

Abstract
Multidrug resistance (MDR) renders cancer cells relatively invulnerable to treatment with many standard cytotoxic anti-cancer agents. Cancer immunotherapy could be an important adjunct other strategies to treat MDR positive cancers, as resistance to immunotherapy generally is unrelated to mechanisms of resistance to cytotoxic agents. Immunotherapy to combat MDR positive tumors could use any of the following strategies: direct immune attack against MDR positive cells, using MDR as an immune target to deliver cytotoxic agents, capitalization on other immune properties of MDR positive cells, or conditional immunotoxins expressed under MDR control. Additional insights into the immunogenic potential of some cytotoxic agents can also be brought to bear on these strategies. This review will highlight key concepts in cancer immunotherapy and illustrate immune principles and strategies that have been or could be used to help destroy MDR positive tumor cells, either alone or in rational combinations.
Keywords: immunotherapy, multidrug resistance, combination therapy, cytokines, immune dysfunction
1. Background
1.1 Brief historical perspective on tumor immunotherapy
When tumor-specific immunity was demonstrated by the mid-1950's (Prehn and Main, 1957), this achievement promised to usher in an era of successful anti-cancer immunotherapy. Unfortunately we have not yet fully attained this goal, but our understanding of the shortcomings of prior approaches to cancer immunotherapy is growing rapidly. Cancer cells display antigens that should make them susceptible to immune attack. These antigens are processed and presented in conjunction with major histocompatibility complex (MHC) molecules and immune co-signaling molecules that together could help mount an effective anti-tumor immune response. Dendritic cells are specialized antigen presenting cells that are key mediators of initiating anti-tumor immunity by processing and presenting tumor antigens to anti-tumor effector cells. Antigens captured by dendritic cells can prime an anti-tumor immune response consisting of tumor antigen-specific CD4+ and CD8+ T cells that ultimately should kill the cancer cells (Fig. 1). Innate (antigen-independent) immune mechanisms that can kill tumor cells include natural killer cells and macrophages. Despite this vast armamentarium of naturally-occurring immune weapons, immunologically-mediated spontaneous rejection of clinically apparent cancers is rare, and meaningful clinical responses to cancer immunotherapy are uncommon (Zitvogel et al., 2006). It is now clear that tumors employ a myriad of active immune escape mechanisms to evade destruction by host immune defenses (Curiel, 2008; Zitvogel et al., 2006) (Fig. 2). In response to these new insights, investigators have developed newer strategies to attempt to overcome cancer-driven immune defenses and allow clinically meaningful anti-tumor immunotherapy.
Figure 1.
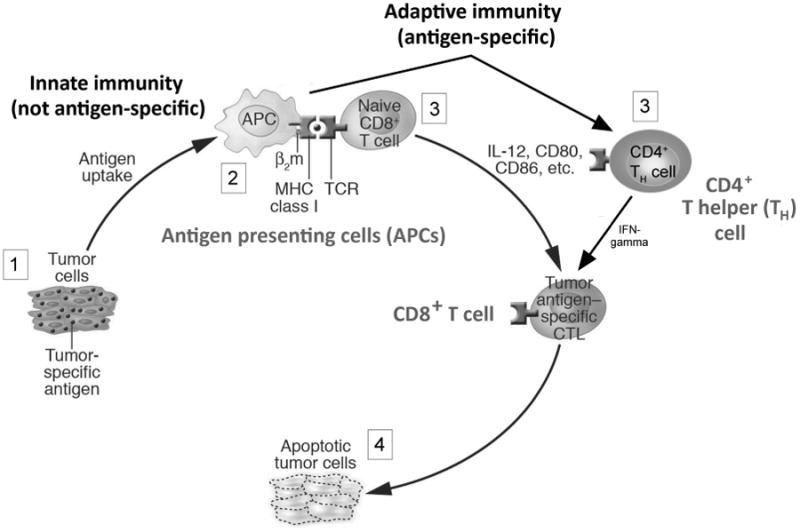
The critical elements of anti-tumor immunity. Tumors express tumor antigens that should be objects of immune attack (1). Antigen presenting cells (2) take up antigen, and process and present them to antigen-specific cells, including CD4+ and CD8+ T lymphocytes (3), that should lead to immune elimination of the tumor (4). Cytokines, such as IL-12, and surface molecules such as CD80 and CD86 provide signals that should promote this tumor-specific immune response. Antigen presenting cells such as dendritic cells, and other non-specific cells suck as natural killer cells and macrophages collectively comprise the innate immune system. Adaptive immunity includes antigen-specific cells such as CD4+ and CD8+ T cells, and B cells (not shown). Despite this sophisticated immune response, which does occur in most cancers, immune elimination does not occur owing to the immune dysfunction shown in figure 2. Figure adapted from (Curiel, 2007).
Figure 2.
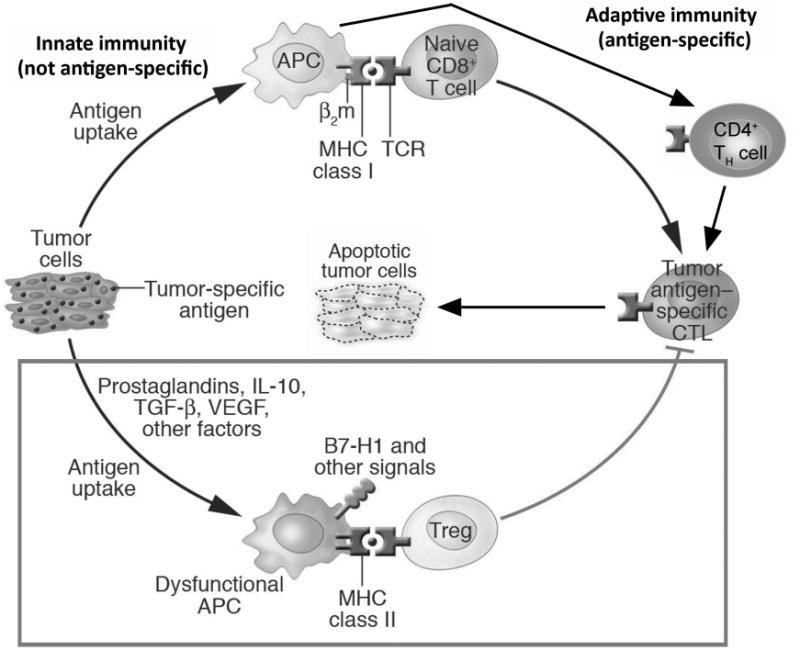
Critical elements of tumor-associated immune dysfunction. Although anti-tumor immunity is elicited according to the scheme outlined in figure 1, and shown in the top half of this figure, active tumor-driven immune dysfunction (boxed portion in bottom half of the figure) thwarts immune cancer elimination. Antigen presenting cells, which in the top half can activate tumor-specific immunity, can also elicit dysfunctional immune cells that turn anti-tumor immunity off, or inhibit it through subversion by tumor factors. Factors responsible for this dysfunction can derive from the tumor itself, or from local stroma or immune cells. These agents include immune suppressive vascular endothelial growth factor (VEGF), transforming growth factor (TGF)-β and interleukin (IL)-10. These molecules can directly inhibit immunity, such as the ability of TGF-β, IL-10 or VEGF to inhibit T cell activation, or can indirectly elicit other dysfunctional cells. In this latter instance, tumor IL-10 or VEGF can promote antigen presenting cells to express B7-H1, an immune molecule that can directly inhibit T cells, or promote generation of regulatory T cells (Tregs) that inhibit anti-tumor immunity. Novel strategies to overcome these complex and potent tumor-driven active defenses against anti-tumor immunity represent major new opportunities to improve the efficacy of anti-tumor immunotherapy. Figure adapted from (Curiel, 2007).
Cytotoxic chemotherapeutic agents have also been developed that could help improve the success rates for cancer therapies. These approaches likewise have limitations, most notably resistance through MDR, which is discussed in detail elsewhere in this special issue.
Most conventional cancer therapies are multi-modal, combining several approaches simultaneously, such as cytotoxic agents plus surgery plus radiotherapy. Despite rational and logical combinations, progress towards effective therapy against most advanced-stage cancers remains frustratingly slow. A significant clinical issue is the development of resistance to anti-cancer cytotoxic agents, and also resistance to the newer anti-growth factor agents and other treatment modalities. Because mechanisms of tumor resistance to cytotoxic agents, growth factor pathway inhibitors and the like are generally distinct from those driving resistance to immunotherapy, it is worthwhile to consider how immunotherapy could be used in conjunction with conventional agents to help improve treatment efficacy. It should be possible to combine specific cytotoxic agents and other anti-cancer strategies with specific immunotherapeutic approaches in rational approaches based on our improved understanding of anti-tumor immunity to help overcome the individual limitations of each individual approach. A second significant cause of cancer treatment failure is relapse from clinically unapparent micrometastatic disease. The exquisite sensitivity of immune responses could help deal with this latter issue. This review will summarize the current state of knowledge in anti-cancer immunotherapy and the approaches that could help overcome the MDR issue by combinations of immune and non-immune approaches.
1.2 Major types of anti-cancer immunotherapy
Cancer immunotherapy can be considered from several perspectives, but one convenient way to categorize immunotherapy is to think of active or passive approaches. An active therapy uses an agent or agents to drive a desired downstream immune response. Examples include vaccines to generate an antigen-specific immune response, or cytokines and interferons to boost activity of immune effector cells. Passive strategies are those that deliver a relatively finished immune product directly to the host. Examples include adoptive transfer of killer T cells or antibody infusions. Any of these approaches potentially could be used to improve the kill rate of MDR-expressing tumor cells when used alone or in combination with other approaches.
1.3 Major mechanisms of anti-tumor immunity
CD8+ T cells recognizing antigens in the context of major histocompatibility complex class I (known as cytotoxic T lymphocytes, CTLs) are important mediators of anti-cancer immunity. Their optimal generation and function could require help from CD4+ T lymphocytes. Both CD4+ and CD8+ T lymphocytes are primed (turned on for the first time in an antigen-specific manner) by professional antigen cells, most notably dendritic cells (Apetoh et al., 2011; Steinman and Banchereau, 2007). Thus, many cancer immunotherapies are attempts to improve the performance of these key immune cells, primarily through vaccinations or adoptive cell transfers. Immune cytokines such as interleukin (IL)-2 (McDermott, 2009) improve the function of T cells and natural killer cells, as could interferon-α. Other agents such as granulocyte-colony stimulating factor (GM-CSF) increase the numbers of, and beneficial functions of dendritic cells, among other mechanisms.
1.4 Major mechanisms of immune dysfunction in cancer
A more recent development in tumor immunology is the understanding that mechanisms of failure of anti-tumor immunity, including attempts at immunotherapy include tumor-associated immune dysfunctions (Curiel, 2007; Zitvogel et al., 2006). Examples include reduced antigen or immune co-signaling expression on tumor and antigen presenting cells, altered effector cell trafficking into the tumor or its draining lymph nodes, outright killing of anti-tumor immune cells by the cancer, resistance of cancer cells to immune apoptosis-inducing factors, or unfavorable pro-inflammatory conditions. Regulatory T cells (Zou, 2006) and myeloid derived suppressor cells (Gabrilovich and Nagaraj, 2009) are immune cells significantly increased in the majority of cancers that actively suppress anti-tumor immunity through a variety of mechanisms. Reversing or mitigating tumor-associated immune dysfunction is a promising new area of development of potentially more effective anti-tumor immunotherapy.
In the following sections we will review how past approaches to cancer immunotherapy have been used or could be tailored to help combat MDR+ cancer, and how the newer approaches fit into the equation.
2. Using cytotoxic chemotherapy in immunotherapy
2.1 Immunochemotherapy
Immunochemotherapy is a strategy combining cytotoxic agents with immune therapy to attempt to capitalize on the best of each modality (Zitvogel and Kroemer, 2009). This approach is most used in cancers that are notoriously refractory to killing by conventional cytotoxic agents. In the United States, immunochemotherapy is most often used to treat renal cell carcinoma, melanoma and some hematologic malignancies including certain lymphomas and leukemias. In Europe, the cytokine tumor necrosis factor (TNF)-α is also used with cytotoxic agents to treat certain sarcomas. Although these cancers all have a high prevalence of MDR expression, little specifically to tailor an immunochemotherapeutic regimen to MDR expression or its consequences has been reported thus far.
Melanoma can be treated with the cytotoxic agent dacarbazine combined with either IL-2, interferon-α or both. Renal cell carcinoma can be treated with 5-fluorocil plus IL-2 or interferon-α, among other combinations of cytotoxic agents and immune modifiers (Bierer et al.; Tarhini et al., 2008). Chemoimmunotherapy for chronic lymphocytic leukemia can include the B cell-targeting anti-CD20 antibody rituximab plus the cytotoxic agents fludarabine and cyclophosphamide (Riches et al., 2011). Because both fludarabine and cyclophosphamide have immunomodulating properties as well, mechanisms of action of these combinations are complex and incompletely understood. Bendamustine can improve the rituximab response in fludarabine-refractory chronic lymphocytic leukemia (Fischer et al., 2011). Some practitioners add the anti-CD52 antibody alemtuzumab to chronic lymphocytic leukemia chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab (Parikh et al., 2011). There is no current immunochemotherapy that is usually curative for any advanced-stage cancers, prompting much additional investigations, of which several illustrative examples are given.
The small molecule multi-tyrosine kinase inhibitors sunitinib and sorafenib increase NKG2D ligands on human nasopharyngeal carcinoma cell lines and make them more sensitive to natural killer cell-mediated cytotoxicity (Huang et al., 2010). Cytokine induced killer cells are T cells cultured ex vivo with activating cytokines such as IL-2 or tumor necrosis factor (TNF)-α to improve their efficacy in adoptive cancer immunotherapy. Cytokine induced killer cells plus oxaliplatin were more effective in vitro and in vivo against oxaliplatin-resistant gastric carcinoma tumors (Zhao et al., 2010). Cytokine induced killer cells plus docetaxel was better than either agent alone in a mouse xenograft model of human lung adenocarcinoma (Liu et al., 2009). Aside from cytotoxic agents and small molecule growth factor inhibitors, the anti-epidermal growth factor receptor antibody cetuximab promotes antibody-dependent cell-mediated cytotoxicity in vitro against primary human rhabdomyosarcoma cell lines (Herrmann et al., 2010).
In a mouse model of mammary carcinoma, combining cyclophosphamide with IL-12 plus granulocyte-colony stimulating factor helped reduce regulatory T cell accumulation and boosted clinical and immune outcomes (Rowswell-Turner et al., 2011). Anti-transferrin receptor antibodies can improve cytotoxic drug efficacy for human glioma tumors in vitro (Xu et al., 2011).
A significant issue to consider in immunochemotherapy is its potential for the cytotoxic agents to reduce immunity as these agents typically target rapidly dividing cells, which include not only the cancer but also cells of hair follicles, gut and bone marrow. As bone marrow is the reservoir from which most immune cells come, it is clear why cytotoxic agents wreak such damage on the immune system. In fact, the term “bone marrow transplant” is commonly misused to describe a high-dose cytotoxic approach to treating cancer. However, the transplant is usually not specifically to treat the cancer itself (although in certain hematologic malignancies that is the case) but is primarily used to rescue the bone marrow after destruction from the high-dose cytotoxic agents, to avoid complications from reduced marrow reserves, including significantly compromised immune function. Thus, a successful immunochemotherapy regimen must provide for adequate timing of immune-degrading agents to ensure that the efficacy of combined immune stimulating agents is not compromised.
In this regard, there have been two interesting developments in the past few years that could help improve rational combinations of cytotoxic agents and immunotherapy for maximal therapeutic effects, including in MDR+ cancers. First, some cytotoxic agents have been shown actually to improve anti-tumor immunity, and second, others have been shown to reduce tumor-associated immune dysfunction. Some of these agents are well-known targets of MDR-mediated resistance, including doxorubicin (Apetoh et al., 2008a; Machiels et al., 2001;Tesniere et al., 2008).
2.2. Using cytotoxic chemotherapy as immunotherapy
As just discussed, cytotoxic anti-cancer drugs kill rapidly dividing cells, including those of the bone marrow. Thus, reduced immune function is frequently an unfortunate and unintended consequence of their use. Nonetheless, certain cytotoxic agents can enhance anti-tumor immunity in specific conditions (Apetoh et al., 2008b). Mechanisms of action to improve anti-tumor immunity for these agents include increasing the immunogenicity of tumor cells, reducing immune dysfunction, or inducing apoptotic cancer cell death that improves anti-tumor immunity (generally through activating antigen presenting cells) (Apetoh et al., 2008a; Apetoh et al., 2008b; Casares et al., 2005; Machiels et al., 2001; Obeid et al., 2007; Shurin et al., 2009; Suzuki et al., 2005; Vincent et al., 2010). Each particular mechanism will be discussed in more detail below.
Some drugs, including the MDR targets anthracyclines, induce preapoptotic exposure of calreticulin by translocation to the plasma membrane and also cause release of non-histone chromatin binding high-mobility group box 1proteins by tumor cells, which mediates dendritic cell antigen uptake and maturation, thereby making the dendritic cells more efficient in priming anti-tumor immunity (Apetoh et al., 2008a; Obeid et al., 2007). Aside from these mechanisms, maximally tolerated doses of the MDR target doxorubicin (as well as intravenous cyclophosphamide and paclitaxel) boosted the performance of a cancer vaccine by breaking self tolerance to tumor antigens (Machiels et al., 2001).
The anthracyclines doxorubicin, idarubicin and mitoxanthrone, as well as oxaliplatin and other drugs can induce apoptotic tumor cell death, which leads to greatly increased antigen uptake by dendritic cells, key inducers of anti-tumor immunity (Apetoh et al., 2011; Steinman and Banchereau, 2007). This capture of apoptotic antigen induces dendritic cell maturation that improves their capacity to prime or activate immune responses against captured tumor antigens as a form of in situ immunization (Apetoh et al., 2008b; Casares et al., 2005). Non-cytotoxic concentrations of the MDR targets doxorubicin and vincristine can boost dendritic cell function in an IL-12-dependent manner that might include mechanisms other than apoptosis induction. This pathway can also be induced by non-MDR targets such as paclitaxel (Shurin et al., 2009).
Thus, appropriate combinations of MDR-targeted cytotoxic agents could help elicit an anti-tumor immune response that could be augmented by specific and complementary immune approaches to help kill MDR+ tumor cells. Since immune effects occur in drug concentrations below those needed to achieve cytotoxicity, these cytotoxic agents can still have anti-cancer effects through boosting anti-tumor immunotherapy even if they are largely ineffective in killing tumor cells directly.
2.3. Cytotoxic agents that can also reduce tumor-driven immune dysfunction
Aside from cytotoxic agents that can directly augment anti-tumor T cell activity, improve dendritic cell function or boost tumor immunogenicity, other agents appear able to mitigate tumor-driven immune dysfunction, which augments anti-tumor immunity indirectly. Perhaps the best-studied example is cyclophosphamide, which was shown decades ago to modulate anti-tumor immunity in mouse models and in humans (Berd and Mastrangelo, 1988). When the importance of tumor-associated regulatory T cells in inhibiting anti-tumor immunity was demonstrated, it was then shown that cyclophosphamide at low doses selectively (but not absolutely specifically) reduced regulatory T cell numbers and function. This reduction in inhibitory regulatory T cells allows more effective function of anti-tumor effect cells such as tumor-specific cytotoxic CD8+ T cells. These results have been replicated in humans, where low dose, metronomic cyclophosphamide reduces regulatory T cells in cancer patients and improves their immune function (Ghiringhelli et al., 2007). More recent work suggests that cyclophosphamide improves anti-tumor immunity through mechanisms in addition to regulatory T cell reduction. For example, after high-dose cyclophosphamide sufficient to cause myelosuppression in a mouse model, regenerated dendritic cells produce more IL-12 that polarizes anti-tumor T cells towards a beneficial Th1 pathway, and less IL-10 that can inhibit T cell responses (Radojcic et al.).
2.4. Timing issues in combining immune and cytotoxic therapies
We have previously demonstrated how timing of immune agents in cancer immunotherapy can affect their immune and clinical efficacy (Litzinger et al., 2007). Interestingly, the timing of cytotoxic agent administration can also alter immune effects. For example, maximally tolerated doses of cyclophosphamide, doxorubicin or paclitaxel given with an experimental cancer vaccine in a mouse model reduced vaccine efficacy. Surprisingly, though, the same doses given prior to vaccination improved vaccine efficacy as evidenced by increased tumor-reactive T cells (Machiels et al., 2001). Based on results of pre-clinical animal models and human observations such as these, it appears possible to combine cytotoxic agents with immunotherapy in effective ways that boost the clinical efficacy of both approaches. Generally speaking, low dose administration and/or metronomic dosing of cytotoxic agents appears to be more effective than giving maximally tolerated doses based on currently available data. However, to optimize such strategies, a better understanding of mechanisms of action of the various doses and schedules of cytotoxic agents, alone and when combined with immune agents is required, as is a better understanding of optimal timing of the combinations.
3. Specific therapies targeting MDR+ cancer cells
3.1 Eradicating MDR+ cancer cells with immunotherapy
Specifics regarding MDR-mediated mechanisms contributing to resistance to conventional cancer therapies are well-discussed elsewhere in this special edition. Progress has also been made regarding using immunotherapy to help treat MDR-expressing cancer cells.
3.1.1 Antibody strategies
MDR expression is a particularly vexing issue in acute myelogenous leukemia. Monoclonal antibodies recognizing specific MDR1 epitopes have been developed, but their ability to detect clinically relevant MDR expression has been questioned (Taylor et al., 2001). Synthetic peptides directed at MDR and made into liposomes after coupling to polyethylene glycol were shown to induce anti-MDR antibodies in mice vaccinated with this construct that augmented doxorubicin sensitivity of multidrug resistant P388R leukemia cells in vitro. Mice vaccinated with these peptides that induced anti-MDR antibodies and challenged with P388R cells survived longer than control mice when given doxorubicin in vivo, demonstrating that targeting MDR gene product function can be chemosensitizing (Gatouillat et al., 2007).
An antibody directing CD8+ cytotoxic T cells to MDR-expressing tumor cells is effective in vitro and can eliminate the P glycoprotein+ MDR leukemia cell line K562/A02 in vivo in xenografted nude mice adoptively transferred with human immune cells, although tumor rapidly recurred. Adding soluble 4-1BBL to agonize the 4-1BB immune co-signaling pathway in conjunction with these MDR-directed CD8+ cytotoxic T cells helped achieve total eradication of K562/A02 in xenografted nude mice in vivo (Guo et al., 2008).
3.1.2 Antibody-directed toxins
Most acute myeloid leukemias express CD33. Gemtuzumab ozogamicin is a conjugate of the cytotoxic agent calicheamicin with humanized anti-CD33 mouse monoclonal antibody. It is widely used to treat acute myeloid leukemia. MDR-mediated P-glycoprotein actively induced calicheamicin efflux from human acute myeloid leukemia cell lines in vitro, which was mitigated using the MDR inhibitors PSC833 or MS209 and improved drug sensitivity in vitro (Matsui et al., 2002).
3.1.3 Antibody-enhanced cytotoxicity
Cripto is an epidermal growth factor family member implicated in drug resistance in a number of cancers. In the MDR+ human leukemia cell line CEM/A7R, an anti-Cripto monoclonal antibody enhanced sensitivity of cells in vitro to the MDR substrates epirubicin and daunorubicin and also to the non-MDR substrate ara-C. Cytotoxicity was associated with augmented c-Jun N-terminal kinase/stress-activated protein kinase and AKT inhibition with associated activation of the mitochondrial apoptosis pathway involving Bad activation. Anti-Cripto monoclonal antibody reduced growth of CEM/A7R cells in a SCID mouse xenograft model, but augmented efficacy with cytotoxic chemotherapy in vivo was not tested in this paper (Hu et al., 2007).
CD147 (also known as extracellular matrix metalloproteinase inducer, basigin or emmprin) is a multifunctional glycoprotein with plieotropic effects including activating proteinases, inducing factors that promote angiogenic tumor and stromal cell angiogenesis, regulating tumor growth, and promoting survival of micrometastatic cancer cells, in addition to promoting the MDR phenotype. CD44, the major hyaluronan receptor, is another protein with pleiotropic effects on cell adhesion, cell trafficking and the MDR phenotype. It is a major component of the extracellular matrix in many cancers that is involved in tumor pathogenesis. Targeting CD44 or CD147 is under consideration to make MDR+ cells more sensitive to cytotoxic agents (Hao et al.).
3.2 Single chain antibody constructs
Single chain antibody scFv constructs have been engineered to recognize the MRP3 MDR epitope in glioblastoma multiforme in vitro (Kuan et al., 2009) but efficacy in drug sensitization was not reported. Patients with hepatocellular carcinoma generate cytotoxic T cell responses against the MDR gene product MRP3 (Mizukoshi et al., 2008) suggesting it as an immune target for immunotherapy.
3.3 Experimental issues with in vitro studies of MDR+ cells in immunotherapy
A complication of some studies of MDR+ cancer cell lines is that they are generated by chronic drug exposure in vitro, and thus their features might not fully or accurately reflect those of de novo MDR+ cells. As an example, it has been postulated that MDR expression renders cancer cells refractory to complement-mediated cytotoxicity. However, careful analysis of MDR contributions versus complement receptor and inhibitor expression in in vitro induced MDR ovarian cancer cell lines demonstrated that complement resistance was due to secondary features acquired during drug exposure in vitro used to generate the MDR phenotype, but MDR gene products did not contribute to complement refractoriness (Odening et al., 2009).
3.4. Exploiting other MDR+ cell properties in immunotherapy
3.4.1 Overcoming apoptosis resistance
The effects of MDR gene products on chemosensitivity are well-described in this special issue. Nonetheless, MDR expression confers additional properties on cancer cells that bear on treatment efficacy. For example, MDR gene products can confer apoptosis protection (Johnstone et al., 2000). As this protection is caspase-dependent but not caspase-independent, a targeted approach to induce caspase-independent apoptosis in MDR+ cells could be useful. In one approach, granzyme A, an immune protein inducing caspase-independent apoptosis, was genetically to make an IL-2/granzyme A fusion protein that delivered granzyme A to IL-2 receptor-expressing malignant cells, killing them in vitro, and improving the doxorubicin sensitivity of the MDR+ lm1-mdr cell line (Grodzovski et al., 2010). MDR-mediated apoptosis resistance can also render leukemia cells less susceptible to immune attack including through natural killer cell-mediated cytotoxicity, which could be granzyme-mediated. N6/ADR is a doxorubicin-resistant MDR+ subline of the human acute lymphoblastic leukemia cell line NALM6. In vitro studies demonstrated reduced tumor cell conjugate formation between tumor cells and natural killer cells, with concomitant reductions in TNF-α release as the major mechanism of MDR-mediated natural killer cell cytotoxicity resistance in MDR+ N6/ADR tumor cells (Treichel et al., 2004). Thus, additional strategies could be developed to overcome resistance to natural killer cell-mediated effects, and to overcome other MDR-mediated anti-cytotoxicity/anti-apoptosis mechanisms.
3.4.2 Cytokine-based strategies
In early studies of immunotherapy to improve cytotoxic agent-mediated tumor cytotoxicity of MDR+ cells, various cytokines were used in vitro. TNF-α, interferon-γ and IL-2 were added to cultures of the human colon carcinoma cell lines Lo Vo, HT 115, SW 480, and LS 174T. Mdr1 expression was reduced although the effect was not seen in all lines and took up to 72 hours to become evident. Notably, vincristine and doxorubicin sensitivity was improved in those cell lines in which mdr1 expression was reduced, but only when added after mdr1 reduction. Cytotoxicity was not enhanced when these drugs were added at the same as the cytokines (Walther and Stein, 1994). These data were an early indication that immunotherapy could be useful to treat MDR+ cancers, and demonstrated early that timing of interventions could have significant effects on treatment outcomes. Timing issues are additionally discussed in section 2.4.
Soon thereafter, in vitro studies of human ovarian and cervical cancer cell lines demonstrated that TNF-α improved topoisomerase II inhibitor-mediated (but not cisplatin-mediated) tumor cytotoxicity in vitro. Augmentation of topoisomerase II inhibitor activity was irrespective of the TNF-α resistance or sensitivity of the tumor cells (Aluigi et al., 1995). Breast cancer cells overexpressing the MDR gene ABCG2 are intrinsically more sensitive to cytotoxicity in vitro by TNF-α (Mosaffa et al., 2011). In other early work, IL-2 gene transfer increased sensitivity of MDR colon carcinoma cell lines to chemotherapy in vitro (Stein et al., 1998), but this approach has not become established clinically.
Chemotherapy-induced MDR expression is extensively discussed elsewhere in this issue. To capitalize on the fact that cytotoxic agents could induce MDR expression, it could be possible to design therapy that is activated in response to MDR upregulation in tumor cells. In one approach, nude mice were xenografted with MCF-7 breast cancer cells engineered to express TNF-α driven off the human mdr1 promoter in response to cytotoxic drugs. Mice bearing MCF-7 cells with the cytotoxic agent-inducible TNF-α cassette exhibited greater tumor reductions in response to doxorubicin treatment compared to mice bearing MCF-7 cells with a cassette constitutively overexpressing TNF-α (Walther et al., 2000).
3.4.3 TNF-related apoptosis-inducing ligand (TRAIL)-related strategies
TNF-related apoptosis-inducing ligand (TRAIL) is a TNF gene superfamily member that can induce apoptosis by engaging specific death receptors. There is great interest in using TRAIL for tumor-specific immunotherapy because it is relatively selective for apoptosis induction in malignant rather than normal cells. TRAIL cytotoxicity also appears to be partially independent of major pathways controlling chemotherapy resistance including those mediated through MDR pathways (Secchiero et al., 2004). Many osteosarcomas are MDR+ and are highly refractory to cytotoxic agents. In a recent study, the MDR-negative human osteosarcoma cell line U2OS was shown to be TRAIL-resistant. By contrast, the MDR+ subline MDR-U2OS was TRAIL sensitive. The mechanism of TRAIL sensitivity was shown to include reduced AKT activation (Cenni et al., 2004). These data demonstrate that improved understanding of MDR signaling alterations could be used to increase anti-tumor therapy, and could be used specifically to boost cytotoxicity of MDR+ tumors (such as by combining treatments with an Akt inhibitor). Naturally-occurring c-MYC over-expression improves TRAIL-mediated killing of MDR+ cancer cells in ovarian and breast cancer (Kim et al., 2011). TRAIL sensitivity can also result from down-regulation of P-glycoprotein by inhibiting the DNA-PKcs/Akt/GSK-3beta pathway and by caspase activation (Seo et al., 2011) or other mechanisms (D'Alessandro et al., 1998; Park et al., 2006), suggesting additional pathways for therapeutic attack.
3.4.4 Inflammation-based strategies
Tumor-mediated inflammation could alter MDR expression. In in vivo studies in naïve, non-tumor-bearing mice (Hartmann et al., 2001), systemic IL-6, or Toll-like receptor ligation with bacterial endotoxin reduced mdr1a, mdr1b, mdr2 and spgp genes in liver whereas IL-1β increased mdr1b gene and protein expression, but reduced gene expression of mdr1a, mdr2 and spgp. TNF-α increased mdr1b gene expression. These data help establish that inflammation can alter mdr gene expression, but specific functional consequences and particulars in individual cancers, where effects are likely to be cell-specific, remain little studied. In humans, TNF-α plus melphalan resulted in good clinical outcomes in isolated limb perfusion for sarcoma and did not induce MDR in tumors (Komdeur et al., 2001). Further work is required to determine if TNF-α (or other cytokines) can suppress MDR development and to determine if their addition to chemotherapy is useful in patients with MDR+ tumors.
4. Miscellaneous approaches and considerations
4.1 Targeting cancer stem cells or cancer initiating cells
A particularly exciting area for future research is investigation into cancer stem cells, or cancer initiating cells as they are also called. Cancer stem cells are similar to non-malignant stem cells in a variety of characteristics such as self-renewal and variable differentiation fates. They also share the features of relative quiescence and in their high resistance to many cytotoxic drugs (Gupta et al., 2009a). Cancer stem cells were first identified in acute myeloid leukemia but have since been putatively identified in a variety of epithelial carcinomas including important cancers such as those of breast, colon and lung (Visvader and Lindeman, 2008) and in important but relatively less common cancers such as melanoma, ovarian cancer, gastric cancer and others (Ji et al., 2009). Strategies aimed at eradicating cancer stem cells thus could be a means to eliminate the root cause of some MDR+ cancers.
Anti-vascular endothelial growth factor antibody synergizes with cyclophosphamide in a xenotransplant glioma model to reduce cancer stem cell like cells with results affected by dose and schedule of cyclophosphamide (Folkins et al., 2007).
A number of labs are employing various strategies to identify agents to treat cancer stem cells (Gupta et al., 2009b). As cancer stem cells express antigens that could make them objects of specific immune attack, they could be particularly amenable to immune approaches. Combining immune and conventional strategies against these cells could be an even more useful strategy. Evidence that immune attacks against cancer stem cells can be mounted (Visus et al.) lends credence and some optimism to this approach.
4.2 Targeting tumor stroma
Tumor-stromal interactions that protect from chemotherapy effects can be interrupted by blocking the chemokine receptor CXCR4 as demonstrated in multiple myeloma (Azab et al., 2009) and other cancers. Tumor stroma is also a key mediator of associated inflammation, which can influence MDR expression and the outcomes of immunotherapy and perhaps certain cytotoxic agents. Additional investigations of tumor stroma in regards to altering the outcomes of treatment of MDR+ tumors, both with conventional and with immunotherapeutic approaches are warranted.
4.3. Assessing gender differences
We recently demonstrated that blockade of the immune co-signaling B7-H1 pathway as cancer immunotherapy has sexually dimorphic outcomes, with females getting a better therapeutic response versus males in a mouse model of B16 melanoma (Lin et al., 2010). Because males and females experience some sexually dimorphic responses to drugs or drug metabolism (Fish, 2008), it is worthwhile to explore whether any combinations of immunotherapy with other approaches will benefit males or females better, or whether any hormonal manipulations will improve treatment effects in a specific sex.
4.4. Age considerations
Although most cancers occur in aged hosts, most research on cancer therapies is done in young hosts. There are no published studies regarding effects of age on combining immunotherapy with other treatment approaches to avoid multidrug resistance. Age is associated with significant alterations in immunity and in generalized inflammation (Komdeur et al., 2001) that could alter mdr gene expression. Thus, understanding age-specific consequences of mdr expression, effects of inflammation and effects of immunotherapy on mdr+ tumors are worthy areas for further studies.
5. Summary
Immunotherapy could be a useful adjunct to help treat multidrug resistant tumors. Numerous approaches are possible, and a number have been attempted in early or limited in vitro and in vivo studies. MDR gene products themselves could be used in targeted therapies for MDR-expressing tumors, or could be used as the principal targets of such attack. MDR-mediated cell signaling alterations, or toxins driven off MDR expression are additional approaches for exploration. Although no immune strategy for treating MDR+ cancers is a clear favorite at this early juncture, immune targeting of MDR+ cancer initiating/cancer stem cells appears promising in theory, even though little has yet been reported. Despite many attempts and approaches, little progress in combining immunotherapy with other agents specifically to combat MDR+ tumors has been reported. With rapid advances in understanding MDR mechanisms and in understanding guiding principles of effective anti-tumor immunotherapies, faster progress towards effective individual and combination approaches in now possible, with work being pursued in labs around the world. Issues of dosing, schedule and timing of individual agents could significantly affect efficacy and toxicities, and bear much additional scrutiny.
Acknowledgments
I thank my colleagues for much productive work, all of which could not be cited here in the interest of space. Thanks to The Journal of Clinical Investigation for permission to use artwork, and to Suzanne R. Thibodeaux, for graphical assistance. Funded by the Voelcker Foundation, the Holly Beach Public Library Association, 1RC2 AG036613, 5P30CA54174, Texas STARS and the Owens Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aluigi M, Debernardis D, Cimoli G, Ottoboni C, Parodi S, Russo P. Tumor-necrosis-factor and DNA topoisomerase-ii inhibitors in human ovarian-cancer - potential role in chemotherapy. Int J Oncol. 1995;7:461–467. [PubMed] [Google Scholar]
- Apetoh L, Locher C, Ghiringhelli F, Kroemer G, Zitvogel L. Harnessing dendritic cells in cancer. Semin Immunol. 2011;23:42–49. doi: 10.1016/j.smim.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Apetoh L, Mignot G, Panaretakis T, Kroemer G, Zitvogel L. Immunogenicity of anthracyclines: moving towards more personalized medicine. Trends Mol Med. 2008a;14:141–151. doi: 10.1016/j.molmed.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Apetoh L, Tesniere A, Ghiringhelli F, Kroemer G, Zitvogel L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. 2008b;68:4026–4030. doi: 10.1158/0008-5472.CAN-08-0427. [DOI] [PubMed] [Google Scholar]
- Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berd D, Mastrangelo MJ. Effect of low dose cyclophosphamide on the immune system of cancer patients: depletion of CD4+, 2H4+ suppressor-inducer T-cells. Cancer Res. 1988;48:1671–1675. [PubMed] [Google Scholar]
- Bierer S, Hoffmeister I, Gerss J, Herrmann E, Wulfing C, Sibrowski W, et al. Combined immunochemotherapy in selected patients with metastatic renal cell carcinoma: HLA class II genotype can help to predict response to therapy. J Immunother. 34:196–201. doi: 10.1097/CJI.0b013e3182027748. [DOI] [PubMed] [Google Scholar]
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenni V, Maraldi NM, Ruggeri A, Secchiero P, Del Coco R, De Pol A, et al. Sensitization of multidrug resistant human ostesarcoma cells to Apo2 Ligand/TRAIL-induced apoptosis by inhibition of the Akt/PKB kinase. Int J Oncol. 2004;25:1599–1608. [PubMed] [Google Scholar]
- Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–246. doi: 10.1016/j.coi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest. 2007;117:1167–1174. doi: 10.1172/JCI31202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessandro N, Flugy A, Tolomeo M, Dusonchet L. The apoptotic signaling of TNF-alpha in multidrug resistant Friend leukemia cells. Anticancer Res. 1998;18:3065–3072. [PubMed] [Google Scholar]
- Fischer K, Cramer P, Busch R, Stilgenbauer S, Bahlo J, Schweighofer CD, et al. Bendamustine Combined With Rituximab in Patients With Relapsed and/or Refractory Chronic Lymphocytic Leukemia: A Multicenter Phase II Trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2011;29:3559–3566. doi: 10.1200/JCO.2010.33.8061. [DOI] [PubMed] [Google Scholar]
- Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol. 2008;8:737–744. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560–3564. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatouillat G, Odot J, Balasse E, Nicolau C, Tosi PF, Hickman DT, et al. Immunization with liposome-anchored pegylated peptides modulates doxorubicin sensitivity in P-glycoprotein-expressing P388 cells. Cancer Lett. 2007;257:165–171. doi: 10.1016/j.canlet.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grodzovski I, Lichtenstein M, Galski H, Lorberboum-Galski H. IL-2-granzyme A chimeric protein overcomes multidrug resistance (MDR) through a caspase 3-independent apoptotic pathway. Int J Cancer. 2010;128:1966–1980. doi: 10.1002/ijc.25527. [DOI] [PubMed] [Google Scholar]
- Guo H, Jiang W, Liu W, Gao Y, Yang M, Zhou Y, et al. Extracellular domain of 4-1BBL enhanced the antitumoral efficacy of peripheral blood lymphocytes mediated by anti-CD3 x anti-Pgp bispecific diabody against human multidrug-resistant leukemia. Cell Immunol. 2008;251:102–108. doi: 10.1016/j.cellimm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009a;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009b;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao JL, Cozzi PJ, Khatri A, Power CA, Li Y. CD147/EMMPRIN and CD44 are potential therapeutic targets for metastatic prostate cancer. Curr Cancer Drug Targets. 2010;10:287–306. doi: 10.2174/156800910791190193. [DOI] [PubMed] [Google Scholar]
- Hartmann G, Kim H, Piquette-Miller M. Regulation of the hepatic multidrug resistance gene expression by endotoxin and inflammatory cytokines in mice. Int Immunopharmacol. 2001;1:189–199. doi: 10.1016/s0162-3109(00)00271-x. [DOI] [PubMed] [Google Scholar]
- Herrmann D, Seitz G, Warmann SW, Bonin M, Fuchs J, Armeanu-Ebinger S. Cetuximab promotes immunotoxicity against rhabdomyosarcoma in vitro. J Immunother. 2010;33:279–286. doi: 10.1097/CJI.0b013e3181c549b0. [DOI] [PubMed] [Google Scholar]
- Hu XF, Li J, Yang E, Vandervalk S, Xing PX. Anti-Cripto Mab inhibit tumour growth and overcome MDR in a human leukaemia MDR cell line by inhibition of Akt and activation of JNK/SAPK and bad death pathways. Br J Cancer. 2007;96:918–927. doi: 10.1038/sj.bjc.6603641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wang Y, Li Y, Guo K, He Y. Role of sorafenib and sunitinib in the induction of expressions of NKG2D ligands in nasopharyngeal carcinoma with high expression of ABCG2. J Cancer Res Clin Oncol. 2010;137:829–837. doi: 10.1007/s00432-010-0944-2. [DOI] [PubMed] [Google Scholar]
- Ji J, Black KL, Yu JS. Glioma stem cell research for the development of immunotherapy. Neurosurg Clin N Am. 2009;21:159–166. doi: 10.1016/j.nec.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Tainton KM, Smyth MJ. A role for P-glycoprotein in regulating cell death. Leuk Lymphoma. 2000;38:1–11. doi: 10.3109/10428190009060314. [DOI] [PubMed] [Google Scholar]
- Kim DY, Kim MJ, Kim HB, Lee JW, Bae JH, Kim DW, et al. Suppression of multidrug resistance by treatment with TRAIL in human ovarian and breast cancer cells with high level of c-Myc. Biochim Biophys Acta. 2011;1812:796–805. doi: 10.1016/j.bbadis.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Komdeur R, Plaat BE, Hoekstra HJ, Molenaar WM, Hollema H, van den Berg E, et al. Expression of P-glycoprotein, multidrug resistance-associated protein 1, and lung resistance-related protein in human soft tissue sarcomas before and after hyperthermic isolated limb perfusion with tumor necrosis factor-alpha and melphalan. Cancer. 2001;91:1940–1948. [PubMed] [Google Scholar]
- Kuan CT, Srivastava N, McLendon RE, Marasco WA, Zalutsky MR, Bigner DD. Recombinant single-chain variable fragment antibodies against extracellular epitopes of human multidrug resistance protein MRP3 for targeting malignant gliomas. Int J Cancer. 2009;127:598–611. doi: 10.1002/ijc.25062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PY, Sun L, Thibodeaux SR, Ludwig SM, Vadlamudi RK, Hurez VJ, et al. B7-H1-Dependent Sex-Related Differences in Tumor Immunity and Immunotherapy Responses. J Immunol. 2010;185:2747–2753. doi: 10.4049/jimmunol.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood. 2007;110:3192–3201. doi: 10.1182/blood-2007-06-094615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Chen L, Huang X. The antitumor effects of CIK cells combined with docetaxel against drug-resistant lung adenocarcinoma cell line SPC-A1/DTX in vitro and in vivo. Cancer Biother Radiopharm. 2009;24:91–98. doi: 10.1089/cbr.2008.0533. [DOI] [PubMed] [Google Scholar]
- Machiels JP, Reilly RT, Emens LA, Ercolini AM, Lei RY, Weintraub D, et al. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001;61:3689–3697. [PubMed] [Google Scholar]
- Matsui H, Takeshita A, Naito K, Shinjo K, Shigeno K, Maekawa M, et al. Reduced effect of gemtuzumab ozogamicin (CMA-676) on P-glycoprotein and/or CD34-positive leukemia cells and its restoration by multidrug resistance modifiers. Leukemia. 2002;16:813–819. doi: 10.1038/sj.leu.2402459. [DOI] [PubMed] [Google Scholar]
- McDermott DF. The application of high-dose interleukin-2 for metastatic renal cell carcinoma. Med Oncol. 2009;26(1):13–17. doi: 10.1007/s12032-008-9152-1. [DOI] [PubMed] [Google Scholar]
- Mizukoshi E, Honda M, Arai K, Yamashita T, Nakamoto Y, Kaneko S. Expression of multidrug resistance-associated protein 3 and cytotoxic T cell responses in patients with hepatocellular carcinoma. J Hepatol. 2008;49:946–954. doi: 10.1016/j.jhep.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Mosaffa F, Kalalinia F, Parhiz BH, Behravan J. Tumor necrosis factor alpha induces stronger cytotoxicity in ABCG2-overexpressing resistant breast cancer cells compared with their drug-sensitive parental line. DNA Cell Biol. 2011;30:413–418. doi: 10.1089/dna.2010.1143. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- Odening KE, Li W, Rutz R, Laufs S, Fruehauf S, Fishelson Z, et al. Enhanced complement resistance in drug-selected P-glycoprotein expressing multi-drug-resistant ovarian carcinoma cells. Clin Exp Immunol. 2009;155:239–248. doi: 10.1111/j.1365-2249.2008.03817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SA, Keating MJ, O'Brien S, Wang X, Ferrajoli A, Faderl S, et al. Frontline chemoimmunotherapy with fludarabine, cyclophosphamide, alemtuzumab, and rituximab for high-risk chronic lymphocytic leukemia. Blood. 2011;118:2062–2068. doi: 10.1182/blood-2011-01-329177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Wu CH, Choi MR, Najafi F, Emami A, Safa AR. P-glycoprotein enhances TRAIL-triggered apoptosis in multidrug resistant cancer cells by interacting with the death receptor DR5. Biochem Pharmacol. 2006;72:293–307. doi: 10.1016/j.bcp.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Prehn RT, Main JM. Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst. 1957;18:769–778. [PubMed] [Google Scholar]
- Radojcic V, Bezak KB, Skarica M, Pletneva MA, Yoshimura K, Schulick RD, et al. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother. 59:137–148. doi: 10.1007/s00262-009-0734-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riches JC, Ramsay AG, Gribben JG. Chronic lymphocytic leukemia: an update on biology and treatment. Curr Oncol Rep. 2011;13:379–385. doi: 10.1007/s11912-011-0188-6. [DOI] [PubMed] [Google Scholar]
- Rowswell-Turner RB, Harden JL, Nair RE, Gu T, Kilinc MO, Egilmez NK. Chronic Chemoimmunotherapy Achieves Cure of Spontaneous Murine Mammary Tumors via Persistent Blockade of Posttherapy Counter-Regulation. J Immunol. 2011;187:4109–4118. doi: 10.4049/jimmunol.1101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secchiero P, Vaccarezza M, Gonelli A, Zauli G. TNF-related apoptosis-inducing ligand (TRAIL): a potential candidate for combined treatment of hematological malignancies. Curr Pharm Des. 2004;10:3673–3681. doi: 10.2174/1381612043382747. [DOI] [PubMed] [Google Scholar]
- Seo SB, Hur JG, Kim MJ, Lee JW, Kim HB, Bae JH, et al. TRAIL sensitize MDR cells to MDR-related drugs by down-regulation of P-glycoprotein through inhibition of DNA-PKcs/Akt/GSK-3beta pathway and activation of caspases. Mol Cancer. 2011;9:199. doi: 10.1186/1476-4598-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. 2009;183:137–144. doi: 10.4049/jimmunol.0900734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein U, Walther W, Shoemaker RH, Schlag PM. IL-2 gene transfer for chemosensitization of multidrug-resistant human colon carcinoma cells. Adv Exp Med Biol. 1998;451:145–149. doi: 10.1007/978-1-4615-5357-1_23. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- Tarhini AA, Kirkwood JM, Gooding WE, Moschos S, Agarwala SS. A phase 2 trial of sequential temozolomide chemotherapy followed by high-dose interleukin 2 immunotherapy for metastatic melanoma. Cancer. 2008;113:1632–1640. doi: 10.1002/cncr.23791. [DOI] [PubMed] [Google Scholar]
- Taylor BJ, Olson DP, Ivy SP. Detection of P-glycoprotein in cell lines and leukemic blasts: failure of select monoclonal antibodies to detect clinically significant Pgp levels in primary cells. Leuk Res. 2001;25:1127–1135. doi: 10.1016/s0145-2126(01)00085-6. [DOI] [PubMed] [Google Scholar]
- Tesniere A, Apetoh L, Ghiringhelli F, Joza N, Panaretakis T, Kepp O, et al. Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol. 2008;20:504–511. doi: 10.1016/j.coi.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Treichel RS, Bunuan M, Hahn N, Wee K. Altered conjugate formation and altered apoptosis of multidrug-resistant human leukemia cell line affects susceptibility to killing by activated natural killer (NK) cells. Int J Cancer. 2004;108:78–85. doi: 10.1002/ijc.11555. [DOI] [PubMed] [Google Scholar]
- Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- Visus C, Wang YY, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8+ T cells. Clin Cancer Res. 17:6174–6184. doi: 10.1158/1078-0432.CCR-11-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Walther W, Stein U. Influence of cytokines on mdr1 expression in human colon carcinoma cell lines: increased cytotoxicity of MDR relevant drugs. J Cancer Res Clin Oncol. 1994;120:471–478. doi: 10.1007/BF01191800. [DOI] [PubMed] [Google Scholar]
- Walther W, Stein U, Fichtner I, Alexander M, Shoemaker RH, Schlag PM. Mdr1 promoter-driven tumor necrosis factor-alpha expression for a chemotherapy-controllable combined in vivo gene therapy and chemotherapy of tumors. Cancer Gene Ther. 2000;7:893–900. doi: 10.1038/sj.cgt.7700196. [DOI] [PubMed] [Google Scholar]
- Xu G, Wen X, Hong Y, Du H, Zhang X, Song J, et al. An anti-transferrin receptor antibody enhanced the growth inhibitory effects of chemotherapeutic drugs on human glioma cells. Int Immunopharmacol. 2011;11:1844–1849. doi: 10.1016/j.intimp.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Zhang H, Li Y, Liu J, Hu X, Fan L. Anti-tumor effects of CIK combined with oxaliplatin in human oxaliplatin-resistant gastric cancer cells in vivo and in vitro. J Exp Clin Cancer Res. 2010;29:118. doi: 10.1186/1756-9966-29-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Kroemer G. Anticancer immunochemotherapy using adjuvants with direct cytotoxic effects. J Clin Invest. 2009;119:2127–2130. doi: 10.1172/JCI39991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–727. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]