

Significance
Coenzyme Q (CoQ) is a requisite component of the mitochondrial oxidative phosphorylation machinery that produces more than 90% of cellular ATP. Despite the discovery of CoQ more than 50 years ago, many aspects of its biosynthesis remain obscure. These include the functions of uncharacterized CoQ-related proteins whose disruption can cause human diseases. Our work reveals that one such protein, COQ9, is a lipid-binding protein that enables CoQ biosynthesis through its physical and functional interaction with COQ7, and via its stabilization of the entire CoQ biosynthetic complex. Unexpectedly, COQ9 achieves these functions by repurposing an ancient bacterial fold typically used for transcriptional regulation. Collectively, our work adds new insight into a core component of the CoQ biosynthesis process.
Keywords: COQ9, coenzyme Q, ubiquinone, COQ7, TFR family
Abstract
Coenzyme Q (CoQ) is an isoprenylated quinone that is essential for cellular respiration and is synthesized in mitochondria by the combined action of at least nine proteins (COQ1–9). Although most COQ proteins are known to catalyze modifications to CoQ precursors, the biochemical role of COQ9 remains unclear. Here, we report that a disease-related COQ9 mutation leads to extensive disruption of the CoQ protein biosynthetic complex in a mouse model, and that COQ9 specifically interacts with COQ7 through a series of conserved residues. Toward understanding how COQ9 can perform these functions, we solved the crystal structure of Homo sapiens COQ9 at 2.4 Å. Unexpectedly, our structure reveals that COQ9 has structural homology to the TFR family of bacterial transcriptional regulators, but that it adopts an atypical TFR dimer orientation and is not predicted to bind DNA. Our structure also reveals a lipid-binding site, and mass spectrometry-based analyses of purified COQ9 demonstrate that it associates with multiple lipid species, including CoQ itself. The conserved COQ9 residues necessary for its interaction with COQ7 comprise a surface patch around the lipid-binding site, suggesting that COQ9 might serve to present its bound lipid to COQ7. Collectively, our data define COQ9 as the first, to our knowledge, mammalian TFR structural homolog and suggest that its lipid-binding capacity and association with COQ7 are key features for enabling CoQ biosynthesis.
Ubiquinone, also known as coenzyme Q (CoQ), is a lipophilic, redox-active small molecule that is present in nearly every cellular membrane. CoQ is a critical component of the mitochondrial electron transport chain where it shuttles electrons from complexes I and II to complex III. In addition to its vital role in cellular respiration, CoQ is instrumental in cellular antioxidation, extracellular electron transport, and membrane rigidity (1).
The de novo biosynthesis of CoQ in eukaryotes takes place in the mitochondrial matrix via the collective action of at least 10 proteins (COQ1–10; Fig. S1) (2). Mutations in these proteins can cause primary CoQ deficiency—a condition associated with cerebellar ataxia, kidney disease, isolated myopathy, and severe childhood-onset multisystemic disorders (3, 4). Alteration in CoQ levels has also been associated with significant life span extensions in organisms ranging from Saccharomyces cerevisiae to mice (5–7). In S. cerevisiae (2, 8, 9), and potentially in higher eukaryotes (10, 11), most of the COQ proteins form a biosynthetic complex on the matrix face of the inner mitochondrial membrane. Although the majority of these proteins catalyze chemical modifications to CoQ precursors, the biochemical functions for COQ4, 8, and 9 have yet to be elucidated (8, 12, 13).
Recently, García-Corzo et al. developed a mouse harboring a truncated version of Coq9 (Coq9R239X)—modeled after a similar mutation observed in a human patient—that causes an encephalomyopathy associated with CoQ deficiency (11, 14). A hallmark feature of these mice is a decrease in the level of Coq7, a diiron protein that catalyzes the final hydroxylation of CoQ (15–19), and an accumulation of the Coq7 substrate, demethoxy coenzyme Q (DMQ) (11) (Fig. S1). This observation is consistent with the accumulation of DMQ seen in Coq7−/− mice, which fail to survive past embryonic development (20, 21). Together, these observations suggest that a central function of Coq9 is to promote the stability and activity of Coq7.
Despite these recent insights, elucidating the function of COQ9 at the molecular level has remained particularly challenging. Here, we report the first (to our knowledge) crystal structure of a Coq9 ortholog, human COQ9, at 2.4-Å resolution. Our work reveals that COQ9 shares striking structural homology to members of the TetR family of regulators (TFRs)—a small molecule-binding family largely composed of bacterial transcriptional regulators (22, 23). Our COQ9 structure reveals the presence of a lipid molecule bound in a hydrophobic pocket; however, in contrast to canonical TFR proteins, COQ9 likely does not retain DNA-binding capabilities. We demonstrate that COQ9 is essential for the stabilization of the putative CoQ biosynthetic complex in higher eukaryotes and that it physically associates with COQ7. Mutations of residues within a COQ9 surface patch, near the lipid-binding pocket of COQ9, disrupt this interaction and lead to decreased CoQ production in yeast, concomitant with the buildup of the COQ7 substrate. Our work defines the first mammalian TFR structural homolog, to our knowledge, and identifies COQ9 lipid- and protein-binding features central to its role in enabling CoQ biosynthesis.
Results
Coq9 Is Essential for the Stability of Mouse CoQ Biosynthesis Proteins.
Mice homozygous for a truncated version of Coq9 (Coq9R239X) have decreased levels of Coq7 and accumulate the substrate for the Coq7 reaction (Fig. S1) (14); however, the effect of this mutation on the other COQ proteins—and on mitochondrial and cellular proteins in general—has not been evaluated. In S. cerevisiae, complete loss of Coq9p destabilizes the CoQ complex and precludes CoQ production, whereas partial loss, aided by artificial stabilization of the complex, allows minimal CoQ production and accumulation of DMQ (2, 24–26). Here, we sought to determine whether disruption of Coq9, via the Coq9R238X mutation, affected the stability of COQ proteins in mice, where the existence of a CoQ biosynthetic complex has yet to be shown.
To test whether steady-state levels of other proteins involved in CoQ biosynthesis were decreased in the Coq9R239X mice, we performed quantitative analysis of the proteomes from Coq9R239X and wild-type mouse tissue. The proteomes from three tissues (heart, kidney, and cerebellum) were analyzed by liquid chromatography (LC)-MS/MS using tandem mass tagging to measure relative protein abundances (Fig. 1 A and B). Strikingly, our analysis shows that the steady-state levels of all detected members of the putative biosynthetic complex are specifically decreased in Coq9R239X hearts relative to other proteins (Fig. 1C). Interestingly, several other mitochondrial proteins changed significantly in abundance, perhaps suggesting their involvement in CoQ biosynthesis or in compensatory processes invoked during CoQ deficiency (Fig. S1 and Dataset S1). For example, the level of a mitochondrial sulfide-quinone oxidoreductase (Sqrdl) is significantly decreased in Coq9R239X mice hearts. This result, along with reports of sulfide accumulation in CoQ-deficient yeast (27), indicates a connection between CoQ biosynthesis and mitochondrial hydrogen sulfide metabolism.
Fig. 1.
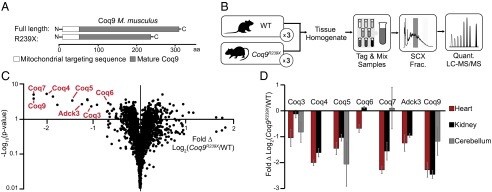
Coq9R239X mice have decreased steady-state levels of CoQ biosynthesis proteins. (A) Coq9R239X truncation scheme designed by García-Corzo et al. (11). (B) Experimental design for proteomic LC-MS/MS analysis of WT and Coq9R239X mice tissue (heart, kidney, cerebellum with n = 3 for each tissue). (C) Volcano plot of protein abundance (•) changes between WT and Coq9R239X mouse heart [Fold Δ Log2(Coq9R239X/WT) versus –Log10(P value)] with COQ proteins labeled. (D) COQ protein abundance changes between WT and Coq9R239X in ( ) heart, (■) kidney, and (
) heart, (■) kidney, and ( ) cerebellum [Fold Δ Log2(Coq9R239X/WT)] with error bars showing standard deviation. See also Fig. S1.
) cerebellum [Fold Δ Log2(Coq9R239X/WT)] with error bars showing standard deviation. See also Fig. S1.
A similar decrease in COQ proteins was observed in the Coq9R239X kidneys and cerebellums (Fig. 1D and Fig. S1). Notably, several COQ proteins (Coq6, Coq4, Adck3) were not detected in our analysis of the cerebellum tissue, and Coq7 levels yielded mixed results, resulting in no significant change (Fig. 1D and Dataset S1). The lack of detection could indicate that these proteins are not present in this tissue, suggesting that a CoQ biosynthetic complex might not be an obligate feature of CoQ biosynthesis in vertebrates. However, only the lack of Coq4 in cerebellum is supported by recent global proteomics analyses (28, 29). The absence of these proteins in our analyses is most likely due to the stochastic nature of peptide sampling combined with the reduced likelihood of detecting low-abundance proteins within heterogeneous cell types from a tissue with lower mitochondrial content than heart and kidney (30). Of all the COQ polypeptides that are detected and whose abundances are significantly changing, only Coq6 in the kidney deviates from the decreasing trend in Coq9R239X mice. This divergence could reflect a tissue-specific regulatory feature of CoQ biosynthesis and CoQ biosynthetic complex formation. Overall, this analysis supports the existence of a CoQ biosynthetic complex in higher eukaryotes and indicates that intact Coq9 is essential for the stability of other CoQ biosynthetic proteins.
COQ9 Crystal Structure Reveals TFR Structural Homology and a Bound Phospholipid.
Little is known about the structure or biochemical function(s) of COQ9, and thus it remains unclear how it can help stabilize the CoQ complex or influence the COQ7 reaction. To gain insight into these features, we sought to solve the crystal structure of a COQ9 ortholog. Ultimately, we were able to purify and crystallize an N-terminally truncated (NΔ79) version of human COQ9 that diffracted at 2.4 Å. This truncation removes 35 hydrophilic and poorly conserved residues downstream of the predicted mitochondrial targeting sequence, thus leaving the majority (87%) of the mature protein intact (Fig. 2 A and B, and Table 1).
Fig. 2.
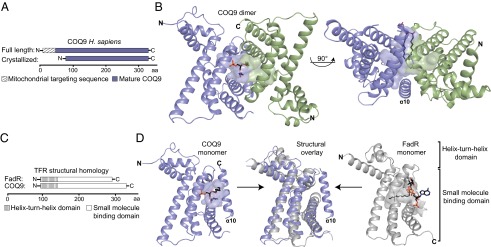
COQ9 crystal structure reveals structural homology to TFR family members. (A) Crystallization construct of COQ9. (B) Crystal structure of the COQ9 dimer with the monomers colored in blue and green, the cavity highlighted, and the cocrystallizing lipid shown. (C) COQ9 structural homology sequence scheme with FadR, a TFR family member (PDB 3anp) (32). (D) Individual and structural overlay of COQ9 (blue) and FadR (gray) monomers with their respective cavities and cocrystallizing ligands.
Table 1.
Data collection and refinement statistics
| Crystal parameters | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c, Å | 38.340, 97.628, 64.109 |
| α, β, γ, ° | 90, 95.766, 90 |
| Matthews coefficient, Å3/Da | 2.2 |
| Data quality | |
| Wavelength, Å | 0.97937 |
| Resolution, Å | 48.8–2.4 (2.49–2.4) |
| Rmerge, %* | 8.9 (51.4) |
| No. of unique reflections | 35,868 |
| No. of reflections in Rfree set† | 3,587 |
| Mean redundancy | 3.6 (3.7) |
| Completeness, overall, % | 99.8 (100) |
| Mean I/σI | 15.8 (2.5) |
| Refinement statistics | |
| Rfree, % | 17.9 |
| Rwork, % | 24.6 |
| Completeness, % | 99.38 (87.7) |
| Model quality | |
| Rmsd bond lengths, Å | 0.008 |
| Rmsd bond angles, ° | 1.1 |
| Ramachandran plot, % | |
| Most favored | 96.1 |
| Additional allowed | 3.9 |
| B factor, Å2 | 41.7 |
| Model contents | |
| Protomers in ASU | 2 |
| Protein residues | A: 95–283 |
| B: 93–233, 237–284, 293–311 | |
| Ligand | 1 PEF |
| No. atoms | 3,263 |
| Protein | 3,168 |
| Ligand | 34 |
| Water molecules | 61 |
Values in parentheses are for the highest resolution shell.
10% of the reflections were selected for free R calculation.
Our crystal structure reveals that COQ9 is structurally homologous to members of the TFRs. By comparing the structure of COQ9 to all structures in the Protein Data Bank (PDB), we found that the top 600 homologous proteins are all TFR or predicted TFR family members (31). The COQ9 structure aligns to TFR family members with z scores of as high as 16.2; however, its sequence identity with these family members is quite low, with only 14% sequence identity shared between the most similar structures. TFR family members are generally bacterial transcriptional regulators that form homodimers and bind small molecule ligands and DNA. Members contain nine α-helices that form two domains: a DNA-binding domain with a helix-turn-helix (HTH) motif (helices α1–3) and a ligand-binding domain with a central triangle of helices (helices α4–9) (22). COQ9 shares these structural features, but also has a 10th α-helix at its C terminus (Fig. 2 C and D). This helix is ordered in only one of the two COQ9 molecules in the asymmetric unit, and the loop connecting it to helix 9 is disordered. As such, this 10th helix might assume a different conformation in vivo. Additionally, although the dimers observed in TFR family members form a four-helix bundle consisting of the C-terminal two helices (α8–9) from each monomer (22), the corresponding helices do not interact in the COQ9 dimer, which instead has the single 10th helix at the dimer interface.
The two COQ9 molecules in the asymmetric unit form a dimer, in agreement with static light-scattering analysis of recombinant COQ9 in solution (Fig. S2), with a total of 1,250 Å2 of the surface area of each monomer buried in the dimer interface. This interface consists of mostly hydrophobic residues, including many noninteracting residues and a single hydrogen bond contained in the 10th helix. Intriguingly, COQ9 crystallized with a bound lipid at the dimer interface, consistent with the small molecule-binding capabilities of the TFR proteins. The partial electron density suggests that this phospholipid has at least 10 carbons in each acyl group, which extend into a predominantly hydrophobic pocket comprised from both monomers (Fig. 3 A–C). This pocket aligns well with that of a protein in the FadR subclass of TFRs that cocrystallized with its ligand, acyl CoA (Fig. 2 C and D) (32). The COQ9 pocket is a 20-Å–deep cavity enriched in aliphatic, neutral, and aromatic amino acids. There are also two charged residues (R189 and E255) residing at the bottom of the pocket, which may play a role in ligand recognition. Although the full acyl tails of the COQ9 ligand were not resolved, there is sufficient space and unassigned electron density to suggest that COQ9 has the capacity to bind a larger hydrophobic molecule than modeled. Although the bound lipid may not represent a bona fide endogenous ligand, the pocket properties suggest that a hydrophobic ligand for COQ9 is likely (Fig. 3B).
Fig. 3.

COQ9 has the ability to bind lipid. (A) Open-book view of the COQ9 dimer interface with the cocrystallizing lipid shown at the lipid-binding site. (B) Electrostatic surface (positive in blue and negative in red) of the COQ9 monomer cavity showing residues on the lipid-binding surface, with hydrophobic residues in black. (C) Electron density map of the cocrystallizing, partially resolved phospholipid, phosphatidic acid (10:0/10:0). (D) Copurifying lipids (in picomoles) with recombinant COQ9 purified from (■) E. coli or () wheat germ extract (CoQ, coenzyme Q; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol) with the picomolar amount of recombinant COQ9 from E. coli indicated (red dashed line). See also Fig. S2.
To attempt to determine the identity of the cocrystallized lipid, we performed LC-MS/MS on lipids extracted from recombinant COQ9 expressed in both Escherichia coli and wheat germ extract. We detected phosphatidylglycerol and phosphatidylethanolamine with various acyl chain lengths and saturations as the most abundant phospholipid species copurifying with COQ9 made from E. coli (Fig. 3D and Fig. S2). However, these phospholipids are highly abundant in E. coli and the calculated lipid/protein stoichiometries suggest that the COQ9/phospholipid stoichiometries exceed the 2:0.5 ratio expected for a COQ9 dimer with 50% lipid occupancy (Fig. S2). Overall, these data, along with the lack of clear electron density for the head group, suggest that the cocrystallizing lipid is a mixture of phospholipid species that help to solubilize the hydrophobic regions of COQ9 when overexpressed recombinantly. Interestingly, in each case, we also detected the host organism’s version of CoQ—CoQ8 in E. coli and CoQ9 in wheat germ extract—copurifying with COQ9, albeit in lower stoichiometric ratios (Fig. 3D and Fig. S2).
Despite the similarity of COQ9’s ligand binding domain to those of other TFR proteins, its HTH motif is quite distinct. In particular, the COQ9 HTH largely lacks the positive charges required for DNA binding by other TFR proteins, including FadR, suggesting that human COQ9 is unlikely to bind DNA itself. To assess this possibility more formally, we applied the DNA binding site identifier (DBSI) (33), which identifies amino acids with a propensity to bind DNA and chemically related molecules such as heparin (34). Calculations performed on the COQ9 monomer failed to identify the COQ9 HTH domain as a DNA-binding site (Fig. 4). In contrast, DBSI analyses correctly identified the known DNA binding sites of FadR (Fig. 4 and Fig. S3). Additionally, the position of the HTH motifs within the COQ9 dimer differs from other TFR proteins. TFR dimers are typically arranged such that the DNA-binding domains are adjacent and poised to interact with palindromic sequences (22). The dimer orientation of COQ9 within the crystal structure, however, places the HTH motifs on opposite sides of the dimer. Importantly, additional DBSI analyses on FadR and COQ9 dimers in their crystallographic orientations, along with a theoretical model of COQ9 threaded onto the FadR dimer using MODELLER (35), also failed to identify the COQ9 HTH domain as a DNA-binding site (Fig. S3). Overall, although it is difficult to prove the complete lack of DNA-binding capacity, our analyses suggest that COQ9 has shed the DNA-binding function of typical TFR proteins.
Fig. 4.
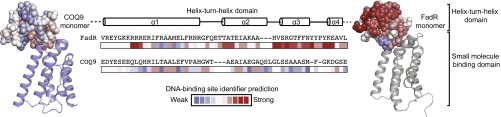
COQ9 HTH domain is not predicted to bind DNA. Residue predictions made by DBSI (33) for residues within the HTH domain in COQ9 and FadR monomers are shown and colored on a scale from weak (blue) to strong (red) likelihood of interacting with DNA. See also Fig. S3.
COQ9 Conserved Residues and Functional Domains Are Distinct from Archetypal TFR Proteins.
The crystal structure reveals that COQ9 retains the two main structural domains of TFR family proteins: a HTH and a small molecule-binding domain. We reasoned that regions within these domains that are highly conserved in primary sequence among COQ9 homologs are likely to be important for COQ9 function. We identified regions of high conservation using the Consurf server and mapped the overall pattern of conservation onto the COQ9 crystal structure (Fig. 5A and Fig. S4) (36–38). Two regions are highlighted: one within the HTH motif that is typically involved in DNA binding in TFR family members (23), and one within the small molecule-binding domain, which is the most highly conserved region of COQ9 (Fig. 5A).
Fig. 5.
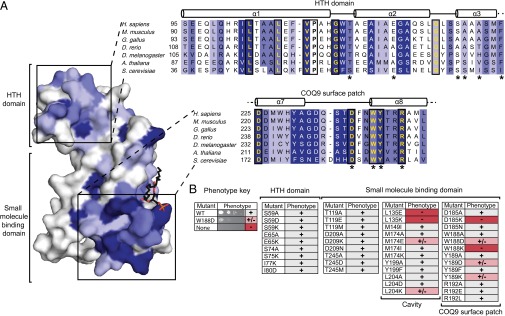
COQ9 has a conserved surface patch with key functional residues. (A) COQ9 residue conservation using 104 homologs mapped onto the monomer structure with the most conserved residues shown in increasing shades of blue. The HTH domain and COQ9 surface patch are highlighted in a subset sequence alignment with the most conserved residues colored in increasing shades of blue and invariant residues boxed and in yellow. Residues tested in the yeast genetic screen are indicated (*). (B) Coq9p point mutants, assayed by S. cerevisiae genetic complementation, with respiration competent (+), diminished competence (+/−), or incompetent (−) phenotypes. See also Figs. S4 and S5.
To begin identifying regions within these domains that are important for the role of COQ9 in CoQ biosynthesis, we conducted a yeast genetic complementation assay using single point mutations in coq9 from S. cerevisiae. We designed conservative and disruptive point mutations for 18 Coq9p residues that we hypothesized to be functionally important based on their location within the structure, their evolutionary conservation, and on the established importance of corresponding residues from other TFR proteins. Previous work has shown that Δcoq9 yeast have an inability to grow on nonfermentable carbon sources, which can be rescued by ectopic expression of wild-type coq9 (25). Here, we tested the functionality of the yeast Coq9p mutants by measuring their capacity to rescue this Δcoq9 growth phenotype (Fig. 5B and Fig. S5). For instance, the expression of WT or S59A Coq9p rescues Coq9p function, whereas expression of other mutants resulted in midrange (e.g., W188D) or severely hindered (e.g., W188K) growth phenotypes.
The results of this mutant screen reveal specific regions of sequence that are likely important for the biological function of Coq9p (Fig. 5B). First, multiple point mutations within the hydrophobic cavity of the small molecule-binding domain impair rescue of the Δcoq9 phenotype. This result suggests that binding a hydrophobic molecule is indeed important for Coq9p function. Second, a portion of the overall small molecule binding domain, which we term the “COQ9 surface patch (CSP),” is likewise very sensitive to point mutations. This region, which is solvent exposed in the crystal structure, is also highly conserved among COQ9 homologs, suggesting that it is a particularly important functional area of COQ9. In contrast, point mutations in residues throughout the N-terminal HTH motif that structurally align with the DNA-binding domain of TFR family members do not appear to disrupt Coq9p function. Consistently, the residues that align structurally with DNA contacts in TFR proteins are not particularly well conserved between COQ9 orthologs (Fig. S4). Coupled with our DNA-binding prediction analyses, these results lend further weight to our hypothesis that DNA binding is not a key feature of COQ9 function.
COQ9 Physically and Functionally Interacts with COQ7.
Given the requirement of full-length Coq9 for the stability of the mouse CoQ complex shown in this work, we hypothesized that the highly conserved COQ9 surface patch may be crucial for a protein–protein interaction with another member of the CoQ biosynthetic complex. Because the mice harboring the truncated Coq9 gene accumulate the substrate for Coq7 (11), and as Coq7p is reported to coprecipitate with Coq9p when isolated from yeast (24), we chose to test whether there is a protein–protein interaction between human COQ9 and COQ7. To do so, we coexpressed these two proteins using a cell-free wheat germ extract protein expression system. Physical association between COQ9 and COQ7 was assayed by coexpressing strep-tag II (SII)-tagged COQ9 (SII-NΔ45-COQ9) and six-histidine (6H)-tagged COQ7 (6H-NΔ38-COQ7), purifying the proteins by their tag affinity, and visualizing copurifying proteins by SDS/PAGE. Our results show that COQ7 copurified with COQ9, and vice versa, indicating that human COQ9 and COQ7 can form a complex in vitro (Fig. 6A and Fig. S6).
Fig. 6.

COQ9 physically and functionally associates with COQ7. (A) Tagged SII-COQ9 and H6-COQ7 cotranslated in wheat germ extract, purified using the SII or 6H affinity tag, or purified sequentially by both tags as indicated by arrows. (B) Point mutants of SII-COQ9 coexpressed with 6H-COQ7 and purified using the SII affinity tag on SII-COQ9. (C) The elution from the SII purification purified using the 6H affinity tag on 6H-COQ7. (D) Percentage of COQ9–COQ7 complex formed with each COQ9 point mutant relative to WT and measured by relative tryptophan fluorescence from the sequential purification shown in C. (E) Melting temperature of COQ9NΔ79 point mutants compared with WT [ΔTm,°C (COQ9MT – COQ9WT)] measured by differential scanning fluorimetry. Error bars show standard deviation (n = 3) and the absence of an observable melt curve is indicated (*). (F) COQ9 monomer with residues assayed for interaction with COQ7 colored by whether point mutants maintain the interaction (green), disrupt the interaction (red), or disrupt the interaction and the melting temperature of COQ9 (yellow). (G) Coq7p reaction efficiency measured by LC-MS/MS detection of DMQ6/(DMQ6 + CoQ6) in Δcoq9 strains harboring Coq9p point mutants, with error bars showing 95% confidence intervals and statistical significance using a two-tailed Student's t-test annotated by *P value < 0.001 (n = 6). See also Figs. S6 and S7.
To define the regions of COQ9 that contribute to the human COQ9–COQ7 interaction, we attempted to copurify COQ7 with point mutants of COQ9. Formation of the COQ9–COQ7 complex was assayed by cell-free coexpression of SII-NΔ45-COQ9 and 6H-NΔ38-COQ7, followed by sequential StrepTactin and nickel affinity purifications. The large excess of COQ9 relative to COQ7 during the first round of purification suggests that noncomplexed COQ9 was purified along with the COQ9–COQ7 complex (Fig. 6B and Fig. S6). When the product of the first purification was subjected to a second purification using the tag on COQ7, we were able to separate the complex from free COQ9 and visualize the amount of COQ9–COQ7 protein complex formed with each COQ9 mutant (Fig. 6C and Fig. S6). We used the relative tryptophan fluorescence of COQ9 and COQ7 after two rounds of purification to quantify whether the COQ9–COQ7 complex was intact, diminished, or absent in the case of each COQ9 mutant (Fig. 6D). These analyses identify a number of COQ9 residues that perturb the COQ9–COQ7 interaction (Fig. 6 B–D and Fig. S6). Several of the corresponding human COQ9 mutants that disrupted the human COQ9–COQ7 complex in vitro also disrupted S. cerevisiae Coq9p function in vivo. In general, these mutations occurred primarily in two regions—within the hydrophobic cavity of COQ9 and along the COQ9 surface patch.
The mutations identified above can be disruptive because they are either necessary for the COQ9–COQ7 interaction or because they result in general destabilization of COQ9. To distinguish between these possibilities, we purified mutant versions of recombinant human NΔ79-COQ9 from E. coli. Each COQ9 mutant was tested for thermal stability using differential scanning fluorimetry (39). A large decrease in the melting temperature (Tm) of the COQ9 mutants relative to wild-type, or the absence of an observable melt curve, would indicate that the mutant is likely unfolded or drastically misfolded. These analyses revealed that multiple mutations in or around the COQ9 lipid-binding cavity, such as L190E, M227E, and L256K, resulted in severely less thermally stable (ΔTm > 5 °C) protein, suggesting that the disrupted COQ9–COQ7 interaction for these mutants is at least partially due to improperly folded COQ9 (Fig. 6 D–F). However, several mutants along the COQ9 surface patch, including W240K, W240D, Y241K, and D237K, have melt temperatures similar to WT (ΔTm < 5 °C) and yet still severely disrupt the COQ9–COQ7 complex (Fig. 6 D–F). These results suggest that the COQ9 surface patch is an important functional domain because of its contribution to the COQ9–COQ7 protein interaction.
Because the substrate for Coq7 (DMQ) accumulates in the Coq9R239X mice, we hypothesized that the protein–protein interaction identified between COQ9 and COQ7 affects the efficiency of the Coq7 reaction. To test whether the folded COQ9 mutants that disrupt the human COQ9–COQ7 interaction in vitro are sufficient to diminish the yeast Coq7p reaction, we monitored CoQ6 production in Δcoq9 yeast containing these respective Coq9p mutants. Specifically, we measured the levels of DMQ6 and CoQ6 produced by actively respiring yeast with LC-MS/MS, and used the DMQ6/(DMQ6 + CoQ6) ratio as a proxy for Coq7p activity. As expected, the CoQ6 levels corresponded well with the respiratory competency of the yeast strains described above (Fig. S7). However, in the three strains harboring a Coq9p variant expected to disrupt the Coq9p–Coq7p interaction, the Coq7p reaction was less efficient than the strain rescued by WT Coq9p (Fig. 6G and Fig. S7). No DMQ6 or CoQ6 production was observed in the control (empty vector) strain that completely lacked Coq9p; this is consistent with previous reports and is likely due to complete disruption of the entire CoQ biosynthetic complex (25, 26). Together, these results suggest that the conserved residues on the COQ9 surface patch are essential for its physical and functional interaction with COQ7.
Discussion
The discovery of CoQ completed the final missing piece of the electron transport chain (ETC) machinery postulated by Warburg and Keilin (13). Following its isolation by Crane and Green in the 1950s, CoQ was established as a requisite gateway for electrons passing from CI and CII to CIII in the ETC, and has also been linked to other key antioxidant and electron transfer functions in membranes throughout the cell (40, 41). Decreased levels of CoQ lead to CoQ deficiency, which has heterogeneous clinical manifestations with presentations ranging from adult-onset isolated myopathy to fatal infantile multisystemic disease (4). CoQ deficiency is also common in mitochondrial disease patients, and, although CoQ supplementation has been popular, the success of this treatment is variable (42, 43).
Despite the vital role for CoQ in human health and disease, many core aspects of its endogenous biosynthesis in eukaryotes remain obscure, thereby preventing pathway manipulation as a therapeutic option. In particular, the functions of three proteins remain almost completely uncharacterized, despite being essential for CoQ production: COQ4, COQ8 (ADCK3), and COQ9. Here, we describe the structure of COQ9 along with its physical and functional interaction with COQ7.
We discovered that human COQ9 is structurally homologous to members of the TFRs. The TFRs are a bacterial family of proteins containing two distinct domains: a DNA-binding domain and a small molecule-binding domain. Characterized TFR family members bind a variety of ligands—from small molecules such as tetracycline to lipids such as stearoyl-CoA (22, 23, 32). This binding event affects the TFRs’ ability to bind DNA and, in turn, affects transcription of target genes (22, 23, 32). Notably, COQ9 is the first reported nonbacterial protein to display the TFR fold (rcsb.org as of August 27, 2014). We suggest that, in contrast to its structural counterparts, COQ9 is unlikely to act as a transcription factor. Although COQ9 exhibits the HTH motif found in the DNA-binding domain of TFRs, DBSI calculations suggest that this domain, which is correctly predicted in other TFRs, is absent in COQ9. Furthermore, there is a lack of positive charge in this region, and residues that structurally map to residues traditionally involved in DNA recognition are not highly conserved in COQ9. Additionally, Coq9p function is not impaired by point mutations within this region. These observations suggest that the TFR structural fold has been repurposed in COQ9 for a function other than transcriptional regulation.
A second function of the TFR fold is to enable binding to small molecules. In addition to having a small molecule-binding domain with a predominately hydrophobic cavity, the COQ9 crystal structure reveals a bound phospholipid. However, because the recombinant COQ9 was not purified from its native, mitochondrial location, we suspect that these abundant copurifying and cocrystallizing phospholipids might not be the endogenous COQ9 ligands. Although we likely have not yet identified a bona fide endogenous ligand for COQ9, its structural homology, along with its cocrystallizing lipid, suggest that COQ9 binds lipid as part of its role in CoQ biosynthesis. Because COQ9 is a member of the CoQ complex, and because CoQ and CoQ intermediates are thought to be part of the complex, it is tempting to speculate that COQ9 binds either CoQ or a CoQ precursor (2, 8). Unambiguous identification of an endogenous ligand for COQ9 will be challenging, because this protein exists natively in the CoQ biosynthetic protein complex and the presence of other members may affect the protein–ligand interaction (2, 24). This is especially true if the ligand is an intermediate in the CoQ biosynthesis pathway, as might be expected in light of the Coq7 reaction deficit in Coq9R239X mice. Regardless, the interaction of COQ9 with a CoQ precursor would be consistent with a previous report of a CoQ species being part of the mature CoQ complex (8, 17, 44) and would present a straightforward model for how COQ9 might functionally interact with other COQ proteins.
In addition to identifying an unexpected lipid-binding domain in COQ9, we show that human COQ9 engages in a protein–protein interaction with COQ7. COQ7 catalyzes the penultimate step in CoQ biosynthesis—the hydroxylation of demethoxy CoQ (15, 16, 18, 19, 45). We identify a highly conserved CSP that is important for maintaining the interaction with COQ7. Furthermore, we show that mutations in the CSP cause a decrease in the Coq7p reaction efficiency in yeast. A truncation of COQ9, observed first in a human patient with CoQ deficiency and then modeled in mice, truncates this CSP as well as a major portion of the cavity, presumably precluding lipid binding (Fig. S7). In both mice and humans, the COQ7 reaction is impeded and the late intermediate demethoxy CoQ accumulates (11, 14). Together, these results strongly suggest that the CSP is crucial for both the physical and functional interaction with COQ7. Furthermore, the vicinity of the CSP to the cocrystallizing lipid and to the hydrophobic cavity indicates that the COQ7 interaction and the lipid-binding capacity of COQ9 are related.
Our work might help resolve certain unexplained aspects of CoQ biosynthesis. For instance, it is known that, due to its hydrophobicity, mature CoQ resides within the bilayer interface (46). Presumably, the precursors to CoQ, which have similar chemical properties, would need to be sequestered out of the membrane while being modified by other COQ proteins. Given COQ9’s ability to bind lipid, and its interaction with at least one CoQ-modifying enzyme (COQ7), we speculate that COQ9 interacts with and presents the immature CoQ species to other proteins in the biosynthetic complex before releasing the final product back into the membrane. Another possibility for COQ9 function is that it catalyzes one of the missing reaction steps in CoQ biosynthesis; however, this possibility seems less likely for multiple reasons. First, TFR proteins are not known to possess catalytic functions. Second, one would expect the active site to be in the most highly conserved region of COQ9, the CSP; yet, when this region is truncated in mice, low levels of CoQ are still produced. Collectively, the work presented here provides the first biochemical and structural analysis of COQ9, to our knowledge, and provides a foundation for future work aimed at establishing its endogenous ligand(s) and elucidating its relationship with the other COQ proteins.
Materials and Methods
Full experimental procedures and associated references are available in SI Materials and Methods. Detailed methods for cloning, protein purification, differential scanning fluorimetry, yeast genetic complementation and homology modeling, as well as full methods on LC-MS/MS proteomic and lipidomic analysis, crystallization and structure determination, and computational modeling, can be found in SI Materials and Methods.
Proteomics Analysis of Mice Tissue by LC-MS/MS.
Mice experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the University of Granada (procedure CEEA 2010-275) and were in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (CETS # 123) and the Spanish laws (32/2007 and R.D. 1201/2005). Protein was extracted by probe sonification and quantified by bicinchoninic acid assay. Proteins were reduced, alkylated, and digested with trypsin. The resulting peptides were labeled with six-plex tandem mass tag (TMT) isobaric labels, mixed in equal amounts by mass, and fractionated by strong cation exchange chromatography. Protein fractions were analyzed by nano reverse-phase liquid chromatography coupled to an Orbitrap Elite (Thermo). Spectra were searched using the open mass spectrometry search algorithm (47), and results were filtered to 1% false discovery rate at the unique peptide level using the COMPASS software suite. TMT quantification and protein grouping were performed according to previously reported rules (48).
Lipidomics Analysis by LC-MS/MS.
Lipids from either mouse tissue, purified recombinant COQ9, or Δcoq9 yeast harboring Coq9p constructs were extracted and submitted to discovery-type targeted lipidomics analyses on an Ascentis Express C18 column held at 35 °C (150 mm × 2.1 mm × 2.7 µm particle size; Supelco) using an Accela LC Pump (500 μL/min flow rate; Thermo Scientific). Full extraction methods, LC-MS/MS data collection, and analysis methods are available in SI Materials and Methods.
Crystallization, Data Collection, and Structure Determination.
The selenomethionine-labeled human COQ9NΔ79 was crystallized by microbatch method at 18 °C. Crystals of COQ9NΔ79 were cryoprotected by 25% (vol/vol) ethylene glycol and flash-frozen in liquid N2 for data collection at 100 K. The crystals of human COQ9NΔ79 belong to space group P21 with cell parameters of a = 38.3 Å, b = 97.6 Å, and c = 64.1 Å and β = 95.8°. There are two molecules of COQ9NΔ79 in the crystallographic asymmetric unit of the protein. A single-wavelength anomalous diffraction dataset to 2.4-Å resolution was collected at the peak absorption wavelength of selenium at the X4A beamline of the National Synchrotron Light Source. The diffraction images were processed with the HKL package (49), and 17 of 22 possible selenium sites were located with the program Shelx (50). SOLVE/RESOLVE (51) was used for phasing the reflections and automated model building, which correctly placed 49% of the residues with side chains in each protomer of ASU. The entire model was built with the program XtalView (52) and refined by PHENIX (53). Noncrystallographic symmetry restraint was applied for most stages of the refinement of the structure, but it was released at the final refinement stage.
Computational Modeling of DNA-Binding Residues.
DBSI (33) is a structure-based model that accurately predicts binding sites for DNA on the 3D surface of a protein. To generate electrostatic features within DBSI, solutions to the Poisson–Boltzmann equation were generated using PBEQ (54) via the CHARMM-GUI (55) website (www.charmm-gui.org). The CHARMM-GUI parameters that differ from default (33) are as follows: dielectric constant for the protein interior (2.0); coarse finite-difference grid spacing (1.0 Å); fine finite-difference grid spacing (0.5 Å). The calculations were repeated independently for these structures in both monomeric and dimeric forms. The DBSI program was then run with these structures and their respective electrostatic potential maps as inputs.
Supplementary Material
Acknowledgments
We thank the following members of the University of Wisconsin–Madison-based Mitochondrial Protein Partnership (MPP) and the Northeast Structural Genomics (NeSG) Consortium for technical and managerial assistance: Fabian Suchy, David Aceti, John Primm, Brian Fox, and John Markley (MPP), and Thomas Acton and Gaetano Montelione (NeSG). We thank Andrew Reidenbach for technical assistance and critical reading of the manuscript, as well as Aseem Ansari for helpful suggestions. This work was supported by a Searle Scholars Award, a Shaw Scientist Award, and NIH Grant U01GM094622 (to D.J.P.), NIH Grant U54GM094597 (to L.T.), National Institute of General Medical Sciences Biotechnology Training Grant 5T32GM08349 (to D.C.L.), NIH Ruth L. Kirschstein National Research Service Award F30AG043282 (to J.A.S.), National Library of Medicine (NLM) Training Grant to the Computation and Informatics in Biology and Medicine Training Program NLM T15LM007359 (to C.E.M.), and by the Ramón y Cajal Programme RYC-2011-07643 and the Junta de Andalucía P10-CTS-6133 (to L.C.L. and M.L.-S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4RHP).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413128111/-/DCSupplemental.
References
- 1.Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660(1-2):171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 2.He CH, Xie LX, Allan CM, Tran UC, Clarke CF. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim Biophys Acta. 2014;1841(4):630–644. doi: 10.1016/j.bbalip.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiMauro S, Quinzii CM, Hirano M. Mutations in coenzyme Q10 biosynthetic genes. J Clin Invest. 2007;117(3):587–589. doi: 10.1172/JCI31423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emmanuele V, et al. Heterogeneity of coenzyme Q10 deficiency: Patient study and literature review. Arch Neurol. 2012;69(8):978–983. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lakowski B, Hekimi S. Determination of life-span in Caenorhabditis elegans by four clock genes. Science. 1996;272(5264):1010–1013. doi: 10.1126/science.272.5264.1010. [DOI] [PubMed] [Google Scholar]
- 6.Larsen PL, Clarke CF. Extension of life-span in Caenorhabditis elegans by a diet lacking coenzyme Q. Science. 2002;295(5552):120–123. doi: 10.1126/science.1064653. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, et al. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19(20):2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran UC, Clarke CF. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion. 2007;7(Suppl):S62–S71. doi: 10.1016/j.mito.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.González-Mariscal I, et al. Regulation of coenzyme Q biosynthesis in yeast: A new complex in the block. IUBMB Life. 2014;66(2):63–70. doi: 10.1002/iub.1243. [DOI] [PubMed] [Google Scholar]
- 10.Ashraf S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123(12):5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.García-Corzo L, et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum Mol Genet. 2013;22(6):1233–1248. doi: 10.1093/hmg/dds530. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Hekimi S. Molecular genetics of ubiquinone biosynthesis in animals. Crit Rev Biochem Mol Biol. 2013;48(1):69–88. doi: 10.3109/10409238.2012.741564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagliarini DJ, Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013;27(24):2615–2627. doi: 10.1101/gad.229724.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan AJ, et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84(5):558–566. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marbois BN, Clarke CF. The COQ7 gene encodes a protein in Saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J Biol Chem. 1996;271(6):2995–3004. doi: 10.1074/jbc.271.6.2995. [DOI] [PubMed] [Google Scholar]
- 16.Stenmark P, et al. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J Biol Chem. 2001;276(36):33297–33300. doi: 10.1074/jbc.C100346200. [DOI] [PubMed] [Google Scholar]
- 17.Tran UC, et al. Complementation of Saccharomyces cerevisiae coq7 mutants by mitochondrial targeting of the Escherichia coli UbiF polypeptide: Two functions of yeast Coq7 polypeptide in coenzyme Q biosynthesis. J Biol Chem. 2006;281(24):16401–16409. doi: 10.1074/jbc.M513267200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behan RK, Lippard SJ. The aging-associated enzyme CLK-1 is a member of the carboxylate-bridged diiron family of proteins. Biochemistry. 2010;49(45):9679–9681. doi: 10.1021/bi101475z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu TT, Lee SJ, Apfel UP, Lippard SJ. Aging-associated enzyme human clock-1: Substrate-mediated reduction of the diiron center for 5-demethoxyubiquinone hydroxylation. Biochemistry. 2013;52(13):2236–2244. doi: 10.1021/bi301674p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakai D, et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem Biophys Res Commun. 2001;289(2):463–471. doi: 10.1006/bbrc.2001.5977. [DOI] [PubMed] [Google Scholar]
- 21.Levavasseur F, et al. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J Biol Chem. 2001;276(49):46160–46164. doi: 10.1074/jbc.M108980200. [DOI] [PubMed] [Google Scholar]
- 22.Cuthbertson L, Nodwell JR. The TetR family of regulators. Microbiol Mol Biol Rev. 2013;77(3):440–475. doi: 10.1128/MMBR.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramos JL, et al. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev. 2005;69(2):326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh EJ, et al. Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q biosynthetic complex. Arch Biochem Biophys. 2007;463(1):19–26. doi: 10.1016/j.abb.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson A, et al. COQ9, a new gene required for the biosynthesis of coenzyme Q in Saccharomyces cerevisiae. J Biol Chem. 2005;280(36):31397–31404. doi: 10.1074/jbc.M503277200. [DOI] [PubMed] [Google Scholar]
- 26.Xie LX, et al. Overexpression of the Coq8 kinase in Saccharomyces cerevisiae coq null mutants allows for accumulation of diagnostic intermediates of the coenzyme Q6 biosynthetic pathway. J Biol Chem. 2012;287(28):23571–23581. doi: 10.1074/jbc.M112.360354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang M, Wakitani S, Hayashi K, Miki R, Kawamukai M. High production of sulfide in coenzyme Q deficient fission yeast. Biofactors. 2008;32(1-4):91–98. doi: 10.1002/biof.5520320111. [DOI] [PubMed] [Google Scholar]
- 28.Kim MS, et al. A draft map of the human proteome. Nature. 2014;509(7502):575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 30.Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134(1):112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(web server issue):W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agari Y, Agari K, Sakamoto K, Kuramitsu S, Shinkai A. TetR-family transcriptional repressor Thermus thermophilus FadR controls fatty acid degradation. Microbiology. 2011;157(Pt 6):1589–1601. doi: 10.1099/mic.0.048017-0. [DOI] [PubMed] [Google Scholar]
- 33.Zhu X, Ericksen SS, Mitchell JC. DBSI: DNA-binding site identifier. Nucleic Acids Res. 2013;41(16):e160. doi: 10.1093/nar/gkt617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Ericksen SS, Demerdash ON, Mitchell JC. Data-driven models for protein interaction and design. Proteins. 2013;81(12):2221–2228. doi: 10.1002/prot.24405. [DOI] [PubMed] [Google Scholar]
- 35.Eswar N, et al. 2006. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chap 5:Unit 5.6.
- 36.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(web server issue):W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Celniker G, et al. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr J Chem. 2013;53(3-4):199–206. [Google Scholar]
- 38.Berezin C, et al. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20(8):1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- 39.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 40.Crane FL. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion. 2007;7(Suppl):S2–S7. doi: 10.1016/j.mito.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 41.Crane FL. Discovery of plastoquinones: A personal perspective. Photosynth Res. 2010;103(3):195–209. doi: 10.1007/s11120-010-9537-9. [DOI] [PubMed] [Google Scholar]
- 42.Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16(2):183–188. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinzii CM, Hirano M. Primary and secondary CoQ(10) deficiencies in humans. Biofactors. 2011;37(5):361–365. doi: 10.1002/biof.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marbois B, et al. Coq3 and Coq4 define a polypeptide complex in yeast mitochondria for the biosynthesis of coenzyme Q. J Biol Chem. 2005;280(21):20231–20238. doi: 10.1074/jbc.M501315200. [DOI] [PubMed] [Google Scholar]
- 45.Jonassen T, et al. Yeast Clk-1 homologue (Coq7/Cat5) is a mitochondrial protein in coenzyme Q synthesis. J Biol Chem. 1998;273(6):3351–3357. doi: 10.1074/jbc.273.6.3351. [DOI] [PubMed] [Google Scholar]
- 46.Kagan VE, Quinn PJ. 2001. Coenzyme Q: Molecular Mechanisms in Health and Disease (CRC, Boca Raton, FL)
- 47.Geer LY, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3(5):958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 48.Phanstiel DH, et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat Methods. 2011;8(10):821–827. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 50.Sheldrick GM. Phase annealing in Shelx-90—direct methods for larger structures. Acta Crystallogr A. 1990;46:467–473. [Google Scholar]
- 51.Terwilliger TC. SOLVE and RESOLVE: Automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 52.McRee DE. XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125(2-3):156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jo S, Vargyas M, Vasko-Szedlar J, Roux B, Im W. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res. 2008;36(web server issue):W270–W275. doi: 10.1093/nar/gkn314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.