruginosa treatment failure is associated with exacerbations in cystic fibrosis. Pediatr Pulmonol. 2012;47:125–134. doi: 10.1002/ppul.21525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, Stokes DC, Wohl ME, Wagener JS, Regelmann WE, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis J Pediatr 2007151134–139.139 e131 [DOI] [PubMed] [Google Scholar]
- 33.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 34.Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Andersen GL, Brown R, Fujimura KE, et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE. 2010;5:e11044. doi: 10.1371/journal.pone.0011044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donaldson SH, Corcoran TE, Laube BL, Bennett WD. Mucociliary clearance as an outcome measure for cystic fibrosis clinical research. Proc Am Thorac Soc. 2007;4:399–405. doi: 10.1513/pats.200703-042BR. [DOI] [PubMed] [Google Scholar]
- 36.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241–250. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 37.Quinton PM. Role of epithelial HCO3⁻ transport in mucin secretion: lessons from cystic fibrosis. Am J Physiol Cell Physiol. 2010;299:C1222–C1233. doi: 10.1152/ajpcell.00362.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafsson JK, Ermund A, Ambort D, Johansson ME, Nilsson HE, Thorell K, Hebert H, Sjövall H, Hansson GC. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J Exp Med. 2012;209:1263–1272. doi: 10.1084/jem.20120562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013;3:a009753. doi: 10.1101/cshperspect.a009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe SM, Borowitz DS, Burns JL, Clancy JP, Donaldson SH, Retsch-Bogart G, Sagel SD, Ramsey BW. Progress in cystic fibrosis and the CF therapeutics development network. Thorax. 2012;67:882–890. doi: 10.1136/thoraxjnl-2012-202550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest. 2009;135:1610–1618. doi: 10.1378/chest.08-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]